

Abstract
Paclitaxel is one of the most effective and widely used agents in treating a variety of cancers, including endometrial cancer. Because of its poor solubility in water, the current intravenous pharmaceutical paclitaxel is formulated in Cremophor EL and dehydrated in ethanol in equal volumes. Cremophor EL is capable of causing complement activation, which can trigger an immediate hypersensitivity reaction. SPR064 is a pro-drug of paclitaxel which has a much higher solubility as compared to the parent drug; hence, SPR064 can be conveniently formulated in non-cremapor based medium, reducing the risk of cremaphor-related hypersensitivity reactions. The pharmacokinetics and solubility of SPR064 were evaluated in rats. The anti-tumorigenic potential of SPR064 was compared to paclitaxel in endometrial cancer cell lines and a genetically engineered mouse model (Lkbfl/flp53fl/fl) of endometrial cancer. Overall, SPR064 exhibited improved solubility and better exposure to drug in rats when compared to paclitaxel. SPR064 and paclitaxel inhibited cell proliferation, induced apoptosis, enhanced cellular stress and caused cell cycle G1 arrest in endometrial cancer cell lines, with similar potency. Both SPR064 and paclitaxel reduced tumor weight in the Lkbfl/flp53fl/fl mouse model under obese and lean conditions compared to their respective controls. Immunohistochemical staining demonstrated that SPR064 and paclitaxel significantly reduced the expression of Ki-67 and BCL-xL in the endometrial tumors of both obese and lean mice. In summary, SPR064 has anti-tumorigenic effects that are equivalent to paclitaxel in endometrial cancer cell lines and a genetically engineered mouse model of endometrial cancer. Thus, SPR064 may be a promising therapy for endometrial cancer without the significant risk of hypersensitivity reactions seen with paclitaxel.
Keywords: Paclitaxel, SPR064, endometrial cancer, cell proliferation, apoptosis
Introduction
Endometrial cancer (EC) is the fourth most common malignancy in women and the most common gynecologic cancer in the US, with 61,880 estimated new cases and 12,160 deaths expected in the United States in 2019 [1]. Multiple risk factors are involved in the carcinogenesis of EC including obesity, unopposed estrogen, early menarche, late menopause, exposure to tamoxifen and nulliparity [2]. Most women are diagnosed with early stage disease and have a good prognosis with 5-year overall survival rates of nearly 90%. However, women who are diagnosed with stage III or IV disease have a poor prognosis with 5-year overall survival rates of 60% and 20%, respectively [3]. Thus, it is imperative to develop novel therapies for advanced stage and recurrent EC [3,4].
Paclitaxel (PTX), a tubulin stabilizer, is one of the most effective and extensively used chemotherapeutic agents in the treatment of different types of cancers, including EC [5]. Due to PTX’s poor solubility in water, the most common pharmaceutical formulation of PTX is made by dissolving it in 50% Cremophor EL and 50% dehydrated ethanol (1:1, v/v). This formulation is used to enhance the solubility of PTX in water for systemic, intravenous administration [6,7]. The disadvantages associated with this formulation have been well-documented, most notably, hypersensitivity reactions to Cremaphor EL. Severe hypersensitivity reactions leading to discontinuation of treatment has been noted in up to 15% patients receiving PTX, despite premedication with both anti-histamines and a corticosteroids and slowing of the infusion [8,9]. Therefore, the development of new alternative formulations without Cremophor EL may overcome hypersensitivity reactions or other side effects caused by this solvent and increase the number of patients successfully treated with PTX [7,10].
SPR064 is a modified drug of PTX that has much improved solubility in saline compared to its parent compound. The modifying agent of SPR064 is a natural compound imparts pH independent solubility. SPR064 does not require Cremophor® or albumin, thus eliminating issues associated with Cremophor® or albumin. Given this, we expect that SPR064 will be more cost effective than liposomal and albumin-based formulations. In the present study, the pharmacokinetics and solubility of SPR064 in rats was evaluated. Furthermore, SPR064’s anti-tumorigenic potential was investigated and compared to PTX in EC using cell lines and a genetically engineered mouse model (Lkbfl/flp53fl/fl) of endometrioid EC.
Materials and methods
Cell culture and reagents
Five EC cell lines, ECC-1, Hec-1A, KLE, Ishikawa and RL-952, were used. The ECC-1 cells were grown in RPMI 1640 medium supplemented with 5% bovine. The Hec-1A cells were grown in McCoy’s 5A supplemented with 10% fetal bovine serum. The Ishikawa cells were grown in MEM supplemented with 5% fetal bovine serum. The KLE and RL-952 cells were grown in DMEM/F12 with 10% FBS. All medium was supplemented with 100 U/ml of penicillin and 100 ug/ml of streptomycin. The cells were cultured in humidified 5% CO2 at 37°C. SPR064 was obtained from Sphaera Pharma. MTT and PTX were purchased from Sigma (St. Louis, MO). The Annexin V FITC kit was purchased from BioVision (Mountain View, CA). All antibodies were purchased from Cell Signaling (Beverly, MA). Enhanced chemiluminescence western blotting detection reagents were purchased from Thermo (Rockford, IL). Caspase 3 was purchased from AAT Bioquest (Sunnyvale, CA). All other chemicals were purchased from Sigma.
Conversion of modified drug of SPR064 to PTX in rat plasma
SPR064 was incubated in rat plasma for 3 hours, and the level of PTX and SPR64 was monitored using high-performance liquid chromatography (HPLC). 450 µl Sprague Dawleyrat plasma with a final concentration of compound of 200 ug/ml was incubated at 37°C. 50 µl of reaction mixture was taken out at different time points (0, 5, 10, 30, 60 and 120 and 180 min), and the reaction was stopped with 200 ul of 100% Acetonitrile. The solution was vortexed for 5 mins and then spun for an additional 10 mins. A standard calibration curve was generated with ACN at a range of concentrations (200 ug/ml to 6.25 ug/ml). The supernatants were analyzed by HPLC (Waters, Milford, MA).
