

Abstract
This study tested the hypothesis that early implantation of mitochondria (Mito) into left myocardium could effectively protect heart against doxorubicin/12 mg/kg-induced dilated cardiomyopathy (DCM) in rat. Adult-male SD rats (n = 18) were equally categorized into group 1 (sham control), group 2 (DCM) and group 3 [DCM + Mito (500 μg/rat intramyocardial injection by day-21 after DCM induction)] and euthanized by day 60. In vitro studies showed that exogenously-transferred Mito was abundantly identified in H9C2 cells. The q-PCR showed significant increase in relative number of mitDNA in Mito-transferred H9C2 cells than in control group (P<0.001). The mRNA-gene and protein expressions of NRF1/NRF2/Tfam/PGC-1α/ERRα/Mfn2 were significantly increased in low-dose Mito-transferred and more significantly increased in high-dose Mito-transferred H29C2 cells than in control group (all P<0.01). Day-60 left-ventricular-ejection-fraction (LVEF) was significantly lower in group 2 than in groups 1 and 3, and significantly lower in group 3 than in group 1 (P<0.0001). The ratios of lung and heart weights to tibial length and myocardial histopathological findings of fibrotic area/myocardial injured score/γ-H2AX+ cells exhibited an opposite pattern to LVEF among the three groups (all P<0.0001). The myocardial protein expressions of oxidative-stress (NOX-1/NOX-2/oxidized protein/p22phox), autophagic (beclin-1/Atg-5/ratio of CL3B-II/CL3B-I), and apoptotic/mitochondrial-damaged (cleaved-caspase-3/mitochondrial Bax/cleaved-PARP/cytosolic-cytochrome-C/DRP1/cyclophilin D1) biomarkers exhibited an opposite pattern, whereas the protein expressions of mitochondrial integrity (mitochondrial-cytochrome-C/mitochondrial-complex I/II/III/IV and Mfn2/PGC-1) exhibited an identical pattern to LVEF among the groups (all P<0.001). In conclusion, early Mito therapy effectively preserved LVEF and myocardial integrity in DCM rat.
Keywords: Mitochondria, dilated cardiomyopathy, doxorubicin, molecular-cellular perturbations
Introduction
Dilated cardiomyopathy (DCM) represents a heterogeneous group of diseases and is a common cause of heart failure (HF) worldwide [1-4]. DCM is characterized by dilation and impaired contraction of one or both ventricles causing impaired systolic function [1-4]. Clinical studies have revealed that mortality from DCM is usually from sudden or progressive pump failure, which represents the end stage of DCM [1,2,5]. Due to its prevalence [6,7] and high rates of hospitalization, morbidity, and mortality [2,8], DCM is a major economic burden and global health concern [1,9,10]. Despite continuous improvements in medications, therapeutic modalities and treatment guidelines for DCM in the past decade [1,9-13], clinical outcomes following onset of HF have not substantially changed and mortality remains unacceptably high [1,9,10,13]. Accordingly, to find a new, safe and efficacious therapeutic option is important for patients [14,15].
DCM is a primary myocardial disease of unknown etiology characterized mainly by cardiac chamber dilatation, impairing systolic and diastolic function [1,2]. The underlying mechanisms of worsening systolic function followed by LV chamber dilatation/remodeling and development of overt HF have been investigated and found to be resultant from increased inflammation, oxidative stress, generation of mitochondrial reactive oxygen species (ROS) [16-19], mitochondrial dysfunction and apoptosis with exhaustion of energy [20-22], as a consequence extracellular fibrosis [14,21], cardiomyocyte death and scar formation [14,23]. These findings [16,23] raise the hypothesis that early exogenous mitochondrial transfer to mitochondrially dysfunctional cardiomyocytes may have therapeutic potential by inhibiting cardiomyocyte apoptosis and death and thereby preserving LV function.
Our previous studies showed that mitochondrial transfusion significantly protected liver [24] and heart [15] from ischemia-reperfusion injury, effectively protected the lung from acute respiratory distress syndrome (ARDS) [25,26] and pulmonary arterial hypertension [27] induced injury mainly through inhibiting the generation of oxidative stress/ROS, inflammation and apoptosis and refreshment of mitochondria in ischemic organs. Accordingly, we tested the hypothesis that early intramyocardial implantation of exogenous mitochondria (Mito) might effectively preserve left ventricular ejection fraction (LVEF) and myocardium integrity in doxorubicin-induced DCM in rat.
Materials and methods
Ethics statement
All animal experimental procedures were approved by the Institutional Animal Care and Use Committee at Kaohsiung Chang Gung Memorial Hospital (Affidavit of Approval of Animal Use Protocol No. 2016111501) and performed in accordance with the Guide for the Care and Use of Laboratory Animals, 8th edition (NIH publication No. 85-23, National Academy Press, Washington, DC, USA, revised 2011). Animals were housed in an Association for Assessment and Accreditation of Laboratory Animal Care International-approved animal facility in our hospital, with controlled temperature and light cycles (24°C and 12/12 light/dark cycle).
Procedure and protocol of DCM induction by doxorubicin and animal grouping
Pathogen-free, adult male Sprague-Dawley (SD) rats (n = 18) weighing 320-350 g (Charles River Technology, BioLASCO Taiwan Co. Ltd., Taiwan) were used in the present study. Dilated cardiomyopathy (DCM) induction was based on our previous report [1] with minimal modification. Briefly, doxorubicin (Dox) was administered intraperitoneally with an accumulated dosage of 12.5 mg/kg given to the animals at 4 separated time points within 20 days (i.e., once every 5 days).
Adult-male SD rats (n = 18) were equally categorized into group 1 (sham control), group 2 (DCM) and group 3 [DCM + Mito (500 μg/rat intramyocardial injection at four sites on the anterior wall of LV myocardium by day-21 after DCM induction)].
The animals in each group were euthanized by day 60 after their final transthoracic echocardiogram. The dosage of mitochondria was based on our previous report [25-27].
Mitochondrial isolation and MitoTracker staining for mitochondria
The procedure and protocol for isolating liver mitochondria from rats have previously been described [25]. Additional six rats were euthanized and the gallbladder and liver removed. Immediately, the liver (3 g) was immersed in 50 mL of ice-cold IBc (10 mM Tris-MOPS, 5 mM EGTA/Tris and 200 mM sucrose, pH 7.4.) in a small beaker, followed by rinsing the liver free of blood with ice-cold IBc. The liver was then minced into small pieces using scissors in a beaker surrounded by ice. IBc was discarded during mincing and replaced with 18 mL of ice-cold fresh IBc. The liver was then be homogenized using a Teflon pestle. The homogenate was transferred to a 50 mL polypropylene Falcon tube and centrifuged at 600 g for 10 minutes at 4°C. The supernatants were transferred to centrifuge tubes for centrifugation at 7,000 g for 10 minutes at 4°C. The supernatants were discarded and the pellets were washed with 5 mL ice-cold IBc. Again, the supernatants from pellets were centrifuged at 7,000 g for 10 minutes at 4°C. The supernatants were discarded and the pellets containing the mitochondria were resuspended again. The concentrations of mitochondrial suspensions were measured using the Biuret method. Each 10 mg of isolated mitochondria were labeled with 1 μM of MitoTracker Red CMXRos (Invitrogen, Carlsbad, CA) through incubation at 37°C for 30 minutes. Mitochondrial administration was performed for the study animals immediately after labeling (i.e., <3 hrs after the isolation procedure).
