

Abstract
Lead (Pb), a highly poisonous heavy metal and an important occupational hazard, is currently a widespread environmental pollutant. The kidney is especially susceptible to the toxic effects of Pb because of its major role in Pb excretion. Heme oxygenase-1 (HO-1) is an inducible antioxidant enzyme that can mitigate cellular injury. However, its role in Pb-elicited nephrotoxicity remains uncertain. This study was designed to examine the role of HO-1 in lead acetate (PbAc)-induced renal tubular cell injury in vitro. PbAc injury was found to suppress HO-1 expression and impair cell viability, with concomitant depletion of the autophagy proteins LC3-II and Beclin 1. Overexpression of HO-1 dramatically restored autophagy and protected cells against PbAc-induced apoptosis. In addition, pretreatment with 3-methyladenine, an inhibitor of autophagy, aggravated apoptosis and abolished renoprotection by HO-1, suggesting that the anti-apoptotic effect of HO-1 in Pb-induced nephrotoxicity is dependent on enhanced autophagy. Furthermore, HO-1 overexpression abrogated the inhibitory effect of PbAc on the adenosine monophosphate-activated protein kinase (AMPK)/mammalian target of rapamycin (mTORC1) signaling pathway. Pretreatment with an AMPK agonist, 5-aminoimidazole-4-carboxamide-1-β-D ribofuranoside, markedly enhanced autophagic activity and diminished apoptosis. Conversely, inhibition of AMPK phosphorylation abolished the pro-autophagic and anti-apoptotic effects of HO-1 in PbAc-injured cells. Our findings suggest that HO-1 alleviates Pb-induced nephrotoxicity via enhanced autophagy, which involves activation of the AMPK/mTORC1 signaling pathway.
Keywords: Heme oxygenase-1, lead acetate, nephrotoxicity, autophagy, AMPK, mTORC1
Introduction
Lead (Pb) is one of the most important environmental pollutants, and ingestion of Pb-contaminated food or water or the inhalation of polluted air in areas with heavy traffic or industrial emissions may cause malfunction of a number of organ systems [1,2]. Although the incidence of occupational lead exposure has been greatly reduced, other sources of lead exposure that have been identified include cosmetics, paints, solders, hair dyes, shielding for X-ray machines, etc. [3]. With a biological half-life of about 10 years, Pb is excreted extremely slowly, and tends to accumulate in the body. Because Pb circulating in the blood accumulates in the kidney via glomerular filtration and tubular reabsorption, the kidney is a major target organ for lead toxicity. Pb-induced kidney toxicity results in reduced glomerular filtration rate (GFR), tubular atrophy, interstitial fibrosis, and chronic renal failure [4,5]. In patients with chronic kidney disease (CKD), exposure to even low doses of Pb may aggravate kidney dysfunction and accelerate progressive renal insufficiency [5,6]. Pb may exert potent toxic effects by inducing oxidative stress [7], activating inflammatory signaling cascades [8], and promoting the mitochondrion-mediated apoptosis pathway [9]. Although previous studies have revealed the mechanisms underlying Pb-induced tissue injury, there is still no effective treatment.
Heme oxygenase-1 (HO-1) (EC 1.14.99.3), an inducible enzyme involved in the degradation of heme, also has potent antioxidant, anti-inflammatory, anti-fibrotic, and anti-apoptotic properties in diverse human and animal diseases [10-14]. Previous studies confirmed that upregulation of HO-1 attenuated nephropathy, whereas HO-1 knockout aggravated renal toxicity induced by methotrexate or Cyclosporine-A [15-17]. In mouse models of diabetic nephropathy, the promotion of HO-1 signaling eliminated reactive oxygen species and reduced apoptosis, thereby alleviating renal pathology [18]. HO-1 also mediates renoprotection in acute kidney injury (AKI) via regulation of several different pathways [19]. In rats that were given a single injection of lead acetate (PbAc), HO-1 expression increased rapidly. Lipid peroxide levels increased in the kidney cortex following the injection, and were enhanced by an HO-1 inhibitor [20]. In contrast, HO-1 concentration was reduced in rats chronically exposed to oral PbAc for three months, and the decrease was associated with oxidative stress, inflammation, and apoptosis, as well as histopathological changes in the kidney. Co-administration of agents, which upregulated HO-1, alleviated Pb-induced nephrotoxicity [21]. While these findings suggest that HO-1 plays a renoprotective role after Pb exposure, biphasic changes in HO-1 expression occur following acute and chronic Pb exposure.
Autophagy is a highly conserved cellular process to recycle cellular components, and to eliminate damaged organelles, proteins, and lipids, in order to maintain homeostasis and cellular integrity. A large number of studies have demonstrated the renoprotective effects of autophagy in AKI and CKD [22]. However, a recent study showed that inhibition of autophagic flux may contribute to Pb-induced injury in primary rat proximal tubular cells [28]. Activating the HO-1 signaling pathway has been found to increase the autophagy marker proteins light chain 3 II/I (LC3-II/I) and Beclin 1, and to inhibit apoptosis in H2O2-injured renal tubular epithelial cells [23]. Pretreatment of podocytes with HO-1 agonist induced autophagy and protected the cells from injury due to high glucose in vitro, whereas HO-1 knockout repressed activation of autophagy and aggravated high glucose-elicited podocyte injury [24,25]. In contrast, Xu et al. suggested that HO-1 protected glomerular mesangial cells against H2O2-induced apoptosis by reducing excessive autophagy [26]. Similarly, upregulation of HO-1 attenuated autophagy-related necrosis induced in lung epithelial cells by airborne fine particulate matter 2.5 [27]. Thus, the relationship between HO-1 and autophagy may differ depending on the disease model, and the role of HO-1-modulated autophagy in Pb-induced nephrotoxicity is poorly understood.