Kinetic solubility of SPR064 and PTX by nephelometry
Nephelometry is a light scattering method to detect particles in liquid samples. The principle behind nephelometry is to measure forwarded scattered light when a laser beam passes through a sample, and the light is deflected by the particles. A stock solution of SPR064 and PTX was prepared in DMSO (20X). Serial dilutions (1:1) were prepared in DMSO in a 96 well plate. 95 µl of MQ was taken from the 96 well UV star plate (Greiner-catalogue no. 655801). 5 µl of compound dilutions in DMSO from the stock plate was added to the 96 well templates (in triplicates) so that the final DMSO concentration was 5%. Plates were kept on a shaker for 2 hr at room temperature. Samples were read in BMG NEPHELOSTAR (Cary, NC). The raw data obtained was averaged. The averaged values were plotted against concentration of drug.
Pharmacokinetics study of SPR064 in rats
Pharmacokinetics studies were carried out to evaluate the plasma exposure of PTX and SPR064 in rats when dosed intravenously (IV). The results were compared with exposure to PTX using the same dosing vehicle. As previously stated, SPR064 has improved aqueous solubility as compared to PTX, leading to easier IV dosing without using Cremophor. SPR064 and PTX were dosed at 3 mpk by IV mode of administration in female SD rat. Blood was collected at different time points in the presence of heparin as an anti-coagulant. 40 ul of blood was collected at each time point. Plasma was collected by centrifugation, and plasma was stored at -80°C for analysis. PK analysis was done using ABI 3200 LCMS (Arcade, NY), and the data analysis was performed using Graphpad Prism (San Diego, CA).
MTT assay
The ECC-1, Hec-1A, KLE, Ishikawa and RL-952 cells were plated and grown in 96-well plates at a concentration of 4000 cells/well for 24 hours. The cells were subsequently treated with varying doses of SPR064 and PTX for 72 hours. MTT (5 mg/ml) was added to the 96-well plates at 5 μl/well, followed by an additional hour of incubation. The MTT reaction was terminated through the addition of 100 μl of DMSO. The results were read by measuring absorption at 575 nm with a microplate reader (Tecan, Morrisville, NC). The effect of SPR064 and PTX was calculated as a percentage of control cell growth obtained from DMSO treated cells grown in the same 96-well plates. Each experiment was performed in triplicate to assess for consistency of results.
Colony formation assay
The ECC-1 and Hec-1A cells were seeded in 6-well plates (400 cells/dish and 200 cells/dish, respectively) in their standard growth media. Cells were allowed to adhere for 24 hours, and then treated with SPR064 for 24 hours. The cells were cultured for 14 days with media changes every third or fourth day. The plates were stained with 0.5% crystal violet, and colonies were counted under the microscope. Each experiment was repeated in triplicate to assess for consistency of results.
Cell cycle assay
The ECC-1 and Hec-1A cells were plated in their appropriate media at 2.5 × 105 cells/well in a 6-well plate for 24 hrs. The cells were subsequently treated with SPR064 or PTX at varying concentrations for 48 hrs. Cells were collected, washed with PBS, fixed in a 90% methanol solution and then stored at -20°C until flow cytometric analysis was performed. On the day of analysis, cells were washed and centrifuged using cold PBS, suspended in 100 µl PBS and 10 µl of RNase A solution (250 µg/ml) followed by incubation for 30 min at 37°C. After incubation, 110 µl of PI stain (100 µg/ml) was added to each tube and kept at 4°C for at least 30 min prior to analysis. Flow cytometric analysis was performed using the CyAn machine (Beckman Coulter, Miami, FL). Cell cycle analysis was performed by FCS4 express software (De Novo Software, Los Angeles, CA). All experiments were performed in triplicate to assess for consistency of response.
Annexin V assay
Apoptosis was detected with the Annexin V FITC kit on Cellometer (Nexcelom, Lawrence, MA). Briefly, 3.0 × 105 cells/well were seeded into 6-well plates, incubated overnight and then treated with SPR064 or PTX at different doses for 16 hrs. The cells were then collected, washed with PBS and resuspended in 100 μl binding buffer. Subsequently, 1 μl of Annexin V-FITC (100 μg/ml) and 0.5 μl of propidium iodide (2 mg/ml) were added in the binding buffer, and the plates placed in the dark for 15 min. The samples were immediately measured by Cellometer. The results were analyzed by FCS4 express software. All experiments were performed in triplicate to assess for consistency of response.
Caspase 3 activity assay
The ECC-1 and Hec-1A cells were plated in their appropriate media at 3.0 × 105 cells/well in a 6-well plate for 24 hours and then treated with SPR064 or PTX at varying concentrations for 16 hrs. Cell lysates were prepared in lysis buffer. Equal amounts of protein were mixed with cleaved caspase 3 substrate at 37°C. The analysis was performed by a microplate reader (Tecan, Morrisville, NC) at an excitation wavelength of 400 nm and an emission wavelength of 505 nm. All experiments were performed in triplicate and repeated twice to assess for consistency of response.
Reactive oxygen species (ROS) assay
The alteration of total production of reactive oxygen species caused by SPR064 or PTX was measured using a DCFH-DA fluorescent dye. The ECC-1 and Hec-1A cells (4000 cells/well) were seeded in black 96-well plates. After 24 hours, the cells were treated with SPR064 and Paclitaxel for 4 hours to induce ROS generation. After the cells were incubated with DCFH-DA (20 μM) for 30 min, the fluorescence was monitored at an excitation wavelength of 485 nm and an emission wavelength of 530 nm using a plate reader (Tecan). All experiments were performed at least twice to assess for consistency of results.