Procedure and protocol for quantification of oxygen consumption rate (OCR) of isolated mitochondria (Seahorse Method)
Functional activity of isolated Mito from rat liver were determined by an Extracellular Flux Analyzer (XFe24, Seahorse Bioscience, MA, USA) by assessing the degree of coupling between the electron transport chain (ETC) and the oxidative phosphorylation machinery (OXPHOS). The procedure and protocol have been described in our previous report [25].
Measurement of left ventricular ejection fraction by echocardiography
Transthoracic echocardiography was performed in each group prior to and on days 35 and 60 after DCM induction. The procedure was performed by an animal cardiologist blinded to the experimental design using an ultrasound machine (Vevo 2100, Visualsonics). M-mode standard two-dimensional (2D) left parasternal-long axis echocardiographic examinations were conducted. Left ventricular (LV) internal dimensions [end-systolic diameter (ESD) and end-diastolic diameter (EDD)] were measured at the mitral valve level of the left ventricle, according to the American Society of Echocardiography leading-edge method using at least three consecutive cardiac cycles. LVEF was calculated as follows: LVEF (%) = [(LVEDD3-LVEDS3)/LVEDD3] × 100%.
Western blot analysis of heart tissues
Western blot analysis was performed as described previously [15,25]. Equal amounts (50 µg) of protein extracts were loaded and separated by SDS-PAGE. Separated proteins were transferred to PVDF membranes, and nonspecific sites were blocked by incubation in blocking buffer [5% nonfat dry milk in T-TBS (TBS containing 0.05% Tween 20)] overnight. The membranes were incubated with the indicated primary antibodies [nuclear respiratory factor 1 (NRF1) (1:1000, Cell Signaling), NRF2 (1:1000, Abcam), mitochondrial transcription factor (TFAM) (1:1000, Abcam), estrogen-related receptor alpha (ERRα) (1:1000, Cell Signaling), mitofusin-2 (Mfn2) (1:1000, Cell Signaling), peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), mitochondrial Bax (1:1000, Abcam), cleaved poly (ADP-ribose) polymerase (PARP) (1:1000, Cell Signaling), caspase 3 (1:1000, Cell Signaling), Bcl-2 (1:200, Abcam), NADPH oxidase (NOX)-1 (1:1500, Sigma), NOX-2 (1:500, Sigma), p22phox (1:1000, Abcam), beclin 1 (1:1000, Cell Signaling), Atg 5 (1:1000, Cell Signaling), LC3B-I (1:1000, Cell Signaling), LC3B-II (1:1000, Cell Signaling), cyclophilin D1 (1:1000, Abcam), dynamin-related protein 1 (Drp1) (1:1000, Cell Signaling), cytosolic cytochrome C (1:2000, BD), mitochondrial cytochrome C (1:2000, BD), mitochondrial complexes I (1:2000, Abcam), II (1:2000, Abcam), IV (1:2000, Abcam) and V (1:2000, Abcam) for 1 hour at room temperature. Horseradish peroxidase-conjugated anti-rabbit IgG (1:2000, Cell Signaling) was used as a secondary antibody. Immuno-reactive bands were visualized by enhanced chemiluminescence (ECL; Amersham Biosciences), and digitized using Labwork software (UVP).
Oxidative stress reaction in LV myocardium
Expression of oxidative stress proteins has been described previously [15,24,25]. Oxyblot Oxidized Protein Detection Kit was purchased from Chemicon (S7150). DNPH derivatization was carried out using 6 μg of protein for 15 minutes according to the manufacturer’s instructions. One-dimensional electrophoresis was conducted on 12% SDS/polyacrylamide gel after DNPH derivatization. Proteins were transferred to nitrocellulose membranes, which were then incubated in the primary antibody solution (anti-DNP 1:150) for 2 hours, followed by incubation in secondary antibody solution (1:300) for 1 hour at room temperature. Immuno-reactive bands were visualized by ECL (Amersham Biosciences) and digitized using Labwork software (UVP).
Immunofluorescent (IF) staining
The procedure and protocol have been described in our previous reports [15,24,25]. Re-hydrated paraffin sections were treated with 3% H2O2 for 30 minutes and incubated with Immuno-Block reagent (BioSB, Santa Barbara, CA, USA) for 30 minutes at room temperature. Sections were then incubated with primary antibodies against γ-H2AX (1:500, Abcam), brain natriuretic peptide (BNP) (1:750, Abcam), connexin43 (Cx43) (1:100, Cell Signaling Technology) and troponin for sarcomere length (1:500, Bioss). Three sections of kidney specimens from each rat were analyzed. For quantification, three randomly selected HPFs (200 × or 400 × for IHC and IF studies) were analyzed in each section. The mean number of positively-stained cells per HPF for each animal was determined by summation of all numbers divided by 9.
Histological quantification of myocardial fibrosis
The procedure and protocol have been described in detail in our previous reports [15,24,25]. Briefly, hematoxylin and eosin (H & E) and Masson’s trichrome staining were used to identify the infarct area and fibrosis of LV myocardium, respectively. Three serial sections of LV myocardium in each animal were prepared at 4 µm thickness by Cryostat (Leica CM3050S). The integrated area (µm2) of infarct area and fibrosis on each section were calculated using the Image Tool 3 (IT3) image analysis software (University of Texas, Health Science Center, San Antonio, UTHSCSA; Image Tool for Windows, Version 3.0, USA). Three randomly selected high-power fields (HPFs) (100 ×) were analyzed in each section. After determining the number of pixels in each infarct and fibrotic area per HPF, the numbers of pixels obtained from three HPFs were calculated. The procedure was repeated in two other sections for each animal. The mean pixel number per HPF for each animal was then determined by calculating all pixel numbers and dividing by 9. The mean integrated area (µm2) of fibrosis in LV myocardium per HPF was obtained using a conversion factor of 19.24 (since 1 µm2 represents 19.24 pixels). This method was also applied for identification of Cx43 expression in myocardium.
RNA extraction and reverse transcription qPCR assessment for mRNA expression relative to that of β actin in LV myocardium
The procedure and protocol of RNA extraction reverse transcription qPCR analysis were performed according to our previous report [28] and the manufacturer’s instructions.
Briefly, the quantitative reverse transcription polymerase chain reaction (RT-qPCR) was performed using LightCycler TaqMan Master (Roche, Germany) in a single capillary tube according to the manufacturer’s guidelines for individual component concentrations. The forward and reverse primers (Supplementary Table 1) were each designed based on individual exons of the target gene sequence to avoid amplifying genomic DNA.
