

Abstract
This study aimed to the role of insulin-like growth factor 1 (IGF-1) in Duchenne muscular dystrophy (DMD), the inflammatory response and the potential mechanism of the effect hIGF1 exerted in muscle inflammation were also been explored. In this study, AAV9, a carrier of the human IGF-1 gene, was injected into mdx mice to observe the role of IGF-1 in DMD. Routine histopathological staining, immunofluorescence and western blot were used to detect the inflammatory response. In addition, we also explored the potential mechanism of the role of hIGF1 in muscle inflammation. The expression of AAV9 in myocardium and muscle tissue of AAV9-GFP group was detected by GFP method. GFP was expressed in different tissues of mdx mice, especially in anterior tibial muscle, triceps muscle and other tissues. The percentage of anterior tibial muscle inflammation area in CD68 and AAV9-hIGF-1 group was lower than that in AAV-GFP group, and the percentage of anterior tibial muscle inflammation area in AAV9-hIGF-1 group (1.78 ± 0.47%) was significantly lower than that in AAV GFP group (3.4 ± 1.22%) (P < 0.05). Western-blot showed that AAV-hIGF-1 group (0.45 + 0.07%) was lower than that of AAV-GFP group (0.76 + 0.13%), higher than the normal group (0.38 + 0.06%). The difference was statistically significant (P < 0.05). In conclusion, this study confirmed that hIGF-1 can reduce the inflammatory response and macrophage infiltration in mdx mice, and further proved that hIGF-1 can down regulate the expression of NF-κB signal pathway, which has anti-inflammatory effect.
Keywords: AAV9, IGF1, inflammatory, mice
Introduction
Duchenne muscular dystrophy (DMD) is the most common X-linked incurable recessive disease in progressive muscular dystrophy, caused by the deletion, nonsense, or repeated mutations of DMD gene (encodes the protein dystrophin) on X chromosome (Xp21.2) [1]. Dystrophin located on the inner surface of myocytes is the basis of myocytes. The deficiency of dystrophin protein causes the chronic damage to muscle fibers during contraction. The main clinical manifestations of DMD are progressive myasthenia, gastrocnemius atrophy and pseudohypertrophy. The main pathological features of DMD include unequal diameter of muscle fiber, progressive necrosis of muscle fiber, inflammation, and fibrosis [2]. Chronic inflammation plays an important role in DMD muscle degeneration [3]. Inflammatory muscle damage increases the inflammatory response of immune cell ectopics, and abnormal activation of inflammatory signaling pathways occurs even before myofibre necrosis occurs [4,5]. In recent years, there has been growing evidence that inflammatory response and activation of inflammatory pathway play an essential role in the occurrence and development of DMD [6,7]. The regulation of inflammatory response is a potential treatment for DMD [8].
As a way to treat DMD by correcting gene defects or playing a role in the treatment, gene therapy can introduce normal genes or therapeutic genes into target cells [9,10]. Adeno-associated virus (AAV) is one of the most commonly used gene therapy vectors, which can express efficiently and for a long time without toxicity, immune response and pathogenicity [11]. Systematic evaluation showed that AAV9 can be effectively transfect skeletal muscle and myocardium [2,12].
Insulin-like growth factor 1 (IGF-1) is an essential nutritional factor in cell proliferation, differentiation, and maturation. In addition, the enhancement of IGF-1 expression in skeletal muscle significantly reduces fibrosis, myonecrosis, and serum CK levels, thereby improving muscle regeneration and muscle strength [13]. The activation of protein kinase B (PKB or Akt) has been shown to be an important part of IGF-1 mediated cell survival and growth [14].
Interestingly, IGF-1 can inhibit the expression and activity of macrophage migration inhibitory factor (MIF), high migration group protein box 1 (HMGB1), and nuclear factor-kappa B (NF-κB). Previous studies have focused on the effects of IGF-1 on muscle fiber regeneration [15-17], however, there has been little research on the role of IGF-1 in muscle inflammation. AAV9 shows the highest expression in muscle and cardiac in the serotypes of AAV, then AAV9 is selected as the carrier in this study [18]. Considering the muscle dystrophy of DMD, systematic injection such as tail vein injection is the best choice in this experiment.
In this study, AAV9, a carrier of the human IGF-1 gene, was injected into mdx mice to observe the role of IGF-1 in DMD. The inflammatory reaction was detected by routine histopathological staining, immunofluorescence and western blot. In addition, the potential mechanism of the role of hIGF1 in muscle inflammation was discussed.
