

Abstract
Reactive oxygen species (ROS) have been implicated in mechanisms of heart development and regenerative therapies such as the use of pluripotent stem cells. The roles of ROS mediating cell fate are dependent on the intensity of stimuli, cellular context, and metabolic status. ROS mainly act through several targets (such as kinases and transcription factors) and have diverse roles in different stages of cardiac differentiation, proliferation, and maturation. Therefore, further detailed investigation and characterization of redox signaling will help the understanding of the molecular mechanisms of ROS during different cellular processes and enable the design of targeted strategies to foster cardiac regeneration and functional recovery. In this review, we focus on the roles of ROS in cardiac differentiation as well as transdifferentiation (direct reprogramming). The potential mechanisms are discussed in regard to ROS generation pathways and regulation of downstream targets. Further methodological optimization is required for translational research in order to robustly enhance the generation efficiency of cardiac myocytes through metabolic modulations. Additionally, we highlight the deleterious effect of the host's ROS on graft (donor) cells in a paracrine manner during stem cell-based implantation. This knowledge is important for the development of antioxidant strategies to enhance cell survival and engraftment of tissue engineering-based technologies. Thus, proper timing and level of ROS generation after a myocardial injury need to be tailored to ensure the maximal efficacy of regenerative therapies and avoid undesired damage.
1. Introduction
Myocardial infarction (MI) is an anemic infarct disease associated with cell death of myocardium and frequently causes heart failure or cardiac arrest [1]. Recently, the promising therapeutic strategies have emerged for regeneration of cardiomyocytes (CMs) or remuscularization of the myocardium in MI [2], including induction of endogenous CM proliferation, direct reprogramming of nonmyocytes to CMs, and transplantation of pluripotent stem cell- (PSC-) derived CMs. Although these studies have demonstrated substantial potentials of in vitro and in vivo CM regeneration, several notable challenges remain to be addressed before translation to a clinical setting. For instance, insufficient long-term engraftment and integration with host tissue after transplantation remains a critical hurdle for using PSC-CMs in regenerative therapy [3]. Other issues including low regeneration efficiency, immaturity, and tumorigenic risk would compromise the therapeutic effects of new regenerative approaches [2, 4]. Therefore, it is important to converge various biochemical strategies with methods developed for regeneration of functional CM to overcome these challenges [5].
Current protocols of CM regeneration have been developed based on activating the embryonic cardiomyogenesis-induced signaling pathways and gene regulatory networks [6]. Most studies of CM regeneration are focusing on the contributions of transcriptional mechanisms including gene programming, epigenetic chromatin modifications, and biochemical differentiation cues [7]. Energy metabolism is central to mammalian heart development and function, and metabolic processes can be modulated to support the contractile apparatus of regenerated CMs [8]. The change in energy metabolism impacts the ability of stem cell self-renewal, differentiation, and cell fate decision [9]. Although the coordination of genetic networks with developmental bioenergetics is critical to CM phenotype specification, the underlying metabolic mechanisms that drive cardiac differentiation are not fully known.
The metabolic processes in heart development and disease are regulated by redox signaling through the direct effects of O2 levels and the byproduct-reactive oxygen species (ROS) [10]. Emerging evidence shows that the production and signaling of ROS plays an important role in heart development and pathogenesis of cardiovascular disease [11, 12]. ROS serve as an important driver of cell cycle arrest in postnatal CMs, and the mechanisms of CM proliferation have been summarized comprehensively [13, 14]. In this review, we discuss the current state of the art in effect of redox signaling on the strategies of myocardial regeneration including PSC-CM differentiation and cardiac reprogramming. In addition, we focus on the effect of ROS on PSC-CM engraftment in the host environment and highlight the importance of antioxidant approaches for enhancing efficacy of cell therapy.
2. Generation and Function of ROS
Here, we briefly outline the sources, forms, and functions of ROS related to cardiac biology.
2.1. Main Sources of Cellular ROS
Oxidation and reduction (redox signaling) induce changes in structural and functional characteristics of molecules or proteins by loss or gain of an electron, thus mediating transmission and amplification of metabolic signals. The major molecules that participate in redox signaling are ROS that are byproducts of the metabolism of oxygen such as superoxide, hydrogen peroxide, and hydroxyl radical [15]. Cellular ROS mostly originate from superoxide O2·- produced by nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs), the electron transport chain (ETC) in the mitochondria, or the nitric oxide synthases (NOSs).
The NOX family includes seven NOX isoforms with distinct catalytic subunits and they are crucial regulators of redox signaling in multiple body systems and organisms [16]. NOX enzymes can transfer electrons from NADPH to oxygen across biological membranes to produce ROS in both phagocytic and nonphagocytic cells [16, 17]. Mitochondrial ETC transfers electrons from NADH (nicotinamide adenine dinucleotide hydrogen) and succinate along a controlled redox path during respiratory ATP synthesis. However, the electron flow in ETC is an imperfect process, and occasionally oxygen molecules may undergo one- or two-electron reduction reactions to form ROS [18]. Depending on mitochondrial states of respiration, complexes I and III of the ETC may contribute to ROS production through leakage of electrons [18, 19]. NOSs catalyze the conversion of L-arginine to L-citrulline and NO, but can become uncoupled under pathological conditions and switch to ROS production [20].
2.2. ROS Exert Different Physiological and Pathological Functions
ROS can be classified depending on their chemical properties into two groups: one-electron oxidants (e.g., free radical O2·- and HO·) and two-electron oxidants (e.g., nonradical H2O2) [21]. Superoxide O2·- can diffuse within a cell with a relatively longer half-life as compared with other radicals but is neither a strong oxidant nor a powerful reductant [22]. Hydrogen peroxide H2O2 is stable, diffuses within and between cells, and can function as a signaling molecule or second messenger in the regulation of a variety of biological processes [23]. Hydroxyl radical HO· is formed from H2O2via Fenton chemistry in the presence of Fe2+. HO·, the most reactive ROS, is responsible for DNA damage, oxidative stress, and lipid oxidation, but its short half-life (10−9 s) restricts its damaging effects [24, 25]. Therefore, H2O2 appears to be a critical ROS molecule in redox-dependent signal transduction.
It is known that a physiological H2O2 flux activates signaling pathways by reversible oxidation of effector proteins. H2O2 oxidizes the thiol side chain of cysteine residues of the targeted functional motifs [26]. The cysteine residues are modified with highly susceptible thiolate anions under physiological condition, while oxidation of these anions into sulfenic forms can change the activity and function of proteins such as protein tyrosine kinases and transcription factors (TFs) [27, 28], thereby modulating the downstream gene expression and cell behaviors.
The borderline between “oxidative eustress” (beneficial responses) and “oxidative distress” (deleterious responses) in different pathophysiological settings is highly context dependent and remains to be clearly characterized in health and disease [29]. When ROS concentrations remain at physiological levels, they are indispensable in maintaining cell signaling and redox homeostasis. However, excessive production of ROS or oxidative stress has been associated with disease pathogenesis including cardiovascular disease and cancer [30]. ROS regulate diverse processes such as cell death, calcium handling, and cardiac hypertrophy involved in the pathophysiology of heart failure [31].
ROS levels are influenced not only by their generation rate but also by ROS-scavenging systems or antioxidants. Endogenous antioxidant defense system exists to detoxify ROS, repair oxidative damage, and maintain redox homeostasis [32]. Specific endogenous antioxidants such as catalase, peroxiredoxins, thioredoxin, and glutathione peroxidases can prevent potential damage of overoxidation by H2O2 [33, 34]. Our previous study also demonstrated that H2O2-induced CM hypertrophy was improved by activation of antioxidant heme oxygenase-1 (HO-1) [35].
In addition, the compartmentalization and temporal profiles of ROS need to be considered to interpret the consequences of downstream signaling cascades. For instance, elevated mitochondrial ROS is a principal source of oxidative stress leading to arrhythmias and contractile dysfunction in heart failure, and reduction of mitochondrial ROS (rather than cytoplasmic ROS) can prevent and reverse electrical instability and sudden cardiac death [36]. Thus, the physiological roles of ROS and their toxic effects are complicated, which are influenced by a multitude of factors including concentration, source, distribution, and type of ROS. We discuss the complex roles of ROS, H2O2 in particular, in CM differentiation and heart regenerative therapy below.
3. ROS Mediate Cardiac Differentiation of PSCs
PSCs including iPSCs (induced pluripotent stem cells) and ESCs (embryonic stem cells) have emerged as one of the promising cell resources used to differentiate into functional CMs for heart regeneration [37]. Activation of embryonic signaling pathways including Activin, TGF-β, Wnt, and BMP is essential for development of CM lineage [38]. Multiple complex interactions between these conserved signaling pathways control the initial differentiation, proliferation, and maturation of myocardium to establish the cardiovascular system [38]. The delineation of specific redox-sensitive pathways and mechanisms that contribute to different components of CM regeneration processes may facilitate to fine-tune existing protocols or devise novel strategies in heart disease modeling and therapy.
New CMs can be generated from mesodermal progenitors during spontaneous differentiation (embryoid body (EB) formation or a monolayer induction) of PSCs by using growth factors and small molecules mimicking developmental signals [39, 40]. For stem cell culture and maintenance, ROS scavengers or antioxidant supplements are extensively used to prevent cellular oxidative stress [41]. However, β-mercaptoethanol and other thiol-based antioxidant supplements may cause changes to cellular redox state and then reduce the cardiogenic potential of stem cells [42]. The molecular mechanisms involved in metabolism and ROS regulation of PSC differentiation are still poorly understood and merit further investigation to optimize stem cell culture methods.
3.1. Generation of ROS in Early Differentiation Stage
Accumulating evidence shows that intracellular ROS are a critical signal to trigger CM differentiation of stem cells (Figure 1). The intracellular ROS level was increasing in early stage of mouse ESC differentiation [43]. The differentiation cues (e.g., growth factors, small molecules, mechanical stimulus, and electrical fields) were found to increase ROS level in ESCs, while cardiac lineage formation would be impaired by inhibition of ROS-generating pathways or ROS activity [44–46].
Figure 1.
