

Abstract
Background: Chronic heart failure (CHF) is a common and serious complication of patients with ischemic heart disease that may eventually lead to the development of pulmonary fibrosis. While other forms of pulmonary fibrosis have been studied extensively, little is known about the mechanisms that lead to heart failure associated with pulmonary fibrosis. The purpose of our study was to develop a rat pulmonary edema/fibrosis model induced by chronically elevated left atrial pressure (LAP), simulating CHF pathophysiology. Methods: In adult rats, LAP was elevated by 15-20 mmHg through mechanical restriction of left ventricular diastolic filling with a maximum effect occurring at 7 days. Sham rats were surgically operated without LAP elevation. Lung tissues were analyzed for wet-to-dry ratio, hydroxyproline content, cellular invasion, and tissue integrity. Lung compliance and airway resistance served as pulmonary mechanical parameters. Hemodynamic parameters, including arterial pressure, heart rate, and cardiac output, were recorded in Sham and LAP elevated rats for 7 days. Results: With increased LAP, pulmonary water content was significantly elevated accompanied by a decrease in lung compliance. Hydroxyproline markedly increased with chronic left atrial pressure elevation, suggesting fibrosis development. Simultaneously, heart failure induced a decrease in cardiac function. Conclusions: LAP elevation resulted in chronic pulmonary edema and tissue fibrosis formation associated with pulmonary dysfunction as measured by decreased dynamic lung compliance.
Keywords: Heart failure, pulmonary fibrosis, chronic pulmonary edema
Introduction
More than 5 million people suffer from heart failure (HF) in the United States with approximately 600,000 more cases per year at an estimated cost of 39 billion, including health care services, medications, and lost productivity [1]. Furthermore, the incidence of congestive heart failure is expected to increase over the next several decades. Pulmonary dysfunction associated with HF continues to challenge the physicians caring for these patients. Substantial pulmonary complications, including decreased lung compliance and arterial hypoxemia, occur because of left-sided HF, in particular. These complications correlate with the severity of heart failure [2-7]. In addition, cardiogenic pulmonary fibrosis often develops with congestive heart failure; however; the mechanism of pulmonary fibrosis development due to congestive heart failure has not been widely researched and is the focus of the present study.
Pulmonary fibrosis has been implicated in several models of congestive HF [2,8-10]. In a myocardial infarction HF model, Ahmed [2] found both increased collagen I mRNA levels and increased fibrosis associated with increased TGF-β and connective tissue growth factor levels in lung tissues [2]. Jasmin [8] showed structural remodeling of the lung, including increased collagen deposition, with experimental HF. In a chronic aortic banding model, structural remodeling in the lungs was also demonstrated [10].
Although chronic HF leads to irreversible changes in the lung architecture, the mechanisms of tissue remodeling and its relationship to pulmonary fibrosis development in chronic HF are poorly understood. During HF, left atrial pressure (LAP) increases significantly, causing increased pulmonary capillary pressures and leading to excess fluid filtration and, subsequently, pulmonary edema development [11]. Though poorly understood, fibrosis is a typical response to the persistence of edema fluid in almost any organ [12,13]. Pulmonary edema represents a serious clinical manifestation in many patients suffering from congestive heart failure [6]. While acute pulmonary edema does not result in permanent pulmonary dysfunction if treated immediately, chronic pulmonary edema can cause gradual but irreversible long-term lung impairment.
Presently, much research has focused on the contribution of inflammation and cytokine secretion to idiopathic pulmonary fibrosis (IPF) [14]. However, little research has been directed to understanding the role of inflammation in cardiogenic pulmonary fibrosis [15]. Thus, while many cytokines are implicated as early mediators of fibrosis development in the lung in IPF, little is known about which cytokines participate in the development of pulmonary fibrosis after HF [16]. Although the role of TNF-α in fibrosis development is not well-understood, it is likely to have proinflammatory and profibrotic effects [17,18]. The role of pro-inflammatory cytokines in the development of cardiogenic pulmonary fibrosis is not clear.