Materials and methods
Animals
6-week-old mdx and wild-type C57BL/10 (WT) mice were obtained from Nanjing University and raised in our animal facility. Considering the inflammatory reaction mode and injection site of mdx mice, 6-week old mice [19] were injected with hIGF-1 via tail vein. The mice were housed in a temperature-controlled, quiet, germ-free environment and had free access to food and water. The experiment followed the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of health. This study was approved by the institutional review board of second hospital of hebei medical university (Approval letter No.2015-P001). All of the experimental procedures were performed in accordance with protocols approved by the Institutional Animal Care and Research Advisory Committee, the Second Hospital of Hebei Medical University.
Systemic injection: Two groups of mdx mice were injected into tail vein within 6 weeks, but one group (n = 4) used AAV9-hIGF1 virus 200 uL, the other group (n = 4) used AAV9-GFP 200 uL. WT mice (n = 4) were treated with normal saline in the control group. At the end of the experiment, we anesthetized them (10% chloral hydrate (400 mg/kg body weight, intraperitoneally)) and took western, pathohistology staining 6-weeks after.
Muscle injection: 6-week-old mdx mice and C57BL/10 mice with similar body weight were divided into three groups. AAV9-GFP and AAV9-hIGF1 were injected into the right tibial anterior muscle of mdx mice. C57BL/10 mice were used as the control group without any intervention.
There were two humane endpoints in our experiment. The intramuscular injection group was injected 8 weeks after the injection. The whole body injection group ends 6 weeks after injection. The mice were suffocated by carbon dioxide (20% full rate) and then dislocated by the cervical spine.
Creatine kinase (CK) analysis
The mice were anesthetized well with 10% chloral hydrate (400 mg/kg body weight, intraperitoneally) and clamped the mice’s feet and tails with pliers to keep them unresponsive. No animal showed signs of peritonitis. Western blot and immunohistochemistry were used to detect the tibial anterior muscle. Since all of these mice were well anesthetized, no follow-up dose was required in this study. Blood was taken from the eye for about 1.0 ml, left for 2 hours (to have a better separation between the blood cell and the serum) and placed in a refrigerator at 4°C for 15 minutes, then centrifuged at 3000 rpm for 10 minutes with a high-speed low-temperature centrifuge and the supernatant was stored at -80°C. Serum CK value was measured by the United States BECKMAN COULTER automatic biochemical analysis. After the injection experiment, mice are sacrifice to collect their blood, muscle and internal organ.
Lengthening contraction
10% chloral hydrate (400 mg/kg body weight) was injected intraperitoneally to keep mice unresponsive to pain stimulation, and chloral hydrate was supplemented when necessary. The anterior tibial muscles of mice were exposed to extensor digitorum longus and separated. The tibialis anterior muscle and its blood supply vessels were not damaged. Non absorbable surgical suture was used to prevent the distal tendon from slipping, and then the tendon was cut at the distal end of the surgical node. The separation operation was completed within 30 minutes, during which a small amount of warm (about 37°C) soda lactate ringer injection could be used to keep the muscles moist. On the platform of the muscle strength test system, the mice were placed in the supine position and the knees were fixed to prevent slipping. The surgical line of the distal tendon is connected with the horizontal arm of the sensor system in a ring to keep the tibialis anterior muscle perpendicular to the horizontal arm. The surface of tibialis anterior muscle were stimulated (each measurement is conducted by the same person and placed in the same position to reduce system error).
During the measurement, the stimulation electric flow was kept constant, and the electrical stimulation pulse time was fixed at 0.2 ms. The optimal initial length (Lo) of muscle was adjusted by the micromanipulator to produce the maximum tetanic contraction force, and Lo was recorded by vernier caliper. The stimulation frequency of tibialis anterior muscle gradually increased (10, 30, 40, 50, 80, 100, 120, 150, 180 and 200 Hz), and the force frequency curve of the maximum stiffness contraction force (Po) was drawn at the interval of 1 minute. The tibialis anterior muscle was relaxed for 5 minutes, and then 10 eccentric contractions were performed at the frequency of obtaining the maximum tetanic contractile force 5 minutes later. The maximal isometric contractile force was obtained at Lo for 175 Ms. Then, at the same time of stimulation, stretch 0.2lo at the speed of 2lf/S (TA fiber length (LF; cm) = 0.6lo), the stimulation was finished at 350 ms, and the stretching was finished, at which time it is returned to Lo at the same speed. Each cycle is 1 minute apart. The maximum isometric contraction force measured after each eccentric contraction was recorded, and the rate of decline of each eccentric contraction was calculated with the first time of 100%. During the measurement, the linger injection was added to the exposed area for 1-2 minutes, and the mice were irradiated with heat source to keep the core temperature at 37°C. After all measurements are completed, the tibialis anterior muscle was removed and the fluid was sucked dry and weighed for further biochemical and histological analysis equipment: ARORA 1200A.