During PCR, the probe was hybridized to its complementary single-strand DNA sequence within the PCR target. As amplification occurred, the probe was degraded due to the exonuclease activity of Taq DNA polymerase, thereby separating the quencher from reporter dye during extension. During the entire amplification cycle, light emission increased exponentially. A positive result was determined by identifying the threshold cycle value at which reporter dye emission appeared above the background.
Mitochondrial DNA copy number in ADMSCs, H9C2 cells and LV myocardium
Total DNA was extracted from adipose derived mesenchymal stem cells (ADMSCs), H9C2 cells and LV myocardium using the DNeasy Blood and Tissue kit (Qiagen) with proteinase K and RNase treatment, according to the manufacturer’s instructions. For the copy number of mitochondrial DNA (mtDNA forward and reverse primers refer to Supplementary Table 1) were quantified by QuantiNOVA SYBR Green PCR assay (Qiagen) and normalized by rat genomic DNA (GAPDH: refer to Table). Triplicate assays were performed for each sample on Step One-Plus system (Applied Biosystems, Life Technologies).
Statistical analysis
Quantitative data are expressed as means ± SD. Statistical analysis was performed by ANOVA followed by Bonferroni multiple-comparison post hoc test. SAS statistical software for Windows version 8.2 (SAS institute, Cary, NC) was utilized. A probability value <0.05 was considered statistically significant.
Results
Oxygen consumption rate and mitochondrial transfusion refreshed the intracellular Mito in H9C2 cells (Figure 1)
Figure 1.

Oxygen consumption rate, and mitochondrial transfusion refreshed the intracellular Mito and TR-PCR determined the gene expressions in H9C2 cells. (A-A2) Illustrating the DAPI stain for identification of nuclei of hepatocytes. (B-B2) Illustrating immunofluorescent microscopic finding (400 ×) for identifying the endogenous mitochondria (Mito) (green color) (C1, C2), exogenous transferred mitochondria (orange color) [i.e., Mito (50 µg and 200 µg) were transfused into 4.0 × 105 hepatocytes, respectively] (D1, D2) abundant mitochondria being transferred into the rat hepatocytes (merge image) (pink color). (E) Analytical result of number of exogenous mitochondrial transfer into hepatocytes, * vs. †, P<0.01. (F) Satisfactory activity (i.e., high oxygen consumption rate) of isolated mitochondria (determined by the Mito stress test kit and the XFe24 Analyzer) (n = 4). OCR = oxygen consumption rate; (G) The relative mitochondrial DNA gene expression in H9C2, * vs. other groups with different symbols (†, ‡), P<0.001. (H) The relative mitochondrial DNA gene expression in ADMSC, * vs. other groups with different symbols (†, ‡), P<0.001. All statistical analyses were performed by one-way ANOVA, followed by Bonferroni multiple comparison post hoc test (n = 4 for each group). Symbols (*, †, ‡) indicate significance (at 0.05 level). Cln = control; L-Mito = low (50 µg) mitochondrial transfer; H-Mito = high (200 µg) mitochondrial transfer.
In vitro, IF microscopy demonstrated that mitochondrial transfusion into H9C2 cells upregulated the expression of exogenic Mito in the H9C2 cells (Figure A1-D3). Additionally, the expression of exogenic Mito in H9C2 cells was significantly higher in the high-dose mitochondrial transfusion group than in the low-dose group (Figure A1-D3). These findings suggest that exogenous mitochondrial refreshment occurred in the recipient cells.
Furthermore, we used the Seahorse method to verify the oxygen consumption rate in the isolated mitochondria. The result showed a satisfactory activity (i.e., high oxygen consumption rate) of isolated Mito (determined by the Mito stress test kit and the XFe24 Analyzer) (Figure 1E, 1F).
We conducted RT-PCR for H9C2 cells and adipose-derived mesenchymal stem cells (ADMSCs) and found that the relative number of mitDNA was significantly increased in the high-dose mitochondrial transfusion group than in the low-dose mitochondrial transfusion group, and significantly higher in low-dose mitochondrial transfusion group than in control group among H9C2 cells and ADMSCs (Figure 1G, 1H).
Protein expressions of mitochondrial integrity in H9C2 cells (Figure 2)
Figure 2.
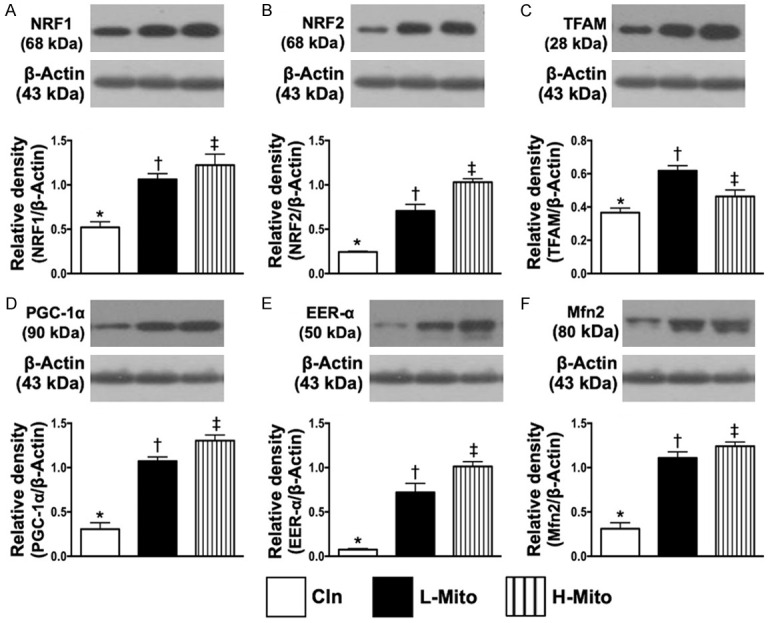
Protein expressions of mitochondrial integrity and antioxidant in H9C2 cells. A. Protein expression of nuclear factor erythroid 2-related factor 1 (NRF1), * vs. other groups with different symbols (†, ‡), P<0.001. B. Protein expression of NRF2, * vs. other groups with different symbols (†, ‡), P<0.001. C. Protein expression of mitochondrial transcription factor (TFAM), * vs. other groups with different symbols (†, ‡), P<0.001. D. Protein expression of peroxisome proliferator activated the receptor-gamma coactivator (PGC)-1α, * vs. other groups with different symbols (†, ‡), P<0.001. E. Protein expression of estrogen-related receptor alpha (ERRα), * vs. other groups with different symbols (†, ‡), P<0.001. F. Protein expression of mitofusin-2 (Mfn2), * vs. other groups with different symbols (†, ‡), P<0.001. All statistical analyses were performed by one-way ANOVA, followed by Bonferroni multiple comparison post hoc test (n = 4 for each group). Symbols (*, †, ‡) indicate significance (at 0.05 level). Cln = control; L-Mito = low (750 µg) mitochondrial transfer; H-Mito = high (1500 µg) mitochondrial transfer.