The highly conserved energy-sensing kinase, AMPK, regulates a wide variety of metabolic processes, including the autophagic pathway [29,48]. AMPK antagonizes mTORC1, thus promoting recruitment of autophagy-related proteins to form the autophagosome. Phosphorylation of AMPK increased after treatment with HO-1 agonist in podocytes exposed to high glucose. Moreover, inhibition of AMPK reversed the upregulation of autophagy by HO-1 and aggravated cellular injury [24], suggesting that renoprotection by HO-1 likely involves enhancement of autophagy via activation of the AMPK signaling pathway.
In this study, HO-1 expression and autophagic activity were examined in HK-2 human renal tubular cells after PbAc injury. In addition, we evaluated whether HO-1 attenuated PbAc-induced cell injury via modulation of autophagy and examined whether the AMPK pathway was involved in HO-1-regulated autophagy.
Materials and methods
Reagents and antibodies
Dulbecco’s modified Eagle’s medium (DMEM/F12) (1:1), fetal bovine serum (FBS), and antibiotic-antimycotic solution were purchased from Gibco. PbAc, 3-methyladenine (3-MA), 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR), and Compound C were purchased from Sigma-Aldrich. Anti-HO-1 and anti-GAPDH were purchased from Santa Cruz, anti-LC3 and anti-Beclin 1 were from Sigma-Aldrich, and anti-cleaved caspase-3, anti-phospho-AMPK, anti-AMPK, anti-phospho-mTOR and anti-mTOR were from Cell Signaling Technology.
Cell culture
Immortalized human renal proximal tubular cells (HK-2) were purchased from American Type Culture Collection. Cells were grown in DMEM/F-12 medium supplemented with 10% FBS containing 1:100 dilution of antibiotic-antimycotic solution at 37°C in a 5%-CO2 incubator, and treated with PbAc after 24 hours (h). To determine the optimal concentration and duration of PbAc treatment, cells were exposed to varying concentrations (0, 5, 50, or 200 μmol/L) for varying periods of time (0, 24, 48, 72 h). In subsequent experiments, cells were incubated with 50 μmol/L PbAc for 48 h. Autophagy inhibitor 3-MA, AMPK agonist AICAR, or AMPK inhibitor Compound C were added 1 h before PbAc treatment.
HO-1 plasmid transfection
Plasmid pcDNA3-smURFP-IRES-HO-1, encoding human HO-1 (hHO-1), was a gift from Erik Rodriguez & Roger Tsien [30]. Cells (2 × 105) were transfected using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s protocol, cultured for 24 h, and processed for immunoblot assay to evaluate transfection efficiency.
Cell viability assay
Cell viability was detected using a Cell Counting Kit-8 (Dojindo Molecular Technologies) according to the manufacturer’s instructions. Absorbance at 450 nm was measured using a microplate reader. Mean optical densities of treated cells were calculated after subtracting the absorbance of blank (medium) from the total absorbance value.
Immunoblot analysis
Protein concentrations of whole cell lysates were determined using Pierce bicinchoninic acid reagent (Thermo Fisher). Equal amounts of protein samples were electrophoresed in sodium dodecyl sulfate-polyacrylamide gradient (5-15%) gels and transferred to nitrocellulose membranes (Millipore). After blocking for 1 h with 5% bovine serum albumin at room temperature, membranes were incubated overnight with anti-HO-1, anti-LC3, anti-Beclin 1, anti-cleaved caspase-3, anti-phospho-AMPK, anti-AMPK, anti-phospho-mTOR, anti-mTOR, or anti-GAPDH antibodies at 4°C with gentle shaking. Subsequently, the membranes were incubated with peroxidase-linked secondary antibody for 1 h at room temperature. Immunoreactive bands were detected using chemiluminescent substrate (Thermo Fisher). Bands were scanned using a densitometer, and the integrated pixel densities were determined using ImageJ analysis software. The data are expressed as mean ± standard deviation.
Evaluation of autophagy by electron microscopy
Cells were fixed overnight in 2% glutaraldehyde at 4°C, and post-fixed with 2% OsO4 for 2 h. The blocks, which had been embedded in araldite, were cut into ultrathin sections (50 nm) using a microtome and were stained with uranyl acetate and lead citrate. Cell ultrastructure was observed using a JEM-1010 electron microscope (JEOL, Tokyo, Japan). Autophagosome numbers were counted in ten randomly selected high-power fields per section.
Statistical analysis
Statistical analysis was performed using the GraphPad Prism software (version 6.0). Comparison of data from multiple groups was performed using one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc tests. Cell viability data were analyzed by two-way ANOVA followed by Bonferroni’s test. Data from two groups were analyzed by Student’s t test. P < 0.05 was considered statistically significant.
Results
PbAc treatment impaired HK-2 cell viability and suppressed HO-1 expression in a time- and dose-dependent manner
Kidney, as a key organ of excretion of toxicants, appears to be a primary site for the accumulation of Pb [31], whose toxic effects in the kidney are mainly manifested as renal tubular damage. The viability of HK-2 cells exposed to PbAc was impaired significantly by treatment with 50 or 200 μmol/L PbAc, compared with untreated cells at the same time points (P < 0.05, Figure 1A). Viability at 200 μmol/L PbAc was more diminished than that at 50 μmol/L PbAc at the same time point (P < 0.05). In addition, cell viability decreased progressively over time at 50 μmol/L PbAc (P < 0.01).
Figure 1.