We recently developed a rat model to study the mechanism of pulmonary fibrosis development in HF in which restricted left ventricular filling causes chronically elevated left atrial pressure (LAP). We utilized this model to explore the hypothesis that chronic pulmonary edema due to elevated LAP leads to an inflammatory response with the subsequent release of profibrogenic cytokines and, eventually, pulmonary fibrosis development. Furthermore, in our model of elevated LAP, there is a link between chronic edema formation and chronic inflammation resulting in elevated TNF-α levels and ultimately fibrosis development.
Material and methods
All procedures were approved by the University of Texas Animal Welfare Committee and were consistent with the National Institutes of Health “Guide for the Care and Use of Laboratory Animals” (NIH publication 85-23, revised 1985).
Animal model of heart failure
Adult Sprague Dawley rats (250-300 g) were chronically instrumented under isoflurane anesthesia. A Tygon catheter was introduced into the left ventricle through the apex of the heart to record left ventricular end diastolic pressure as a surrogate for LAP. A wire was placed through the left ventricle to restrict left ventricular filling during cardiac diastole. This procedure causes an increase in LAP of about 15-20 mmHg above baseline. At the end of the surgical procedure, the catheter was removed, the lungs were carefully inflated, and the thorax was closed. Intraoperative care was facilitated by the infiltration of bupivacaine 0.25% into the wounds. Suction was applied to the thoracic drain before removal. Finally, the administration of isoflurane was discontinued, and animals were allowed to recover from surgery. Buprenorphine was administered at 0.02 mg/kg on one occasion after completion of the surgery.
To measure cardiac function, an additional group of animals subjected to HF was instrumented with a Tygon catheter introduced into the abdominal aorta via the femoral artery to simultaneously record heart rate and arterial blood pressure. A 20-MHz Doppler flow probe was positioned around the aorta to record cardiac output [19]. Intraoperative care and post-operative care were carried out as previously described for the HF group.
Experimental design
Rats were randomized into either the heart failure (HF) group (with elevated LAP) or the sham operated group (sham control). To assess the role of the lung inflammatory process in prolonged heart failure, additional groups of animals subjected to elevated LAP were studied. Macrophages and neutrophils were depleted with platelet-activating factor acetylhydrolase (PAF-AH) and cyclophosphamide (CTX), respectively. At the time of surgery to induce heart failure, an osmotic minipump was subcutaneously inserted to deliver PAF-AH (30 µg/kg/hr) or CTX (0.6 µg/kg/hr). After completion of all protocols, animals (HF and sham control) were sacrificed on day 7 and lung tissues were harvested in bloc, immediately freeze-clamped, and stored at -70°C until studied.
Wet-to-dry lung weight ratios
Lungs were rinsed briefly in PBS, blotted, and then weighed to obtain the wet weight. Lungs were dried in an oven at 80°C for 5 days to obtain the dry weights.
Lung fixation and histology in HF and sham control rats at 0 and 7 days
Lungs were fixed by perfusion with neutral buffered formaldehyde before routine processing and paraffin embedding. Multiple sections from each lobe were stained with hematoxylin. Histological assessment of neutrophils and macrophages was performed on Days 0 (surgery day) and 7 in rats with elevated LAP and compared to sham control rats on Days 0 and 7.
Determination of cytokine production and MPO activity in bronchoalveolar fluid (BALF) as markers of inflammation in HF
BALF was collected by flushing the lung four times with 5 ml of sterile, pyrogen-free, physiologic saline via a tracheal cannula. The recovered BALF samples were pooled and stored at -20°C for later use. BALF concentrations of IL-1β, TNF-α, and TGF-β1 were determined using ELISAs specific for each cytokine under investigation. MPO activity was determined by a dianisidine-H2O2 method [20].