To verify the impact of exogenous mitochondrial transfusion on H9C2 cells, Western blotting was performed. The analyses demonstrated that the protein expression of NRF1 (Figure 2A), NRF2 (Figure 2B), TFAM (Figure 2C), PGC-1α (Figure 2D), ERRα (Figure 2E) and Mfn2 (Figure 2F), six indices of mitochondrial integrity/energy biogenesis biomarkers, were significantly higher in the low-dose mitochondrial transfusion group and significantly even higher in the high-dose mitochondrial transfusion group than in the control group (Supplementary Figure 1, illustrating the raw materials of Western blot image).
Time courses of LVEF, ratio of heart and lung weight to tibial length (Figure 3)
Figure 3.
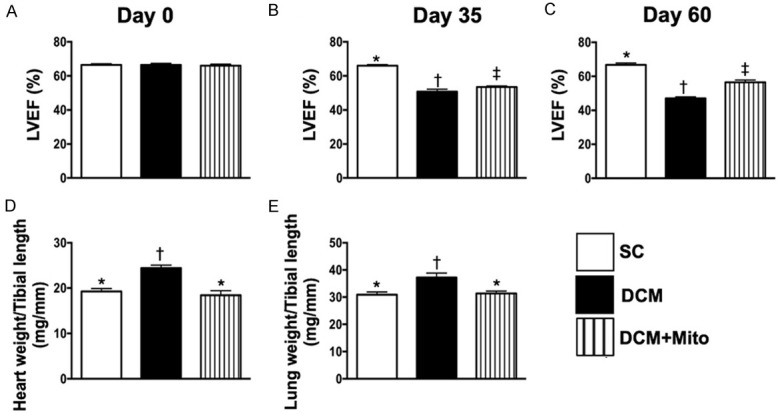
Time courses of LVEF, and the ratios of heart and lung weight to tibial length by day 60 after DCM induction. A. By day 0, analytical result of left ventricular ejection fraction (LVEF), P>0.5. B. By day 35 after DCM induction, analytical result of LVEF, * vs. other groups with different symbols (†, ‡), P<0.001. C. By day 60 after the DCM induction, analytical result of LVEF, * vs. other groups with different symbols (†, ‡), P<0.001. D. The ratio of total heart weight to tibial length by 60 after DCM induction, * vs. other groups with different symbols (†, ‡), P<0.001. E. The ratio of total lung weight to tibial length by 60 after DCM induction, * vs. other groups with different symbols (†, ‡), P<0.001. All statistical analyses were performed by one-way ANOVA, followed by Bonferroni multiple comparison post hoc test (n = 6 for each group). Symbols (*, †, ‡) indicate significance (at 0.05 level). SC = sham-operated control; DCM = dilated cardiomyopathy; Mito = mitochondria.
At day 0, LVEF did not differ among the three groups (Figure 3A). However, by days 35 and 60 after DCM induction, LVEF was significantly higher in group 1 (SC) than in group 2 (DCM only) and group 3 (DCM + Mito) and significantly higher in group 3 than in group 2 (Figure 3B, 3C).
Additionally, the ratio of heart weight to tibial length (Figure 3D) and ratio of lung weight to tibial length (Figure 3E) showed an opposite pattern to day-60 LVEF among the three groups.
LV myocardial scar area and cardiomyocyte size by day 60 after DCM induction (Figure 4)
Figure 4.

LV myocardium injury score and cardiomyocyte size by day 60 after DCM induction. A-C. Illustrating the microscopic finding (100 ×) of H&E stain for identification of LV myocardial injury area (i.e., yellow dotted line). Scale bars in right lower corner represent 100 µm. D. Analytical result of LV myocardial injury score, * vs. other groups with different symbols (†, ‡), P<0.0001. E-G. Illustrating the microscopic finding (400 ×) of H&E stain for identification of cardiomyocyte size. Yellow dotted line indicated one cardiomyocyte. H. Analytical result of cardiomyocyte size at day 60 after DCM induction, * vs. other groups with different symbols (†, ‡), P<0.0001. Scale bars in right lower corner represent 20 µm. All statistical analyses were performed by one-way ANOVA, followed by Bonferroni multiple comparison post hoc test (n = 6 for each group). Symbols (*, †, ‡) indicate significance (at 0.05 level). SC = sham-operated control; DCM = dilated cardiomyopathy; Mito = mitochondria.
To elucidate the therapeutic impact of early administration of mitochondria on protecting LV myocardium against DCM injury, histopathological light microscopy examinations were performed. As expected, H.E. stain showed that the LV myocardial scar area (Figure 4A-C) was significantly increased in group 2 than in groups 1 and 3, and significantly increased in group 3 than in group 1 (Figure 4D). Cardiomyocyte size (Figure 4E-G) exhibited a similar pattern to LV myocardial injury area among the three groups (Figure 4H).
The fibrotic area and DNA-damaged biomarker by day 60 after DCM induction (Figure 5)
Figure 5.
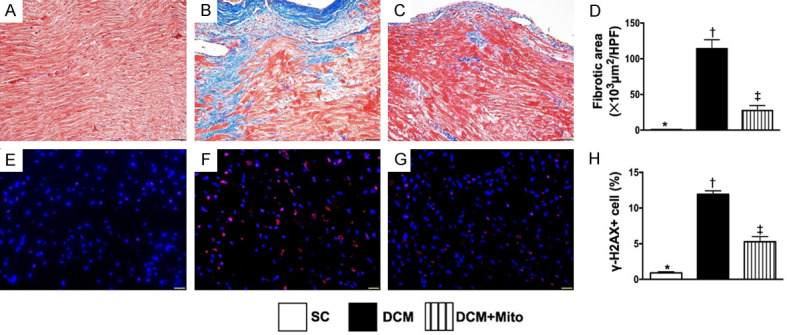
Fibrotic area and DNA-damaged biomarker in LV myocardium by day 60 after DCM induction. A-C. Illustrating the microscopic finding (200 ×) of Masson’s trichrome for identification of fibrotic area (blue color). D. Analytical result of fibrotic area, * vs. other groups with different symbols (†, ‡), P<0.0001. Scale bars in right lower corner represent 50 µm. E-G. Illustrating the immunofluorescent microscopic finding (400 ×) for identification of γ-H2AX+ cells (green color). H. Analytical result of number of γ-H2AX+ cells, * vs. other groups with different symbols (†, ‡), P<0.0001. Scale bars in right lower corner represent 20 µm. All statistical analyses were performed by one-way ANOVA, followed by Bonferroni multiple comparison post hoc test (n = 6 for each group). Symbols (*, †, ‡) indicate significance (at 0.05 level). SC = sham-operated control; DCM = dilated cardiomyopathy; Mito = mitochondria.
Next, the Masson’s trichrome staining demonstrated that the fibrotic area of LV myocardium (Figure 5C) was significantly increased in group 2 than in groups 1 and 3, and significantly increased in group 3 than in group 1 (Figure 5D). Additionally, IF microscopy showed that the cellular expression of γ-H2AX (Figure 5E-G), an indicator of DNA-damage, displayed an identical pattern to the fibrotic area among the groups (Figure 5H).