Western immunoblotting
The ECC-1 and Hec-1A cells were plated at 3.0 × 105 cells/well in 6-well plates in their corresponding media and then treated with SPR064 or PTX overnight. Cell lysates were prepared in RIPA buffer. Equal amounts of protein were separated by gel electrophoresis and transferred onto a PVDF membrane. The membrane was blocked with 5% nonfat dry milk and then incubated with a 1:1500 dilution of primary antibody overnight at 4°C. The membrane was then washed and incubated with a secondary peroxidase-conjugated antibody for 1 hour after washing. Antibody binding was detected using an enhanced chemiluminescence detection system on the Alpha Innotech Imaging System (Protein Simple, Santa Clara, CA). After developing, the membrane was stripped and re-probed using antibodies against β-actin to confirm equal loading. Intensity for each band was measured and normalized to β-actin as an internal control. Each experiment was repeated two times to assess for consistency of results.
Genetically engineered mouse model of endometrioid endometrial cancer
The homozygous Lkb1fl/flp53fl/fl double conditional knockout mice were used [11]. All experimental animals were maintained in accordance with the Institutional Animal Care and Use Committee (IACUC) and the NIH guide for the Care and Use of Laboratory Animals. To maximize weight gain, mice were provided a high-fat diet (HFD, obese group) (60% kcal from fat, Research Diets, New Brunswick, NJ) and control mice (non-obese group) were provided a low-fat diet (LFD) (10% kcal from fat) ad libitum, beginning at 3 weeks of age after birth. Recombinant adenovirus Ad5-CMV-Cre (AdCre) was purchased from the University of Iowa Transfer Vector Core at a titer of 1011-1012 infectious particles/ml. Ad-Cre injection into the right uterine horn of Lkb1fl/flp53fl/fl mice was performed using a syringe with a 30-gauge needle at 6-8 weeks of age to induce EC. At 8 weeks after AdCre infection, the obese and lean mice were randomly divided into six groups (15 mice/per group) including control and PTX and SPR064 treatment groups. PTX was dissolved in DMSO and SPR064 was dissolved in PEG400-40%, 10% ethanol and 50% normal saline. The obese and lean mice were treated with PTX (6.25 mg/kg, IP) and SPR064 (8.06 mg/kg, IP) every 5 days for 4 weeks, using molecular weight correction for SPR064 versus PTX to determine equivalent dosing of drugs. All mice were euthanized after 4 weeks of SPR064 and PTX treatments. Endometrial umor tissue were collected for immunohistochemical staining.
Immunohistochemical staining (IHC)
Endometrial tumor tissues from mice were formalin-fixed and paraffin-embedded. After rehydration and antigen retrieval, the slides (thickness = 4 μm) were incubated with primary antibodies: anti-Ki-67 (1:300), anti-bcl-x l(1:1200). The staining was visualized using DAB (Invitrogen, CA). The slides were scanned by Motic and analyzed by Image Pro (Warrendale, PA).
Statistical analysis
Results for experiments were normalized to the mean of the control and analyzed using the Student t-test. Differences were considered significant if the p value was less than 0.05 (P<0.05) with a confidence interval of 95%.
Results
Characterization of SPR064
The solubility of SPR064 and PTX were compared at different concentrations in 5% DMSO solution using Nephelometry. As shown in Figure 1A, the solubility of SPR064 was comparable to PTX at less than 50 ug/ml. With increasing concentrations, SPR064 consistently exhibited lower nephelometric turbidity units (NTUs) as compared to PTX, suggesting that SPR064 has much better solubility than PTX.
Figure 1.

Comparison of pharmacokinetics profile and solubility between SPR064 and Paclitaxel. SPR064 shows significant improvement in solubility over parent drug PTX using Nephelometry. SPR064, with increasing concentrations, consistently showed lower nephelometric turbidity units (NTUs) when compared to PTX (A). Modified PTX analog SPR064 shows quantitative conversion to PTX at the first time point in rat plasma, no SPR064 is observed by HPLC (B). SPR064 was found to have much better exposure and a more favorable PK profile as compared to the PTX when dosed in saline based formulation in SD rats (C).
Since SPR064 is the modified drug of PTX, we next examined the conversion of SPR064 to PTX in plasma using HPLC. Incubation SPR064 in rat plasma for 3 hours showed complete conversion of SPR064 to PTX at different time points (Figure 1B). We also compared the pharmacokinetics profile of SPR064 and PTX in SD rats. Since SPR064 has better aqueous solubility as compared to PTX, SPR064 was formulated in a saline-based formulation without using Cremophor. The pharmacokinetic parameters of SPR064 and PTX after intravenous administration are shown in Figure 1C. SPR064 was found to have much better exposure and a more favorable PK profile compared to PTX when dosed in a saline-based formulation.
Comparison of SPR064 with PTX in the inhibition of cell proliferation in EC cells
The effect of SPR064 on EC cell proliferation was assessed by MTT assay. Five EC cell lines, Hec-1A, ECC-1, Ishikawa, RL-95-2 and KLE, were exposed to varying doses of SPR064 and PTX for 72 hours. Treatment of five EC cell lines with various concentrations of SPR064 and PTX caused a dose-dependent inhibitory effects on cell viability, with no significant difference in potency between SPR064 and PTX (Figure 2A). The mean IC50 value for SPR064 and PTX in each of the five cell lines is shown in Figure 2B. Next, we performed colony formation assays to investigate the long-term effect of SPR064 on proliferation in the Hec-1A and ECC-1 cells. As shown in Figure 2C, the colony-forming ability of both cell lines was reduced after exposure to SPR064 for 14 days. These results indicate that SPR064 and PTX showed similar efficacy in the inhibition of EC cell proliferation.
Figure 2.