Lung collagen
Hydroxyproline content was measured as described by Bergman and Loxley [21]. Briefly, a sample of dried homogenate was placed in 6N HCL and hydrolyzed at 100°C for 12 hrs, followed by neutralization with HCL. Hydroxyproline was oxidized to a pyrrole, and then condensed with Ehrlich’s reagent. The absorption at 558 nm was then measured to quantitate the hydroxyproline content.
Fluorescence Immunostaining of endothelial nitric oxide synthase (eNOS) and inducible (iNOS)
Immunohistochemical staining for eNOS and iNOS was performed on post-fixed sections of lungs in 3.7% formaldehyde for 5 min and non-specific staining was blocked by incubating sections with 10% goat serum for 30 min at room temperature. Stock antibody was diluted 1:1000 in 10% goat serum and specimens were incubated for 30 to 45 min at 37ºC. Coverslips were rinsed 2 to 3 times with 0.05% Tween 20. Then, we incubated samples in diluted fluorescent secondary antibody 1:500 in 0.05% Tween 20 and 10% goat serum for 30 min at 37°C. Coverslips were examined on an inverted Nikon Optiphot microscope (Nikon, New York, NY) and scanned with a Fluorescence Microscope (Issaquah, WA) fitted with an Olympus IX70 microscope with deconvolution capabilities. Images were captured at the maximum fluorescence [22].
Data analysis
Data were analyzed with a one-way analysis of variance (ANOVA) to assess overall significance. When differences were significant, multiple within-comparisons were performed using Dunnett’s t test. When changes were significant, the magnitude of changes in each experimental condition was compared using an unpaired t-test. A p value less than 0.05 was considered significant. Data are expressed as Mean ± SEM.
Results
Although measurements were also made at 2 and 4 days, the maximum effects of elevated LAP were seen at 7 days. Consequently, only data collected for 7 days are represented in the following figures. Heart failure was induced by restricting left ventricular filling as described above. Seven days later the lungs were removed for measurement of pulmonary edema development, as measured by wet to dry weight ratios. As shown in Figure 1, lungs harvested 7 days after LAP elevation contained significantly more fluid compared to lungs from sham controls (3.94 ± 0.57 vs. 3.21 ± 0.17 ml/kg, P<0.05). Hydroxyproline content was significantly higher in heart failure (HF) rats as compared to sham control rats (19.2 ± 3.4 vs. 16.3 ± 2.3 mg/kg) at 7 days. In addition, dynamic lung compliance was significantly lower in HF rats as compared to sham controls (0.13 ± 0.05 vs. 0.23 ± 0.05 ml/cm H2O, P<0.05) and airway resistance was markedly increased in HF rats compared to sham operated controls (LAP rat resistance = 0.23 ± 0.08, sham operated control rat resistance = 0.12 ± 0.02 cm H2O/ml/s; P<0.05; data not shown in Figure 1. As depicted in Table 1, our data show that blood pressure remained essentially unchanged despite significant decreases in heart rate and cardiac output in HF animals as compared to sham animals. The present study shows evidence that cardiac performance is impaired in HF rats. It is well known that cardiac contractility may be positively correlated with heart rate, preload, and afterload. Although not recorded in the present study, our data indicate that changes in hemodynamics were not compensated by the increase in systemic vascular resistance, suggesting a hidden cardiac depression.
Figure 1.