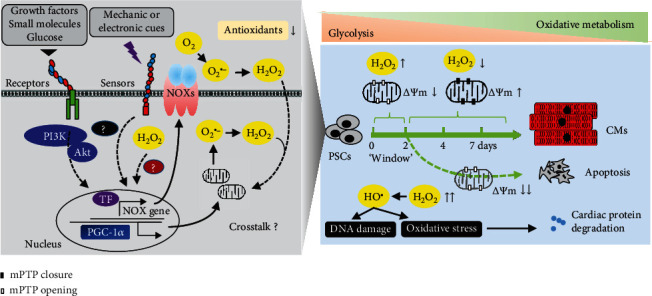
ROS are important for initial stage of differentiation but dispensable for the late stage. ROS are generated by multiple pathways and involved in differentiation of PSCs in response to developmental cues. After closure of mPTP, ROS are decreased and redox signaling is set for further differentiation and functional maturation, while excessive ROS levels would inhibit this process through increased oxidative stress and degradation of structural proteins, eventually leading to apoptotic cell death. NOXs: NADPH oxidases; TF: transcription factor; PGC-1α: peroxisome proliferator-activated receptor γ coactivator 1α; mPTP: mitochondrial permeability transition pore; PSCs: pluripotent stem cells; CMs: cardiomyocytes.
Compared to differentiated cells, PSCs have few immature mitochondria (that are globular in shape with poor cristae structure) and mostly rely on glycolysis to meet their energy demands [47, 48]. Therefore, cardiac specification and excitation-contraction coupling require a switch of glycolytic metabolism towards more efficient mitochondrial oxidative metabolism in PSCs. The energetic switch during differentiation of ESCs was programmed by rearrangement of the metabolic transcriptome (encoding enzymes of glycolysis, fatty acid oxidation, the Krebs cycle, and the ETC) and development of a mature mitochondrial network [49]. ROS are subsequently generated during oxidative metabolism in redox regulation of mitochondrial biogenesis and promote cardiac differentiation and maturation [50]. Thus, ROS generation is potential crosstalk between genetic and metabolic signaling in directing cell fate.
The mechanisms underlying ROS generation remain poorly known in current studies of initiating cardiac differentiation of PSCs. A cytokine-PI3-kinase-NOXs cascade was reported as an initial signal of ROS upregulation in cardiac differentiation of mouse ESCs [43, 45], suggesting the role of ROS as intracellular second messengers. Additionally, stimulation of fatty acid metabolism by activation of peroxisome proliferator-activated receptor-α may be an upstream signal of NOX4-induced ROS generation in mouse ESCs, while mitochondrial electron transport was not involved in this process [51]. Mechanical strain-NOXs, metabotropic glutamate receptor 5, and the PI3K/AKT pathway may also contribute to ROS generation in cardiomyogenesis of ESCs [52–54]. Other studies showed a high expression level of NOX4 in mouse ESCs and demonstrated it as an important source of ROS signals involved in cardiomyogenesis by using siRNA approach [55]. NOX4-induced ROS was also an important signal of differentiating cardiac progenitors under stimulation of magnetic fields [56].
While most of the above studies involve activation of NOX4, ROS derived from mitochondria also play an important signaling role in differentiation and maturation. Specific antagonists had been used to demonstrate an essential role of complex III activity of the mitochondrial ETC in cardiac differentiation and calcium oscillations [57]. In mitochondria of cardiac myocytes, complex III is the principal site for ROS production during the oxidation of complex I substrates [58]. Importantly, a high glucose concentration had been shown to promote cardiac differentiation of ESCs via mitochondrial ROS generation [59]. Temporally reduced antioxidant activity of peroxiredoxin-2 via nitrosylation can cause transient endogenous ROS accumulation and promote ESC-derived cardiomyogenesis [60]. During cardiac differentiation of human ESCs, PGC-1α-dependent mitochondrial biogenesis was associated with increased ROS levels in the CM population [61]. Therefore, cellular ROS are tightly regulated by a variety of proteins involved in the redox regulation of PSCs undergoing a metabolic switch when they differentiate.
ROS may be differently generated in multiple subcellular compartments in targeted cells. Communications between these distinct sites of ROS generation are also functionally relevant to cardiac differentiation. NOX4 can be activated by mitochondrial ROS in differentiated ESCs under the high glucose condition, suggesting an integrated signal between NOXs and mitochondrial ETC [59]. Moreover, a feed-forward regulation of ROS generation was shown by H2O2-induced NOX4 gene expression in cardiac differentiation [62]. Intriguingly, an increasing level of ROS can lead to further release of mitochondrial ROS, termed ROS-induced ROS release, which propagates and amplifies ROS production and effects in cardiac myocytes [63], although this remains undetermined in cardiac differentiation.
The location of ROS generation should be considered when interpreting their effects. Although instructive, the antioxidant compounds do not readily identify the source of ROS due to low specificity. The dynamics of H2O2 metabolism can be assessed by the use of fluorescent probes and other redox-sensitive tools [64]. H2O2 release and cell distribution can be visualized by new ratiometric reporters that have been targeted to subcellular compartments [65]. These molecular tools will be a more specific system for in vivo monitoring of cardiac redox signaling and heterogeneity of individual cell responses to oxidants.
3.2. Continuous Exposure to ROS Inhibits Cardiomyogenesis
The physiological range of H2O2 concentrations was estimated to be between 1 and 10 nM, but it depends on several parameters including cell type and developmental stage [66]. Exogenous H2O2 is a useful tool to determine the direct contribution of ROS in CM differentiation. Stimulation of cardiomyogenesis by exogenous H2O2 (10 nM) was showed to increase the number of beating EB containing CMs and the expression of cardiac genes at 2-3 induction days [43, 55, 62]. Several cardiogenic TFs and cytokines were upregulated by addition of H2O2 in ESCs [67].
In addition to ROS sources, the role of ROS in cardiac differentiation is dependent on metabolism phases and redox balance. Continuous exposure to ROS at a high concentration may overwhelm the antioxidative capacity of cells, thereby exerting a detrimental effect on cell differentiation. Indeed, exogenous H2O2 (100 nM) was showed to inhibit the beating activity of EBs from day 5 to 12 [68]. Excessive H2O2 levels (1 μM) can reduce and degrade Gata4 protein in P19 stem cells [69]. Moreover, increase of intracellular ROS level was responsible for inhibitory effect of valproic acid on cardiomyogenesis [70]. The enforced expression of the pyruvate dehydrogenase phosphatase catalytic subunit 1 gene increased mitochondrial ROS levels in ESCs and inhibited cardiac differentiation [71].
These data suggest that a particular window of “cardiopoietic programming” [72] may exist where a proper level of ROS is important for cardiac differentiation during early stages. During the early period of cardiac differentiation, a high ROS level and low ATP production from immature mitochondria of PSCs may help themselves (or regenerative cells) to adapt to the stress of metabolic switch. After metabolic demand is fulfilled, activation of endogenous antioxidant defense will decrease ROS level to avoid excessive oxidative stress on genetic programming of further CM differentiation and maturation (Figure 1).
Accumulating evidence points out that the redox signaling is associated with mitochondrial permeability transition (MPT) regulating myocyte differentiation and maturation. MPT is caused by the opening of mitochondrial permeability transition pores (mPTP) in the inner mitochondrial membrane. mPTP opening can couple to mitochondrial ETC-dependent ROS production in unstressed cells [73], while mechanisms by which mPTP regulates ROS remain to be determined. Importantly, a study of heart development showed that mPTP opening was nonpathologic in embryonic cardiac myocytes (E9.5) with immature mitochondrial structure and function, low ATP production, and high ROS levels [74]. Differentiation of embryonic CMs was accelerated after closure of mPTP companied with decreased ROS levels, whereas concurrent treatment with oxidant and mPTP blocker inhibited differentiation [74]. Therefore, the beneficent effect of ROS in the window of “cardiopoietic programming” would be offset after closure of mPTP.
Recently, some mPTP inhibitors have been assessed for inducing cardiac differentiation. mPTP inhibition by cyclosporine-A increased ROS generation, but addition of antioxidants rather than prooxidant can enhance cardiomyogenesis [75]. Prolonged closure of mPTP with cyclosporine-A in human iPSC-derived endothelial cells resulted in more mature mitochondria, prevention of ROS leakage, and functional improvements [76]. These studies suggested that the redox signaling is a cardiogenic regulatory factor lying the downstream of mPTP inhibition. The approaches relying on manipulation of redox status should be dependent on monitoring the mode of mPTP.
There are several common features (e.g., cytochrome c release and caspase activation) that govern cell differentiation and apoptosis [77, 78]. MPT and ROS are known to involve in the etiology of several pathological conditions related to necrosis and apoptosis [79], while they can trigger cell differentiation as discussed above. Basic ROS activity contributes to cell differentiation but can induce caspase-dependent apoptosis once the oxidative stress exceeds a certain threshold [80]. Lower levels of ROS, loss of one p53 isoform, and reversible loss of the mitochondrial membrane potential were observed in the differentiating cells as compared to the apoptotic cells that were induced by doxorubicin treatment (an antitumor agent or useful tool with cardiotoxicity), although these features were absent in undifferentiated ESCs [81]. This study indicated that the timing, intensity, and reversibility of activation of mitochondrion-dependent apoptotic pathway may determine whether a cell dies or differentiates.
3.3. ROS Regulate Cardiac Gene Transcription and Expression
ROS have been considered as critical small-molecule messengers in cell signaling transduction. Several signal transducers are redox-sensitive and can be reversibly or irreversibly modified by ROS, providing a link with the control of gene expression [82]. Principal modifications are selective oxidation or nitrosylation of key redox-sensitive cysteine residues in kinases with low ionization pKa (4-5 vs. 8.5 in nonreactive cysteines of most other proteins) [83]. Cysteine oxidation results in either inhibition or activation of targeted molecules depending on the tertiary structure [83]. Furthermore, ROS have been implicated in modulating epigenetic pathways including histone modifications, DNA modifications, expression of noncoding RNAs, and ATP-dependent chromatin remodeling in cardiovascular diseases [84]. Herein, we discuss the direct targets of ROS involved in the mechanisms of cardiac differentiation and heart regeneration (Figure 2).
Figure 2.
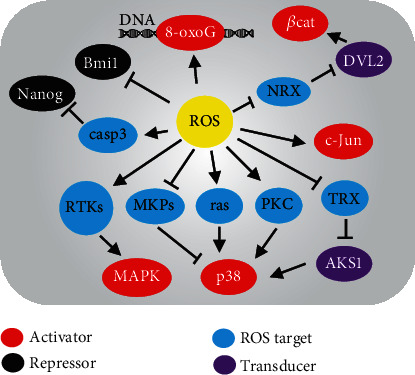
A possible network of molecular events targeted by ROS related to cardiac differentiation pathways. The activators or repressors of cardiac gene expression can be directly or indirectly regulated through ROS modifying the redox-sensitive molecules. 8-oxoG: 8-oxoguanine; βcat: β-catenin; casp3: caspase3; DVL2: dishevelled segment polarity protein 2; NRX: nucleoredoxin; TRX: thioredoxin; ASK1: apoptosis signal-regulating kinase 1; PKC: protein kinase C; RTKs: receptor tyrosine kinases; MAPKs: mitogen-activated protein kinases; MKPs: MAPK phosphatases.