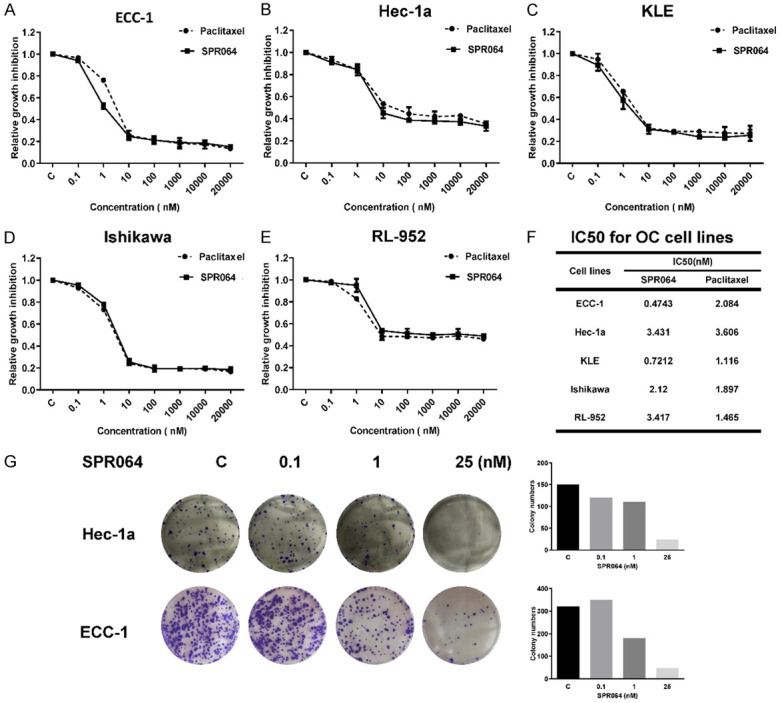
SPR064 inhibited the proliferation of EC cells. The ECC-1, Hec-1a, KLE, Ishikawa and RL-952 cells were cultured for 24 hours and then treated with varying concentrations of SPR064 and PTX in 96 well plates for 72 hours. Cell proliferation was assessed by MTT assay. SPR064 has similar anti-proliferative effects in the five cell lines compared with PTX (A-E). The IC50 value for SPR064 and PTX was calculated for each cell line (F). The ECC-1 and HEC-1a cells were treated with SPR064 for 24 hours and then the cells were cultured for 14 days with medium changes every third or fourth day. Colonies were visualized by crystal violet staining and counted under the microscope (G).
SPR064 induced cell cycle G1 arrest in EC cells
To determine whether SPR064 regulates cell cycle, we used flow cytometry to compare the effect of SPR064 and PTX on the cell cycle in the Hec-1A and ECC-1 cells. Treatment with 25 µM SPR064 for 48 hours significantly increased the number of Hec-1A and ECC-1 cells in the G1 phase from 33.93% to 61.9% and 29.93% to 60.07%, respectively. Cells treated with PTX at the same dose also arrested the cell cycle at G1 phase, causing an increase in cells in the G1 phase from 35.74% to 72.41% in the Hec-1A cells and 42.25% to 80.07% in the ECC-1 cells, respectively (Figure 3A). To further determine the cell cycle inhibitory effects of SPR064, we examined the effect of SPR064 on cell cycle checkpoint proteins in both cell lines. Western blotting results showed that SPR064 significantly reduced the expression of cyclin D1 and CDK4 after 14 hours of treatment (Figure 3B). These results indicate that SPR064 has similar effects on the cell cycle in EC cells when compared to PTX treatment.
Figure 3.

SPR064 induced cell cycle G1 arrest in EC cells. The ECC-1 and Hec-1a cell lines were treated with SPR064 and PTX for 48 hours. Cell cycle progression was analyzed by flow cytometry. SPR064 and PTX induced G0/G1 cell cycle arrest and reduced S phase in a dose-dependent manner in both cell lines (A and B). The effect of SPR064 on cell cycle-related proteins (cyclin D1 and CDK4) was assessed by Western blotting after 14 hours of treatment. The results showed that SPR064 reduced cyclinD1 and CDK4 expression in both cell lines (C).
Comparison of SPR064 with PTX in induction of apoptosis in EC cells
Since PTX is known to induce apoptosis in cancer cells [12], we further examined the effect of SPR064 on apoptosis in EC cells. Treatment of cell lines with SPR064 or PTX at different concentrations for 16 hours resulted in a significant induction in apoptosis relative to the control as determined by Annexin V assay. In a similar fashion, SPR064 and PTX at a dose of 25 nM increased the percentage of apoptotic cells by 10-17% (Figure 4A). Furthermore, cleaved caspase 3 activity increased in both cell lines treated with SPR064 or PTX for 16 hours in a dose-dependent manner (Figure 4B). Western blotting results further confirmed that SPR064 decreased the expression of anti-apoptotic genes, BCL-1 and MCL-1 in both cells, after 14 hours of treatment (Figure 4C).
Figure 4.

SPR064 induced apoptosis in EC cells. The ECC-1 and Hec-1a cell lines were treated with varying concentrations of SPR064 and PTX for 16 hours. Apoptosis was detected using the Annexin-V FITC assay and cleaved caspase 3 ELISA assay in both cell lines. SPR064 and PTX increased Annexin V expression and cleaved caspase 3 activity in a dose dependent manner in the ECC-1 and Hec-1a cells (A and B). Western blotting demonstrated that treatment with SPR064 for 24 hours decreased the expression of MCL-1 and BCL-XL in both cell lines (C).
SPR064 increased cellular stress in EC cells
As cellular stress can lead to activation of cell apoptosis, we then investigated the effect of SPR064 and PTX on ROS production in EC cells. PTX and SPR064 both induced a statistically significant dose-dependent increase in ROS production after 4 hours of treatment. When the cells were treated with SPR064 or PTX at the same doses, an equivalent increase in ROS production was observed in the Hec-1A and ECC-1 cells (Figure 5A). Furthermore, we examined the expression of cellular stress proteins following treatment with SPR064 using Western immunoblotting. As expected from our ROS findings, SPR064 increased the expression of the PERK, BIP and calnexin proteins in a dose-dependent fashion after 8 hours of treatment (Figure 5B). These results suggested that, similar to PTX, the inhibition of cell proliferation by SPR064 also involves induction of cellular stress in EC cells.