Histopathology
8-um thick cryostat sections were obtained from diaphragm, myocardium, tibialis anterior muscle, quadriceps femoris, musculus gastrocnemius, and then stained with hematoxylin-eosin (H&E), ACP staining, immunohistochemistry staining and immunofluorescent staining. In the muscle injection group, the tibialis anterior muscle used in the contraction protocol was also used in the western and histology. While in the tail injection group, the tibialis anterior muscle as the contraction mode was used for western and the contralateral side was used for histology.
Western blot analysis
Samples from tibialis anterior muscle were homogenized in the dissolved buffer. Protein samples were denatured by the reduced barrier, and SDS (12%) polyacrylamide gel electrophoresis was isolated. The isolated protein was transferred to nitrocellulose membrane in 180-300 mA transfer buffer for 2 h. The membrane was blocked with 5% non-fat milk or 0.01 mol/L BSA at room temperature for 1 h, washed with TBS (0.01 M) for three times, and incubated with the PP65 polyclonal antibody (1:500; Abcam, Cambridge, UK) and GAPDH antibody (1:5000; santa) overnight at 4°C. After washing three times in TBS-0.01 M Tween for 10 min each, the membranes were incubated with IRDye700DX, IRDye800DX for 1 h at room temperature. After washing, the membranes were imaged on the Odyssey Infrared Imaging System at a wavelength of 700,800 nm.
Supplementary experiment: triceps were collected and incubated overnight with TNFα antibody (1:1000; Sanying, Wuhan, China) and GAPDH antibody (1:1000; Servicebio) at 4°C. After washing three times in TBS-0.01 M Tween for 10 minutes, the membrane was incubated with IRDye700DX, IRDye800DX for 1 hour at room temperature. After cleaning, the film was imaged on the Odyssey infrared imaging system at a wavelength of 700,800 nm.
Drug
AAV9 virus vector carrying the GFP gene or the hIGF-1 gene was produced by Beijing Wujiahe Company. The promoter is CMV. The virus was dialyzed against hydroxyethylpiperazineethanesulfonic acid (HEPES) after twice centrifugation with a gradient of cesium chloride. The virus was stored in an ultra-low temperature refrigerator at -80°C, and thawed to room temperature before use. The titer of the virus was determined by real-time quantitative PCR in units of viral genomes per milliliter (1 × 1012 vg/ml). The expression of GFP protein in tibialis anterior muscle was observed 8 weeks after virus injection, and green fluorescence was observed under fluorescent microscope.
Statistical analysis
Results were expressed as mean ± SD or median. Two independent sample t-test was used to analyze difference among groups, and Kruskal wallis test was used for the analysis of mean among groups. Statistical evaluation was performed by SPSS 13.0. P values < 0.05 were considered significant.
Results
The expression of hIGF-1 in tibialis anterior muscle
In the systemic injection group, about 30.9% of muscle cells expressed GFP protein, which confirmed its transfection and expression efficiency (Figure 1A-F). In the muscle injection group, AAV9-hIGF-1 was highly expressed in the tibial anterior muscle of mdx mice, and the number of positive cells accounted for about 13.5%, but there was no expression of muscle fibers (Figure 1G).
Figure 1.
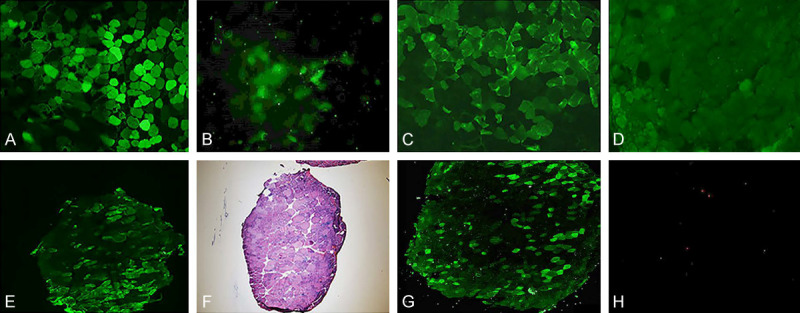
(A, B) The expression of GFP in systemic injection group. (A) is a piece of muscle with AAV9-GFP, (B) is a piece of muscle without any injection [The green fluorescence as protein expression]. (C-F) The expression of hIGF-1 when mice at 12 weeks in systemic injection group. (C) is the immunofluorescence result of the muscle injected with AAV9-hIGF-1; (D) is the control group without primary antibody; (E) is the expression of a cross section; (F) is the serial section with C staining HE. (C is compounded by software). (G) is the expression of hIGF1 protein in the anterior muscle of mdx mice in AAV9-hIGF-1 group with muscle injection. (H) is the negative control.