In response to differentiation cues such as growth factors, the downstream cell signaling pathways will be activated before the gene transcription determining cardiac lineage [85]. Tightly controlling phosphorylation of mitogen-activated protein kinase (MAPK) is important for early mesoderm and subsequent CM formation [86]. ROS were shown to enhance differentiation of human ESCs into bipotent mesendoderm via the activation of MAPK family [87]. The phosphorylation of p38 MAPK was inhibited by knockdown of NOX4 and nuclear translocation of Mef2c was prevented, thereby reducing cardiac differentiation [55]. Activation of p38 MAPK was eliminated by an antioxidant in ESCs, and p38 phosphorylation may provide a checkpoint during mesodermal differentiation to the cardiac lineage [59]. These studies suggested that activation of p38 MAPK was closely related to high ROS levels.
In contrast, activation of p38 MAPK mediated by ROS was involved in inhibiting cardiac differentiation of murine ESCs [88], suggesting that the effects of p38 MAPK may be different in distinct timing of differentiation. It remains unknown how ROS can interact with p38 MAPK signaling during cardiogenesis. Oxidative modifications of upstream signaling proteins or receptor kinases by ROS may be a plausible mechanism for activation of the MAPK pathways [89]. Apoptosis signal-regulating kinase 1 (ASK1) was an upstream protein of p38 MAPK and bound to reduced thioredoxin in unstressed HEK293A cells, while thioredoxin can be oxidized upon oxidative stress and disassociate from ASK1, thereby leading to p38 phosphorylation via oligomerization of ASK1 [90]. Alternatively, degradation or inactivation of MAPK phosphatase by ROS-related ubiquitin-proteasome system may contribute to activation of the MAPK pathways in ESCs and other cells [91, 92]. Therefore, MAPKs might not be directly redox-sensitive but instead rely on ROS-mediated upstream proteins such as ras and PKC [93]. These potential mechanisms of ROS-related pathways need to be further determined in the setting of cardiac differentiation.
Cardiac commitment of PSCs is controlled by the regulatory network of TFs such as Nkx2.5, Gata4, and Tbx5 [6, 85]. Although these TFs might not be directly targeted by ROS, their transcription can be regulated by other epigenetic modulators or constitutively active TFs (e.g., AP1 and HIF1α) that are ROS-sensing in vascular cells [94]. Expression of earliest cardiogenic TFs such as Gata4 and Mef2c was dependent upon Nox4-generated ROS that activate redox-sensitive TFs including c-Jun in P19 stem cells [95]. Moreover, extrinsic ROS can enhance the redox-sensitive caspase-mediated degradation of Oct4 and Nanog (pluripotent factors), thereby activating Gata4 and Nkx2.5 promoters that were repressed by Nanog/Hdac4 complex in P19 stem cells [69]. Interestingly, an increase of ROS due to removal of antioxidant in medium can induce epigenetic DNA modifications (such as 8-oxoG) on Tbx5 promoter, leading to Tbx5 activation that enhanced cardiac differentiation of ESCs [96]. Bmi1 is an epigenetic repressor silencing cardiac genes in steady state of cardiac progenitors, while ROS and oxidative damage induced Bmi1 delocalization from canonical DNA targets, therefore triggering an imbalance toward upregulation of differentiation-related genes and downregulation of stemness-related genes in cardiac progenitors [97]. In neural progenitor cell lines, ROS may induce dissociation of redox-sensitive targets such as nucleoredoxin from dishevelled complex that was responsible for activation of the Wnt/β-catenin cascade in transcription of differentiation-related genes [98]. Although different cell models including ESCs have been tested, ROS may regulate downstream gene expression through a common mechanism targeting the transcription-related factors.
The above mechanism studies suggest that identification of redox-sensitive targets helps to delineate how ROS or oxidative stress contributes to cell fate decision. New methods are therefore needed to screen ROS targets and verify their redox functions in various models of cardiomyogenesis and heart regeneration on a global scale. For instance, cysteine reactivity in response to oxidative modifications can be labeled using chemical probes and further assessed by quantitative mass spectrometry in targeted proteins or in a whole proteome scale [99, 100]. In addition to protein assays, several methods have been developed using next-generation sequencing to assess the genome wide distribution of oxidative DNA modifications [101]. Importantly, several computational tools and databases have been developed for analysis of redox-sensitive cysteines and annotation of ROS-related proteins and peroxidase families [102, 103]. Thus, these chemical-genetic methods enable detailed characterization of protein or DNA modifications that are targeted by ROS in the redox environment related to CM regeneration.
4. Unexploited Role of ROS in Direct Cardiac Reprogramming
Transdifferentiation is a new paradigm that has been devised to generate cardiac lineage-specific cells directly from somatic cells, by combining transient overexpression of the cardiac specific TFs. The retroviral transfections of Gata4, Mef2c, and Tbx5 (or with Hand2) reprogramed mouse postnatal cardiac or skin fibroblasts directly into CM-like cells (termed induced CMs (iCMs)), but with low efficiency [104–106]. The TF overexpression was an inefficient method to induce cardiac reprogramming, and the infected cells lacked some molecular and electrophysiological phenotypes of mature CMs [107]. Therefore, researchers are exhibiting tremendous enthusiasm and interest in the quest to elucidate the mechanisms of iCM generation and further enhance reprogramming efficiency. The current progress in this field has been summarized in other reviews [108, 109].
Yet, it remains unknown whether ROS are involved in the process of direct cardiac reprogramming. A preliminary study showed that the treatment of vitamin E nicotinate (an antioxidant) facilitated application of direct cardiac reprogramming approach to repair heart damage in vivo [110]. Further investigation should determine whether the observed effects were related to the elimination of ROS or redox imbalance in iCMs or injured host CMs. Exogenous ROS incubation, use of redox-sensitive probes, treatment of antioxidants in different induction timing, and loss-of-function studies of ROS-associated genes would be helpful strategies to address the unexplored role of ROS in both in vitro and in vivo direct cardiac reprogramming.
The studies of ROS in induced pluripotency reprogramming may bring new insights into genetic resetting during direct cardiac lineage conversion. NOX expression and ROS generation were increased in the early stage of iPSC reprogramming, whereas antagonism of ROS using antioxidants or knockdown of NOXs decreased reprogramming efficiency [111]. Excessive ROS generation impaired iPSC generation, and antioxidant enzymes such as Gpx2 and Nrf2 were upregulated in the late phase of reprogramming [111, 112]. Therefore, these data indicate that the kinetics and intensity of redox signaling is critical for efficient cell reprogramming. Importantly, short-term opening of mPTP has been found during the early stage of somatic cell reprogramming into iPSCs, as companied with activation of mitochondrial ROS [113]. Furthermore, ROS generation triggered by activation of innate immune signaling is required for pluripotent reprogramming and lineage transdifferentiation [114].
The precise mechanisms of direct cardiac reprogramming are not well understood. Recently, next-generation sequencing techniques have been employed not only to decipher the transcriptional mechanisms of cardiac TFs but also to uncover the dynamic process of cell fate reprogramming in a genome-wide scale or at a single-cell level [115–117]. These data suggest that innate immune signaling is critical for cardiac fate acquisition at early stage and cell cycle exit is essential for successful reprogramming. It is conceivable that immune response genes can be activated due to the common use of viral vectors for reprogramming gene delivery [118, 119]. For instance, expression of Toll-like receptor 3 (an immune regulatory gene) contributed to human cardiac reprogramming through impacting DNA methylation status of cardiac loci [115]. Given that ROS can interact with innate immune receptors including Toll-like receptors and NOD-like receptors [120], it is likely that ROS are an important signal during the early stage of cardiac reprogramming and redox balance ensures the further functional maturation of iCMs, which is similar to cardiac differentiation as discussed above. However, it is unknown whether the innate immune pathways are still reactivated in alternative, nonviral reprogramming approaches such as chemically induced CM-like cells [121]. Despite the complexity, the ultimate goal of cardiac TFs or reprogramming factors is to convert the fibroblasts to contracting muscle cells with a high metabolic demand. Based on gene expression of metabolic enzymes, iCMs utilized fatty acid oxidation as the main pathway, which was distinguishable from iPSC-CMs primarily using glycolysis [122]. All above findings encourage further investigation of ROS in the mechanisms of cardiac reprogramming with respect to chromatin accessibility changes, innate immune response, cell cycle regulation, and metabolic switch.
5. ROS Affect Regenerative Therapy in the Infarcted Heart
Currently, PSCs are the main cell sources that can definitively generate cardiovascular cells (seed cells) in high quantities for MI therapy using cardiac tissue engineering [123]. However, insufficient integration of transplanted cells with ischemic tissue remains a major hurdle for clinical translation of using engineered heart tissues (EHTs) in regenerative therapy. Understanding of the healing process of MI, including inflammatory, proliferative, and maturation phases, is important for design and timing selection of cell transplantation in patients. There exists a potential feedback loop (cell-cell interaction) between the host infarcted myocardium and the engraftment of implanted cells, as discussed by us [124]. In this section, we integrate the current evidence to speculate how ROS affect the cell survival and functional engraftment of implanted or regenerated CMs in the infarcted heart (Figure 3).
Figure 3.
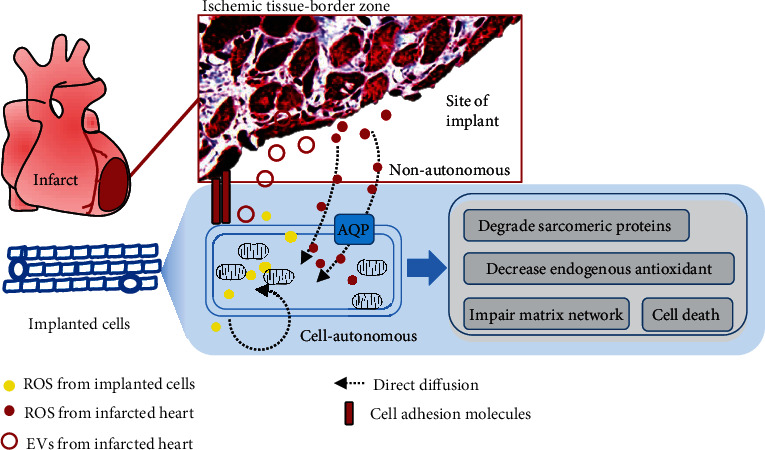
Potential interactions between the ischemic heart and implanted cells contribute to low engraftment efficiency. When stem cells or PSC-CMs are implanted, intracellular ROS would be increased and induce cell death in a cell-autonomous manner in response to the hypoxic microenvironment. Paracrine effects of host's ROS are involved in regulation of the graft cell fate and may lead to engrafted cell death in a nonautonomous manner. EVs: extracellular vesicles; AQP: aquaporin.