Figure 5.
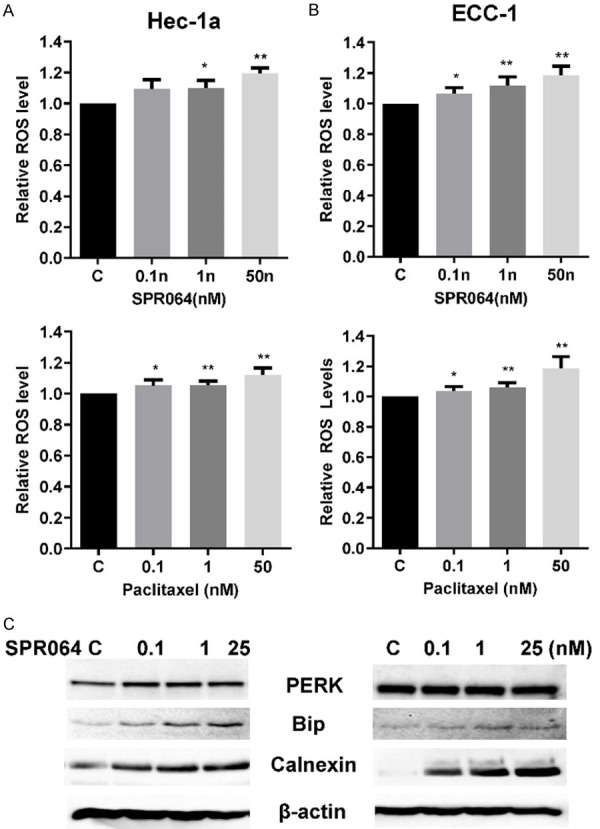
SPR064 induced cellular stress in EC cells. The ECC-1 and Hec-1a cells were treated with SPR064 and PTX at the indicated doses for 4 hours, and ROS production was determined using the DCFH-DA assay. SPR064 and paclitaxel increased the ROS levels in a dose-dependent manner in both cell lines (A and B). The expression of the cellular stress proteins (PERK, Bip, and Calnexin) was detected by Western blotting after treatment with SPR064 for 16 hours. SPR064 increased the expression of PERK, Bip and Calnexin in ECC-1 and Hec-1a cells (C).
SPR064 inhibits tumor growth in the Lkb1fl/flp53fl/fl mouse model of endometrial cancer
The anti-tumorigenic effect of the SPR064 and PTX was analyzed in a genetically engineered mouse model of endometrioid EC (Lkb1fl/flp53fl/fl) [11]. Since obesity is linked with EC pathogenesis and potentially response to PTX treatment [13], Lkb1fl/flp53fl/fl mice were fed a LFD or HFD at 3 weeks of age and were divided into six treatment groups. These groups included LFD (non-obese) and HFD (obese) groups treated with either placebo, PTX (6.25 mg/kg) or SPR064 (8.06 mg/kg). The mice were treated with either SPR064 or PTX approximately 8 weeks after AdCre was injected into the right uterine horn. During treatment, tumor growth was monitored by palpation twice a week. Regular twice-weekly measurements yielded no changes in body weight or normal activity during SPR064 and PTX treatment. Obesity accelerated tumor growth with a 2.33-fold increase in tumor weight at sacrifice compared to mice fed a LFD. A substantial reduction in tumor weight was found in the SPR064 and PTX groups in comparison with the placebo groups (Figure 6A and 6B). In the obese mice, tumor weight decreased by 77.63% with SPR064 and 81.25% with PTX treatment when compared with control mice. Among the non-obese mice, tumor weight decreased by 65.93% in SPR064 group and 63.16% in PTX group when compared with control-treated animals. These results suggest that obesity promotes endometrial tumor growth, and SPR064 and PTX exhibited the similar efficacy in inhibition of tumor growth in both obese and lean mice (P<0.05).
Figure 6.
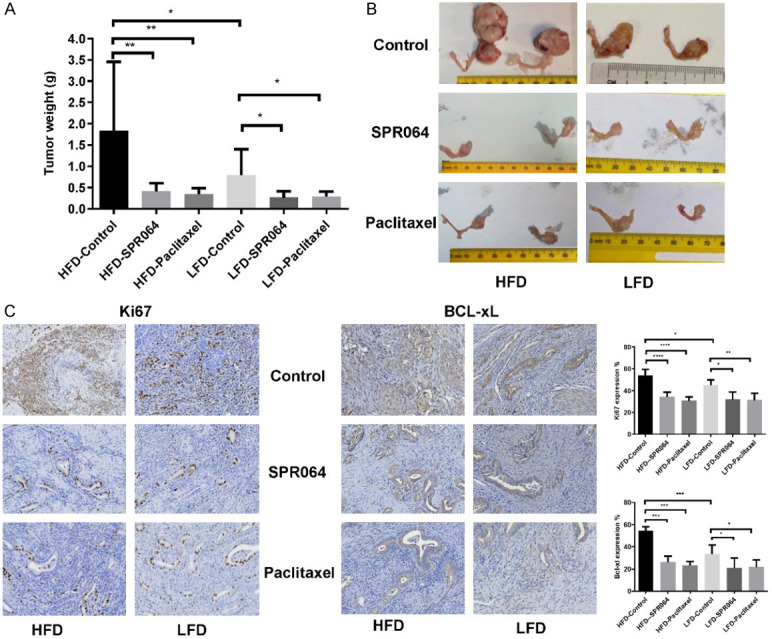
The effect of SPR064 and PTX on tumor growth in the Lkb1fl/flp53fl/fl endometrioid EC mouse model. The mice were fed with either a HFD or LFD starting at 3 weeks of age. The HFD- (obese) and LFD- (non-obese) fed mice were injected AdCre after 6-8 weeks of birth and treated with PTX (6.25 mg/kg, IP), SPR064 (8.06 mg/kg, IP) or placebo every 5 days for 4 weeks after 8 weeks of injections. Obesity significantly increased tumor weights in HFD control group as compared to LFD group. SPR064 and PTX significantly inhibited tumor growth in both obese and lean mice (A and B). The impact of SPR064 and PTX treatment on Ki-67 and Bcl-xl were assessed by immunohistochemistry in the endometrial cancer tissues. Treatment with SPR064 and PTX significantly decreased the expression of Ki-67 and Bcl-xl in obese and lean mice as compared to control groups (C).