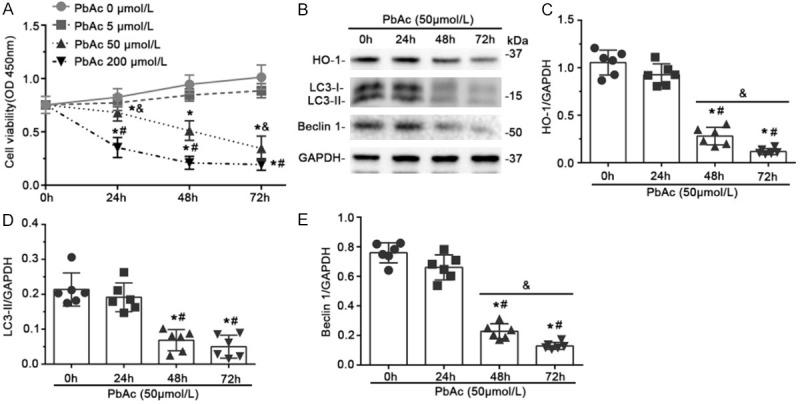
Expression of HO-1, viability, and autophagy activity were reduced in PbAc-injured HK-2 cells. (A) Cells were treated with the indicated concentrations of PbAc, and cell viability was measured after 0, 24, 48 or 72 h. Viability decreased versus time and in a concentration-dependent manner. *P < 0.05 versus no PbAc at the same time point; #P < 0.05 versus 50 μmol/L PbAc at the same time point; &P < 0.01 versus 50 μmol/L PbAc for 48 h (n = 6); (B) Cells were treated with 50 μmol/L PbAc, and expression of HO-1, LC3-II, and Beclin 1 were evaluated by immunoblotting. GAPDH served as a loading control. Abundance of HO-1 (C), LC3-II (D), and Beclin 1 (E) were determined by densitometric analysis of immunoblots, and normalized to GAPDH levels. The expression of HO-1 and LC3-II, as well as Beclin 1, decreased persistently following 48 or 72 h of PbAc treatment. *P < 0.01 versus 0 h; #P < 0.01 versus 24 h treatment; &P < 0.05 versus 48 h treatment (n = 6).
HO-1 plays an important role in regulating oxidative stress, inflammation, and apoptosis, and is protective in a variety of human and animal models of kidney diseases. However, the role of HO-1 in Pb-induced kidney injury is still unclear. Levels of HO-1 in HK-2 cells after PbAc injury were determined by immunoblot assay (Figure 1B, 1C). HO-1 expression was reduced from initial levels after PbAc treatment for 24 h, although the difference was not statistically significant. This effect persisted following 48 h and 72 h treatment (Figure 1B, 1C). Collectively, these data suggest that PbAc exposure impaired cell viability and reduced HO-1 levels in a dose- and time-dependent manner. Treatment with 50 μmol/L PbAc for 48 h was used in subsequent experiments.
Overexpression of HO-1 attenuated PbAc-induced apoptosis by enhancing autophagy
Autophagy is an essential cellular process involving degradation and recycling of cytoplasmic components, misfolded proteins, and damaged organelles to maintain intracellular homeostasis [32-34]. Although many studies suggest that autophagy is involved in the pathogenesis of kidney disease, the role of autophagy in Pb-induced renal tubular cell apoptosis remains to be elucidated. We found that LC3-II and Beclin 1, two markers of autophagy, were expressed basally in HK-2 cells and that levels of both proteins decreased significantly after PbAc treatment for 48 or 72 h (Figure 1B, 1D, 1E). To determine the relationship between HO-1 and autophagy in PbAc-induced renal tubular cell injury, cells were transfected with a plasmid encoding human HO-1 or with empty vector (EV), followed by PbAc treatment for 48 h. HO-1 transfection improved autophagic activity after PbAc injury, as indicated by increased LC3-II and Beclin 1 expression detected by immunoblot assay (Figure 2A, 2C, 2D). This was accompanied by marked improvement in apoptosis (Figure 2A, 2E).
Figure 2.

Overexpression of HO-1 enhanced autophagy and diminished apoptosis after PbAc injury. Cells were transfected with plasmid encoding human HO-1 (hHO-1) or empty vector (EV) 24 h before treatment with 50 μmol/L PbAc or vehicle for 48 h. (A) Expression of HO-1, LC3-II, Beclin 1, and cleaved caspase-3 were evaluated by immunoblot analysis, with GAPDH as a loading control. Levels of HO-1 (B), LC3-II (C), Beclin 1 (D), and cleaved caspase-3 (E) were estimated by densitometric analysis. HO-1 transfection abrogated the reduction of LC3-II and Beclin 1, and increased caspase-3 cleavage induced by PbAc treatment. *P < 0.05 versus EV and vehicle treatment; #P < 0.05 versus EV and PbAc treatment (n = 6). (F) Transmission electron microscopic demonstration of autophagosomes (black arrowheads); scale bar = 2 μm. (G) In cells overexpressing HO-1, autophagosome counts per high-power field increased significantly following PbAc injury. *P < 0.05 versus EV and vehicle; #P < 0.01 versus EV and PbAc treatment (n = 6).
To validate the immunoblotting results, transmission electron microscopy was employed to measure autophagic vacuole formation. In cells transfected with EV, PbAc treatment diminished the number of autophagosomes relative to vehicle (P < 0.05, Figure 2F, 2G). Autophagic vacuole formation increased significantly in cells overexpressing HO-1 following PbAc injury (P < 0.01). These results suggest that overexpression of HO-1 improved cellular apoptosis, enhanced autophagic activity, and played a protective role in PbAc-induced renal tubular cell injury.