5.1. A High Level of Intracellular or Extracellular ROS Harms Graft Cell Survival
Clinical application of stem cell therapies requires large-scale cell culture technologies such as bioreactors that allow for conditional manipulations of the survival, differentiation, and maturation of PSC-CMs [125]. Maintenance of low cellular H2O2 concentration may facilitate in vitro maturation of PSC-CMs [126]. However, PSC-CMs appear to be particularly sensitive to hypoxia and nutrient deprivation-induced cell death associated with increased ROS formation and modulation of key nutrient sensors [127]. A gradual cessation of contractility with increased intracellular ROS and loss of calcium transients was found in mouse PSC-CMs after short-term exposure to monochromatic light [128]. It is likely that ROS-induced protein glutathionylation contributes to a loss of myofibril integrity and degradation of sarcomeric proteins in CMs [129]. Cellular ROS are substantially elevated in cardiovascular cells during ischemia and reperfusion procedure and also involved in the post-MI remodeling of heart failure [36, 130]. Excessive ROS generation depletes endogenous antioxidant defenses in the ischemic heart and primes the cell for oxidative damage at reperfusion [130]. ROS also can persistently impair myocardial matrix network by nonenzymatic protein degradation and modification or activating specific proteolytic enzymes [130]. Therefore, maintenance of redox homeostasis through reduced intrinsic ROS generation and increased antioxidant defense mechanism may promote therapeutic efficacy of cardiac cell replacement approaches (see later).
Extracellular ROS and oxidative stress are critical components of harsh conditions in the infarcted myocardium. Despite the short half-lives, extracellular ROS likely participate in cell-cell communications at the site of ischemia. NOX isoforms are responsible for generation of superoxide (O2·-) toward intracellular or extracellular space and its autocrine or paracrine-like action [131]. Unlike superoxide free radicals (O2·-) with a negative charge, H2O2 is known as a membrane permeable molecule which can diffuse through the mitochondrial and cell membranes. Therefore, ROS can serve as a paracrine-diffusible signal to mediate nearby cells. For instance, H2O2 increased in the infarct core can diffuse into circulating cells and produce 3-nitrotyrosine that was cytotoxic and contributed to decreased recruitment of endogenous progenitor cells to the site of injury [132]. ROS generated in the infarcted heart can hinder the adhesiveness of injected cells via interference of focal adhesion molecules [133]. Interestingly, several aquaporins (water channels) have been identified to facilitate movement of H2O2 across cell membranes at a much higher rate than passive diffusion [134], but their roles remain largely unknown in cardiac cells or regenerated CMs.
5.2. New Insights of ROS in Intercellular Communications
New lines of evidence show that ROS signaling can be transferred in a diffusion-independent fashion from donor cells to nearby cells [135]. For instance, pericardial ROS were shown to directly modulate the expression of cell adhesion and cytoskeleton molecules that facilitate interaction between the pericardial cells and cardiac myocytes [136]. In addition, extracellular vesicles such as exosomes that derived from CMs in response to H2O2 were shown to exacerbate apoptosis of transplanted stem cells [137]. ROS were contained in microvesicles isolated from endothelial cells after hypoxia-reoxygenation, leading to apoptosis and oxidative stress in myoblasts [138]. A study of spinal injury brought a novel sight from finding of exosomal delivery of ROS-producing NOX2 to the injury site and triggering inflammation [139]. The potential mechanisms of ROS and their producers transferring through exosomes or microvesicles require further research in the setting of MI and cell therapies. Knowledge obtained from these studies helps to interpret a possibility that ischemic myocardium-derived ROS target engrafted stem cells or PSC-CMs in a paracrine manner via extracellular vesicles or free diffusion.
6. Convergences of Antioxidants and Cell-Based Therapy
When the oxidative insult overwhelms the endogenous antioxidant defense system during MI, a prolonged elevation in ROS levels leads to chronic inflammation with scarring and tissue dysfunction [140]. Therefore, proper timing and level of ROS generation after MI injury need to be tailored to ensure maximal efficacy in order to avoid undesired damage. Nevertheless, pharmacological interventions using nonselective antioxidants (e.g., vitamin C, vitamin E, and β-carotene) failed to show a significant impact on prevention or treatment of cardiovascular disease in trials [140]. Noneffective or harmful outcome of these antioxidants is likely owing to low drug specificity or disturbed the redox balance signaling. Moreover, systemic delivery of antioxidants might be limited by low bioavailability or low effective levels in the site of injury. To this end, stable materials are being developed for localized antioxidant activity. We could also take advantage of novel biomaterials using in cardiac tissue engineering to scavenge ROS, enhance graft survival, and achieve replenishment of the lost myocardium (Figure 4).
Figure 4.
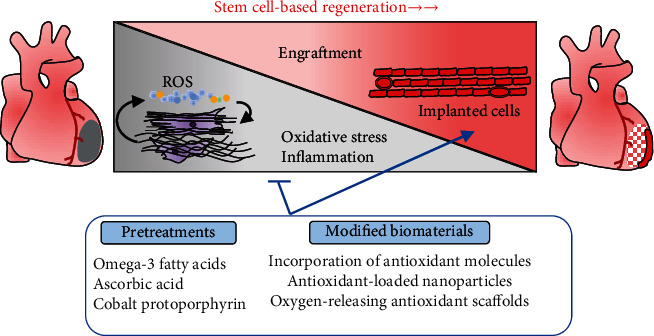
Overview of antioxidant approaches to enhance stem cell-based regeneration. Antioxidant strategies including pretreatments and modified biomaterials targeting the ROS signaling can be applied to enhance the engraftment of implanted stem cells or PSC-CMs.
To obtain functional EHT for cell therapy, natural biomaterials or synthetic nanomaterials have been used to provide mechanical, electroactive support and generate 2D or 3D cardiac sheets [141, 142]. Nonetheless, oxidative stress would be generated due to a detrimental immune response to biomaterials at the site of implantation [143]. It remains challenging to identify biocompatible, biodegradable scaffolds that allow cell migration into infarct zone and protect cells against the oxidative stress. Antioxidants function in different mechanisms, such as free radical scavengers, singlet oxygen quenchers, inactivators of peroxides, chelators of redox metal ion, and quenchers of secondary oxidation products and inhibitors of prooxidative enzymes [144]. We focus on the antioxidative biomaterials that are cardiac-compatible and also show application potential to enhance graft cell survival in preclinical studies.
Recently, incorporation of small antioxidant molecules into polymeric scaffold is a straightforward means to retain the antioxidant activity. For instance, a degradable polyurethane backbone conjugating with ascorbic acid was shown to provide sustainable antioxidant properties and robust mechanical support for CM growth, which rescued CM death under oxidative stress [145]. Interestingly, incorporation of calcium peroxide into an antioxidant hydrophobic polymer can yield a 3D scaffold with a sustained oxygen release as well as attenuation of free radicals [146]. The development of scaffolds with oxygen-releasing and antioxidant properties will offer a unique solution to protect graft from hypoxia-induced cell death by providing sufficient oxygen and attenuating the oxidative stress during oxygen generation, leading to better survival of the critically perfused tissues [146]. Additionally, the antioxidant property of injectable hydrogel can be enhanced by structural introduction of antioxidants such as citric acid and glutathione, and their protective potential effects on graft cells have been determined in MI or oxidative stress models [147, 148]. Antioxidant-loaded nanoparticles can be embedded in hydrogel that possesses a highly porous structure, and this system might have an excellent biocompatibility to support the adhesion and survival of CMs for injectable cardiac tissue engineering [149, 150].
The development of scaffold-based cell delivery techniques is in the early stages for cardiac tissue engineering, and there are still opportunities to incorporate additional treatments to modulate the antioxidative and anti-inflammatory process. Pharmacological pretreatments (such as omega-3 fatty acids and cobalt protoporphyrin) have beneficent effects on survival of ESC-CMs as evidenced by upregulation of HO-1 and decreased ROS levels under oxidative or hypoxic conditions [151, 152]. Future study will reveal new targets and pharmacological compounds to enhance cell engraftment of EHTs after delineating the mechanisms by which the fate of transplanted cells is mediated by increased ROS or downregulated endogenous antioxidant system.
7. Perspectives and Conclusion
In light of the extensive impact of ROS on different aspects of cell differentiation and metabolic homeostasis, there has been continued interest in targeting ROS for therapeutic benefit in the development of heart regenerative medicine. The potential of redox signaling to promote or inhibit CM differentiation may depend upon the ROS source, cell context, and probably the magnitude of ROS generation. It should be noted that the beneficial or detrimental roles of ROS in this scenario do not necessarily need to be mutually exclusive. Cellular ROS may act through several targets and have diverse roles in different stages of cardiac differentiation, proliferation, and maturation. Stem cells are thought to maintain a low basal level of ROS for preserving their functions in quiescence, while increased ROS after differentiation can be countered by the antioxidant defense system to avoid sustained oxidative stress. Although mouse or human ESCs provide a unique experimental model to study the role of ROS and ROS-generating enzymes in the regulation of CM differentiation in vitro, it remains further investigation in human iPSCs to refine the methodologies regulating cellular redox states via metabolic modulations for translational research. Improvement in omic technologies, including genetic screening, single-cell approach, and large-scale profiling of redox-sensitive targets, will undoubtedly advance the understanding of the complexities of ROS and antioxidant pathways during cardiac differentiation and heart development. Additionally, the detailed role of ROS has not been determined in direct cardiac transdifferentiation (reprogramming). Further investigation of epigenetic mechanisms, innate immune response, and mitochondrial regulation will bring new insights into the field of metabolic reprogramming in order to enhance the CM conversion efficiency.
