

Abstract

Quantifying peptides based on unique peptide fragment ions avoids the issue of ratio distortion that is commonly observed for reporter ion-based quantification approaches. Herein, we present a collision-induced dissociation-cleavable, isobaric acetyl-isoleucine-proline-glycine (Ac-IPG) tag, which conserves the merits of quantifying peptides based on unique fragments while reducing the complexity of the b-ion series compared to conventional fragment ion-based quantification methods thus facilitating data processing. Multiplex labeling is based on selective N-terminal dimethylation followed by derivatization of the ε-amino group of the C-terminal Lys residue of LysC peptides with isobaric Ac-IPG tags having complementary isotope distributions on Pro-Gly and Ac-Ile. Upon fragmentation between Ile and Pro, the resulting y ions, with the neutral loss of Ac-Ile, can be distinguished between the different labeling channels based on different numbers of isotope labels on the Pro-Gly part and thus contain the information for relative quantification, while b ions of different labeling channels have the same m/z values. The proteome quantification capability of this method was demonstrated by triplex labeling of a yeast proteome spiked with bovine serum albumin (BSA) over a 10-fold dynamic range. With the yeast proteins as the background, BSA was detected at ratios of 1.14:5.06:9.78 when spiked at 1:5:10 ratios. The raw mass data is available on the ProteomeXchange with the identifier PXD 018790.
Keywords: isobaric labeling, tandem mass spectrometry, fragment ion, quantitative proteomics, stable isotope
Introduction
Quantitative information at the proteome level aids in revealing the roles of proteins in biological, physiological, and pathological processes.1 The most widely used strategies in mass spectrometry-based proteome-wide quantitative approaches, other than label-free quantification, are based on stable isotope labeling, which allows us to analyze multiple samples simultaneously in a single LC-MS run.2−4 The major advantage of multiplexed labeling is that it allows quantifying peptides/proteins under the same experimental conditions, which circumvents signal variation due to, among others, variable sample loss during work-up and changing ionization efficiency between injections.5 As reviewed by Arul et al.,4 the currently most widely used multiplexing methods, like iTRAQ6,7 and TMT,8−10 are based on isobaric labeling. Peptides from different samples are quantified relative to each other based on isotopically distinct reporter ions in the MS2 spectra. In that way, the complexity of MS1 and MS2 spectra is not increased by the number of labeled samples. However, reporter ion-based quantification methods suffer from the issue of ratio distortion,5,11−14 which is caused by co-fragmentation of multiple precursor ions having similar m/z values and retention times. When more than one peptide is fragmented concurrently, the produced reporter ions from different peptides are indistinguishable in the MS2 spectrum, resulting in intensities that no longer reflect the ratios of the individual constituent peptides. To address the issue of ratio distortion, narrowing the width of the precursor ion isolation window,5,13 gas-phase purification12 and MultiNotch MS311 have been reported. These methods alleviate the problem of ratio distortion to some extent but are not widely used as they require sophisticated equipment combined with scan routines that are difficult to optimize and control.
An alternative isobaric labeling strategy, which does not suffer from ratio distortion, is isobaric peptide termini labeling (IPTL), which quantifies peptides based on isotopically labeled peptide fragments (b and y ions). In IPTL,15−20 LysC peptides are isobarically labeled with a pair of complementary tags at their N- and C-termini and fragmented into different sets of b- and y-ion clusters upon MS/MS. The fragment ions not only convey sequence information, but each fragment ion cluster contains information to determine the peptide ratio between samples in contrast to single reporter ions in iTRAQ or TMT. While co-fragmentation occurs also in IPTL, fragment ions can be attributed to the correct peptide. Recently, we reported the SMD-IPTL method,21 in which the multiplexing capacity of IPTL was increased to four-plex with the option to extend it to seven-plex. While SMD-IPTL provides accurate and precise quantitative data, it increases the complexity of MS2 spectra with challenges for subsequent data analysis.20 For example, established protein identification software considers additional peaks in the isotope envelop of fragment ions as unmatched peaks reducing the peptide score.11,22−24
To reduce the complexity of MS2 spectra of IPTL methods and to further improve multiplexing capacity, we propose a novel labeling approach that relies on a collision-induced dissociation (CID)-cleavable isobaric Ac-IPG tag. In this method, quantitative information is concealed in the Ac-IPG tag, which is located on the ε-amino group of the C-terminal Lys residue of LysC peptides and revealed upon tandem MS in the form of y-ion clusters containing different numbers of stable isotopes. The b ions of different labeling channels have the same mass because the N-terminus is not differentially labeled with stable isotopes. This allows multiplexing and relative peptide and protein quantification based on y-ion clusters, while identification is simpler than for conventional IPTL approaches because the MS2 spectra have regular b-ion series.
Experimental Section
Chemicals and Materials
2-Chlorotrityl chloride resin, Fmoc-Gly-OH, Fmoc-13C1-Gly-OH, Fmoc-13C2-Gly-OH, Fmoc-Pro-OH, Fmoc-Ile-OH, sodium cyanoborohydride, formaldehyde solution (36.5–38% in H2O), bovine serum albumin (BSA), acetic anhydride (12C4H6O3), acetic anhydride-1,1′-13C2 (13C212C2H6O3), acetic anhydride-13C4 (13C4H6O3), 1-[bis(dimethylamino) methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl), piperidine, pyridine, N,N-diisopropylethylamine (DIPEA), trifluoroethanol, p-nitrophenol (PNP), tris(2-carboxyethyl)phosphine hydrochloride (TCEP), 2-iodoacetamide, N,N-dimethylformamide (DMF), acetonitrile (ACN), acetone, dichloromethane (DCM), acetic acid, sodium acetate, sodium hydroxide (NaOH), hydroxylamine hydrate, triethylammonium bicarbonate (TEAB), and trifluoroacetic acid (TFA) were purchased from Sigma-Aldrich. LysC and yeast extracts were purchased from Promega. Synthetic peptide GTDWLANK and FDWA was purchased from GL Biochem (Shanghai, China). Solid-phase extraction disk Empore Octadecyl C18 was purchased from 3M.