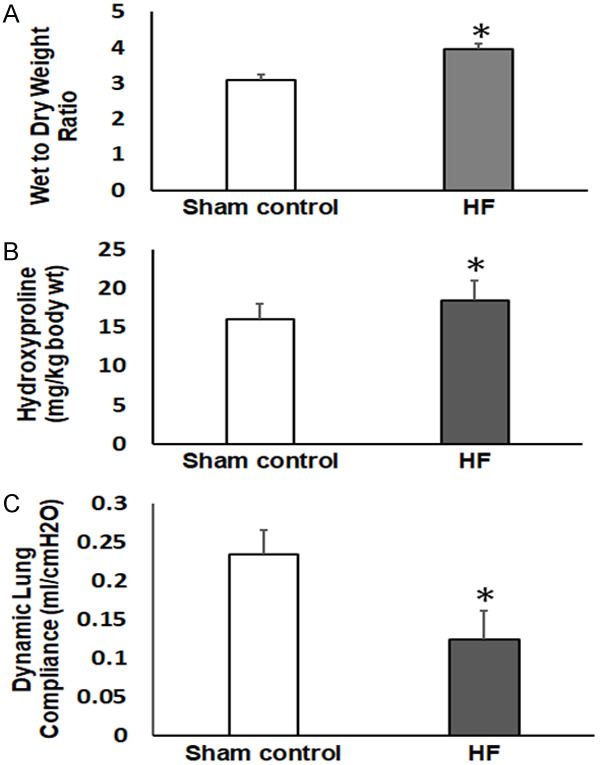
Lung parameters measured 7 days after the induction of heart failure (HF) compared to the sham control. A. Lung wet to dry weight ratios indicating pulmonary edema development. B. Pulmonary hydroxyproline content indicating collagen accumulation/fibrosis development. C. Dynamic lung compliance. *P<0.05; n=6 in the sham control group and n=8 in the HF group.
Table 1.
Measurement of cardiac function in heart failure rats at Day 7 as compared to sham control rats
| Sham Control 7 days | HF 7 Days | |
|---|---|---|
| MAP (mmHg) | 97 ± 8 | 92 ± 7 |
| HR (bts/min) | 350 ± 15 | 295 ± 12* |
| CO (ml/min) | 57 ± 5 | 37 ± 4* |
| SVR (units) | 1.9 ± 0.4 | 2.5 ± 0.3* |
P<0.05, HF vs. sham control (n=5 in each group).
Inflammation in the lungs after heart failure
As compared to sham operated controls, no changes were detected in lung histology in rats subjected to elevated LAP for 2 days (data not shown). However, immuno-histochemical staining of lung tissue 7 days after induction of HF compared to sham controls shows gross distortion of the lung architecture and the development of patchy interstitial fibrogenesis (Figure 2). These morphological changes were accompanied by infiltration of inflammatory cells into the lung tissue, as shown in Figure 2. Specifically, elevated macrophages and neutrophils counts, as well as an increase in myeloperoxidase activity, were recorded in BALF from the HF group as compared to sham controls on Day 7 (Figure 3). Figure 4 shows elevated iNOS expression, indicative of increased macrophage activity, in the HF group compared to sham controls.
Figure 2.
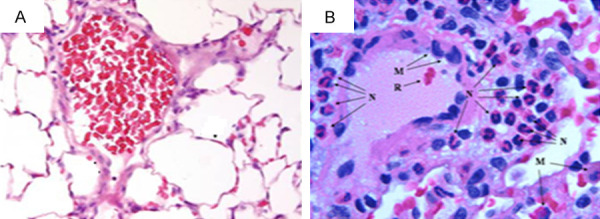
Lung tissue sections stained with hematoxylin and eosin after 7 days of sham control (A) and 7 days of heart failure (HF) (B) in rats. Changes in alveolar architecture and increased inflammatory cell infiltration are shown. (M, macrophage; N, neutrophil; R, red blood cells).
Figure 3.