Inhibition of autophagy sensitized cells to PbAc-induced apoptosis and abolished the protective effect of HO-1
To further delineate the relationship between autophagy and PbAc-induced apoptosis, cells were pretreated with the autophagy inhibitor 3-MA, and apoptosis as well as autophagy were analyzed. As expected, 3-MA application further reduced LC3-II and Beclin 1 expression after PbAc treatment, accompanied by enhanced elevation of caspase-3 cleavage (Figure 3A-D). To clarify whether autophagy contributed to the anti-apoptotic effect of HO-1 in PbAc-injured renal tubular cells, human HO-1 plasmid was transfected prior to treatment with 3-MA and PbAc. As indicated in Figure 3E, 3F, despite HO-1 overexpression, 3-MA, in combination with PbAc, resulted in aggravated caspase-3 cleavage, suggesting that the inhibition of autophagy abolished the anti-apoptotic effect of HO-1 in PbAc-induced renal tubular cell injury.
Figure 3.

Inhibition of autophagy by 3-MA pretreatment abolished anti-apoptotic effects of HO-1 in PbAc-injured renal tubular cells. (A) 3-MA was added 1 h before treatment with 50 μmol/L PbAc for 48 h. LC3-II, Beclin 1, and cleaved caspase-3 were evaluated by immunoblot analysis, with GAPDH as a loading control. Levels of LC3-II (B), Beclin 1 (C), and cleaved caspase-3 (D) were estimated by densitometric analysis. 3-MA pretreatment decreased LC3-II and Beclin 1 levels, and promoted PbAc-induced caspase-3 cleavage. *P < 0.05 versus vehicle; #P < 0.05 versus PbAc treatment (n = 6). (E) Cells were transfected with plasmid encoding human HO-1 (hHO-1) or empty vector (EV) 24 h before addition of 3-MA or vehicle, and then treated after 1 h with PbAc for 48 h. Levels of cleaved caspase-3 were analyzed by immunoblot assay, with GAPDH as loading control. (F) Densitometric analysis of cleaved caspase-3 is shown. 3-MA pretreatment abrogated the anti-apoptotic effect of HO-1 in PbAc-injured renal tubule cells. *P < 0.01 versus EV transfection in combination with vehicle and PbAc treatments; #P < 0.01 versus hHO-1 transfection in combination with 3-MA and PbAc treatments (n = 6).
HO-1 enhanced autophagy by activating the AMPK/mTORC1 signaling pathway in PbAc-induced renal tubular cell injury
Although many signaling pathways may influence autophagy, the two pivotal regulators of autophagic degradation are the closely connected signaling transducers mTORC1 and AMPK. AMPK can negatively regulate mTORC1 activity, leading to increased autophagy. AMPK phosphorylation and phosphorylation of mTOR Ser 2448 mediate the mTORC1 signaling pathway. To determine whether AMPK/mTORC1 was involved in HO-1-promoted autophagy in PbAc-induced renal tubular cell injury, immunoblot assays were used to detect the phosphorylated forms of AMPK and mTOR (Figure 4A-C). Changes in phosphorylation of AMPK and mTOR indicated that the AMPK/mTORC1 signaling pathway was inhibited after PbAc treatment in EV-expressing cells, but was activated with HO-1 overexpression. In PbAc-treated cells, AMPK phosphorylation was activated by AICAR, an established pharmacological agonist of AMPK, and mTOR phosphorylation was decreased (Figure 5A-C). Moreover, treatment with PbAc in the presence of AICAR led to increases in LC3-II and Beclin 1 expression, concomitant with reduced caspase-3 cleavage (Figure 5A, 5D-F). Conversely, pretreatment with Compound C, an AMPK inhibitor, blocked phosphorylation of AMPK and increased phosphorylation of mTOR (Figure 5G-I). In addition, the capacity of HO-1 overexpression to induce LC3-II and Beclin 1 expression was abrogated by Compound C, as was the effect of HO-1 on caspase-3 cleavage (Figure 5G, 5J-L). These results indicated that AMPK/mTORC1 signaling was involved in the modulation of autophagy by HO-1 in Pb-induced nephrotoxicity.
Figure 4.

Overexpression of HO-1 upregulated AMPK phosphorylation and inactivated the mTORC1 signaling pathway after PbAc treatment. Cells were transfected with plasmid encoding human HO-1 (hHO-1) or empty vector (EV) 24 h before treatment with 50 μmol/L PbAc or vehicle for 48 h. (A) Phosphorylated AMPK and Ser 2448-phosphorylated mTOR were examined by immunoblot analysis. GAPDH served as loading control. Levels of phosphorylated AMPK (B) and Ser 2448-phosphorylated mTOR (C) were estimated by densitometry. *P < 0.01 versus EV transfection and vehicle treatment; #P < 0.01 versus EV transfection with PbAc treatment (n = 6).
Figure 5.

HO-1 augmented levels of autophagic factors and ameliorated apoptosis by activating AMPK/mTORC1 signaling in PbAc-induced nephrotoxicity. A. Cells were pretreated with or without AICAR for 1 h, followed by incubation for 48 h with 50 μmol/L PbAc. Expression of p-AMPK, p-mTOR (Ser 2448), LC3-II, Beclin 1, and cleaved caspase-3 were examined by immunoblot analysis, with GAPDH as loading control. B-F. Densitometric analyses of immunoblots are shown. *P < 0.01 versus vehicle; #P < 0.05 versus PbAc treatment (n = 6). G. Cells were transfected with plasmid encoding human HO-1 (hHO-1) 24 h before addition of vehicle or Compound C, followed 1 h later by treatment with 50 μmol/L PbAc for 48 h. Levels of p-AMPK, p-mTOR (Ser 2448), LC3-II, Beclin 1, and cleaved caspase-3 were evaluated by immunoblotting, with GAPDH as loading control. (H-L) Densitometric analyses of immunoblots. Compound C inhibited AMPK phosphorylation and increased Ser 2448 phosphorylation of mTOR, thus abrogating the effects of HO-1 transfection in improving LC3-II and Beclin 1 expression and reducing caspase-3 cleavage. *P < 0.01 versus hHO-1 transfection combined with vehicle and PbAc treatment (n = 6).