To further compare the anti-tumorigenic mechanism of SPR064 in vivo, the expression of Ki-67 and BCL-XL in the endometrial tumor tissues was evaluated by immunohistochemistry. As we expected, the expression of the proliferation marker Ki-67 was significantly reduced in the endometrial tumors following SPR064 and PTX treatment, quantified as a 35.97% and 42.97% decrease in tumors of obese mice and 28.90% and 30.54% in tumors of lean mice compared to controls respectively. Similarly, we observed that SPR064 and PTX significantly reduced BCL-XL expression in endometrial tumors in the mouse model of EC under obese and lean conditions (Figure 6C).
Discussion
In this study, we investigated the efficacy of SPR064 in inhibiting EC tumor growth in vitro and in vivo compared to PTX. SPR064 is a prodrug of PTX with much improved solubility over PTX. Improved solubility provides the advantage of dosing without Cremaphor; and hence, preventing the risk of associated side effects of Cremophor, in particular hypersensitivity reactions. Our results indicate that SPR064 is a potent anti-tumorigenic agent that exhibited the same potency at similar doses in two EC cell lines and a transgenic mouse model of endometrioid EC when compared to PTX. Similar to PTX-induced inhibition of tumor cell growth, SPR064 has been shown to act on multiple targets and pathways in EC cells, including proliferation, apoptosis, cell cycle arrest and cellular stress.
The mechanism of induction of hypersensitivity reactions in response to PTX treatment has not yet been fully elucidated. Given that molecules present in Cremophor EL are similar in structure to certain non-ionic block co-polymers that activate complement proteins, unsolicited complement activation due to Cremophor EL and an IgE-mediated reaction are thought to be the major contributory mechanisms in hypersensitivity reactions from PTX [8,14]. Developing new drug delivery formulations without Cremophor EL, including formation of albumin, nanoparticle, emulsion, prodrug, liposome, porous particle, polymeric micelle, and ionic liquid mediated microspheres of PTX, have recently been investigated [7,15-17]. SPR064 is a modified drug of PTX that has much improved solubility in saline over the parent compound without Cremophor EL and albumin, eliminating the issues associated with Cremophor EL and albumin. SPR064 shows immediate conversion to parent drug in blood; hence, there is no systemic exposure of the modified drug, suggesting it may minimize hypersensitivity reactions during PTX administration. In this study, the concentrations of SPR064 required to induce growth inhibition are similar to PTX in the treatment of EC cells in vitro. Similarly, SPR064 treatment (8.06 mg/kg) led to profound inhibition of endometrial tumor growth compared to an equal dose of PTX (6.25 mg/kg) in obese and non-obese mice. During treatment, SPR064 did not seem to cause adverse effects on normal activities in the mice and did not affect body weight and blood sugar (Data not shown), indicating that SPR064 has a defined safety prolife in obese and non-obese mice.
The underlying mechanism responsible for PTX-induced growth inhibition has not been entirely defined [18]. PTX has been found to induce cytotoxicity in cancer cells through manipulation of stabilizing microtubules, interfering with microtubule disassembly and causing mitotic arrest in cell division, resulting in induction of apoptosis and arrest cell cycle in G0/G1 and G2/M phases [18-20]. The apoptosis induced by PTX is associated with the release of the apoptogenic factors cytochrome c and caspase activation, which is in turn controlled by the pro-apoptotic and anti-apoptotic Bcl-2 family proteins [21-24]. Although the effect of PTX is characterized by the accumulation of cancer cells in the G2/M phase, there is evidence that PTX also results in G1 phase arrest dependent on concentrations of PTX and the status of p53, p21 and p27 [25-28]. Down-regulation of CDK4 caused by PTX has been closely associated with cell cycle G1 arrest in gastric cancer cells [29]. In this study, inhibition of cell proliferation in the Hec-1A and ECC-1 cells at the death-inducing concentration of SPR064 resulted in a similar apoptosis pattern from that of PTX. Both SPR064 and PTX caused the same cell cycle distribution and G1 phase arrest, along with down-regulation of CDK4 and cyclin D1 after treatment of SPR064 in both cells. These results suggested that the ability of SPR064 to inhibit cell growth through cell cycle arrest and apoptosis in vitro is the same as that of PTX in EC cells.
Substantial evidence indicates that the cell cycle G2/M and G1 phases arrest and apoptosis are not the only mechanisms for PTX-induced cytotoxicity in cancer cells. Cellular stress is proposed as another pathway involved in PTX-induced cell death [30,31]. PTX induces oxidative stress, and the activity of PTX is linked to ROS levels [32-34]. Furthermore, the treatment of breast cancer, lung cancer and leukemia K562 cells with the anti-oxidant agent N-acetylcysteine led to impaired susceptibility to PTX [30,35,36], suggesting that the increase in ROS production triggered by PTX has early pro-apoptotic effects [21]. Given this backdrop, we treated EC cells with SPR064 and PTX to compare the effect of both drugs on ROS production at the same doses. ROS levels were gradually increased in a dose-dependent manner in the SPR064 and PTX-treated cells. The difference in the ROS levels was not statistically significant between both drugs when comparing equal drug concentrations. In addition, SPR064 increased the expression of cellular stress proteins in the Hec-1A and ECC-1 cells. These results support that SPR064 induces cellular stress in a similar pattern to that which was observed in PTX-treated cells.