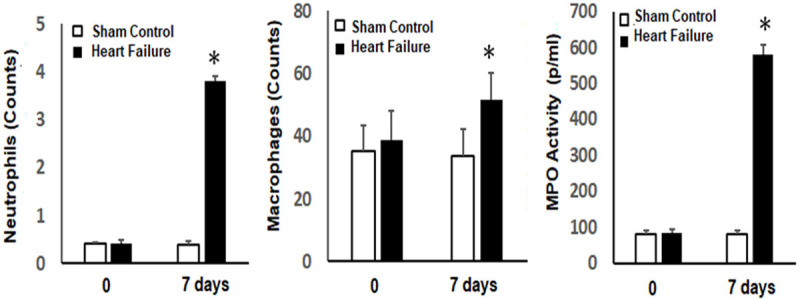
Inflammatory cell numbers in BALF and myeloperoxidase activity (MPO) after induction of heart failure (HF) compared to sham control rats at 0 and 7 days. *P<0.05; n=6 in sham control groups and n=8 in HF groups.
Figure 4.
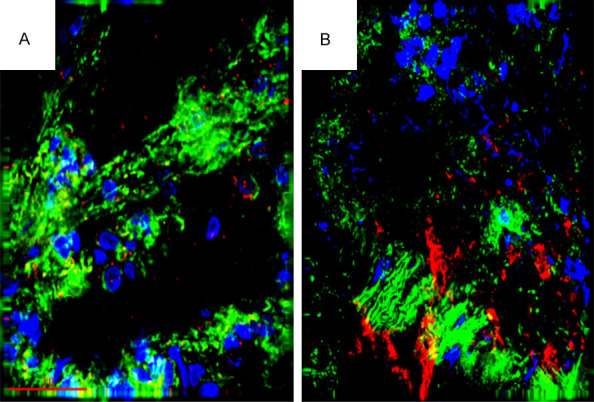
Immunohistochemical staining for iNOS (red) in lung sections 7 days after induction of heart failure (HF) (B) or in the sham control (A). Inducible NOS staining is indicative of increased macrophage activity. (Blue is nuclear staining and green is F-Actin staining).
Alterations in cytokine levels in BALF after induction of HF
As shown in Figure 5, TNFα increased after induction of HF; however, no changes in IL-1 and, surprisingly, no changes in TGF-β were detected. Figure 6 shows increased TNF-α specific staining in the lungs of HF animals 7 days after HF induction compared to sham control animals. Our data support the hypothesis that the presence of prolonged pulmonary edema in our model of HF is associated with infiltration of inflammatory cells, including macrophages and neutrophils and, subsequently, increased levels of proinflammatory cytokines, including TNF-α.
Figure 5.
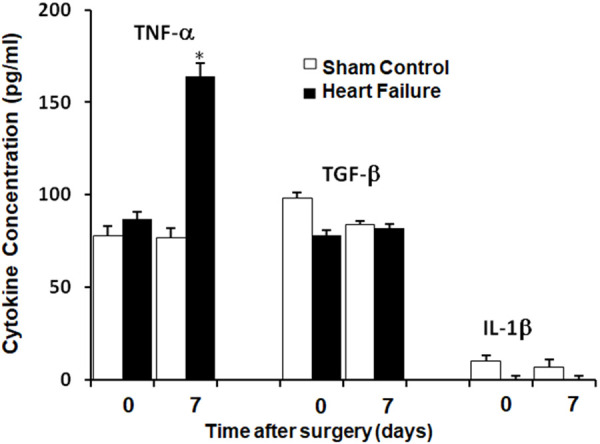
Cytokine Levels in BALF after induction of heart failure (HF) compared to sham control rats at 0 and 7 days. *P<0.05, n=6 in sham control group and n=8 in HF group.
Figure 6.
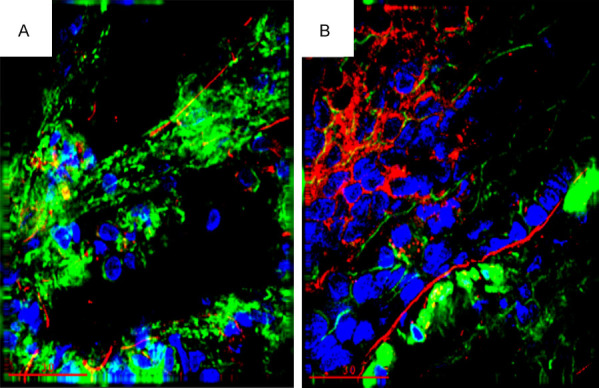
Increased TNF-α expression in lungs from the heart failure (HF) group (B) compared to the sham control group at 7 days (A). (green, F-actin; blue, Nuclei; red, TNF-α).