Histology
There were two types of muscle fibers in the control group: dark cytoplasm, small area, light type I and large cytoplasm. The cells were uniform in size. There was no significant difference in the muscle fiber types between AAV9-GFP mice and normal mice. Compared with AAV9-GFP group, AAV9-hIGF-1 group had a larger proportion of muscle fibers, but the size of muscle fibers and regenerated muscle were different (Figure 2A-C). The ratio of necrosis of tibialis anterior muscle in AAV9-hIGF-1 mdx mice was 2.14 ± 0.43%, which was significantly lower than that of 6.33 ± 2.51% in AAV9-GFP mdx mice. The difference was statistically significant. (P < 0.05) (Figure 2D-F).
Figure 2.

(A-C) HE staining results of the distributions of myocyte areas and mini Feret diameter in systemic injection group. The cells of wild type mice were evenly distributed. Compared with wild-type mice, the cell distribution of GFP mice and hIGF-1 mice was unbalanced. (D) is the control group. The proportion of necrotic area in GFP group (E) was 6.33 (+2.51%) and hIGF-1 group (F) was 2.14 (+0.43%). There was a significant difference between the two groups (P < 0.05).
Serum creatine kinase
In the systemic injection group, the CK level of control group mice was 5076.67 ± 3959.18 U/L, that of AAV9-GFP mdx mice was 26345.64 ± 5762.39 U/L, and that of AAV9-hIGF-1 mdx mice was 39446.28 ± 15905.25 U/L. Compared with normal control group mice, mdx mice had higher CK, but there was no significant difference between GFP group and hIGF-1 group (P > 0.05).
In the muscle injection group, the CK value of control group mice was 5076.67 ± 3959.18 U/L, that of AAV9-GFP mdx mice was 33418.57 ± 11695.54 U/L, and that of AAV9-hIGF-1 mdx mice was 31855.8 ± 16394.13 U/L. Compared with normal control group mice, mdx mice had higher CK, but there was no significant difference between GFP group and hIGF-1 group (P > 0.05).
Muscle force
In the systemic injection group, the mice in the control group got the minimum strength due to muscle weakness. The muscle strength of AAV9-GFP mdx mice without effective intervention was significantly stronger than that of wild-type mice in the control group. After administration of AAV9-hIGF-1, the muscle strength of mice increased significantly (Figure 3A, 3B). The difference between the hIGF-1 group and the control group and the AAV9-GFP group was statistically significant (P < 0.05). But there was no significant difference between the control group and AAV9-GFP group (P > 0.05). The endurance of the anterior tibialis muscle of the hIGF-1 group was compared with the GFP group and C57BL/10 group, respectively, with significant differences (P < 0.05), and no significant difference between the hIGF-1 group and the GFP group (P > 0.05), (Figure 3C).
Figure 3.
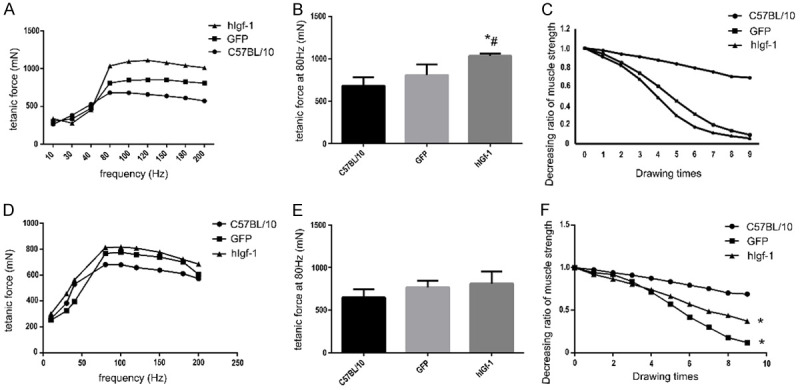
(A-C) The muscle force of different groups in systemic injection group. (A) is the tetanic force condition at different stimulating frequencies. (B) was the force levels of different groups at 80 Hz (* compares with C57BL/10 group, # compares with GFP group, P < 0.05). (C) was the endurance test result which is opposite with the tetanic force result (* compares with C57BL/10 group, P < 0.05). (D-F) The muscle force of different groups in muscle injection group. (D) was the tetanic force condition at different stimulating frequencies. (E) showed the force levels of different groups at 80 Hz, there was no difference between each group (P > 0.05). (F) showed that there was significant difference between GFP group and hIGF-1 group and C57BL/10 group (P < 0.05), but there was no significant difference between GFP group and hIGF-1 group (P > 0.05) (* compares with C57BL/10 group, P < 0.05).