Cellular gap function and pressure overload/heart failure biomarker by day 60 after DCM induction (Figure 6)
Figure 6.
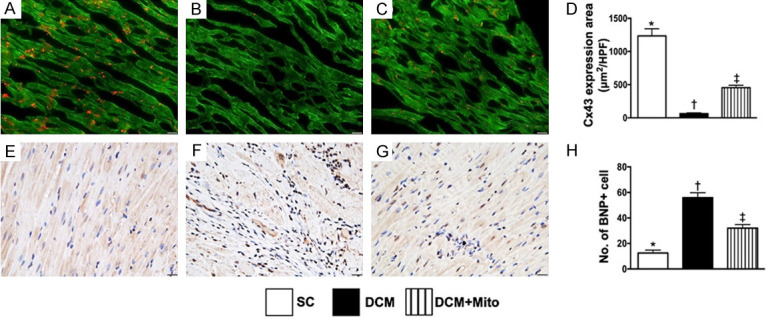
The cellular gap function and pressure overload/heart failure biomarker in LV myocardium by day 60 after DCM induction. A-C. Illustrating the immunofluorescent microscopic finding (400 ×) for identification of expression of connexin43 (Cx43) in LV myocardium (red color). D. Analytical result of Cx43 expression, * vs. other groups with different symbols (†, ‡), P<0.0001. Scale bars in right lower corner represent 20 µm. E-G. Illustrating the microscopic finding (200 ×) of immunohistochemical stain for identification of brain natriuretic peptide (BNP) (gray color) in LV myocardium. H. Analytical result of BNP expression, * vs. other groups with different symbols (†, ‡), P<0.0001. Scale bars in right lower corner represent 20 µm. * vs. other groups with different symbols (†, ‡), P<0.0001. Scale bars in right lower corner represent 50 µm. All statistical analyses were performed by one-way ANOVA, followed by Bonferroni multiple comparison post hoc test (n = 6 for each group). Symbols (*, †, ‡) indicate significance (at 0.05 level). SC = sham-operated control; DCM = dilated cardiomyopathy; Mito = mitochondria.
To clarify the integrity of cell to cell communication within LV myocardium, we utilized IF staining. The results showed that the expression of Cx43 (Figure 6A-C), an indicator of cellular gap junction for cell to cell communication, was significantly reduced in group 2 than in groups 1 and 3, and significantly reduced in group 3 than in group 1 (Figure 6D). Conversely, the expression of BNP in LV myocardium (Figure 6E-G), an indicator of pressure-overload and a heart failure biomarker, displayed an opposite pattern to Cx43 among the three groups (Figure 6H).
RT-PCR measurement of gene expressions of mitochondrial-integrity and biogenesis biomarkers by day 60 after DCM induction (Figure 7)
Figure 7.

RT-PCR measurement of mRNA expressions of mitochondrial-integrity and biogenesis biomarkers in LV myocardium by day 60 after DCM induction. A. mRNA expression of mitochondrial transcription factor (TFAM), * vs. other groups with different symbols (†, ‡), P<0.0001. B. mRNA expression of peroxisome proliferator activated the receptor-gamma coactivator (PGC)-1α, * vs. other groups with different symbols (†, ‡), P<0.001. C. mRNA expression of nuclear respiratory factor 1 (Nrf1), * vs. †, P<0.01. D. mRNA expression of nuclear factor erythroid 2-related factor 2 (Nrf2), * vs. †, P<0.001. E. mRNA expression of estrogen-related receptor alpha (ERR-α), * vs. †, P<0.001. F. The relative mitochondrial DNA gene expression, * vs. other groups with different symbols (†, ‡), P<0.001. All statistical analyses were performed by one-way ANOVA, followed by Bonferroni multiple comparison post hoc test (n = 6 for each group). Symbols (*, †, ‡) indicate significance (at 0.05 level). SC = sham-operated control; DCM = dilated cardiomyopathy; Mito = mitochondria.
Gene expression of mitochondrial transcription factor (TFAM), an essential activator of mitochondrial transcription and a participant in mitochondrial genome replication, was significantly downregulated in group 2 than in groups 1 and 3, and significantly downregulated in group 3 than in group 1, suggesting that Mito therapy preserved TFAM expression in LV myocardium (Figure 7A). Additionally, the gene expression of peroxisome proliferator activated the receptor-gamma coactivator (PGC)-1α (Figure 7B), which is a transcriptional coactivator that regulates genes involved in energy biogenesis and metabolism, also exhibited a pattern identical to that of TFAM among the three groups.
The gene expression of nuclear factor erythroid 2-related factor 1 (Nrf1) (Figure 7C) and Nrf2 (Figure 7D), two emerging regulators of cellular resistance to oxidants, were significantly lower in group 2 than in groups 1 and 3 and significantly lower in group 3 than in group 1.
Estrogen-related receptor alpha (ERRα) is required for the activation of mitochondrial genes as well as increased mitochondrial biogenesis. In the mRNA study, we found that the gene expression of ERRα displayed an identical pattern to that of Nrf1 among the three groups (Figure 7E).
The relative mitochondrial DNA gene expression (i.e., mitochondrial DNA relative to genomic DNA) was significantly reduced in group 2 than in groups 1 and 3, and significantly reduced in group 3 than in group 1 (Figure 7F).
The protein expression of oxidative stress and autophagic biomarkers by day 60 after DCM induction (Figure 8)
Figure 8.

Protein expressions of oxidative stress and autophagic biomarkers in LV myocardium by day 60 after DCM induction. A. Protein expression of NOX-1, * vs. other groups with different symbols (†, ‡), P<0.001. B. Protein expression of NOX-2, * vs. other groups with different symbols (†, ‡), P<0.0001. C. Protein expression of p22phox, * vs. other groups with different symbols (†, ‡), P<0.0001. D. The oxidized protein expression, * vs. other groups with different symbols (†, ‡), P<0.0001 (Note: the right and left lanes shown on the upper panel represent protein molecular weight marker and control oxidized molecular protein standard, respectively). M.W = molecular weight; DNP = 1-3 dinitrophenylhydrazone. E. Protein expression of beclin-1, * vs. other groups with different symbols (†, ‡), P<0.0001. F. Protein expression of atg-5, * vs. other groups with different symbols (†, ‡), P<0.001. G. Protein expression of ratio of CL3B-II to CL3B-I, * vs. other groups with different symbols (†, ‡), P<0.001. All statistical analyses were performed by one-way ANOVA, followed by Bonferroni multiple comparison post hoc test (n = 6 for each group). Symbols (*, †, ‡) indicate significance (at 0.05 level). SC = sham-operated control; DCM = dilated cardiomyopathy; Mito = mitochondria.
In vivo, the protein expression of NOX-1 (Figure 8A), NOX-2 (Figure 8B), p22phox (Figure 8C) oxidized protein (Figure 8D), four indices of oxidative stress, were significantly higher in group 2 than in groups 1 and 3, and significantly higher in group 3 than in group 1.