Synthesis of Isobarically Labeled Acetyl-Isoleucine-Proline-Glycine-p-Nitrophenol Ester
The synthesis scheme of triplex isobaric Ac-IPG-PNP tags can be found in Scheme S1. Thirty milligrams of 2-chlorotrityl chloride (CTC) resin (1.0–1.8 mmol g–1) was swollen with 1.5 mL of DCM/DMF (1/1) on a thermomixer at 800 rpm for 30 min at room temperature. The swollen resin was washed three times with 0.75 mL of DMF. Then, 1 mL of 0.8 M DIPEA in DMF, 0.5 mL of DMF, and 0.2 mmol (60 mg) of Fmoc-Gly-OH, Fmoc-13C1-Gly-OH or Fmoc-13C2-Gly-OH were added. The reaction mixture was shaken for 90 min at room temperature. The solution was filtered out, and the remaining isotopically labeled Fmoc-Gly-CTC resin was washed three times with 0.75 mL of DMF. Afterward, the resin was treated with 1.5 mL of 20% piperidine in DMF for 5 min and 15 min sequentially. Then, 0.5 mL of 0.4 M HATU in DMF, 0.5 mL of 0.8 M DIPEA in DMF, 0.5 mL of DMF, and 0.2 mmol (68 mg) of Fmoc-Pro-OH were added. The reaction mixture was shaken for 90 min at room temperature. The resulting isotopically labeled Fmoc-Pro-Gly-CTC resin was washed three times with 0.75 mL of DMF. Afterward, the resin was treated with 1.5 mL of 20% piperidine in DMF for 5 min and 15 min sequentially. Then, 0.5 mL of 0.4 M HATU in DMF, 0.5 mL of 0.8 M DIPEA in DMF, 0.5 mL of DMF, and 0.2 mmol (71 mg) of Fmoc-Ile-OH were added. The reaction mixture was shaken for 90 min at room temperature. Then, the resin was treated with 1.5 mL of 20% piperidine in DMF for 5 min and 15 min sequentially to afford 3a–c. A 0.5 mL solution of DCM, 5 μL of pyridine, and 5 μL of acetic anhydride were added to 3c; 0.5 mL of DCM, 5 μL of pyridine, and 5 μL of acetic anhydride-1,1′-13C2 were added to 3b; 0.5 mL of DCM, 5 μL of pyridine, and 5 μL of acetic anhydride-13C4 were added to 3a. The reaction mixtures were shaken for 30 min at room temperature and then the isobarically labeled 4a–c were released with 0.75 mL of 20% trifluoroethanol in DCM. Subsequently, the dried isobarically labeled 4a–c were reacted with 80 mg of EDC·HCl and 40 mg of p-nitrophenol (PNP) in 0.7 mL of DCM to generate 5a–c, respectively. The reactions were shaken for 3 h, and after drying, the Ac-Ile-Pro-Gly-PNP esters were purified by RP-HPLC with detection at 254 nm. The collected HPLC fraction was freeze-dried to afford a white fluffy solid, which was stored at −20 °C until use. Around 3 mg was obtained for each tag. The LC-MS/MS analysis of Ac-Ile-Pro-Gly-PNP esters can be found in Figure S2. Five milligrams of non-isotopically labeled Ac-IPG-PNP was synthesized for NMR analysis (Figures S3 and S4).
LC Purification
For LC purification, the HPLC system (Shimadzu, Kyoto, Japan) was equipped with a SIL-20AC autosampler, two LC-20AT pumps, and an SPD-20A absorbance detector. The synthesized Ac-IPG-PNP esters were separated on an XBridge BEH C18 OBD Prep Column (250 mm × 10 mm, 5 μm particles, 130 Å pore size, Waters, Ireland) with a 30 min gradient of 2–90% acetonitrile in water/0.1% formic acid at a flow rate of 2 mL min–1.
NMR
1H NMR and 13C NMR spectra were recorded on a Bruker Ascend 600 MHz NMR spectrometer (operating at 600 and 150 MHz, respectively).
Alkylation and LysC Digestion
One hundred micrograms of BSA or yeast protein was dissolved in 100 μL of 100 mM TEAB. Then, 5 μL of 200 mM TCEP was added and the mixture was incubated for 1 h at 37 °C. Afterward, 5 μL of 375 mM freshly prepared 2-iodoacetamide was added and the mixture was incubated for 30 min in the dark at room temperature. Finally, 800 μL of acetone was added and the mixture was kept at −20 °C overnight for precipitation. The precipitated protein was centrifuged at 20,800g for 30 min. Subsequently, the supernatant was decanted and alkylated proteins were stored at −20 °C until digestion. One hundred micrograms of alkylated BSA or yeast protein was dissolved in 200 μL of 100 mM TEAB, and 2 μg of LysC dissolved in 1% formic acid was added. The pH was adjusted to 7.5–8 with 1 M TEAB, and the digestion was kept overnight at 37 °C. The reaction was terminated by acidification using 10% TFA, and resulting LysC peptide mixtures of BSA and yeast proteins were freeze-dried and stored at −80 °C before conducting the labeling reactions.
Selective N-Terminal Dimethylation of Peptides
One hundred micrograms of peptide GTDWLANK, LysC peptides of BSA, or LysC peptides of yeast proteins was dissolved in 100 μL 1% (aq.) acetic acid. Ten microliters of 4% formaldehyde in H2O was added to the peptide solution, and the mixture was shaken for 2 min before adding 15 μL of 600 mM sodium cyanoborohydride dissolved in 1% (aq.) acetic acid.18,25 The reaction was shaken for 15 min at room temperature. Excess reagents were removed by SPE using the STAGE (STop And Go Extraction) TIPS Desalting Procedure,26 and the collected N-terminally dimethylated peptides were freeze-dried and stored at −80 °C.