In the group with muscle injection, when measuring the maximum contractile force (Figure 3D, 3E), the mdx mice administered AAV9-hIGF-1 produced the strongest muscle, followed by AAV9-GFP mdx mice, and finally the normal control group mice. But there was no significant difference (P > 0.05). In the muscle endurance test (Figure 3F), there was significant difference between the hIGF-1 group and GFP group and C57BL/10 group (P < 0.05), but there was no significant difference between hIGF-1 group and GFP group (P > 0.05).
The expression of AAV9-GFP in mdx mice
The expression of AAV9 in myocardium and muscle tissue of AAV9-GFP group was detected by GFP method. GFP was expressed in different tissues of mdx mice, especially in anterior tibial muscle, triceps muscle and other tissues. It was proved that AAV9 could be successfully expressed in mdx mice. The expression rate of tibialis anterior muscle was the highest (Figure 4A-F).
Figure 4.

(A-F) The results of (A-F) showed the expression (Green fluorescence in the figure) of GFP in diaphragm (A), myocardium (B), triceps (C), quadriceps (D), gastrocnemius (E) and tibialis anterior muscle (F). The tibialis anterior muscles expressed the most. (G-I) were HE staining results of mdx mice anterior cervical muscle in AAV9-hIGF-1 group, AAV9-GFP group and C57BL/10 group. (J-L) were ACP staining results of mdx mouse anterior muscle among AAV9-hIGF-1 group, AAV9-GFP group and C57BL/10 group.
The inflammatory response of mdx mice after AAV9-hIGF-1 therapy
He staining and ACP staining showed mixed inflammatory cell infiltration in AAV9-GFP group and a small amount of inflammatory cell infiltration in AAV9-hIGF-1 group (Figure 4G-L). The percentage of anterior tibial muscle inflammation area in CD68 and AAV9-hIGF-1 group was lower than that in AAV-GFP group (Figure 5A-I), and the percentage of anterior tibial muscle inflammation area in AAV9-hIGF-1 group (1.78 ± 0.47%) was significantly lower than that in AAV GFP group (3.4 ± 1.22%) (P < 0.05).
Figure 5.

The expression of CD68 (Green fluorescence in the figure) and pp65 (A-I). (A-C) were AAV9-hIGF-1, AAV9-GFP and C57BL/10, respectively. The CD68 fluorescence spectra of the anterior muscle of mdx mice and the (D-F) maps were the corresponding nuclear staining of the figure (A-C), (G) was the co-localization of (A) and (D, H) was the co-localization of (B) and (E, I) was the co-localization of (C) and (F), which showed that the fluorescence expression in the AAV9-hIGF-1 group is lower than that of AAV9-GFP group. (G). AAV9-hIGF-1 group, C57/10 group, AAV9-hIGF-1 group NF-κB comparison. Compared with AAV9-GFP group, the CD68 expression in AAV9-hIGF-1 group decreased, the difference was statistically significant (P < 0.05). (J, K) Western blots showed that the levels of pp65 and TNFα in AAV-hIGF-1 mice group were lower than those in AAV-GFP mice group, but higher than those in the C57 mice group (control group).
The change of NF-kappa B signaling pathway
Western-blot results showed that AAV9-hIGF-1 can reduced the expression of PP65. AAV-hIGF-1 group (0.45 + 0.07%) was lower than that of AAV-GFP group (0.76 + 0.13%), higher than the normal group (0.38 + 0.06%). The difference was statistically significant (P < 0.05) (Figure 5J).
Western blots showed that the levels of pp65 and TNFα in AAV-hIGF-1 mice group were lower than those in AAV-GFP mice group, but higher than those in the C57 mice group, (Figure 5J, 5K) suggesting that overexpression of IGF-1 can reduce the expression of pp65 and TNFα. These results also indicated that the overexpression of IGF-1 can inhibit the inflammatory response and NF-κB signaling pathway in mdx mice.