Additionally, the protein expressions of beclin-1 (Figure 8E), atg-5 (Figure 8F) and the ratio of CL3B-II to CL3B-I (Figure 8G), three indicators of autophagic biomarkers, exhibited an identical pattern to oxidative stress among the groups (Supplementary Figure 2, illustrating the raw materials of Western blot image).
The protein expression of apoptotic and mitochondrial-damaged biomarkers by day 60 after DCM induction (Figure 9)
Figure 9.
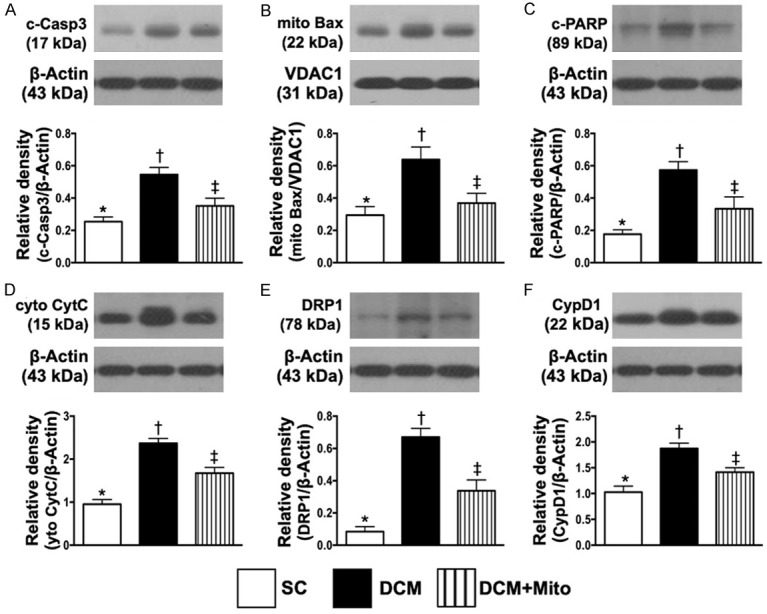
Protein expression of apoptotic and mitochondrial-damaged biomarkers in LV myocardium by day 60 after DCM induction. A. Protein expressions of caspase 3, * vs. other groups with different symbols (†, ‡), P<0.001. B. Protein expression of mitochondrial Bax (Mito-Bax), * vs. other groups with different symbols (†, ‡), P<0.001. C. Protein expression of cleaved (c)-PARP, * vs. other groups with different symbols (†, ‡), P<0.001. D. Protein expressions of cytosolic cytochrome C (cyt-Cyto C), * vs. other groups with different symbols (†, ‡), P<0.001. E. Protein expression of DRP1, * vs. other groups with different symbols (†, ‡), P<0.001. F. Protein expression of cyclophilin D1 (CypD1), * vs. other groups with different symbols (†, ‡), P<0.001. All statistical analyses were performed by one-way ANOVA, followed by Bonferroni multiple comparison post hoc test (n = 6 for each group). Symbols (*, †, ‡) indicate significance (at 0.05 level). SC = sham-operated control; DCM = dilated cardiomyopathy; Mito = mitochondria.
The protein expressions of caspase 3 (Figure 9A), mitochondrial Bax (Figure 9B) and cleaved PARP (Figure 9C), three indicators of apoptosis, were significantly increased in group 2 than in groups 1 and 3, and significantly increased in group 3 than in group 1. Additionally, the protein expressions of cytosolic cytochrome C (Figure 9D), DRP1 (Figure 9E) and cyclophilin D1 (Figure 9F), three indicators of mitochondrial damaged markers, displayed an identical pattern to apoptosis among the groups (Supplementary Figure 3, illustrating the raw materials of Western blot image).
Protein expressions of mitochondrial-integrity biomarkers by day 60 after DCM induction (Figure 10)
Figure 10.

Protein expressions of mitochondrial-integrity biomarkers by day 60 after DCM induction. A. Protein expression of mitochondrial complex I (Mito-CI), * vs. other groups with different symbols (†, ‡), P<0.001. B. Protein expression of Mito-CII, * vs. other groups with different symbols (†, ‡), P<0.001. C. Protein expression of Mito-CIII, * vs. other groups with different symbols (†, ‡), P<0.001. D. Protein expression of Mito-CIV, * vs. other groups with different symbols (†, ‡), P<0.001. E. Protein expression of mitochondrial cytochrome C (mito-Cyt-C), * vs. other groups with different symbols (†, ‡), P<0.001. F. Protein expression of Mfn2, * vs. other groups with different symbols (†, ‡), P<0.001. G. Protein expression of PGC-1, * vs. other groups with different symbols (†, ‡), P<0.001. All statistical analyses were performed by one-way ANOVA, followed by Bonferroni multiple comparison post hoc test (n = 6 for each group). Symbols (*, †, ‡) indicate significance (at 0.05 level). SC = sham-operated control; DCM = dilated cardiomyopathy; Mito = mitochondria.
The protein expressions of mitochondrial complex I, II, III and IV (Figure 10A-D), mitochondrial cytochrome C (Figure 10E), five indices of mitochondrial integrity, were significantly higher in group 1 than in groups 2 and 3, and significantly higher in group 3 than in group 2.
However, the protein expression of Mfn2 (Figure 10F) and PGC-1 (Figure 10G), two mitochondria-related energy biogenesis biomarkers, significantly and progressively increased from group 1 to group 2, suggesting due to the results of refreshment mitochondria after early mitochondrial therapy (Supplementary Figure 4, illustrating the raw materials of Western blot image).
Discussion
This study examined the therapeutic impact of implanted mitochondria into LV myocardium in DCM rat and yielded several striking preclinical implications. First, in vitro study verified that exogenous mitochondria could be transferred to recipient cells, resulting in refreshment of mitochondrial function/activity for energy biogenesis in recipient cells. Second, in vivo study established that early direct implantation of mitochondria into LV myocardium of DCM rats significantly suppressed apoptosis, fibrosis, oxidative stress, autophagy and further mitochondrial damage. Third, LV function was preserved in DCM rats after receiving early exogenous mitochondrial implantation, probably by refreshing mitochondrial supply.
An essential finding in the in vitro study was that the exogenous mitochondria isolated from rat liver could be transfused into the H9C2 cells without difficulty. Another essential finding was that high-dose exogenous mitochondria was preferable to low-dose exogenous mitochondria for transferring more mitochondria into the recipient cells, suggesting that there was a dose-dependent effect for saturation of exogenous mitochondrial in recipient cells. Importantly, exogenous transfer of mitochondria proved effective at upregulating energy biogenesis and mitochondrial integrity in recipient cells. These findings encouraged us to verify whether implantation of exogenous mitochondrial could preserve heart function in DCM rat by early refreshing the mitochondria (i.e., source of energy) in DCM myocardium.