Discussion
Kidney dysfunction due to long-term exposure to environmental Pb represents a serious healthcare concern, but there is still no effective therapeutic strategy for treating Pb-induced kidney injury. Chronic environmental Pb exposure may interfere with urate excretion [35]. Higher serum uric acid levels may be associated with renal injury in Pb-exposed subjects even in the earlier stages of exposure [36]. For each unit increase in blood Pb level, age-adjusted eGFR was reduced by 59.2 mL/min/1.73 m2 in male workers at smelters [37]. Exposure to Pb has been shown to lead to glomerular hypertrophy, renal leukocyte infiltration, interstitial fibrosis, and tubular atrophy [38]. Suggested mechanisms of Pb-induced nephrotoxicity include oxidative stress [4], inflammation [8], and apoptosis [9]. In this study, the role of HO-1 in PbAc-induced renal tubular cell injury was explored.
A large number of studies using experimental models of diseases have demonstrated that HO-1 can function via antioxidant, anti-inflammatory, anti-fibrotic, and anti-apoptotic mechanisms [10-14]. HO-1 can be induced in the kidney as a result of injurious factors such as endotoxins, H2O2, certain growth factors, cytokines, and heavy metals [20,39]. Upregulation of HO-1 expression prevents renal oxidative stress, inflammation, apoptosis, and fibrosis in animal models of nephropathy [15,16,18,40]. However, the role of HO-1 in Pb-induced renal injury is poorly understood. Rats exposed to a single administration of Pb showed increased HO-1 mRNA and protein expression. Lipid peroxide levels also increased in the kidney cortex following Pb treatment, and were further aggravated by HO-1 inhibition, suggesting that HO-1 induction may be an antioxidant response meant to reduce lipid peroxidation, thereby protecting against Pb-induced renal injury [20]. However, another in vivo study has demonstrated that chronic Pb exposure resulted in reduced HO-1 expression, concomitant with impaired renal function and structure. Co-administration of agents which increase HO-1 levels afforded renal protection, evidenced by decreased renal oxidative stress, inflammatory cytokine secretion, and apoptosis, as well as improvements in kidney histopathology [21]. Thus, all data in PbAc-intoxicated rats have consistently indicated a renoprotective role of HO-1, although changes in HO-1 expression differed depending on the mode of administration. In agreement, in the present study, PbAc injury impaired cell viability in a time- and dose-dependent manner. Also, expression of HO-1 declined progressively over time, and this loss may underlie cellular injury. Furthermore, HO-1 overexpression was found to impede apoptosis, suggesting a renoprotective effect, in accordance with the aforementioned in vivo studies.
In autophagy, which is a conserved cellular process to maintain homeostasis in eukaryotic organisms, damaged organelles, proteins, and lipids are engulfed by autophagosomes, and then degraded by lysosomes [41]. Autophagy-deficient aged mice exhibited significant deterioration of kidney function, with fibrosis and concomitant mitochondrial dysfunction, as well as mitochondrial DNA abnormalities and nuclear DNA damage [22]. Inhibition of autophagosome sequestration by inhibitors such as 3-MA resulted in more severe kidney dysfunction in a mouse model of ischemia-reperfusion injury [42]. Lin et al. found impaired activation of autophagy in CKD patients after fasting [43]. Activated autophagy protected renal tubular cells against toxic agents such as cisplatin, cyclosporine, and heavy metals [44-46]. A basal level of autophagy exists in glomerular epithelial cells and proximal tubular cells [44]. Likewise, in this study, constitutive expression of the autophagy protein markers LC3-II and Beclin 1 were observed in renal tubular cells under resting conditions. Marked decreases in LC3-II formation and Beclin 1 expression that were observed 48 and 72 h after PbAc injury suggest impaired autophagy. This finding contrasts with results of a study by Song et al., in which a low-dose treatment with Pb (0.5 μmol/L) for 12 h resulted in the elevation of LC3-II levels in rat proximal tubular cells [28]. They proposed that the increase was due to a blockade of autophagic flux, with the lysosomal degradation of autophagosomes inhibited. In our study, the higher dose of PbAc (50 μmol/L) and longer incubation periods (24 h, 48 h, 72 h) may have exhausted the autophagic processes.
As is well known, HO-1 and autophagy are both inducible under stress. Thus, there might be a causal relationship between HO-1 and autophagy. Zhan et al. provided evidence that HO-1 deficiency repressed the autophagic pathway, and aggravated high glucose-evoked podocyte apoptosis and inflammation [25]. This is consistent with previous reports that HO-1 inactivation inhibited autophagy, whereas HO-1 agonist induced autophagy and protected podocytes from hyperglycemia [24]. Likewise, HO-1 overexpression protected cardiomyocytes from hypoxia/reoxygenation injury by enhancing autophagy and ameliorating apoptosis [47]. In contrast, Xu et al. suggested that HO-1 alleviated H2O2-induced mesangial cell injury by reducing autophagy [26]. This contrasts with our findings that HO-1 overexpression increased LC3-II and Beclin 1 levels and autophagosome formation, and attenuated apoptosis after PbAc injury, while 3-MA abolished these beneficial effects. Our findings indicate that autophagy might be required for renoprotection by HO-1 in Pb-induced nephrotoxicity. The discrepancy between our findings and those of Xu et al. may reflect different roles of autophagy in different types of injuries.