ROS also play a role in applications of cardiac regenerative therapies for MI treatment. Intracellular ROS are increased and induce cell death of implanted stem cells or PSC-CMs in a cell-autonomous manner in the ischemic microenvironment. Paracrine effects of host's cells on the site of implant also likely cause graft cell death in a nonautonomous manner due to uptake of transferred ROS. Yet, this remains to be elucidated to what extent potential paracrine mechanisms contribute to the low engraftment and survival rate of stem cell-based therapies. It will help to address this question by the gain- and loss-of-function studies of the relevant genes in ROS generation pathways in host and donor cells, respectively. This knowledge is important for the design and selection of antioxidant strategies for development of tissue engineering-based technologies. Natural or synthetic biomaterials with antioxidant activity have been used in tissue engineering scaffolds. Further optimization of cardiac tissue engineering needs in-depth evaluation of new biomaterials in regard to donor-host cell coupling, immunogenicity, antioxidant and anti-inflammatory activity, and mechanical and electronic properties. These antioxidant intervention approaches should ensure protecting against infarct expansion, ventricular rupture, and other potentially devastating post-MI complications and avoid disruption of other important signaling of self-healing processes when combining with stem cell-based technology.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (2018YFA0108700 and 2017YFA0105602), the NSFC Projects of International Cooperation and Exchanges (81720108004), the National Natural Science Foundation of China (81974019), the Research Team Project of Natural Science Foundation of Guangdong Province of China (2017A030312007), the Key Program of Guangzhou Science Research Plan (201904020047), and the Special Project of Dengfeng Program of Guangdong Provincial People's Hospital (DFJH201812, KJ012019119, and KJ012019423) to P.Z.
Contributor Information
Jinsong Huang, Email: jasonwong32@hotmail.com.
Ping Zhu, Email: tanganqier@163.com.
Conflicts of Interest
The authors indicated no potential conflicts of interest.
Authors' Contributions
J.L., C.C., M.W., M.M., J.H., and P.Z. did the manuscript writing and the final approval of the manuscript. Jialiang Liang, Min Wu, and Chen Chen contributed equally to this work.
References
- 1.Thygesen K., Alpert J. S., Jaffe A. S., et al. Third universal definition of myocardial infarction. Journal of the American College of Cardiology. 2012;60(16):1581–1598. doi: 10.1016/j.jacc.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 2.Sadek H., Olson E. N. Toward the goal of human heart regeneration. Cell Stem Cell. 2020;26(1):7–16. doi: 10.1016/j.stem.2019.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riegler J., Tiburcy M., Ebert A., et al. Human engineered heart muscles engraft and survive long term in a rodent myocardial infarction model. Circulation Research. 2015;117(8):720–730. doi: 10.1161/CIRCRESAHA.115.306985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oikonomopoulos A., Kitani T., Wu J. C. Pluripotent stem cell-derived cardiomyocytes as a platform for cell therapy applications: progress and hurdles for clinical translation. Molecular Therapy. 2018;26(7):1624–1634. doi: 10.1016/j.ymthe.2018.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kolanowski T. J., Antos C. L., Guan K. Making human cardiomyocytes up to date: derivation, maturation state and perspectives. International Journal of Cardiology. 2017;241:379–386. doi: 10.1016/j.ijcard.2017.03.099. [DOI] [PubMed] [Google Scholar]
- 6.Olson E. N. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313(5795):1922–1927. doi: 10.1126/science.1132292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galdos F. X., Guo Y., Paige S. L., VanDusen N. J., Wu S. M., Pu W. T. Cardiac Regeneration. Circulation Research. 2017;120(6):941–959. doi: 10.1161/CIRCRESAHA.116.309040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lopaschuk G. D., Jaswal J. S. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. Journal of Cardiovascular Pharmacology. 2010;56(2):130–140. doi: 10.1097/FJC.0b013e3181e74a14. [DOI] [PubMed] [Google Scholar]
- 9.Tatapudy S., Aloisio F., Barber D., Nystul T. Cell fate decisions: emerging roles for metabolic signals and cell morphology. EMBO Reports. 2017;18(12):2105–2118. doi: 10.15252/embr.201744816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murray T. V. A., Ahmad A., Brewer A. C. Reactive oxygen at the heart of metabolism. Trends in Cardiovascular Medicine. 2014;24(3):113–120. doi: 10.1016/j.tcm.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Santos C. X. C., Anilkumar N., Zhang M., Brewer A. C., Shah A. M. Redox signaling in cardiac myocytes. Free Radical Biology and Medicine. 2011;50(7):777–793. doi: 10.1016/j.freeradbiomed.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Panth N., Paudel K. R., Parajuli K. Reactive oxygen species: a key hallmark of cardiovascular disease. Advances in Medicine. 2016;2016:12. doi: 10.1155/2016/9152732.9152732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cardoso A. C., Pereira A. H. M., Sadek H. A. Mechanisms of neonatal heart regeneration. Current Cardiology Reports. 2020;22(5):p. 33. doi: 10.1007/s11886-020-01282-5. [DOI] [PubMed] [Google Scholar]
- 14.Puente B. N., Kimura W., Muralidhar S. A., et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157(3):565–579. doi: 10.1016/j.cell.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forman H. J., Ursini F., Maiorino M. An overview of mechanisms of redox signaling. J Mol Cell Cardiol. 2014;73(2-9):2–9. doi: 10.1016/j.yjmcc.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bedard K., Krause K. H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological Reviews. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 17.Lambeth J. D. NOX enzymes and the biology of reactive oxygen. Nature Reviews. Immunology. 2004;4(3):181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 18.Murphy M. P. How mitochondria produce reactive oxygen species. The Biochemical Journal. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sies H. Role of metabolic H2O2 generation: redox signaling and oxidative stress. The Journal of Biological Chemistry. 2014;289(13):8735–8741. doi: 10.1074/jbc.R113.544635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kass D. A., Shah A. M. Redox and nitrosative regulation of cardiac remodeling. Antioxidants & Redox Signaling. 2013;18(9):1021–1023. doi: 10.1089/ars.2012.4942. [DOI] [PubMed] [Google Scholar]
- 21.Bachi A., Dalle-Donne I., Scaloni A. Redox proteomics: chemical principles, methodological approaches and biological/biomedical promises. Chemical Reviews. 2012;113(1):596–698. doi: 10.1021/cr300073p. [DOI] [PubMed] [Google Scholar]
- 22.Xu X., Arriaga E. A. Qualitative determination of superoxide release at both sides of the mitochondrial inner membrane by capillary electrophoretic analysis of the oxidation products of triphenylphosphonium hydroethidine. Free Radical Biology & Medicine. 2009;46(7):905–913. doi: 10.1016/j.freeradbiomed.2008.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Veal E. A., Day A. M., Morgan B. A. Hydrogen peroxide sensing and signaling. Molecular Cell. 2007;26(1):1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 24.Wang Q., Ding F., Zhu N., Li H., He P., Fang Y. Determination of hydroxyl radical by capillary zone electrophoresis with amperometric detection. Journal of Chromatography A. 2003;1016(1):123–128. doi: 10.1016/S0021-9673(03)01294-9. [DOI] [PubMed] [Google Scholar]
- 25.Dizdaroglu M., Jaruga P. Mechanisms of free radical-induced damage to DNA. Free Radical Research. 2012;46(4):382–419. doi: 10.3109/10715762.2011.653969. [DOI] [PubMed] [Google Scholar]
- 26.Rhee S. G. Cell signaling: H2O2, a necessary evil for cell signaling. Science. 2006;312(5782):1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 27.Dickinson B. C., Chang C. J. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nature Chemical Biology. 2011;7(8):504–511. doi: 10.1038/nchembio.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Le Moal E., Pialoux V., Juban G., et al. Redox control of skeletal muscle regeneration. Antioxidants & Redox Signaling. 2017;27(5):276–310. doi: 10.1089/ars.2016.6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sies H., Jones D. P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. 2020;21(7):363–383. doi: 10.1038/s41580-020-0230-3. [DOI] [PubMed] [Google Scholar]
- 30.Koene R. J., Prizment A. E., Blaes A., Konety S. H. Shared risk factors in cardiovascular disease and cancer. Circulation. 2016;133(11):1104–1114. doi: 10.1161/CIRCULATIONAHA.115.020406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hafstad A. D., Nabeebaccus A. A., Shah A. M. Novel aspects of ROS signalling in heart failure. Basic Research in Cardiology. 2013;108(4):p. 359. doi: 10.1007/s00395-013-0359-8. [DOI] [PubMed] [Google Scholar]
- 32.Birben E., Sahiner U. M., Sackesen C., Erzurum S., Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organization Journal. 2012;5(1):9–19. doi: 10.1097/WOX.0b013e3182439613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rhee S. G., Yang K. S., Kang S. W., Woo H. A., Chang T. S. Controlled elimination of intracellular H2O2: regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxidants & Redox Signaling. 2005;7(5-6):619–626. doi: 10.1089/ars.2005.7.619. [DOI] [PubMed] [Google Scholar]
- 34.Yamawaki H., Berk B. C. Thioredoxin: a multifunctional antioxidant enzyme in kidney, heart and vessels. Current Opinion in Nephrology and Hypertension. 2005;14(2):149–153. doi: 10.1097/00041552-200503000-00010. [DOI] [PubMed] [Google Scholar]
- 35.Zhao M., Guo H., Chen J., et al. 5-Aminolevulinic acid combined with sodium ferrous citrate ameliorates H2O2-induced cardiomyocyte hypertrophy via activation of the MAPK/Nrf2/HO-1 pathway. American Journal of Physiology. Cell Physiology. 2015;308(8):C665–C672. doi: 10.1152/ajpcell.00369.2014. [DOI] [PubMed] [Google Scholar]
- 36.Dey S., DeMazumder D., Sidor A., Foster D. B., O’Rourke B. Mitochondrial ROS drive sudden cardiac death and chronic proteome remodeling in heart failure. Circulation Research. 2018;123(3):356–371. doi: 10.1161/CIRCRESAHA.118.312708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tu C., Zoldan J. Moving iPSC-derived cardiomyocytes forward to treat myocardial infarction. Cell Stem Cell. 2018;23(3):322–323. doi: 10.1016/j.stem.2018.08.011. [DOI] [PubMed] [Google Scholar]