Discussion
Duchenne muscular dystrophy (DMD) is a fatal, human muscle wasting disease caused by the mutation of dystrophin gene, which can lead to the deficiency of the complete or partial deletion of the dystrophin protein. In recent years, many researchers have proved that inflammatory reaction and activation of inflammatory signaling pathway play an essential role in the progress of DMD [5,15]. Mendell’s study showed that some patients who participated in the clinical trial of AAV-micro dystrophin injection had dystrophin-special T cells, and increased along with the aggravation of inflammatory reaction [15,18,20]. In this case, the dystrophin-positive fiber may not improve even we inject dystrophin gene for treatment. This indicated that only gene repair can not play a good role, which is why we need to pay attention to monitor cell immunity and start treating it as a therapeutic target [21,22].
From the perspective of exploring the treatment of chronic inflammatory diseases (such as juvenile idiopathic arthritis, chronic kidney disease, inflammatory bowel disease, atherosclerosis), IGF-1 has the effect of inhibiting immune inflammation [23-25]. Studies have shown that IGF-1 in muscle can regulate immune response through MIF, HMGB and NF-κB [12]. In this experiment, IGF-1 gene was introduced into mdx mice by a single injection of tail vein to observe the effect of IGF-1 gene on the immune function of mdx mice and further explore its mechanism. In GFP group, hematoxylin eosin (H&E) had phagocytes around the necrotic tissue and large inflammatory necrosis area. Compared with GFP group, a small amount of inflammatory necrosis area was found in the ACP staining of hIGF-1 group. Compared with GFP group, the inflammatory area of hIGF-1 group was significantly reduced, indicating that hIGF-1 had a certain effect on the inflammation of mdx mice. When tissue is damaged, monocytes migrate to the damaged tissue and enter macrophages to deal with the damage [26,27]. CD68 is a transmembrane glycoprotein, known as the most reliable biomarker of monocyte macrophage system [26-28]. The results showed that the infiltration of CD68 + macrophages decreased, which further confirmed the pathological data.
Previous studies on mdx mice and dogs have shown that gene therapy alone should not be used, which will lead to a decrease in mdx expression in inflammatory response [29,30]. Glucocorticoids have been shown to prolong life. Although it still has a series of side effects, interrupting the whole treatment, it makes the new anti-inflammatory treatment more critical [31]. We hope that hIGF-1 will be a potential combination therapy for DMD. Therefore, further understanding of the inflammatory mechanism regulating fibrogenesis is of great significance for the development of therapeutic methods aimed at inhibiting or reversing the progression of DMD in the future.
Acharyya S and other studies have found that the specifically down-regulated NF-κB pathway in macrophages can significantly reduce the expression of proinflammatory cytokines and myofibrosis, suggesting that the expression of NF-κB signaling pathway in macrophages can significantly aggravate the pathological damage of DMD to a large extent [32]. At the same time, we observed that knockout of mdx mice with p65 subunit can reduce the number of necrotic muscle fibers and macrophage infiltration [33].
NF-κB is an important pro-inflammatory transcription factor, widely involved in the inflammatory process, apoptosis, proliferation and differentiation, protein degradation, stress response, redox response, etc. NF-κB family consists of five subunits, RelA (p65), RelB, c-Rel, p50 and p52, of which p65/p50 is the most effective one, which is found in almost all cells [34]. In the inactivated status, NF-κB is located in the cytoplasm by binding to the inhibitor IkB (Inhibitor kB). Signaling from the cell membrane activates the upstream I-kB kinase (inhibitor of nuclear factor kappa-B kinase, IKK) to phosphorylate, ubiquitinate downstream of IkB, and phosphorylate p50/p65 dimer, especially p65. It was phosphorylated into pp65 and transferred to the nucleus, and then combined with the corresponding homologous DNA sites to start the transcription of a series of downstream target genes [35]. The results of this study showed that the treatment of mdx mice with hIGF-1 could reduce the expression of pp65, suggesting that hIGF-1 down regulated NF-κB pathway, thereby alleviating the inflammation of muscle.
In conclusion, this study confirmed that hIGF-1 can reduce the inflammatory response and macrophage infiltration in mdx mice, and further proved that hIGF-1 can down regulate the expression of NF-κB signal pathway, which has anti-inflammatory effect.
Acknowledgements
This study was funded by grants from the National Natural Science Foundation of China (81471228).
Disclosure of conflict of interest
None.