AMPK regulates autophagy in yeast and mammalian cells [49-51]. AMPK activates autophagy by negatively regulating mTORC1 activity via two complementary actions [29]. mTORC1 can inhibit autophagy by directly or indirectly reducing activity of the ULK1 complex, an essential factor in the recruitment of autophagy-related proteins during autophagosome biogenesis. Whether and how AMPK/mTORC1 signaling pathway involved in the beneficial effects of HO-1 in lead-injuced nephrotoxicity were unclear. In the present study, HO-1 overexpression increased AMPK phosphorylation and reduced Ser 2448 phosphorylation of mTOR, leading to autophagic activation in PbAc-injured cells. In addition, specific activation of AMPK with AICAR amplified the expression of LC3-II and Beclin 1, and alleviated apoptosis. Conversely, inhibition of AMPK with Compound C abolished HO-1-mediated upregulation of autophagy, as well as its anti-apoptotic effect. These results imply that the AMPK/mTORC1 pathway is required for HO-1-promoted autophagy in Pb-injured renal tubular cells.
In summary, PbAc injury of renal tubular cells represses HO-1 and impairs autophagy, resulting in apoptosis. HO-1 overexpression enhances autophagy by activating the AMPK/mTORC1 signaling pathway and attenuates apoptosis during PbAc-induced nephrotoxicity. Although our study demonstrates a renoprotective effect of HO-1 in vitro, further studies are needed to validate the role of HO-1 in Pb-elicited kidney diseases in animal models. In addition, the link between serum or urinary HO-1 levels and the severity of Pb nephrotoxicity in patients is worthy of further exploration.
Acknowledgements
This research was funded in part by “Scientific Research Project” of Shanghai Municipal Commission of Health and Family Program 201740231.
Disclosure of conflict of interest
None.
References
- 1.de Souza ID, de Andrade AS, Dalmolin RJS. Lead-interacting proteins and their implication in lead poisoning. Crit Rev Toxicol. 2018;48:375–386. doi: 10.1080/10408444.2018.1429387. [DOI] [PubMed] [Google Scholar]
- 2.Mitra P, Sharma S, Purohit P, Sharma P. Clinical and molecular aspects of lead toxicity: an update. Crit Rev Clin Lab Sci. 2017;54:506–528. doi: 10.1080/10408363.2017.1408562. [DOI] [PubMed] [Google Scholar]
- 3.Fortoul TI, Moncada-Hernandez S, Saldivar-Osorio L, Espejel-Maya G, Mussali-Galante P, del Carmen Avila-Casado M, Colin-Barenque L, Hernandez-Serrato MI, Avila-Costa MR. Sex differences in bronchiolar epithelium response after the inhalation of lead acetate (Pb) Toxicology. 2005;207:323–330. doi: 10.1016/j.tox.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 4.BaSalamah MA, Abdelghany AH, El-Boshy M, Ahmad J, Idris S, Refaat B. Vitamin D alleviates lead induced renal and testicular injuries by immunomodulatory and antioxidant mechanisms in rats. Sci Rep. 2018;8:4853. doi: 10.1038/s41598-018-23258-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harari F, Sallsten G, Christensson A, Petkovic M, Hedblad B, Forsgard N, Melander O, Nilsson PM, Borne Y, Engstrom G, Barregard L. Blood lead levels and decreased kidney function in a population-based cohort. Am J Kidney Dis. 2018;72:381–389. doi: 10.1053/j.ajkd.2018.02.358. [DOI] [PubMed] [Google Scholar]
- 6.Lin JL, Lin-Tan DT, Li YJ, Chen KH, Huang YL. Low-level environmental exposure to lead and progressive chronic kidney diseases. Am J Med. 2006;119:707, e701–709. doi: 10.1016/j.amjmed.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Gargouri M, Hamed H, Akrouti A, Christian M, Ksouri R, El Feki A. Immunomodulatory and antioxidant protective effect of Sarcocornia perennis L. (swampfire) in lead intoxicated rat. Toxicol Mech Methods. 2017;27:697–706. doi: 10.1080/15376516.2017.1351018. [DOI] [PubMed] [Google Scholar]
- 8.Boskabady M, Marefati N, Farkhondeh T, Shakeri F, Farshbaf A, Boskabady MH. The effect of environmental lead exposure on human health and the contribution of inflammatory mechanisms, a review. Environ Int. 2018;120:404–420. doi: 10.1016/j.envint.2018.08.013. [DOI] [PubMed] [Google Scholar]
- 9.Jin X, Xu Z, Zhao X, Chen M, Xu S. The antagonistic effect of selenium on lead-induced apoptosis via mitochondrial dynamics pathway in the chicken kidney. Chemosphere. 2017;180:259–266. doi: 10.1016/j.chemosphere.2017.03.130. [DOI] [PubMed] [Google Scholar]
- 10.Haines DD, Lekli I, Teissier P, Bak I, Tosaki A. Role of haeme oxygenase-1 in resolution of oxidative stress-related pathologies: focus on cardiovascular, lung, neurological and kidney disorders. Acta Physiol (Oxf) 2012;204:487–501. doi: 10.1111/j.1748-1716.2011.02387.x. [DOI] [PubMed] [Google Scholar]
- 11.Han M, Hu L, Chen Y. Rutaecarpine may improve neuronal injury, inhibits apoptosis, inflammation and oxidative stress by regulating the expression of ERK1/2 and Nrf2/HO-1 pathway in rats with cerebral ischemia-reperfusion injury. Drug Des Devel Ther. 2019;13:2923–2931. doi: 10.2147/DDDT.S216156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.HAS AL, Alotaibi MF, Bin-Jumah M, Elgebaly H, Mahmoud AM. Olea europaea leaf extract up-regulates Nrf2/ARE/HO-1 signaling and attenuates cyclophosphamide-induced oxidative stress, inflammation and apoptosis in rat kidney. Biomed Pharmacother. 2019;111:676–685. doi: 10.1016/j.biopha.2018.12.112. [DOI] [PubMed] [Google Scholar]