Contribution of inflammatory cell infiltration to pulmonary fibrosis development in chronic heart failure: loss of function
To demonstrate the contribution of macrophages to the development of pulmonary fibrosis, macrophages were depleted with PAF-AH. Fibrosis development was attenuated (Figure 7A and 7B) after treatment with PAF-AH in HF rats compared to vehicle-treated HF rats. In addition, wet- to- dry weight ratios in the lung decreased in the HF group treated with PAF-AH compared to the vehicle-treated HF group (Figure 7C). Interestingly, macrophage depletion did not seem to affect TNF-α levels (Figure 7D) implying that at least one other proinflammatory cytokine is involved in cardiogenic pulmonary fibrosis development.
Figure 7.
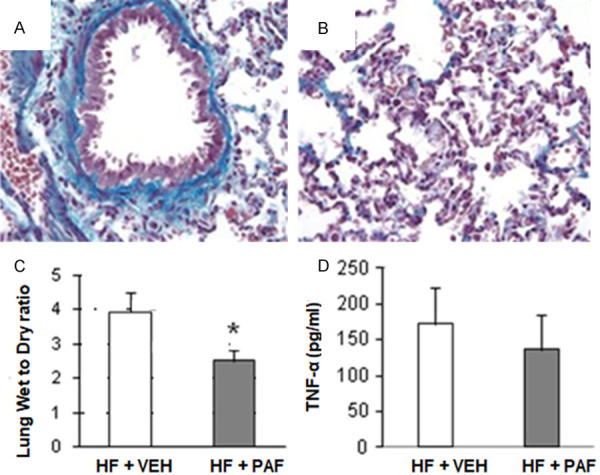
The effects of macrophage depletion with platelet-activating factor acetylhydrolase (PAF-AH). A and B. Show collagen staining (blue) in the lungs of rats with heart failure (HF) treated with vehicle (VEH) or with HF treated with PAF-AH (7 days), respectively. C. PAF-AH treatment attenuated HF-induced edema formation, as indicated by decreased wet-to-dry weight ratio. D. TNF-α levels in the lung were not significantly altered after treatment with PAF-AH in the HF group compared to VEH treated HF group. *, P<0.05, n=6 (HF + VEH) and n=6 (PAF-AH) treated group.
The contribution of neutrophils to pulmonary fibrosis development was assessed by depleting neutrophils with cyclophosphamide (CTX). Neutrophil depletion reduced collagen content and wet- to- dry weight ratios similar to macrophage depletion (Figure 8A-C), In contrast to macrophage depletion, neutrophils also inhibited TNF-α secretion in the lungs (Figure 8D).
Figure 8.
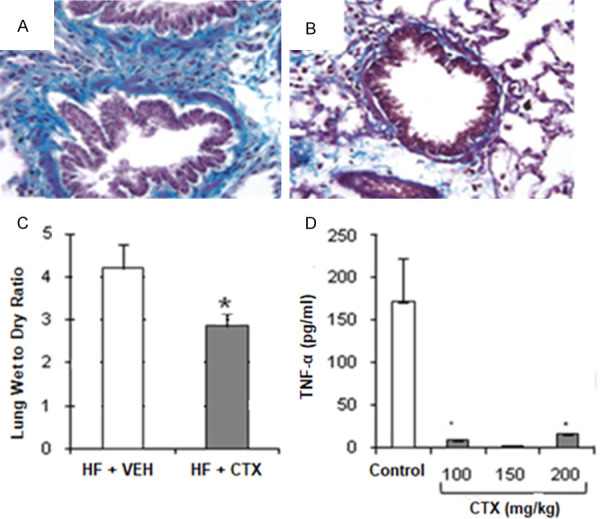
The effects of neutrophil depletion with cyclophosphamide (CTX). A and B. Show collagen staining (blue) in the lungs of rats with heart failure (HF) treated with vehicle (VEH) or with HF treated with CTX (7 days), respectively. C. CTX treatment (150 mg/kg) attenuated HF-induced edema formation, as indicated by decreased wet-to-dry weight ratio. D. Decreased TNF-α levels in the lung after treatment with CTX in the HF group compared to VEH treated HF group. *, P<0.05, n=6 (HF + VEH) and n=6 (CTX) treated groups.