Triplex Labeling of N-Terminally Dimethylated Peptides
Five micrograms of N-terminally dimethylated GTDWLANK or LysC peptides of BSA or LysC peptides of yeast proteins was dissolved in 50 μL of 200 mM TEAB buffer of pH 8.5. Then, 4 μL of 50 mM Ac-IP-13C2-G-PNP, 13C1-Ac-IP-13C1-G-PNP, and 13C2-Ac-IPG-PNP in DMF was added to three peptide solutions, respectively. The reaction mixtures were shaken for 2 h at room temperature. To ensure complete labeling, 2 μL of p-nitrophenol ester was added again and incubated for 1 h more. Any esterification on the hydroxyl groups of Ser, Thr, or Tyr and excess PNP ester were hydrolyzed in the presence of 5% hydroxylamine hydrate at room temperature for 5 min and then desalted by SPE using the STAGE (STop And Go Extraction) TIPS Desalting Procedure prior to LC-MS analysis.26 A 750 μL solution of 2% acetonitrile (0.1% TFA) was added to remove excess Ac-IPG-COOH before eluting peptides from the STAGE tips, as shown in Figure S9.
LC/MS/MS Analysis
All mixed samples of isobarically labeled peptide were analyzed on a Q Exactive Plus, and other samples were analyzed on an Orbitrap Velos Pro as detailed below.
LC-MS/MS analyses were performed on an Orbitrap Velos Pro mass spectrometer (Thermo Fisher Scientific) coupled to an LC system (Shimadzu, Kyoto, Japan) equipped with a SIL-20AC HT autosampler, and two LC-20AD pumps. The sample was separated on an ACQUITY UPLC BEH C18 column (2.1 mm × 50 mm, 1.7 μm particles, Waters) at a column temperature of 60 °C with a 10 min gradient of 2–90% acetonitrile in water/0.1% formic acid at a flow rate of 300 μL min–1.
NanoLC-MS/MS analyses were performed on a Q Exactive Plus mass spectrometer (Thermo Scientific, San José, CA) equipped with an UltiMate 3000 RSLCnano system (Thermo Scientific). The peptides (around 300 ng) were first loaded onto a trap column (Acclaim PepMap 100 pre-column, 75 μm, 2 cm, C18, 3 mm, 100 Å, Thermo Scientific) and then separated on an analytical column (PepMap RSLC C18, 50 cm 75 μm ID, 2 μm, 100 Å, Thermo Scientific). A flow rate of 300 nL min–1 and a column temperature of 40 °C were utilized. Solvent A was 0.1% formic acid in water, and solvent B was 0.1% formic acid in acetonitrile. A gradient was applied from 2% to 50% or 65% B in the first 60 min then 85% B for 5 min and equilibration for 15 min with 2% B. Mass spectrometry data were measured using a data-dependent top 10 method. Full MS scans were acquired between m/z 300 and 1650 with a resolution of 70,000 (at m/z 200); the automatic gain control (AGC) target was 3E6 and maximum injection time was 50 ms. Precursor ions were selected with a window of 2 or 0.8 Th with −0.2 Th as offset. Selected ions were fragmented in the HCD collision cell with stepped normalized collision energies (NCE) of 18, 20, 22, 24, 26, 28, 30, and 32. The tandem (MS2) mass spectra were acquired in the Orbitrap mass analyzer with a resolution of 17,500 (at m/z 200). The AGC target was 1E5, and the maximum injection time was 100 ms. The charge exclusion discards ions with a single charge and charges larger than 4, and the dynamic exclusion time was 20 s.
Database Searching and Quantification
LC-MS/MS raw files were analyzed with the Andromeda search engine in SearchGUI27,28 and searched against the UniProt reference database of yeast (UP000002311, 6049 entries, downloaded on Jan. 20, 2020) into which the BSA entry (P02769) was inserted manually. LysC was selected as the enzyme, digestion mode as specific, and max missed cleavage sites as 0. A tolerance of 20 ppm for the precursor ion and 0.05 Da for the MS/MS fragment ions was applied. Carbamidomethylation (+57.02) on cysteine and dimethylation (+28.03) on N-terminus were set as fixed modification, oxidation (+15.99) on methionine as variable modification. For triplex labeling experiments, variable modifications on Lys were set as triplex isobaric Ac-IPG tags with two 13C isotopes (+311.17) with neutral losses of 13C2-Ac-Ile (−157.10), 13C1-Ac-Ile (−156.10), and Ac-Ile (−155.10) in MS2 spectra. The three labeling channels were searched in one run, and the search results were processed with PeptideShaker.29
The peptide spectrum matches (PSM) were exported from the PeptideShaker identified features. All the following steps were performed using in-house built Python scripts. For every PSM of unique peptides, the corresponding theoretical fragment ions with neutral loss of Ac-Ile were calculated and grouped by retention time. The raw data was converted into a Python readable mgf file with RawConverter30 to circumvent that the precursor ion m/z value is adjusted to the monoisotopic mass by Andromeda in SearchGUI. All MS/MS spectra were extracted from the resulting mgf file and also grouped by retention time. Afterward, based on the retention time, the original peak intensities in the mgf file were related to the corresponding theoretical fragment ions and the matched intensities were used to quantify peptides and ultimately proteins. The y1 ions with neutral loss from the C-terminal lysine and the fragment ions losing H2O and NH3 were excluded.21 The ratios for each PSM, peptide and protein were calculated based on the total peak intensities of the same labeling channel. For fragment ion spectra that were produced from monoisotopic peaks and in which no 13C interference existed, all matched y ions except y1 were used for quantification and the minimum number of matched ions was set to 3. For fragment ion spectra that were not produced from monoisotopic peaks only and in which 13C interference was observed in all y ions, only the small y2 ions were used for peptide quantification. Examples of calculating ratios can be found in Figure S10. Each peptide ratio was calculated from the three spectra with the highest total peak intensity, and each protein ratio was calculated from the three peptides with the highest total intensity. The Python scripts and quantification outputs are available at https://github.com/tianxiaobo002/Ac-IPG-scripts-and-quantification-outputs-at-multiple-levels. The raw mass spectrometry proteomics data of yeast and BSA-yeast samples have been deposited to the ProteomeXchange Consortium via the PRIDE31 partner repository with the dataset identifier PXD 018790.