- 13.Wu CT, Deng JS, Huang WC, Shieh PC, Chung MI, Huang GJ. Salvianolic acid C against acetaminophen-induced acute liver injury by attenuating inflammation, oxidative stress, and apoptosis through inhibition of the Keap1/Nrf2/HO-1 signaling. Oxid Med Cell Longev. 2019;2019:9056845. doi: 10.1155/2019/9056845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zarjou A, Agarwal A. Heme oxygenase-1 as a target for TGF-beta in kidney disease. Semin Nephrol. 2012;32:277–286. doi: 10.1016/j.semnephrol.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aladaileh SH, Hussein OE, Abukhalil MH, Saghir SAM, Bin-Jumah M, Alfwuaires MA, Germoush MO, Almaiman AA, Mahmoud AM. Formononetin upregulates Nrf2/HO-1 signaling and prevents oxidative stress, inflammation, and kidney injury in methotrexate-induced rats. Antioxidants (Basel) 2019;8:430. doi: 10.3390/antiox8100430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu C, Zhu P, Fujino M, Isaka Y, Ito H, Takahashi K, Nakajima M, Tanaka T, Zhuang J, Li XK. 5-aminolaevulinic acid (ALA), enhances heme oxygenase (HO)-1 expression and attenuates tubulointerstitial fibrosis and renal apoptosis in chronic cyclosporine nephropathy. Biochem Biophys Res Commun. 2019;508:583–589. doi: 10.1016/j.bbrc.2018.11.175. [DOI] [PubMed] [Google Scholar]
- 17.Loboda A, Mucha O, Podkalicka P, Sobczak M, Miksza-Cybulska A, Kaczara P, Jozkowicz A, Dulak J. Kidney injury by cyclosporine A is aggravated in heme oxygenase-1 deficient mice and involves regulation of microRNAs. Acta Biochim Pol. 2018;65:613–620. doi: 10.18388/abp.2018_2658. [DOI] [PubMed] [Google Scholar]
- 18.Zhang B, Zhang X, Zhang C, Shen Q, Sun G, Sun X. Notoginsenoside R1 protects db/db mice against diabetic nephropathy via upregulation of Nrf2-mediated HO-1 expression. Molecules. 2019;24:247. doi: 10.3390/molecules24020247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bolisetty S, Zarjou A, Agarwal A. Heme oxygenase 1 as a therapeutic target in acute kidney injury. Am J Kidney Dis. 2017;69:531–545. doi: 10.1053/j.ajkd.2016.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vargas H, Castillo C, Posadas F, Escalante B. Acute lead exposure induces renal haeme oxygenase-1 and decreases urinary Na excretion. Hum Exp Toxicol. 2003;22:237–44. doi: 10.1191/0960327103ht360oa. [DOI] [PubMed] [Google Scholar]
- 21.Abdel-Zaher AO, Abd-Ellatief RB, Aboulhagag NA, Farghaly HSM, Al-Wasei FMM. The interrelationship between gasotransmitters and lead-induced renal toxicity in rats. Toxicol Lett. 2019;310:39–50. doi: 10.1016/j.toxlet.2019.04.012. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto T, Takabatake Y, Kimura T, Takahashi A, Namba T, Matsuda J, Minami S, Kaimori JY, Matsui I, Kitamura H, Matsusaka T, Niimura F, Yanagita M, Isaka Y, Rakugi H. Time-dependent dysregulation of autophagy: implications in aging and mitochondrial homeostasis in the kidney proximal tubule. Autophagy. 2016;12:801–813. doi: 10.1080/15548627.2016.1159376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L, Lin M, Zhang L, Huang S, Hu C, Zheng L, Li L, Zhang C, Yang C, Long Y, Rong R, Zhu T. Cyclic helix B peptide protects HK2 cells from oxidative stress by inhibiting ER stress and activating Nrf2 signalling and autophagy. Mol Med Rep. 2017;16:8055–8061. doi: 10.3892/mmr.2017.7588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong C, Zheng H, Huang S, You N, Xu J, Ye X, Zhu Q, Feng Y, You Q, Miao H, Ding D, Lu Y. Heme oxygenase-1 enhances autophagy in podocytes as a protective mechanism against high glucose-induced apoptosis. Exp Cell Res. 2015;337:146–159. doi: 10.1016/j.yexcr.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 25.Zhan X, Yan C, Chen Y, Wei X, Xiao J, Deng L, Yang Y, Qiu P, Chen Q. Celastrol antagonizes high glucose-evoked podocyte injury, inflammation and insulin resistance by restoring the HO-1-mediated autophagy pathway. Mol Immunol. 2018;104:61–68. doi: 10.1016/j.molimm.2018.10.021. [DOI] [PubMed] [Google Scholar]
- 26.Xu J, Li J, Wang J, Chi Y, Zhang K, Cui R. Heme oxygenase1 protects H2O2insulted glomerular mesangial cells from excessive autophagy. Mol Med Rep. 2016;13:5269–5275. doi: 10.3892/mmr.2016.5177. [DOI] [PubMed] [Google Scholar]
- 27.Zhou W, Yuan X, Zhang L, Su B, Tian D, Li Y, Zhao J, Wang Y, Peng S. Overexpression of HO-1 assisted PM2.5-induced apoptosis failure and autophagy-related cell necrosis. Ecotoxicol Environ Saf. 2017;145:605–614. doi: 10.1016/j.ecoenv.2017.07.047. [DOI] [PubMed] [Google Scholar]