References
- 1.Hoffman EP, Brown RH Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 2.Eckardt L, Harzer W. Facial structure and functional findings in patients with progressive muscular dystrophy (Duchenne) Am J Orthod Dentofacial Orthop. 1996;110:185–190. doi: 10.1016/s0889-5406(96)70107-5. [DOI] [PubMed] [Google Scholar]
- 3.Prins KW, Humston JL, Mehta A, Tate V, Ralston E, Ervasti JM. Dystrophin is a microtubule-associated protein. J Cell Biol. 2009;186:363–369. doi: 10.1083/jcb.200905048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhatnagar S, Kumar A. Therapeutic targeting of signaling pathways in muscular dystrophy. J Mol Med (Berl) 2010;88:155–166. doi: 10.1007/s00109-009-0550-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hodgetts S, Radley H, Davies M, Grounds MD. Reduced necrosis of dystrophic muscle by depletion of host neutrophils, or blocking TNFalpha function with Etanercept in mdx mice. Neuromuscul Disord. 2006;16:591–602. doi: 10.1016/j.nmd.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 6.Baron D, Magot A, Ramstein G, Steenman M, Fayet G, Chevalier C, Jourdon P, Houlgatte R, Savagner F, Pereon Y. Immune response and mitochondrial metabolism are commonly deregulated in DMD and aging skeletal muscle. PLoS One. 2011;6:e26952. doi: 10.1371/journal.pone.0026952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Juban G, Chazaud B. Metabolic regulation of macrophages during tissue repair: insights from skeletal muscle regeneration. FEBS Lett. 2017;591:3007–3021. doi: 10.1002/1873-3468.12703. [DOI] [PubMed] [Google Scholar]
- 8.Bengtsson NE, Seto JT, Hall JK, Chamberlain JS, Odom GL. Progress and prospects of gene therapy clinical trials for the muscular dystrophies. Hum Mol Genet. 2016;25:R9–17. doi: 10.1093/hmg/ddv420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guiraud S, Chen H, Burns DT, Davies KE. Advances in genetic therapeutic strategies for Duchenne muscular dystrophy. Exp Physiol. 2015;100:1458–1467. doi: 10.1113/EP085308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haslett JN, Sanoudou D, Kho AT, Bennett RR, Greenberg SA, Kohane IS, Beggs AH, Kunkel LM. Gene expression comparison of biopsies from Duchenne muscular dystrophy (DMD) and normal skeletal muscle. Proc Natl Acad Sci U S A. 2002;99:15000–15005. doi: 10.1073/pnas.192571199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 13.Barton ER, Morris L, Musaro A, Rosenthal N, Sweeney HL. Muscle-specific expression of insulin-like growth factor I counters muscle decline in mdx mice. J Cell Biol. 2002;157:137–148. doi: 10.1083/jcb.200108071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pelosi L, Giacinti C, Nardis C, Borsellino G, Rizzuto E, Nicoletti C, Wannenes F, Battistini L, Rosenthal N, Molinaro M, Musaro A. Local expression of IGF-1 accelerates muscle regeneration by rapidly modulating inflammatory cytokines and chemokines. FASEB J. 2007;21:1393–1402. doi: 10.1096/fj.06-7690com. [DOI] [PubMed] [Google Scholar]
- 15.Schertzer JD, Gehrig SM, Ryall JG, Lynch GS. Modulation of insulin-like growth factor (IGF)-I and IGF-binding protein interactions enhances skeletal muscle regeneration and ameliorates the dystrophic pathology in mdx mice. Am J Pathol. 2007;171:1180–1188. doi: 10.2353/ajpath.2007.070292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tonkin J, Temmerman L, Sampson RD, Gallego-Colon E, Barberi L, Bilbao D, Schneider MD, Musaro A, Rosenthal N. Monocyte/macrophage-derived IGF-1 orchestrates murine skeletal muscle regeneration and modulates autocrine polarization. Mol Ther. 2015;23:1189–1200. doi: 10.1038/mt.2015.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ye F, Mathur S, Liu M, Borst SE, Walter GA, Sweeney HL, Vandenborne K. Overexpression of insulin-like growth factor-1 attenuates skeletal muscle damage and accelerates muscle regeneration and functional recovery after disuse. Exp Physiol. 2013;98:1038–1052. doi: 10.1113/expphysiol.2012.070722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ohshima S, Shin JH, Yuasa K, Nishiyama A, Kira J, Okada T, Takeda S. Transduction efficiency and immune response associated with the administration of AAV8 vector into dog skeletal muscle. Mol Ther. 2009;17:73–80. doi: 10.1038/mt.2008.