Results and Discussion
Design of Ac-IPG Tag and Labeling Strategy
The CID-cleavable Ac-IPG tag targets the ε-amino group of the C-terminal Lys residues of LysC peptides after blocking their N-termini by dimethylation. The Ac-IPG tag consists of three parts: (1) Ac-Ile, which will form a neutral loss fragment upon CID; (2) the Pro-Gly tag, which will remain at the C-terminal Lys upon CID, and (3) an amine reactive p-nitrophenol ester (PNP) (Figure 1A). With different combinations of commercially available 13C- and 15N-labeled acetic anhydride, isoleucine, proline, and glycine, the Ac-IPG tag has a multiplexing capacity of 3 with an interval of 1 Da with the potential of being extended to the 10-plex level (Figure S1). Although deuterium-labeled amino acids can extend the multiplexing capacity further, these are generally avoided due to the possibility of varying retention times of the same peptide modified with a different number of deuterium atoms.32,33 The peptide bond N-terminal to Pro is known to be fragile upon CID.34−38 This was exploited in the Ac-IPG tag to induce neutral loss of Ac-Ile upon CID in addition to fragmentation along the peptide backbone. The different isotope distributions of the Pro-Gly tag render the fragment ions distinguishable between the labeling channels and the corresponding peak intensities represent the information for relative quantification (inset in Figure 1C).
Figure 1.

Schematic view of the Ac-IPG approach. (A) Functional design of the Ac-IPG-PNP tag (13C isotope locations of the triplex Ac-IPG-PNP tag are marked with asterisks). (B) Triplex isobaric labeling steps. (C) LC-MS/MS for a mixture of triplex-labeled samples.
In order to reduce complexity of the MS2 spectra, the N-termini of peptides were first modified by selective N-terminal dimethylation (Figure 1B). This serves two purposes: (1) it blocks the N-terminus of the LysC peptide to permit modifying the ε-amino group of the C-terminal Lys with a single isobaric tag per peptide and (2) it preserves ionization efficiency and improves completeness of the b-ion series by converting a primary into a tertiary amine.39−42 As there is no isotope label at the N-terminus, b ions originating from different samples will have the same mass, reducing complexity of the MS2 spectra. The differentially labeled samples are mixed prior to LC-MS/MS analysis, as shown in Figure 1C. The monoisotopic peak, selected with an isolation window of 0.8 Th and −0.2 Th offset,5,13,14 is fragmented into sets of b ions and y-ion clusters upon CID from which peak intensities of the y ions are extracted for each labeling channel. Their intensity ratios represent the relative quantification information of each sample in the mixture for that particular peptide.
Peptide Derivatization and Optimization of Fragmentation Energy
To assess the labeling conditions, the fragmentation properties and quantification at the peptide level, we first prepared N-terminally dimethylated GTDWLANK (N-dime-GTDWLANK) and applied FDWA as the control peptide. N-terminal dimethylation by reductive amination was highly selective and complete (Figure S5), and the yield was higher than 98% (Figure 2A). The intensities of both peptides were increased after N-terminal dimethylation, which agrees with the fact that the newly formed tertiary amine ionizes more easily than the original primary amine. N-dime-GTDWLANK was reacted with three isobaric Ac-IPG tags separately in 200 mM TEAB buffer (pH 8.5) at room temperature for 2 h. LC-MS of the products showed that the peptide was efficiently labeled (Figure 2A) with a yield of around 98%. There was only a slight decline in intensity upon labeling with the Ac-IPG tag, but the intensity of N-dime-GTDWLANK-GPI-Ac was still higher than that of the original unmodified peptide. Fragmentation of N-dime-GTDWLANK-GPI-Ac was studied at different normalized collision energies (NCE).43Figure 2C shows the MS2 spectrum at an NCE of 28 (MS2 spectra at NCE values ranging from 18 to 32 are shown in Figure S6). Neutral loss of Ac-Ile was complete at an NCE of 18 without any fragmentation of the peptide backbone. The intensity of fragment ions with neutral loss of Ac-Ile increased from NCE 22 to 28 but dropped again when the NCE was increased to 32. NCEs of 26, 28, and 30 were further evaluated with N-dime-BSA peptides-GPI-Ac and an NCE of 28 resulted in the highest number of PSMs (Figure S15), which is consistent with the results at the peptide level. Having established the optimal NCE at 28, we determined whether the CID-cleavable isobaric Ac-IPG tag allows for accurate quantification by comparing the intensities of y ions of three differentially labeled N-dime-GTDWLANK-GPI-Ac peptides. Separate analyses (Figure S7) of the three forms shows them having identical b ions but different y ions. When the three differentially labeled forms were mixed at ratios 1:1:1, 1:2:5, and 1:10:20 and analyzed by LC-MS/MS (Figure 2B and Figure S8), the corresponding measured ratios were 0.94:0.99:1.06, 1.01:2.04:4.94, and 1.30:9.60:20.11, respectively, which closely match the mixing ratios.
Figure 2.