Taken together, we conclude that the SPR064, a modified and highly soluble form of PTX, exhibits similar efficacy in the inhibition of cell proliferation, induction of cell cycle arrest and apoptosis and activation of cellular stress in EC cell lines and a transgenic mouse model of EC when compared with commercially available PTX. Based on the preliminary data generated by these studies, we believe that SPR064 holds promise as a novel, safe and well-tolerated agent in the treatment of EC.
Acknowledgements
This study was supported by V foundation (VJB).
Disclosure of conflict of interest
Sundeep Dugar is an employee of Sphaera Pharma Singapore Pte Ltd and Somdutta Sen is an employee of Sphaera Pharma Pvt Ltd. The rest of the authors declare no conflict of interest except that the SPR064 drug was provided to us by Sphaera for these studies.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Rizzo S, Femia M, Buscarino V, Franchi D, Garbi A, Zanagnolo V, Del Grande M, Manganaro L, Alessi S, Giannitto C, Ruju F, Bellomi M. Endometrial cancer: an overview of novelties in treatment and related imaging keypoints for local staging. Cancer Imaging. 2018;18:45. doi: 10.1186/s40644-018-0180-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McDonald ME, Bender DP. Endometrial cancer: obesity, genetics, and targeted agents. Obstet Gynecol Clin North Am. 2019;46:89–105. doi: 10.1016/j.ogc.2018.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Tran AQ, Gehrig P. Recent advances in endometrial cancer. F1000Res. 2017;6:81. doi: 10.12688/f1000research.10020.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kogan L, Laskov I, Amajoud Z, Abitbol J, Yasmeen A, Octeau D, Fatnassi A, Kessous R, Eisenberg N, Lau S, Gotlieb WH, Salvador S. Dose dense carboplatin paclitaxel improves progression free survival in patients with endometrial cancer. Gynecol Oncol. 2017;147:30–35. doi: 10.1016/j.ygyno.2017.07.134. [DOI] [PubMed] [Google Scholar]
- 6.Chowdhury MR, Moshikur RM, Wakabayashi R, Tahara Y, Kamiya N, Moniruzzaman M, Goto M. Ionic-liquid-based paclitaxel preparation: a new potential formulation for cancer treatment. Mol Pharm. 2018;15:2484–2488. doi: 10.1021/acs.molpharmaceut.8b00305. [DOI] [PubMed] [Google Scholar]
- 7.Chowdhury MR, Moshikur RM, Wakabayashi R, Tahara Y, Kamiya N, Moniruzzaman M, Goto M. In vivo biocompatibility, pharmacokinetics, antitumor efficacy, and hypersensitivity evaluation of ionic liquid-mediated paclitaxel formulations. Int J Pharm. 2019;565:219–226. doi: 10.1016/j.ijpharm.2019.05.020. [DOI] [PubMed] [Google Scholar]
- 8.Picard M. Management of hypersensitivity reactions to taxanes. Immunol Allergy Clin North Am. 2017;37:679–693. doi: 10.1016/j.iac.2017.07.004. [DOI] [PubMed] [Google Scholar]
- 9.Sendo T, Sakai N, Itoh Y, Ikesue H, Kobayashi H, Hirakawa T, Nakano H, Oishi R. Incidence and risk factors for paclitaxel hypersensitivity during ovarian cancer chemotherapy. Cancer Chemother Pharmacol. 2005;56:91–96. doi: 10.1007/s00280-004-0924-9. [DOI] [PubMed] [Google Scholar]
- 10.Barkat MA, Beg S, Pottoo FH, Ahmad FJ. Nanopaclitaxel therapy: an evidence based review on the battle for next-generation formulation challenges. Nanomedicine (Lond) 2019;14:1323–1341. doi: 10.2217/nnm-2018-0313. [DOI] [PubMed] [Google Scholar]
- 11.Guo H, Kong W, Zhang L, Han J, Clark LH, Yin Y, Fang Z, Sun W, Wang J, Gilliam TP, Lee D, Makowski L, Zhou C, Bae-Jump VL. Reversal of obesity-driven aggressiveness of endometrial cancer by metformin. Am J Cancer Res. 2019;9:2170–2193. [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar A, Hoskins PJ, Tinker AV. Dose-dense paclitaxel in advanced ovarian cancer. Clin Oncol (R Coll Radiol) 2015;27:40–47. doi: 10.1016/j.clon.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Shaw E, Farris M, McNeil J, Friedenreich C. Obesity and endometrial cancer. Recent Results Cancer Res. 2016;208:107–136. doi: 10.1007/978-3-319-42542-9_7. [DOI] [PubMed] [Google Scholar]
- 14.Szebeni J, Muggia FM, Alving CR. Complement activation by Cremophor EL as a possible contributor to hypersensitivity to paclitaxel: an in vitro study. J Natl Cancer Inst. 1998;90:300–306. doi: 10.1093/jnci/90.4.300. [DOI] [PubMed] [Google Scholar]
- 15.Roviello G, Conter FU, Mini E, Generali D, Traversini M, Lavacchi D, Nobili S, Sobhani N. Nanoparticle albumin-bound paclitaxel: a big nano for the treatment of gastric cancer. Cancer Chemother Pharmacol. 2019;84:669–677. doi: 10.1007/s00280-019-03887-2. [DOI] [PubMed] [Google Scholar]
- 16.Du X, Khan AR, Fu M, Ji J, Yu A, Zhai G. Current development in the formulations of non-injection administration of paclitaxel. Int J Pharm. 2018;542:242–252. doi: 10.1016/j.ijpharm.2018.03.030. [DOI] [PubMed] [Google Scholar]
- 17.Picard M, Castells MC. Re-visiting hypersensitivity reactions to taxanes: a comprehensive review. Clin Rev Allergy Immunol. 2015;49:177–191. doi: 10.1007/s12016-014-8416-0. [DOI] [PubMed] [Google Scholar]