- 38.Evans S. M., Yelon D., Conlon F. L., Kirby M. L. Myocardial lineage development. Circulation Research. 2010;107(12):1428–1444. doi: 10.1161/CIRCRESAHA.110.227405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mummery C. L., Zhang J., Ng E. S., Elliott D. A., Elefanty A. G., Kamp T. J. Differentiation of human embryonic stem cells and induced pluripotent stem cells to cardiomyocytes: a methods overview. Circulation Research. 2012;111(3):344–358. doi: 10.1161/CIRCRESAHA.110.227512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karakikes I., Ameen M., Termglinchan V., Wu J. C. Human induced pluripotent stem cell-derived cardiomyocytes: insights into molecular, cellular, and functional phenotypes. Circulation Research. 2015;117(1):80–88. doi: 10.1161/CIRCRESAHA.117.305365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ji J., Sharma V., Qi S., et al. Antioxidant supplementation reduces genomic aberrations in human induced pluripotent stem cells. Stem Cell Reports. 2014;2(1):44–51. doi: 10.1016/j.stemcr.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tu C., Allen A., Deng W., Conroy O., Nambiar M., Zoldan J. Commonly used thiol-containing antioxidants reduce cardiac differentiation and alter gene expression ratios of sarcomeric isoforms. Experimental Cell Research. 2018;370(1):150–159. doi: 10.1016/j.yexcr.2018.06.017. [DOI] [PubMed] [Google Scholar]
- 43.Sauer H., Rahimi G., Hescheler J., Wartenberg M. Role of reactive oxygen species and phosphatidylinositol 3-kinase in cardiomyocyte differentiation of embryonic stem cells. FEBS Letters. 2000;476(3):218–223. doi: 10.1016/S0014-5793(00)01747-6. [DOI] [PubMed] [Google Scholar]
- 44.Serena E., Figallo E., Tandon N., et al. Electrical stimulation of human embryonic stem cells: cardiac differentiation and the generation of reactive oxygen species. Experimental Cell Research. 2009;315(20):3611–3619. doi: 10.1016/j.yexcr.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sauer H., Neukirchen W., Rahimi G., Grünheck F., Hescheler J., Wartenberg M. Involvement of reactive oxygen species in cardiotrophin-1-induced proliferation of cardiomyocytes differentiated from murine embryonic stem cells. Experimental Cell Research. 2004;294(2):313–324. doi: 10.1016/j.yexcr.2003.10.032. [DOI] [PubMed] [Google Scholar]
- 46.Wo Y. B., Zhu D. Y., Hu Y., Wang Z. Q., Liu J., Lou Y. J. Reactive oxygen species involved in prenylflavonoids, icariin and icaritin, initiating cardiac differentiation of mouse embryonic stem cells. Journal of Cellular Biochemistry. 2008;103(5):1536–1550. doi: 10.1002/jcb.21541. [DOI] [PubMed] [Google Scholar]
- 47.Varum S., Rodrigues A. S., Moura M. B., et al. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS One. 2011;6(6, article e20914) doi: 10.1371/journal.pone.0020914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu X., Duan S., Yi F., Ocampo A., Liu G. H., Izpisua Belmonte J. C. Mitochondrial regulation in pluripotent stem cells. Cell Metabolism. 2013;18(3):325–332. doi: 10.1016/j.cmet.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 49.Chung S., Dzeja P. P., Faustino R. S., Perez-Terzic C., Behfar A., Terzic A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nature Clinical Practice Cardiovascular Medicine. 2007;4(S1):S60–S67. doi: 10.1038/ncpcardio0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suliman H. B., Zobi F., Piantadosi C. A. Heme oxygenase-1/carbon monoxide system and embryonic stem cell differentiation and maturation into cardiomyocytes. Antioxidants & Redox Signaling. 2016;24(7):345–360. doi: 10.1089/ars.2015.6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sharifpanah F., Wartenberg M., Hannig M., Piper H. M., Sauer H. Peroxisome proliferator-activated receptor α agonists enhance cardiomyogenesis of mouse ES cells by utilization of a reactive oxygen species-dependent mechanism. Stem Cells. 2008;26(1):64–71. doi: 10.1634/stemcells.2007-0532. [DOI] [PubMed] [Google Scholar]
- 52.Schmelter M., Ateghang B., Helmig S., et al. Embryonic stem cells utilize reactive oxygen species as transducers of mechanical strain-induced cardiovascular differentiation. The FASEB Journal. 2006;20(8):1182–1184. doi: 10.1096/fj.05-4723fje. [DOI] [PubMed] [Google Scholar]
- 53.Heo J. S., Lee J. C. β-catenin mediates cyclic strain-stimulated cardiomyogenesis in mouse embryonic stem cells through ROS-dependent and integrin-mediated PI3K/Akt pathways. Journal of Cellular Biochemistry. 2011;112(7):1880–1889. doi: 10.1002/jcb.23108. [DOI] [PubMed] [Google Scholar]
- 54.Zhou L., Huang Y., Zhang Y., et al. mGluR5 stimulating Homer-PIKE formation initiates icariin induced cardiomyogenesis of mouse embryonic stem cells by activating reactive oxygen species. Experimental Cell Research. 2013;319(10):1505–1514. doi: 10.1016/j.yexcr.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 55.Li J., Stouffs M., Serrander L., et al. The NADPH oxidase NOX4 drives cardiac differentiation: role in regulating cardiac transcription factors and MAP kinase activation. Molecular Biology of the Cell. 2006;17(9):3978–3988. doi: 10.1091/mbc.e05-06-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bekhite M. M., Figulla H. R., Sauer H., Wartenberg M. Static magnetic fields increase cardiomyocyte differentiation of FLK-1+ cells derived from mouse embryonic stem cells via Ca2+ influx and ROS production. International Journal of Cardiology. 2013;167(3):798–808. doi: 10.1016/j.ijcard.2012.02.020. [DOI] [PubMed] [Google Scholar]
- 57.Spitkovsky D., Sasse P., Kolossov E., et al. Activity of complex III of the mitochondrial electron transport chain is essential for early heart muscle cell differentiation. The FASEB Journal. 2004;18(11):1300–1302. doi: 10.1096/fj.03-0520fje. [DOI] [PubMed] [Google Scholar]
- 58.Chen Q., Vazquez E. J., Moghaddas S., Hoppel C. L., Lesnefsky E. J. Production of reactive oxygen species by mitochondria: central role of complex III. The Journal of Biological Chemistry. 2003;278(38):36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 59.Crespo F. L., Sobrado V. R., Gomez L., Cervera A. M., McCreath K. Mitochondrial reactive oxygen species mediate cardiomyocyte formation from embryonic stem cells in high glucose. Stem Cells. 2010;28(7):1132–1142. doi: 10.1002/stem.441. [DOI] [PubMed] [Google Scholar]
- 60.Wu B., Yu H., Wang Y., et al. Peroxiredoxin-2 nitrosylation facilitates cardiomyogenesis of mouse embryonic stem cells via XBP-1s/PI3K pathway. Free Radical Biology and Medicine. 2016;97:179–191. doi: 10.1016/j.freeradbiomed.2016.05.025. [DOI] [PubMed] [Google Scholar]
- 61.Birket M. J., Casini S., Kosmidis G., et al. PGC-1α and Reactive Oxygen Species Regulate Human Embryonic Stem Cell-Derived Cardiomyocyte Function. Stem Cell Reports. 2013;1(6):560–574. doi: 10.1016/j.stemcr.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buggisch M., Ateghang B., Ruhe C., et al. Stimulation of ES-cell-derived cardiomyogenesis and neonatal cardiac cell proliferation by reactive oxygen species and NADPH oxidase. Journal of Cell Science. 2007;120(5):885–894. doi: 10.1242/jcs.03386. [DOI] [PubMed] [Google Scholar]
- 63.Zorov D. B., Filburn C. R., Klotz L. O., Zweier J. L., Sollott S. J. Reactive oxygen species (ROS-induced) ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. The Journal of Experimental Medicine. 2000;192(7):1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bilan D. S., Belousov V. V. In vivo imaging of hydrogen peroxide with hyper probes. Antioxidants & Redox Signaling. 2018;29(6):569–584. doi: 10.1089/ars.2018.7540. [DOI] [PubMed] [Google Scholar]
- 65.Mishina N. M., Bogdanova Y. A., Ermakova Y. G., et al. Which antioxidant system shapes intracellular H2O2Gradients? Antioxidants & Redox Signaling. 2019;31(9):664–670. doi: 10.1089/ars.2018.7697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lyublinskaya O., Antunes F. Measuring intracellular concentration of hydrogen peroxide with the use of genetically encoded H2O2 biosensor hyper. Redox Biology. 2019;24(24):p. 101200. doi: 10.1016/j.redox.2019.101200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Law S. K., Leung C. S.-L., Yau K. L., et al. Regulation of multiple transcription factors by reactive oxygen species and effects of pro-inflammatory cytokines released during myocardial infarction on cardiac differentiation of embryonic stem cells. International Journal of Cardiology. 2013;168(4):3458–3472. doi: 10.1016/j.ijcard.2013.04.178. [DOI] [PubMed] [Google Scholar]
- 68.Pucéat M., Travo P., Quinn M. T., Fort P. A dual role of the GTPase Rac in cardiac differentiation of stem cells. Molecular Biology of the Cell. 2003;14(7):2781–2792. doi: 10.1091/mbc.e02-09-0562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li T., Zhang X., Jiang K., Liu J., Liu Z. Dural effects of oxidative stress on cardiomyogenesis via Gata4 transcription and protein ubiquitination. Cell Death & Disease. 2018;9(2):p. 246. doi: 10.1038/s41419-018-0281-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Na L., Wartenberg M., Nau H., Hescheler J., Sauer H. Anticonvulsant valproic acid inhibits cardiomyocyte differentiation of embryonic stem cells by increasing intracellular levels of reactive oxygen species. Birth defects research Part A, Clinical and molecular teratology. 2003;67(3):174–180. doi: 10.1002/bdra.10030. [DOI] [PubMed] [Google Scholar]