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee SA, Lee SH, Kim JY, Lee WS. Effects of glycyrrhizin on lipopolysaccharide-induced acute lung injury in a mouse model. J Thorac Dis. 2019;11:1287–1302. doi: 10.21037/jtd.2019.04.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McGreevy JW, Hakim CH, McIntosh MA, Duan D. Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis Model Mech. 2015;8:195–213. doi: 10.1242/dmm.018424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Al-Zaidy S, Rodino-Klapac L, Mendell JR. Gene therapy for muscular dystrophy: moving the field forward. Pediatr Neurol. 2014;51:607–618. doi: 10.1016/j.pediatrneurol.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kayali R, Bury F, Ballard M, Bertoni C. Site-directed gene repair of the dystrophin gene mediated by PNA-ssODNs. Hum Mol Genet. 2010;19:3266–3281. doi: 10.1093/hmg/ddq235. [DOI] [PubMed] [Google Scholar]
- 23.Lapidos KA, Kakkar R, McNally EM. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ Res. 2004;94:1023–1031. doi: 10.1161/01.RES.0000126574.61061.25. [DOI] [PubMed] [Google Scholar]
- 24.Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S, Bowles D, Gray S, Li C, Galloway G, Malik V, Coley B, Clark KR, Li J, Xiao X, Samulski J, McPhee SW, Samulski RJ, Walker CM. Dystrophin immunity in Duchenne’s muscular dystrophy. N Engl J Med. 2010;363:1429–1437. doi: 10.1056/NEJMoa1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Z, Kuhr CS, Allen JM, Blankinship M, Gregorevic P, Chamberlain JS, Tapscott SJ, Storb R. Sustained AAV-mediated dystrophin expression in a canine model of Duchenne muscular dystrophy with a brief course of immunosuppression. Mol Ther. 2007;15:1160–1166. doi: 10.1038/sj.mt.6300161. [DOI] [PubMed] [Google Scholar]
- 26.Heller KN, Montgomery CL, Janssen PM, Clark KR, Mendell JR, Rodino-Klapac LR. AAV-mediated overexpression of human alpha7 integrin leads to histological and functional improvement in dystrophic mice. Mol Ther. 2013;21:520–525. doi: 10.1038/mt.2012.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Z, Tapscott SJ, Chamberlain JS, Storb R. Immunity and AAV-mediated gene therapy for muscular dystrophies in large animal models and human trials. Front Microbiol. 2011;2:201. doi: 10.3389/fmicb.2011.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Back K, Islam R, Johansson GS, Chisalita SI, Arnqvist HJ. Insulin and IGF1 receptors in human cardiac microvascular endothelial cells: metabolic, mitogenic and anti-inflammatory effects. J Endocrinol. 2012;215:89–96. doi: 10.1530/JOE-12-0261. [DOI] [PubMed] [Google Scholar]
- 29.Evans NP, Misyak SA, Robertson JL, Bassaganya-Riera J, Grange RW. Immune-mediated mechanisms potentially regulate the disease time-course of duchenne muscular dystrophy and provide targets for therapeutic intervention. PM R. 2009;1:755–768. doi: 10.1016/j.pmrj.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang B, Li J, Fu FH, Chen C, Zhu X, Zhou L, Jiang X, Xiao X. Construction and analysis of compact muscle-specific promoters for AAV vectors. Gene Ther. 2008;15:1489–1499. doi: 10.1038/gt.2008.104. [DOI] [PubMed] [Google Scholar]
- 31.Liaskou E, Zimmermann HW, Li KK, Oo YH, Suresh S, Stamataki Z, Qureshi O, Lalor PF, Shaw J, Syn WK, Curbishley SM, Adams DH. Monocyte subsets in human liver disease show distinct phenotypic and functional characteristics. Hepatology. 2013;57:385–398. doi: 10.1002/hep.26016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGuinness PH, Painter D, Davies S, McCaughan GW. Increases in intrahepatic CD68 positive cells, MAC387 positive cells, and proinflammatory cytokines (particularly interleukin 18) in chronic hepatitis C infection. Gut. 2000;46:260–269. doi: 10.1136/gut.46.2.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wehling M, Spencer MJ, Tidball JG. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J Cell Biol. 2001;155:123–131. doi: 10.1083/jcb.200105110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schultze JL, Schmieder A, Goerdt S. Macrophage activation in human diseases. Semin Immunol. 2015;27:249–256. doi: 10.1016/j.smim.2015.07.003. [DOI] [PubMed] [Google Scholar]