Interestingly, our previous studies have shown that exogenous mitochondrial administration effectively protected the lung and liver against ARDS [25] and ischemia-reperfusion injury [24], respectively. As yet, there is no available data to address the impact of exogenous mitochondrial administration in DCM. The most important finding in the present study was that, as compared with SC animals, LVEF was substantially reduced in DMC animals. However, by the end of study, LVEF was substantially preserved in DCM animals after receiving exogenous mitochondrial therapy. Intriguingly, the therapeutic effect was even observed at an early phase (i.e., by day 35 after DCM induction) of mitochondrial therapy. Our novel finding, in addition to extending the findings of our previous studies [24,25], suggest that this therapy may be useful for DCM patients in advanced stages of decompensated HF that is refractory to conventional therapy.
Our previous experimental studies have clearly identified that myocardial fibrosis was markedly increased whereas the expressions of Cx4 and small vessel density was remarkably reduced in the settings of DCM and cardiorenal syndrome [25,28,29]. A principal finding in the present study was that the Masson’s trichrome stain demonstrated a significant increase in fibrotic area (i.e., histopathological finding) in LV myocardium of DCM rats. In addition, IF staining further demonstrated that expressions of Cx43 was consistently reduced in LV myocardium in DCM rat. Thus, our findings were consistent with previous investigations [25,28,29]. However, these parameters were notably reversed in DMC animals after receiving mitochondrial therapy. Our results could, at least in part, explain why LVEF was preserved in DCM after treatment with mitochondria.
Intriguingly, our experimental studies [25,28,29] also found that the apoptotic, fibrotic, oxidative-stress and autophagic biomarkers were substantially augmented in the setting of DCM. Consistently, molecular-cellular levels of these parameters were significantly enhanced in LV myocardium of DCM rat. However, these cellular-molecular perturbations were ameliorated in DCM animals after receiving early mitochondrial therapy. In this way, our findings, in addition to strengthening the findings of our previous studies [25,28,29], could, once again, explain why LVEF was preserved in mitochondrial treated DCM rat.
It is well recognized that mitochondrial dysfunction results in exhaustion of energy [20-22] due to cardiomyocyte death and scar formation [14,23]. In the present study, we found that the mitochondrial-damaged markers were substantially increased in DCM animals than in SC animals. These findings could explain why the myocardial injury score/fibrotic area (i.e., indices of scar formation) was remarkably increased in DCM animals. Accordingly, our findings corroborated those of previous studies [14,20-23]. A crucial finding in the present study was that these aforementioned parameters were significantly altered in DCM animals after receiving early mitochondrial therapy. On the other hand, the protein levels of mitochondrial complex I, II, III and IV, indicators of integrity of electronic transport chain for upregulation of oxygen consumption in rat, and effective energy generation for supplying myocardium were notably reduced in DCM animals but were significantly reversed in DCM animals after receiving mitochondrial therapy. These, could, once more, explain why LVEF was preserved whereas the myocardial score was notably suppressed in mitochondrial treated DCM animals.
Study limitation
This study has limitations. First, despite extensive works both in vitro and in vivo, the precise mechanisms of how mitochondrial therapy improves heart function in DMC myocardial injury remains unconfirmed. Although the results were attractive and promising, this study did not assess the stepwise and optimal dosage of mitochondrial therapy for DCM.
Conclusions
The present study demonstrated that early exogenous mitochondrial therapy was safe and feasible for improving heart function in DCM rats.
Acknowledgements
This study was funded by research grants from both the National Science Council, Taiwan, Republic of China (MOST 106-2314-B-182A-108-MY3; NMRPG8G6111, NMRPG8G6112, NMRPG8G6113). All animal experimental procedures were approved by the Institutional Animal Care and Use Committee at Kaohsiung Chang Gung Memorial Hospital (Affidavit of Approval of Animal Use Protocol No. 2017110201) and performed in accordance with the Guide for the Care and Use of Laboratory Animals, 8th edition (NIH publication No. 85-23, National Academy Press, Washington, DC, USA, revised 2011). Animals were housed in an Association for Assessment and Accreditation of Laboratory Animal Care International-approved animal facility in our hospital, with controlled temperature and light cycles (24°C and 12/12 light/dark cycle).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Manolio TA, Baughman KL, Rodeheffer R, Pearson TA, Bristow JD, Michels VV, Abelmann WH, Harlan WR. Prevalence and etiology of idiopathic dilated cardiomyopathy (summary of a National Heart, Lung, and Blood Institute workshop. Am J Cardiol. 1992;69:1458–1466. doi: 10.1016/0002-9149(92)90901-a. [DOI] [PubMed] [Google Scholar]
- 2.Dec GW, Fuster V. Idiopathic dilated cardiomyopathy. N Engl J Med. 1994;331:1564–1575. doi: 10.1056/NEJM199412083312307. [DOI] [PubMed] [Google Scholar]
- 3.Ishida J, Konishi M, von Haehling S. The appropriate dose of angiotensin-converting-enzyme inhibitors or angiotensin receptor blockers in patients with dilated cardiomyopathy. The higher, the better? ESC Heart Fail. 2015;2:103–105. doi: 10.1002/ehf2.12073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gigli M, Stolfo D, Merlo M, Barbati G, Ramani F, Brun F, Pinamonti B, Sinagra G. Insights into mildly dilated cardiomyopathy: temporal evolution and long-term prognosis. Eur J Heart Fail. 2017;19:531–539. doi: 10.1002/ejhf.608. [DOI] [PubMed] [Google Scholar]
- 5.Codd MB, Sugrue DD, Gersh BJ, Melton LJ 3rd. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975-1984. Circulation. 1989;80:564–572. doi: 10.1161/01.cir.80.3.564. [DOI] [PubMed] [Google Scholar]
- 6.Rakar S, Sinagra G, Di Lenarda A, Poletti A, Bussani R, Silvestri F, Camerini F. Epidemiology of dilated cardiomyopathy. A prospective post-mortem study of 5252 necropsies. The Heart Muscle Disease Study Group. Eur Heart J. 1997;18:117–123. doi: 10.1093/oxfordjournals.eurheartj.a015092. [DOI] [PubMed] [Google Scholar]
- 7.Felker GM, Thompson RE, Hare JM, Hruban RH, Clemetson DE, Howard DL, Baughman KL, Kasper EK. Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N Engl J Med. 2000;342:1077–1084. doi: 10.1056/NEJM200004133421502. [DOI] [PubMed] [Google Scholar]