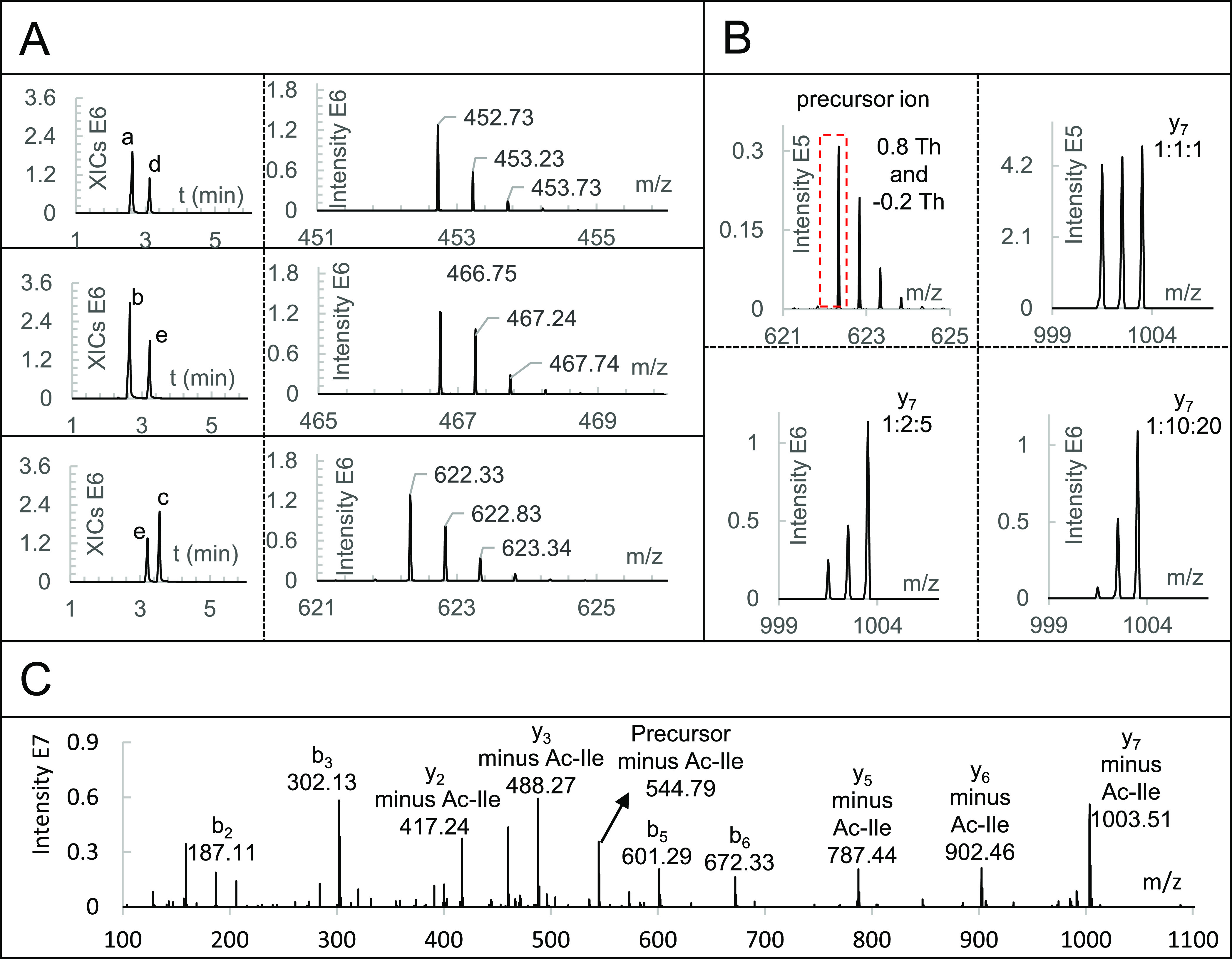
Derivatization and quantification of GTDWLANK. (A) LC-MS of selective N-terminal dimethylation and derivatization of the C-terminal Lys with isobaric Ac-IPG tags. From top to bottom: extracted ion chromatogram (XIC) of m/z 452.73 and 538.23, the combined XICs of m/z 452.73, 466.74, 480.75, and 566.26, and the combined XICs of m/z 466.74, 566.26, and 622.33. Peak a is unmodified GTDWLANK. Peak b is N-dime-GTDWLANK. Peak c is N-dime-GTDWLANK-13C1-GPI-Ac-13C1. Peak d is unmodified FDWA. Peak e is N-dime-FDWA. Mass spectra are shown to the right of the chromatograms. (B) Precursor ion selection of triplex-labeled N-dime-GTDWLANK-GPI-Ac and the corresponding y7 ion in the MS2 spectra at ratios 1:1:1, 1:2:5, and 1:10:20. (C) MS2 spectrum of N-dime-GTDWLANK-13C2-GPI-Ac with an NCE of 28.
Peptide and Protein Identification and Quantification
Peptide and protein identification and quantification were assessed with N-terminally dimethylated LysC peptides of bovine serum albumin (BSA) that were labeled with triplex isobaric Ac-IPG tags and mixed at different ratios prior to LC-MS/MS analysis. As shown in Figure 3A, the log2-normalized intensity ratios of y ions of triplex-labeled BSA peptides mixed at a 1:1:1 ratio cluster around 0 with better convergence for more intense ions. The overall ratios between different labeling channels at the PSM level were calculated from the sums of all y-ion intensities from the same labeling channel. Since multiple quantification ions, in the form of y ions, are present in each PSM, random fluctuations of peak intensities of single ions have less influence on the overall ratio (see Figure S10 for details). Ratios at the peptide level were calculated from the three most intense PSMs, and ratios at the protein level were calculated based on the three most intense peptides. Examples of calculations at the PSM, peptide, and protein levels are shown in Figure S10. As a result of averaging multiple data points, the log2-normalized ratio distribution at the PSM level (Figure S11) and at the peptide level (Figure 3B) show good convergence on 0. Subsequently, the triplex-labeled LysC peptides of BSA were mixed at a ratio of 1:5:10 (Figure 3C), and the medians of log2-normalized peptide ratios were found to be 0.12:2.32:3.30 (measured ratio = 1.09:4.99:9.85).
Figure 3.

Analysis of Ac-IPG triplex tagged BSA-derived LysC peptides. (A) Log2-normalized ratio distribution at the fragment ion level at a mixing ratio of 1:1:1. (B) Log2-normalized ratio distribution at the peptide level at a mixing ratio of 1:1:1. (C) Log2-normalized ratios at the peptide level at a mixing ratio of 1:5:10. Expected values for log2-normalized mixing ratios are shown as dashed lines.
Labeling Efficiency and Reproducibility at the Level of a Complex Proteomics Sample
The efficiency and reproducibility of the isobaric labeling steps and the quantification accuracy in complex samples were assessed by performing triplicate experiments on LysC peptides from a yeast proteome sample. The yield of selective N-terminal dimethylation was determined as the percentage of intensity of the N-terminally dimethylated form of each identified peptide to the total intensity of all possible forms (unmodified, N-terminally dimethylated, C-terminally dimethylated, and both N- and C-terminally dimethylated peptides). As shown in Figure S12, more than 98% of all peptides had a labeling yield exceeding 95% with an average coefficient of variation (CV) of 12.4% (n = 3). The yield of labeling the C-terminal Lys residues with isobaric Ac-IPG tags was calculated as the ratio of the intensity of the Ac-IPG-labeled form to the sum of the Ac-IPG-labeled form and the unmodified form. More than 93% of Ac-IPG-labeled peptides had a labeling yield higher than 95% in the triplex LysC yeast peptides mixed at a ratio of 1:1:1 (Figure S13). The average CV of the labeling yield of the three channels of 13C2-Ac-IPG, 13C1-Ac-IP-13C1-G, and Ac-IP-13C2-G was 8.7% (n = 3) (Figure 4A).