- 18.Weaver BA. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell. 2014;25:2677–2681. doi: 10.1091/mbc.E14-04-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barbuti AM, Chen ZS. Paclitaxel through the ages of anticancer therapy: exploring its role in chemoresistance and radiation therapy. Cancers (Basel) 2015;7:2360–2371. doi: 10.3390/cancers7040897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bharadwaj R, Yu H. The spindle checkpoint, aneuploidy, and cancer. Oncogene. 2004;23:2016–2027. doi: 10.1038/sj.onc.1207374. [DOI] [PubMed] [Google Scholar]
- 21.Andre N, Carre M, Brasseur G, Pourroy B, Kovacic H, Briand C, Braguer D. Paclitaxel targets mitochondria upstream of caspase activation in intact human neuroblastoma cells. FEBS Lett. 2002;532:256–260. doi: 10.1016/s0014-5793(02)03691-8. [DOI] [PubMed] [Google Scholar]
- 22.Noh KT, Cha GS, Kang TH, Cho J, Jung ID, Kim KY, Ahn SC, You JC, Park YM. Enhancement of paclitaxel-induced breast cancer cell death via the glycogen synthase kinase-3beta-mediated B-cell lymphoma 2 regulation. BMB Rep. 2016;49:51–56. doi: 10.5483/BMBRep.2016.49.1.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bah N, Maillet L, Ryan J, Dubreil S, Gautier F, Letai A, Juin P, Barille-Nion S. Bcl-xL controls a switch between cell death modes during mitotic arrest. Cell Death Dis. 2014;5:e1291. doi: 10.1038/cddis.2014.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Srivastava RK, Mi QS, Hardwick JM, Longo DL. Deletion of the loop region of Bcl-2 completely blocks paclitaxel-induced apoptosis. Proc Natl Acad Sci U S A. 1999;96:3775–3780. doi: 10.1073/pnas.96.7.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giannakakou P, Robey R, Fojo T, Blagosklonny MV. Low concentrations of paclitaxel induce cell type-dependent p53, p21 and G1/G2 arrest instead of mitotic arrest: molecular determinants of paclitaxel-induced cytotoxicity. Oncogene. 2001;20:3806–3813. doi: 10.1038/sj.onc.1204487. [DOI] [PubMed] [Google Scholar]
- 26.Das GC, Holiday D, Gallardo R, Haas C. Taxol-induced cell cycle arrest and apoptosis: dose-response relationship in lung cancer cells of different wild-type p53 status and under isogenic condition. Cancer Lett. 2001;165:147–153. doi: 10.1016/s0304-3835(01)00404-9. [DOI] [PubMed] [Google Scholar]
- 27.Sakashita F, Osada S, Takemura M, Imai H, Tomita H, Nonaka K, Takahashi T, Seishima M. The effect of p53 gene expression on the inhibition of cell proliferation by paclitaxel. Cancer Chemother Pharmacol. 2008;62:379–385. doi: 10.1007/s00280-007-0614-5. [DOI] [PubMed] [Google Scholar]
- 28.Chang YF, Li LL, Wu CW, Liu TY, Lui WY, P’Eng FK, Chi CW. Paclitaxel-induced apoptosis in human gastric carcinoma cell lines. Cancer. 1996;77:14–18. doi: 10.1002/(SICI)1097-0142(19960101)77:1<14::AID-CNCR4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 29.Yoo YD, Park JK, Choi JY, Lee KH, Kang YK, Kim CS, Shin SW, Kim YH, Kim JS. CDK4 down-regulation induced by paclitaxel is associated with G1 arrest in gastric cancer cells. Clin Cancer Res. 1998;4:3063–3068. [PubMed] [Google Scholar]
- 30.Meshkini A, Yazdanparast R. Involvement of oxidative stress in taxol-induced apoptosis in chronic myelogenous leukemia K562 cells. Exp Toxicol Pathol. 2012;64:357–365. doi: 10.1016/j.etp.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 31.Moos PJ, Fitzpatrick FA. Taxane-mediated gene induction is independent of microtubule stabilization: induction of transcription regulators and enzymes that modulate inflammation and apoptosis. Proc Natl Acad Sci U S A. 1998;95:3896–3901. doi: 10.1073/pnas.95.7.3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Panis C, Herrera AC, Victorino VJ, Campos FC, Freitas LF, De Rossi T, Colado Simao AN, Cecchini AL, Cecchini R. Oxidative stress and hematological profiles of advanced breast cancer patients subjected to paclitaxel or doxorubicin chemotherapy. Breast Cancer Res Treat. 2012;133:89–97. doi: 10.1007/s10549-011-1693-x. [DOI] [PubMed] [Google Scholar]
- 33.Tanimukai H, Kanayama D, Omi T, Takeda M, Kudo T. Paclitaxel induces neurotoxicity through endoplasmic reticulum stress. Biochem Biophys Res Commun. 2013;437:151–155. doi: 10.1016/j.bbrc.2013.06.057. [DOI] [PubMed] [Google Scholar]
- 34.Yang JC, Lu MC, Lee CL, Chen GY, Lin YY, Chang FR, Wu YC. Selective targeting of breast cancer cells through ROS-mediated mechanisms potentiates the lethality of paclitaxel by a novel diterpene, gelomulide K. Free Radic Biol Med. 2011;51:641–657. doi: 10.1016/j.freeradbiomed.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 35.Fukui M, Yamabe N, Zhu BT. Resveratrol attenuates the anticancer efficacy of paclitaxel in human breast cancer cells in vitro and in vivo. Eur J Cancer. 2010;46:1882–1891. doi: 10.1016/j.ejca.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lyle PA, Mitsopoulos P, Suntres ZE. N-acetylcysteine modulates the cytotoxic effects of Paclitaxel. Chemotherapy. 2011;57:298–304. doi: 10.1159/000329510. [DOI] [PubMed] [Google Scholar]