- 71.Heo H. J., Kim H. K., Youm J. B., et al. Mitochondrial pyruvate dehydrogenase phosphatase 1 regulates the early differentiation of cardiomyocytes from mouse embryonic stem cells. Experimental & Molecular Medicine. 2016;48(8, article e254) doi: 10.1038/emm.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Behfar A., Perez-Terzic C., Faustino R. S., et al. Cardiopoietic programming of embryonic stem cells for tumor-free heart repair. The Journal of Experimental Medicine. 2007;204(2):405–420. doi: 10.1084/jem.20061916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang W., Fang H., Groom L., et al. Superoxide flashes in single mitochondria. Cell. 2008;134(2):279–290. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hom J. R., Quintanilla R. A., Hoffman D. L., et al. The permeability transition pore controls cardiac mitochondrial maturation and myocyte differentiation. Developmental Cell. 2011;21(3):469–478. doi: 10.1016/j.devcel.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cho S. W., Park J. S., Heo H. J., et al. Dual modulation of the mitochondrial permeability transition pore and redox signaling synergistically promotes cardiomyocyte differentiation from pluripotent stem cells. Journal of the American Heart Association. 2014;3(2, article e000693) doi: 10.1161/JAHA.113.000693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tiemeier G. L., Wang G., Dumas S. J., et al. Closing the mitochondrial permeability transition pore in hiPSC-derived endothelial cells induces glycocalyx formation and functional maturation. Stem Cell Reports. 2019;13(5):803–816. doi: 10.1016/j.stemcr.2019.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Akbari-Birgani S., Hosseinkhani S., Mollamohamadi S., Baharvand H. Delay in apoptosome formation attenuates apoptosis in mouse embryonic stem cell differentiation. The Journal of Biological Chemistry. 2014;289(24):16905–16913. doi: 10.1074/jbc.M113.536730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fernando P., Kelly J. F., Balazsi K., Slack R. S., Megeney L. A. Caspase 3 activity is required for skeletal muscle differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(17):11025–11030. doi: 10.1073/pnas.162172899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hausenloy D. J., Yellon D. M. The mitochondrial permeability transition pore: its fundamental role in mediating cell death during ischaemia and reperfusion. Journal of Molecular and Cellular Cardiology. 2003;35(4):339–341. doi: 10.1016/S0022-2828(03)00043-9. [DOI] [PubMed] [Google Scholar]
- 80.Nugud A., Sandeep D., El-Serafi A. T. Two faces of the coin: minireview for dissecting the role of reactive oxygen species in stem cell potency and lineage commitment. Journal of Advanced Research. 2018;14:73–79. doi: 10.1016/j.jare.2018.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ghiasi P., Hosseinkhani S., Ansari H., et al. Reversible permeabilization of the mitochondrial membrane promotes human cardiomyocyte differentiation from embryonic stem cells. Journal of Cellular Physiology. 2018;234(1):521–536. doi: 10.1002/jcp.26758. [DOI] [PubMed] [Google Scholar]
- 82.Schieber M., Chandel N. S. ROS function in redox signaling and oxidative stress. Current biology : CB. 2014;24(10):R453–R462. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Janssen-Heininger Y. M. W., Mossman B. T., Heintz N. H., et al. Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radical Biology & Medicine. 2008;45(1):1–17. doi: 10.1016/j.freeradbiomed.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kietzmann T., Petry A., Shvetsova A., Gerhold J. M., Görlach A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. British Journal of Pharmacology. 2017;174(12):1533–1554. doi: 10.1111/bph.13792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lewandowski J., Kolanowski T. J., Kurpisz M. Techniques for the induction of human pluripotent stem cell differentiation towards cardiomyocytes. Journal of Tissue Engineering and Regenerative Medicine. 2017;11(5):1658–1674. doi: 10.1002/term.2117. [DOI] [PubMed] [Google Scholar]
- 86.Kempf H., Lecina M., Ting S., Zweigerdt R., Oh S. Distinct regulation of mitogen-activated protein kinase activities is coupled with enhanced cardiac differentiation of human embryonic stem cells. Stem Cell Research. 2011;7(3):198–209. doi: 10.1016/j.scr.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 87.Ji A.-R., Ku S.-Y., Cho M. S., et al. Reactive oxygen species enhance differentiation of human embryonic stem cells into mesendodermal lineage. Experimental & Molecular Medicine. 2010;42(3):175–186. doi: 10.3858/emm.2010.42.3.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fang H., Cong L., Zhi Y., Xu H., Jia X., Peng S. T-2 toxin inhibits murine ES cells cardiac differentiation and mitochondrial biogenesis by ROS and p-38 MAPK-mediated pathway. Toxicology Letters. 2016;258:259–266. doi: 10.1016/j.toxlet.2016.06.2103. [DOI] [PubMed] [Google Scholar]
- 89.Son Y., Cheong Y. K., Kim N. H., Chung H. T., Kang D. G., Pae H. O. Mitogen-activated protein kinases and reactive oxygen species: how can ROS activate MAPK pathways? J Signal Transduct. 2011;2011, article 792639:1–6. doi: 10.1155/2011/792639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fujino G., Noguchi T., Matsuzawa A., et al. Thioredoxin and TRAF family proteins regulate reactive oxygen species-dependent activation of ASK1 through reciprocal modulation of the N-terminal homophilic interaction of ASK1. Molecular and Cellular Biology. 2007;27(23):8152–8163. doi: 10.1128/MCB.00227-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Smiley R., Naik P., McCallum R., Showkat Ali M. Reactive oxygen species overproduction and MAP kinase phosphatase-1 degradation are associated with gastroparesis in a streptozotocin-induced male diabetic rat model. Neurogastroenterology & Motility. 2018;30(3, article e13218) doi: 10.1111/nmo.13218. [DOI] [PubMed] [Google Scholar]
- 92.Demasi M., Simoes V., Bonatto D. Cross-talk between redox regulation and the ubiquitin-proteasome system in mammalian cell differentiation. Biochimica et Biophysica Acta. 2015;1850(8):1594–1606. doi: 10.1016/j.bbagen.2014.10.031. [DOI] [PubMed] [Google Scholar]
- 93.Knock G. A., Ward J. P. T. Redox regulation of protein kinases as a modulator of vascular function. Antioxidants & Redox Signaling. 2011;15(6):1531–1547. doi: 10.1089/ars.2010.3614. [DOI] [PubMed] [Google Scholar]
- 94.Kohlgrüber S., Upadhye A., Dyballa-Rukes N., McNamara C. A., Altschmied J. Regulation of transcription factors by reactive oxygen species and nitric oxide in vascular physiology and pathology. Antioxidants & Redox Signaling. 2017;26(13):679–699. doi: 10.1089/ars.2016.6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Murray T. V. A., Smyrnias I., Shah A. M., Brewer A. C. NADPH oxidase 4 regulates cardiomyocyte differentiation via redox activation of c-Jun protein and the cis-regulation of GATA-4 gene transcription. The Journal of Biological Chemistry. 2013;288(22):15745–15759. doi: 10.1074/jbc.M112.439844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Park J., Park J. W., Oh H., Maria F. S., Kang J., Tian X. Gene-specific assessment of guanine oxidation as an epigenetic modulator for cardiac specification of mouse embryonic stem cells. PLoS One. 2016;11(6, article e0155792) doi: 10.1371/journal.pone.0155792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Herrero D., Tomé M., Cañón S., et al. Redox-dependent BMI1 activity drives in vivo adult cardiac progenitor cell differentiation. Cell Death and Differentiation. 2018;25(4):809–822. doi: 10.1038/s41418-017-0022-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rharass T., Lemcke H., Lantow M., Kuznetsov S. A., Weiss D. G., Panáková D. Ca2+-mediated mitochondrial reactive oxygen species metabolism augments Wnt/β-catenin pathway activation to facilitate cell differentiation. The Journal of Biological Chemistry. 2014;289(40):27937–27951. doi: 10.1074/jbc.M114.573519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.van der Reest J., Lilla S., Zheng L., Zanivan S., Gottlieb E. Proteome-wide analysis of cysteine oxidation reveals metabolic sensitivity to redox stress. Nature Communications. 2018;9(1):p. 1581. doi: 10.1038/s41467-018-04003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Weerapana E., Wang C., Simon G. M., et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468(7325):790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Poetsch A. R. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Computational and Structural Biotechnology Journal. 2020;18:207–219. doi: 10.1016/j.csbj.2019.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Savelli B., Li Q., Webber M., et al. RedoxiBase: a database for ROS homeostasis regulated proteins. Redox Biol. 2019;26:p. 101247. doi: 10.1016/j.redox.2019.101247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sun M. A., Zhang Q., Wang Y., Ge W., Guo D. Prediction of redox-sensitive cysteines using sequential distance and other sequence-based features. BMC Bioinformatics. 2016;17(1):p. 316. doi: 10.1186/s12859-016-1185-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ieda M., Fu J. D., Delgado-Olguin P., et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010;142(3):375–386. doi: 10.1016/j.cell.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Efe J. A., Hilcove S., Kim J., et al. Conversion of mouse fibroblasts into cardiomyocytes using a direct reprogramming strategy. Nature Cell Biology. 2011;13(3):215–222. doi: 10.1038/ncb2164. [DOI] [PubMed] [Google Scholar]
- 106.Song K., Nam Y. J., Luo X., et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature. 2012;485(7400):599–604. doi: 10.1038/nature11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen J. X., Krane M., Deutsch M. A., et al. Inefficient reprogramming of fibroblasts into cardiomyocytes using Gata4, Mef2c, and Tbx5. Circulation Research. 2012;111(1):50–55. doi: 10.1161/CIRCRESAHA.112.270264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sadahiro T., Ieda M. Direct cardiac reprogramming for cardiovascular regeneration and differentiation. The Keio Journal of Medicine. 2019 doi: 10.2302/kjm.2019-0008-oa. [DOI] [PubMed] [Google Scholar]
- 109.Keepers B., Liu J., Qian L. What's in a cardiomyocyte - And how do we make one through reprogramming? Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2020;1867(3):p. 118464. doi: 10.1016/j.bbamcr.2019.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Suzuki Y. J., Shults N. V. Antioxidant regulation of cell reprogramming. Antioxidants. 2019;8(8):p. 323. doi: 10.3390/antiox8080323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhou G., Meng S., Li Y., Ghebre Y. T., Cooke J. P. Optimal ROS signaling is critical for nuclear reprogramming. Cell Reports. 2016;15(5):919–925. doi: 10.1016/j.celrep.2016.03.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hawkins K. E., Joy S., Delhove J. M. K. M., et al. Nrf2 orchestrates the metabolic shift during induced pluripotent stem cell reprogramming. Cell Reports. 2016;14(8):1883–1891. doi: 10.1016/j.celrep.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ying Z., Xiang G., Zheng L., et al. Short-term mitochondrial permeability transition pore opening modulates histone lysine methylation at the early phase of somatic cell reprogramming. Cell Metabolism. 2018;28(6):935–945.e5. doi: 10.1016/j.cmet.2018.08.001. [DOI] [PubMed] [Google Scholar]