Figure 4.

Analysis of Ac-IPG tagged yeast-derived LysC peptides. (A) Log2-normalized protein ratio distribution of triplex-labeled LysC yeast peptides mixed at a ratio of 1:1:1. (B) Log2-normalized protein ratio distribution of triplex-labeled LysC yeast peptides mixed with a ratio of 1:2:5. (C) Log2-normalized protein ratio of triplex-labeled LysC BSA peptides mixed with a ratio of 1:5:10. Expected values for log2-normalized mixing ratios are shown as dashed lines.
Quantification of a Specific Protein within a Complex Proteomics Background
The quantification capability and dynamic range of the Ac-IPG approach in a complex background were assessed by preparing a BSA-yeast proteomics sample consisting of triplex-labeled LysC-digested BSA mixed at 1:5:10 and triplex-labeled LysC yeast peptides mixed at 1:2:5, as shown in Figure S14. A total of 485 yeast proteins were quantified, and the medians of the log2-normalized protein ratios of the three labeling channels were determined as 0.00:1.09:2.29, which is close to the expected values of 0.00:1.00:2.32 (Figure 4B). For triplex-labeled BSA mixed at 1:5:10, the log2-normalized protein ratios were 0.19:2.34:3.29, respectively, which is close to the expected values of 0.00:2.32:3.32 (Figure 4C).
In conclusion, we propose a novel quantification approach relying on a CID-cleavable Ac-IPG tag and selective N-terminal dimethylation. This approach has the advantage that MS2 spectra are less complex than for conventional IPTL while avoiding the issue of ratio distortion observed for reporter ion-based quantification methods, such as TMT and iTRAQ. The proteome quantification capability of the CID-cleavable Ac-IPG tag method was demonstrated by triplex labeling of a yeast proteome spiked with BSA over a 10-fold dynamic range. The multiplexing capacity can be readily expanded to 10-plex with commercially available 13C- and 15N-labeled acetic anhydride, isoleucine, proline, and glycine via the reported synthesis route.
Acknowledgments
We gratefully acknowledge the China Scholarship Council (CSC) for a Ph.D. fellowship to X.T. X.T. thanks Jos Hermans for help with the LC-MS analyses and C. Ye for help with the NMR analyses. This work is partially funded by the Open Technology Program of Toegepaste en Technische Wetenschappen (TTW) with project number 15230, which is financed by the Netherlands Organization for Scientific Research (NWO).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.0c00371.
(Scheme S1) Synthesis of the triplex Ac-IPG tag, (Figure S1) schematic view of the 10-plex Ac-IPG labeling, (Figure S2) LC-MS/MS of triplex Ac-IPG-PNP tags, (Figure S3) 1H NMR of Ac-IPG-PNP, (Figure S4) 13C NMR of Ac-IPG-PNP, (Figure S5) selective N-terminal dimethylation of peptide GTDWLANK, (Figure S6) MS2 spectra of N-dime-GTDWLANK-13C2-GPI-Ac under various NCE energies, (Figure S7) separate analyses of three different forms of isobarically labeled N-dime-GTDWLANK-GPI-Ac, (Figure S8) MS2 spectra of triplex-labeled N-dime-GTDWLANK-GPI-Ac mixed at various ratios, (Figure S9) removing excess Ac-IPG-COOH by five times flushing with 2% AcCN, (Figure S10) Examples of calculating ratios at the PSM, peptide, and protein level for triplex-labeled LysC peptides of BSA mixed at a ratio of 1:5:10, (Figure S11) the log2-normalized ratio distribution at the PSM level of BSA at a mixing ratio of 1:1:1, (Figure S12) yield distribution of triplicate samples of selective N-terminal dimethylation of the LysC yeast peptides, (Figure S13) Ac-IPG labeling yield in the triplex LysC yeast peptides mixed at a ratio of 1:1:1, (Figure S14) scheme of preparing the BSA-yeast proteomics sample, and (Figure S15) the influence of NCE on fragmentation and identification of LysC peptides of BSA (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Zhang Y.; Fonslow B. R.; Shan B.; Baek M.-C.; Yates J. R. III Protein Analysis by Shotgun/Bottom-up Proteomics. Chem. Rev. 2013, 113, 2343–2394. 10.1021/cr3003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauniyar N.; Yates J. R. III Isobaric Labeling-Based Relative Quantification in Shotgun Proteomics. J. Proteome Res. 2014, 13, 5293–5309. 10.1021/pr500880b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour O.; Cobice D.; Malone J. Stable isotope labelling methods in mass spectrometry-based quantitative proteomics. J. Pharm. Biomed. Anal. 2015, 113, 2–20. 10.1016/j.jpba.2015.04.013. [DOI] [PubMed] [Google Scholar]