- 28.Song XB, Liu G, Liu F, Yan ZG, Wang ZY, Liu ZP, Wang L. Autophagy blockade and lysosomal membrane permeabilization contribute to lead-induced nephrotoxicity in primary rat proximal tubular cells. Cell Death Dis. 2017;8:e2863. doi: 10.1038/cddis.2017.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tamargo-Gomez I, Marino G. AMPK: regulation of metabolic dynamics in the context of autophagy. Int J Mol Sci. 2018;19:3812. doi: 10.3390/ijms19123812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez EA, Tran GN, Gross LA, Crisp JL, Shu X, Lin JY, Tsien RY. A far-red fluorescent protein evolved from a cyanobacterial phycobiliprotein. Nat Methods. 2016;13:763–769. doi: 10.1038/nmeth.3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fowler BA, DuVal G. Effects of lead on the kidney: roles of high-affinity lead-binding proteins. Environ Health Perspect. 1991;91:77–80. doi: 10.1289/ehp.919177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 34.Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol. 2010;12:831–835. doi: 10.1038/ncb0910-831. [DOI] [PubMed] [Google Scholar]
- 35.Lin JL, Yu CC, Lin-Tan DT, Ho HH. Lead chelation therapy and urate excretion in patients with chronic renal diseases and gout. Kidney Int. 2001;60:266–271. doi: 10.1046/j.1523-1755.2001.00795.x. [DOI] [PubMed] [Google Scholar]
- 36.Baki AE, Ekiz T, Ozturk GT, Tutkun E, Yilmaz H, Yildizgoren MT. The effects of lead exposure on serum uric acid and hyperuricemia in young adult workers: a cross-sectional controlled study. Arch Rheumatol. 2016;31:71–75. doi: 10.5606/ArchRheumatol.2016.5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reilly R, Spalding S, Walsh B, Wainer J, Pickens S, Royster M, Villanacci J, Little BB. Chronic environmental and occupational lead exposure and kidney function among african americans: dallas lead project II. Int J Environ Res Public Health. 2018;15:2875. doi: 10.3390/ijerph15122875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Orr SE, Bridges CC. Chronic kidney disease and exposure to nephrotoxic metals. Int J Mol Sci. 2017;18:1039. doi: 10.3390/ijms18051039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.So KY, Oh SH. Cadmium-induced heme-oxygenase-1 expression plays dual roles in autophagy and apoptosis and is regulated by both PKC-delta and PKB/Akt activation in NRK52E kidney cells. Toxicology. 2016;370:49–59. doi: 10.1016/j.tox.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 40.Wahdan SA, Azab SS, Elsherbiny DA, El-Demerdash E. Piceatannol protects against cisplatin nephrotoxicity via activation of Nrf2/HO-1 pathway and hindering NF-kappaB inflammatory cascade. Naunyn Schmiedebergs Arch Pharmacol. 2019;392:1331–1345. doi: 10.1007/s00210-019-01673-8. [DOI] [PubMed] [Google Scholar]
- 41.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 42.Guan X, Qian Y, Shen Y, Zhang L, Du Y, Dai H, Qian J, Yan Y. Autophagy protects renal tubular cells against ischemia/reperfusion injury in a time-dependent manner. Cell Physiol Biochem. 2015;36:285–298. doi: 10.1159/000374071. [DOI] [PubMed] [Google Scholar]
- 43.Chen WT, Hung KC, Wen MS, Hsu PY, Chen TH, Wang HD, Fang JT, Shie SS, Wang CY. Impaired leukocytes autophagy in chronic kidney disease patients. Cardiorenal Med. 2013;3:254–264. doi: 10.1159/000356212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kume S, Uzu T, Maegawa H, Koya D. Autophagy: a novel therapeutic target for kidney diseases. Clin Exp Nephrol. 2012;16:827–832. doi: 10.1007/s10157-012-0695-2. [DOI] [PubMed] [Google Scholar]
- 45.Kimura A, Ishida Y, Nosaka M, Kuninaka Y, Hama M, Kawaguchi T, Sakamoto S, Shinozaki K, Iwahashi Y, Takayasu T, Kondo T. Exaggerated arsenic nephrotoxicity in female mice through estrogen-dependent impairments in the autophagic flux. Toxicology. 2016;339:9–18. doi: 10.1016/j.tox.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 46.Kaushal GP, Shah SV. Autophagy in acute kidney injury. Kidney Int. 2016;89:779–791. doi: 10.1016/j.kint.2015.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meng X, Yuan Y, Shen F, Li C. Heme oxygenase-1 ameliorates hypoxia/reoxygenation via suppressing apoptosis and enhancing autophagy and cell proliferation though Sirt3 signaling pathway in H9c2 cells. Naunyn Schmiedebergs Arch Pharmacol. 2019;392:189–198. doi: 10.1007/s00210-018-1575-4. [DOI] [PubMed] [Google Scholar]
- 48.Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19:121–135. doi: 10.1038/nrm.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, Meijer AJ. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem. 2006;281:34870–34879. doi: 10.1074/jbc.M605488200. [DOI] [PubMed] [Google Scholar]
- 50.Wang Z, Wilson WA, Fujino MA, Roach PJ. Antagonistic controls of autophagy and glycogen accumulation by Snf1p, the yeast homolog of AMP-activated protein kinase, and the cyclin-dependent kinase Pho85p. Mol Cell Biol. 2001;21:5742–5752. doi: 10.1128/MCB.21.17.5742-5752.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Russell RC, Yuan HX, Guan KL. Autophagy regulation by nutrient signaling. Cell Res. 2014;24:42–57. doi: 10.1038/cr.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]