- 8.Nikolaidis LA, Doverspike A, Huerbin R, Hentosz T, Shannon RP. Angiotensin-converting enzyme inhibitors improve coronary flow reserve in dilated cardiomyopathy by a bradykinin-mediated, nitric oxide-dependent mechanism. Circulation. 2002;105:2785–2790. doi: 10.1161/01.cir.0000017433.90061.2e. [DOI] [PubMed] [Google Scholar]
- 9.Chan YK, Tuttle C, Ball J, Teng TK, Ahamed Y, Carrington MJ, Stewart S. Current and projected burden of heart failure in the Australian adult population: a substantive but still ill-defined major health issue. BMC Health Serv Res. 2016;16:501. doi: 10.1186/s12913-016-1748-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jennings DL. Heart failure therapy in 2016: SHIFTing the PARADIGM from antiquated therapies toward novel agents. Ann Pharmacother. 2017;51:79–82. doi: 10.1177/1060028016672245. [DOI] [PubMed] [Google Scholar]
- 11.Pitt B, Poole-Wilson PA, Segal R, Martinez FA, Dickstein K, Camm AJ, Konstam MA, Riegger G, Klinger GH, Neaton J, Sharma D, Thiyagarajan B. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: randomised trial--the Losartan Heart Failure Survival Study ELITE II. Lancet. 2000;355:1582–1587. doi: 10.1016/s0140-6736(00)02213-3. [DOI] [PubMed] [Google Scholar]
- 12.Dandel M, Weng Y, Siniawski H, Potapov E, Lehmkuhl HB, Hetzer R. Long-term results in patients with idiopathic dilated cardiomyopathy after weaning from left ventricular assist devices. Circulation. 2005;112:I37–45. doi: 10.1161/CIRCULATIONAHA.104.525352. [DOI] [PubMed] [Google Scholar]
- 13.Taylor MR, Carniel E, Mestroni L. Cardiomyopathy, familial dilated. Orphanet J Rare Dis. 2006;1:27. doi: 10.1186/1750-1172-1-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oh H, Ito H, Sano S. Challenges to success in heart failure: cardiac cell therapies in patients with heart diseases. J Cardiol. 2016;68:361–367. doi: 10.1016/j.jjcc.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 15.Lee FY, Shao PL, Wallace CG, Chua S, Sung PH, Ko SF, Chai HT, Chung SY, Chen KH, Lu HI, Chen YL, Huang TH, Sheu JJ, Yip HK. Combined therapy with SS31 and mitochondria mitigates myocardial ischemia-reperfusion injury in rats. Int J Mol Sci. 2018;19:2782. doi: 10.3390/ijms19092782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kotamraju S, Konorev EA, Joseph J, Kalyanaraman B. Doxorubicin-induced apoptosis in endothelial cells and cardiomyocytes is ameliorated by nitrone spin traps and ebselen. Role of reactive oxygen and nitrogen species. J Biol Chem. 2000;275:33585–33592. doi: 10.1074/jbc.M003890200. [DOI] [PubMed] [Google Scholar]
- 17.Simeunovic D, Seferovic PM, Ristic AD, Nikolic D, Risimic D, Seferovic J, Maksimovic R, Nedeljkovic I, Karan R, Bajcetic M. Evaluation of oxidative stress markers and catecholamine changes in patients with dilated cardiomyopathy before and after cardiopulmonary exercise testing. Hellenic J Cardiol. 2015;56:394–401. [PubMed] [Google Scholar]
- 18.Kurian GA, Rajagopal R, Vedantham S, Rajesh M. The role of oxidative stress in myocardial ischemia and reperfusion injury and remodeling: revisited. Oxid Med Cell Longev. 2016;2016:1656450. doi: 10.1155/2016/1656450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang S, Ding L, Ji H, Xu Z, Liu Q, Zheng Y. The role of p38 MAPK in the development of diabetic cardiomyopathy. Int J Mol Sci. 2016;17:1037. doi: 10.3390/ijms17071037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galloway CA, Yoon Y. Mitochondrial dynamics in diabetic cardiomyopathy. Antioxid Redox Signal. 2015;22:1545–1562. doi: 10.1089/ars.2015.6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kubli DA, Gustafsson AB. Unbreak my heart: targeting mitochondrial autophagy in diabetic cardiomyopathy. Antioxid Redox Signal. 2015;22:1527–1544. doi: 10.1089/ars.2015.6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Westermeier F, Navarro-Marquez M, Lopez-Crisosto C, Bravo-Sagua R, Quiroga C, Bustamante M, Verdejo HE, Zalaquett R, Ibacache M, Parra V, Castro PF, Rothermel BA, Hill JA, Lavandero S. Defective insulin signaling and mitochondrial dynamics in diabetic cardiomyopathy. Biochim Biophys Acta. 2015;1853:1113–1118. doi: 10.1016/j.bbamcr.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Won S, Davies-Venn C, Liu S, Bluemke DA. Noninvasive imaging of myocardial extracellular matrix for assessment of fibrosis. Curr Opin Cardiol. 2013;28:282–289. doi: 10.1097/HCO.0b013e32835f5a2b. [DOI] [PubMed] [Google Scholar]
- 24.Chen HH, Chen YT, Yang CC, Chen KH, Sung PH, Chiang HJ, Chen CH, Chua S, Chung SY, Chen YL, Huang TH, Kao GS, Chen SY, Lee MS, Yip HK. Melatonin pretreatment enhances the therapeutic effects of exogenous mitochondria against hepatic ischemia-reperfusion injury in rats through suppression of mitochondrial permeability transition. J Pineal Res. 2016;61:52–68. doi: 10.1111/jpi.12326. [DOI] [PubMed] [Google Scholar]
- 25.Sun CK, Lee FY, Kao YH, Chiang HJ, Sung PH, Tsai TH, Lin YC, Leu S, Wu YC, Lu HI, Chen YL, Chung SY, Su HL, Yip HK. Systemic combined melatonin-mitochondria treatment improves acute respiratory distress syndrome in the rat. J Pineal Res. 2015;58:137–150. doi: 10.1111/jpi.12199. [DOI] [PubMed] [Google Scholar]
- 26.Lin KC, Wallace CG, Yin TC, Sung PH, Chen KH, Lu HI, Chai HT, Chen CH, Chen YL, Li YC, Shao PL, Lee MS, Sheu JJ, Yip HK. Shock wave therapy enhances mitochondrial delivery into target cells and protects against acute respiratory distress syndrome. Mediators Inflamm. 2018;2018:5425346. doi: 10.1155/2018/5425346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang TH, Chung SY, Chua S, Chai HT, Sheu JJ, Chen YL, Chen CH, Chang HW, Tong MS, Sung PH, Sun CK, Lu HI, Yip HK. Effect of early administration of lower dose versus high dose of fresh mitochondria on reducing monocrotaline-induced pulmonary artery hypertension in rat. Am J Transl Res. 2016;8:5151–5168. [PMC free article] [PubMed] [Google Scholar]
- 28.Sheu JJ, Lin PY, Sung PH, Chen YC, Leu S, Chen YL, Tsai TH, Chai HT, Chua S, Chang HW, Chung SY, Chen CH, Ko SF, Yip HK. Levels and values of lipoprotein-associated phospholipase A2, galectin-3, RhoA/ROCK, and endothelial progenitor cells in critical limb ischemia: pharmaco-therapeutic role of cilostazol and clopidogrel combination therapy. J Transl Med. 2014;12:101. doi: 10.1186/1479-5876-12-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chua S, Lee FY, Chiang HJ, Chen KH, Lu HI, Chen YT, Yang CC, Lin KC, Chen YL, Kao GS, Chen CH, Chang HW, Yip HK. The cardioprotective effect of melatonin and exendin-4 treatment in a rat model of cardiorenal syndrome. J Pineal Res. 2016;61:438–456. doi: 10.1111/jpi.12357. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.