- 114.Meng S., Chanda P., Thandavarayan R. A., Cooke J. P. Transflammation: how innate immune activation and free radicals drive nuclear reprogramming. Antioxidants & Redox Signaling. 2018;29(2):205–218. doi: 10.1089/ars.2017.7364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhou Y., Liu Z., Welch J. D., et al. Single-cell transcriptomic analyses of cell fate transitions during human cardiac reprogramming. Cell Stem Cell. 2019;25(1):149–164.e9. doi: 10.1016/j.stem.2019.05.020. e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Stone N. R., Gifford C. A., Thomas R., et al. Context-specific transcription factor functions regulate epigenomic and transcriptional dynamics during cardiac reprogramming. Cell Stem Cell. 2019;25(1):87–102.e9. doi: 10.1016/j.stem.2019.06.012. e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hashimoto H., Wang Z., Garry G. A., et al. Cardiac reprogramming factors synergistically activate genome-wide cardiogenic stage-specific enhancers. Cell Stem Cell. 2019;25(1):69–86.e5. doi: 10.1016/j.stem.2019.03.022. e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lee J., Sayed N., Hunter A., et al. Activation of innate immunity is required for efficient nuclear reprogramming. Cell. 2012;151(3):547–558. doi: 10.1016/j.cell.2012.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sayed N., Wong W. T., Ospino F., et al. Transdifferentiation of human fibroblasts to endothelial cells: role of innate immunity. Circulation. 2015;131(3):300–309. doi: 10.1161/CIRCULATIONAHA.113.007394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chen Y., Zhou Z., Min W. Mitochondria, oxidative stress and innate immunity. Frontiers in Physiology. 2018;9 doi: 10.3389/fphys.2018.01487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cao N., Huang Y., Zheng J., et al. Conversion of human fibroblasts into functional cardiomyocytes by small molecules. Science. 2016;352(6290):1216–1220. doi: 10.1126/science.aaf1502. [DOI] [PubMed] [Google Scholar]
- 122.Zhou Y., Wang L., Liu Z., et al. Comparative gene expression analyses reveal distinct molecular signatures between differentially reprogrammed cardiomyocytes. Cell Reports. 2017;20(13):3014–3024. doi: 10.1016/j.celrep.2017.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yoshida Y., Yamanaka S. Induced pluripotent stem cells 10 years later: for cardiac applications. Circulation Research. 2017;120(12):1958–1968. doi: 10.1161/CIRCRESAHA.117.311080. [DOI] [PubMed] [Google Scholar]
- 124.Liang J., Huang W., Jiang L., Paul C., Li X., Wang Y. Concise review: reduction of adverse cardiac scarring facilitates pluripotent stem cell-based therapy for myocardial infarction. Stem Cells. 2019;37(7):844–854. doi: 10.1002/stem.3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ronaldson-Bouchard K., Yeager K., Teles D., et al. Engineering of human cardiac muscle electromechanically matured to an adult-like phenotype. Nature Protocols. 2019;14(10):2781–2817. doi: 10.1038/s41596-019-0189-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Momtahan N., Crosby C. O., Zoldan J. The Role of Reactive Oxygen Species in In Vitro Cardiac Maturation. Trends in Molecular Medicine. 2019;25(6):482–493. doi: 10.1016/j.molmed.2019.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Brodarac A., Šarić T., Oberwallner B., et al. Susceptibility of murine induced pluripotent stem cell-derived cardiomyocytes to hypoxia and nutrient deprivation. Stem Cell Res Ther. 2015;6(1) doi: 10.1186/s13287-015-0057-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Singh G., Sridharan D., Khan M., Seshagiri P. B. Mouse embryonic stem cell-derived cardiomyocytes cease to beat following exposure to monochromatic light: association with increased ROS and loss of calcium transients. American Journal of Physiology. Cell Physiology. 2019;317(4):C725–C736. doi: 10.1152/ajpcell.00188.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Munkanatta Godage D. N. P., VanHecke G. C., Samarasinghe K. T. G., et al. SMYD2 glutathionylation contributes to degradation of sarcomeric proteins. Nature Communications. 2018;9(1):p. 4341. doi: 10.1038/s41467-018-06786-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Raedschelders K., Ansley D. M., Chen D. D. Y. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacology & Therapeutics. 2012;133(2):230–255. doi: 10.1016/j.pharmthera.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 131.Zhang G., Zhang F., Muh R., et al. Autocrine/paracrine pattern of superoxide production through NAD(P)H oxidase in coronary arterial myocytes. American Journal of Physiology. Heart and Circulatory Physiology. 2007;292(1):H483–H495. doi: 10.1152/ajpheart.00632.2006. [DOI] [PubMed] [Google Scholar]
- 132.Moldovan N. I., Anghelina M., Varadharaj S., et al. Reoxygenation-derived toxic reactive oxygen/nitrogen species modulate the contribution of bone marrow progenitor cells to remodeling after myocardial infarction. Journal of the American Heart Association. 2014;3(1, article e000471) doi: 10.1161/JAHA.113.000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Song H., Cha M. J., Song B. W., et al. Reactive oxygen species inhibit adhesion of mesenchymal stem cells implanted into ischemic myocardium via interference of focal adhesion complex. Stem Cells. 2010;28(3):555–563. doi: 10.1002/stem.302. [DOI] [PubMed] [Google Scholar]
- 134.Tamma G., Valenti G., Grossini E., et al. Aquaporin membrane channels in oxidative stress, cell signaling, and aging: recent advances and research trends. Oxidative medicine and cellular longevity. 2018;2018:14. doi: 10.1155/2018/1501847.1501847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hervera A., Santos C. X., De Virgiliis F., Shah A. M., Di Giovanni S. Paracrine mechanisms of redox signalling for postmitotic cell and tissue regeneration. Trends in Cell Biology. 2019;29(6):514–530. doi: 10.1016/j.tcb.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 136.Lim H. Y., Bao H., Liu Y., Wang W. Select Septate Junction Proteins Direct ROS-Mediated Paracrine Regulation of Drosophila Cardiac Function. Cell Reports. 2019;28(6):1455–1470.e4. doi: 10.1016/j.celrep.2019.07.004. e1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hu M., Guo G., Huang Q., et al. The harsh microenvironment in infarcted heart accelerates transplanted bone marrow mesenchymal stem cells injury: the role of injured cardiomyocytes-derived exosomes. Cell Death & Disease. 2018;9(3):p. 357. doi: 10.1038/s41419-018-0392-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhang Q., Shang M., Zhang M., et al. Microvesicles derived from hypoxia/reoxygenation-treated human umbilical vein endothelial cells promote apoptosis and oxidative stress in H9c2 cardiomyocytes. BMC Cell Biology. 2016;17(1):p. 25. doi: 10.1186/s12860-016-0100-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hervera A., De Virgiliis F., Palmisano I., et al. Reactive oxygen species regulate axonal regeneration through the release of exosomal NADPH oxidase 2 complexes into injured axons. Nature Cell Biology. 2018;20(3):307–319. doi: 10.1038/s41556-018-0039-x. [DOI] [PubMed] [Google Scholar]
- 140.Sugamura K., Keaney J. F. Reactive oxygen species in cardiovascular disease. Free Radical Biology and Medicine. 2011;51(5):978–992. doi: 10.1016/j.freeradbiomed.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Liang J., Wang Y. Stem/progenitor cell based therapies for repair of myocardial infarction: current developments in methods of cell delivery. Surgery: Current Research. 2013;3(2):p. 131. [Google Scholar]
- 142.Baheiraei N., Yeganeh H., Ai J., Gharibi R., Azami M., Faghihi F. Synthesis, characterization and antioxidant activity of a novel electroactive and biodegradable polyurethane for cardiac tissue engineering application. Materials Science and Engineering: C. 2014;44:24–37. doi: 10.1016/j.msec.2014.07.061. [DOI] [PubMed] [Google Scholar]
- 143.Franz S., Rammelt S., Scharnweber D., Simon J. C. Immune responses to implants - a review of the implications for the design of immunomodulatory biomaterials. Biomaterials. 2011;32(28):6692–6709. doi: 10.1016/j.biomaterials.2011.05.078. [DOI] [PubMed] [Google Scholar]
- 144.Pisoschi A. M., Pop A. The role of antioxidants in the chemistry of oxidative stress: a review. European Journal of Medicinal Chemistry. 2015;97:55–74. doi: 10.1016/j.ejmech.2015.04.040. [DOI] [PubMed] [Google Scholar]
- 145.Shiekh P. A., Singh A., Kumar A. Engineering bioinspired antioxidant materials promoting cardiomyocyte functionality and maturation for tissue engineering application. ACS Applied Materials & Interfaces. 2018;10(4):3260–3273. doi: 10.1021/acsami.7b14777. [DOI] [PubMed] [Google Scholar]
- 146.Shiekh P. A., Singh A., Kumar A. Oxygen-releasing antioxidant cryogel scaffolds with sustained oxygen delivery for tissue engineering applications. ACS Applied Materials & Interfaces. 2018;10(22):18458–18469. doi: 10.1021/acsami.8b01736. [DOI] [PubMed] [Google Scholar]
- 147.Li J., Shu Y., Hao T., et al. A chitosan-glutathione based injectable hydrogel for suppression of oxidative stress damage in cardiomyocytes. Biomaterials. 2013;34(36):9071–9081. doi: 10.1016/j.biomaterials.2013.08.031. [DOI] [PubMed] [Google Scholar]
- 148.Yang J., van Lith R., Baler K., Hoshi R. A., Ameer G. A. A thermoresponsive biodegradable polymer with intrinsic antioxidant properties. Biomacromolecules. 2014;15(11):3942–3952. doi: 10.1021/bm5010004. [DOI] [PubMed] [Google Scholar]
- 149.Hao T., Li J., Yao F., et al. Injectable fullerenol/alginate hydrogel for suppression of oxidative stress damage in brown adipose-derived stem cells and cardiac repair. ACS Nano. 2017;11(6):5474–5488. doi: 10.1021/acsnano.7b00221. [DOI] [PubMed] [Google Scholar]
- 150.Qu Y., Tang J., Liu L., Song L. L., Chen S., Gao Y. α-Tocopherol liposome loaded chitosan hydrogel to suppress oxidative stress injury in cardiomyocytes. International Journal of Biological Macromolecules. 2019;125:1192–1202. doi: 10.1016/j.ijbiomac.2018.09.092. [DOI] [PubMed] [Google Scholar]
- 151.Shabani P., Ghazizadeh Z., Gorgani-Firuzjaee S., et al. Cardioprotective effects of omega-3 fatty acids and ascorbic acid improve regenerative capacity of embryonic stem cell-derived cardiac lineage cells. BioFactors. 2019;45(3):427–438. doi: 10.1002/biof.1501. [DOI] [PubMed] [Google Scholar]
- 152.Luo J., Weaver M. S., Cao B., et al. Cobalt protoporphyrin pretreatment protects human embryonic stem cell-derived cardiomyocytes from hypoxia/reoxygenation injury in vitro and increases graft size and vascularization in vivo. Stem Cells Translational Medicine. 2014;3(6):734–744. doi: 10.5966/sctm.2013-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]