- Arul A. B.; Robinson R. A. S. Sample Multiplexing Strategies in Quantitative Proteomics. Anal. Chem. 2019, 91, 178–189. 10.1021/acs.analchem.8b05626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnett M.; Yeung E.; Wühr M. Accurate, Sensitive, and Precise Multiplexed Proteomics Using the Complement Reporter Ion Cluster. Anal. Chem. 2018, 90, 5032–5039. 10.1021/acs.analchem.7b04713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross P. L.; Huang Y. N.; Marchese J. N.; Williamson B.; Parker K.; Hattan S.; Khainovski N.; Pillai S.; Dey S.; Daniels S.; Purkayastha S.; Juhasz P.; Martin S.; Bartlet-Jones M.; He F.; Jacobson A.; Pappin D. J. Multiplexed Protein Quantitation in Saccharomyces cerevisiae Using Amine-reactive Isobaric Tagging Reagents. Mol. Cell. Proteomics 2004, 3, 1154–1169. 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- Ow S. Y.; Cardona T.; Taton A.; Magnuson A.; Lindblad P.; Stensjö K.; Wright P. C. Quantitative Shotgun Proteomics of Enriched Heterocysts from Nostoc sp. PCC 7120 Using 8-Plex Isobaric Peptide Tags. J. Proteome Res. 2008, 7, 1615–1628. 10.1021/pr700604v. [DOI] [PubMed] [Google Scholar]
- Thompson A.; Schäfer J.; Kuhn K.; Kienle S.; Schwarz J.; Schmidt G.; Neumann T.; Hamon C. Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- Dayon L.; Hainard A.; Licker V.; Turck N.; Kuhn K.; Hochstrasser D. F.; Burkhard P. R.; Sanchez J. C. Relative Quantification of Proteins in Human Cerebrospinal Fluids by MS/MS Using 6-Plex Isobaric Tags. Anal. Chem. 2008, 80, 2921–2931. 10.1021/ac702422x. [DOI] [PubMed] [Google Scholar]
- Thompson A.; Wölmer N.; Koncarevic S.; Selzer S.; Böhm G.; Legner H.; Schmid P.; Kienle S.; Penning P.; Höhle C.; Berfelde A.; Martinez-Pinna R.; Farztdinov V.; Jung S.; Kuhn K.; Pike I. TMTpro: Design, Synthesis, and Initial Evaluation of a Proline-Based Isobaric 16-Plex Tandem Mass Tag Reagent Set. Anal. Chem. 2019, 91, 15941–15950. 10.1021/acs.analchem.9b04474. [DOI] [PubMed] [Google Scholar]
- Ting L.; Rad R.; Gygi S. P.; Haas W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 2011, 8, 937–940. 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger C. D.; Lee M. V.; Hebert A. S.; McAlister G. C.; Phanstiel D. H.; Westphall M. S.; Coon J. J. Gas-phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nat. Methods 2011, 8, 933–935. 10.1038/nmeth.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wühr M.; Haas W.; McAlister G. C.; Peshkin L.; Rad R.; Kirschner M. W.; Gygi S. P. Accurate Multiplexed Proteomics at the MS2 Level Using the Complement Reporter Ion Cluster. Anal. Chem. 2012, 84, 9214–9221. 10.1021/ac301962s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter S. V.; Meier F.; Wichmann C.; Cox J.; Mann M.; Meissner F. EASI-tag enables accurate multiplexed and interference-free MS2-based proteome quantification. Nat. Methods 2018, 15, 527–530. 10.1038/s41592-018-0037-8. [DOI] [PubMed] [Google Scholar]
- Koehler C. J.; Strozynski M.; Kozielski F.; Treumann A.; Thiede B. Isobaric Peptide Termini Labeling for MS/MS-Based Quantitative Proteomics. J. Proteome Res. 2009, 8, 4333–4341. 10.1021/pr900425n. [DOI] [PubMed] [Google Scholar]
- Koehler C. J.; Arntzen M. O̷.; Strozynski M.; Treumann A.; Thiede B. Isobaric Peptide Termini Labeling Utilizing Site-Specific N-Terminal Succinylation. Anal. Chem. 2011, 83, 4775–4781. 10.1021/ac200229w. [DOI] [PubMed] [Google Scholar]
- Nie A. Y.; Zhang L.; Yan G. Q.; Yao J.; Zhang Y.; Lu H. J.; Yang P. Y.; He F. C. In Vivo Termini Amino Acid Labeling for Quantitative Proteomics. Anal. Chem. 2011, 83, 6026–6033. 10.1021/ac201035f. [DOI] [PubMed] [Google Scholar]
- Koehler C. J.; Arntzen M. O̷.; de Souza G. A.; Thiede B. An Approach for Triplex-Isobaric Peptide Termini Labeling (Triplex-IPTL). Anal. Chem. 2013, 85, 2478–2485. 10.1021/ac3035508. [DOI] [PubMed] [Google Scholar]
- Liu J.; Zhou Y.; Shan Y.; Zhao B.; Hu Y.; Sui Z.; Liang Z.; Zhang L.; Zhang Y. A Multiplex Fragment-Ion-Based Method for Accurate Proteome Quantification. Anal. Chem. 2019, 91, 3921–3928. 10.1021/acs.analchem.8b04806. [DOI] [PubMed] [Google Scholar]
- Jiang H.; Yin H.; Xie L.; Zhang Y.; Zhang L.; Yang P.-Y.; Lu H. A novel triplex isobaric termini labeling quantitative approach for simultaneously supplying three quantitative sources. Anal. Chim. Acta 2018, 1001, 70–77. 10.1016/j.aca.2017.11.004. [DOI] [PubMed] [Google Scholar]
- Tian X.; de Vries M. P.; Visscher S. W. J.; Permentier H. P.; Bischoff R. Selective maleylation-directed isobaric peptide termini labeling for accurate proteome quantification. Anal. Chem. 2020, 92, 7836–7844. 10.1021/acs.analchem.0c01059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arntzen M. O̷.; Koehler C. J.; Barsnes H.; Berven F. S.; Treumann A.; Thiede B. IsobariQ: Software for Isobaric Quantitative Proteomics using IPTL, iTRAQ, and TMT. J. Proteome Res. 2011, 10, 913–920. 10.1021/pr1009977. [DOI] [PubMed] [Google Scholar]
- Xie L.-Q.; Zhang L.; Nie A.-Y.; Yan G.-Q.; Yao J.; Zhang Y.; Yang P.-Y.; Lu H.-J. ITMSQ: A software tool for N- and C-terminal fragment ion pairs based isobaric tandem mass spectrometry quantification. Proteomics 2015, 15, 3755–3764. 10.1002/pmic.201400513. [DOI] [PubMed] [Google Scholar]
- Zheng N. R.; Shan Y. C.; Deng Y. L.; Zhang Y. K. Novel Algorithm for Identification and Quantification of Proteins Based on Strategy of Isobaric Peptide Termini Labelling. Chin. J. Anal. Chem 2017, 45, 1441–1447. 10.11895/j.issn.0253-3820.170250. [DOI] [Google Scholar]
- Qin H.; Wang F.; Zhang Y.; Hu Z.; Song C.; Wu R.; Ye M.; Zou H. Isobaric cross-sequence labeling of peptides by using site-selective N-terminus dimethylation. Chem. Commun. 2012, 48, 6265–6267. 10.1039/c2cc31705b. [DOI] [PubMed] [Google Scholar]
- Rappsilber J.; Ishihama Y.; Mann M. Stop and Go Extraction Tips for Matrix-Assisted Laser Desorption/Ionization, Nanoelectrospray, and LC/MS Sample Pretreatment in Proteomics. Anal. Chem. 2003, 75, 663–670. 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- Vaudel M.; Barsnes H.; Berven F. S.; Sickmann A.; Martens L. SearchGUI: An open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics 2011, 11, 996–999. 10.1002/pmic.201000595. [DOI] [PubMed] [Google Scholar]
- Barsnes H.; Vaudel M. SearchGUI: A Highly Adaptable Common Interface for Proteomics Search and de Novo Engines. J. Proteome Res. 2018, 17, 2552–2555. 10.1021/acs.jproteome.8b00175. [DOI] [PubMed] [Google Scholar]
- Vaudel M.; Burkhart J. M.; Zahedi R. P.; Oveland E.; Berven F. S.; Sickmann A.; Martens L.; Barsnes H. PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat. Biotechnol. 2015, 33, 22–24. 10.1038/nbt.3109. [DOI] [PubMed] [Google Scholar]
- He L.; Diedrich J.; Chu Y. Y.; Yates J. R. III Extracting Accurate Precursor Information for Tandem Mass Spectra by RawConverter. Anal. Chem 2015, 87, 11361–11367. 10.1021/acs.analchem.5b02721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y.; Csordas A.; Bai J.; Bernal-Llinares M.; Hewapathirana S.; Kundu D. J.; Inuganti A.; Griss J.; Mayer G.; Eisenacher M.; Pérez E.; Uszkoreit J.; Pfeuffer J.; Sachsenberg T.; Yılmaz Ş.; Tiwary S.; Cox J.; Audain E.; Walzer M.; Jarnuczak A. F.; Ternent T.; Brazma A.; Vizcaíno J. A. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; Sioma C. S.; Wang S.; Regnier F. E. Fractionation of Isotopically Labeled Peptides in Quantitative Proteomics. Anal. Chem. 2001, 73, 5142–5149. 10.1021/ac010583a. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Sioma C. S.; Thompson R. A.; Xiong L.; Regnier F. E. Controlling Deuterium Isotope Effects in Comparative Proteomics. Anal. Chem. 2002, 74, 3662–3669. 10.1021/ac025614w. [DOI] [PubMed] [Google Scholar]
- Dongré A. R.; Jones J. L.; Somogyi Á.; Wysocki V. H. Influence of peptide composition, gas-phase basicity, and chemical modification on fragmentation efficiency: Evidence for the mobile proton model. J. Am. Chem. Soc. 1996, 118, 8365–8374. 10.1021/ja9542193. [DOI] [Google Scholar]
- Mák M.; Mezö G.; Skribanek Z.; Hudecz F. Stability of Asp-Pro bond under high and low energy collision induced dissociation conditions in the immunodominant epitope region of Herpes simplex virion glycoprotein D. Rapid Commun. Mass Spectrom. 1998, 12, 837–842. . [DOI] [PubMed] [Google Scholar]
- Hogan J. M.; McLuckey S. A. Charge state dependent collision-induced dissociation of native and reduced porcine elastase. J. Mass Spectrom. 2003, 38, 245–256. 10.1002/jms.458. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Triscari J. M.; Pasa-Tolic L.; Anderson G. A.; Lipton M. S.; Smith R. D.; Wysocki V. H. Dissociation Behavior of Doubly-Charged Tryptic Peptides: Correlation of Gas-Phase Cleavage Abundance with Ramachandran Plots. J. Am. Chem. Soc. 2004, 126, 3034–3035. 10.1021/ja038041t. [DOI] [PubMed] [Google Scholar]
- Tiwary S.; Levy R.; Gutenbrunner P.; Salinas Soto F.; Palaniappan K. K.; Deming L.; Berndl M.; Brant A.; Cimermancic P.; Cox J. High-quality MS/MS spectrum prediction for data-dependent and data-independent acquisition data analysis. Nat. Methods 2019, 16, 519–525. 10.1038/s41592-019-0427-6. [DOI] [PubMed] [Google Scholar]
- Hsu J. L.; Huang S. Y.; Chow N. H.; Chen S. H. Stable-isotope dimethyl labeling for quantitative proteomics. Anal. Chem. 2003, 75, 6843–6852. 10.1021/ac0348625. [DOI] [PubMed] [Google Scholar]
- Guo K.; Ji C.; Li L. Stable-Isotope Dimethylation Labeling Combined with LC–ESI MS for Quantification of Amine-Containing Metabolites in Biological Samples. Anal. Chem. 2007, 79, 8631–8638. 10.1021/ac0704356. [DOI] [PubMed] [Google Scholar]
- Cho K.-C.; Kang J. W.; Choi Y.; Kim T. W.; Kim K. P. Effects of peptide acetylation and dimethylation on electrospray ionization efficiency. J. Mass Spectrom. 2016, 51, 105–110. 10.1002/jms.3723. [DOI] [PubMed] [Google Scholar]
- Fu Q.; Li L. De Novo Sequencing of Neuropeptides Using Reductive Isotopic Methylation and Investigation of ESI QTOF MS/MS Fragmentation Pattern of Neuropeptides with N-Terminal Dimethylation. Anal. Chem. 2005, 77, 7783–7795. 10.1021/ac051324e. [DOI] [PubMed] [Google Scholar]
- Olsen J. V.; Macek B.; Lange O.; Makarov A.; Horning S.; Mann M. Higher-energy C-trap dissociation for peptide modification analysis. Nat. Methods 2007, 4, 709–712. 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.