Discussion
Our model of increased LAP causes pulmonary edema (increased wet to dry ratio), pulmonary fibrosis development as evidenced by increased hydroxyproline content, and decreased lung compliance. Moreover, our data support our hypothesis that proinflammatory cells mediate pulmonary fibrosis development via increased cytokine secretion in HF.
We hypothesize that the initial fluid accumulation in the lungs following elevated LAP induces an inflammatory response with activation of neutrophils and macrophages. Cytokine production by neutrophils and macrophages leads to more edema and neutrophil/macrophage infiltration, setting up a chronic damaging positive feedback loop. Since fibrosis of the alveolar wall is generally an irreversible process, an understanding of the mechanisms modulating the fibrotic state is necessary to develop treatment strategies and drugs to prevent pulmonary fibrosis in patients with chronic heart failure.
Current models for studying the development of pulmonary fibrosis have limited physiological and clinical relevance. Bleomycin-induced lung injury, the most commonly used model for idiopathic pulmonary fibrosis (IPF), is relevant for drug-induced pulmonary fibrosis but not for the study of cardiogenic pulmonary fibrosis development [23]. The mechanisms for the development of pulmonary fibrosis with HF are likely to be somewhat different from the mechanisms for drug- or chemical-induced pulmonary fibrosis. Our animal model of HF is a relevant model that mimics the pulmonary manifestations that occur with left HF in patients [24]. Since pulmonary fibrosis development is a well-known problem in HF, mechanisms of pulmonary fibrosis development should be studied in a physiologically relevant model of HF. Clinically, cardiogenic pulmonary edema is caused by the increase in LAP when the left heart fails. We have demonstrated that 7 days of LAP elevation resulted in significant alteration of cardiac function, including decreases in heart rate and cardiac output similar to previously described models of cardiac failure [24]. Fibrosis is a typical response to the persistence of edema fluid in almost any organ [12,13]. The increased LAP causes rapid fluid accumulation within the lung interstitial spaces in our model of heart failure, mimicking a clinical scenario. To our knowledge, this novel model has not been utilized for studying the development of pulmonary fibrosis development. Of the various forms of left heart failure, mitral valve failure probably most closely approximates pure left atrial pressure elevation [25].
Our data show that chronic LAP elevation leads to increases in lung edema, accompanied by increased hydroxyproline and collagen formation. These changes were accompanied by significant changes in pulmonary dysfunction, as shown by decreased dynamic lung compliance. Thus, our results are consistent with other models of pulmonary fibrosis, including bleomycin silica administration [26]. Furthermore, pulmonary tissue architecture showed significant changes over time in terms of fibrosis development associated with significant deterioration of the mechanical properties of the lung. Specifically, no changes were detected in lung histology in rats subjected to LAP elevation for 2 days; whereas an accumulation of red blood cells in the alveolar occurs at 4 days and was accentuated at 7 days following elevated LAP, suggesting severe congestion. We have further shown gross distortion of the lung architecture that occurs 7 days after LAP elevation. Long term elevation of LAP induced alveolar damage associated with an increase in the thickness of the interstitial space due to cellular infiltration. Increased LAP causes the initial pulmonary edema formation and inflammation of the lungs escalates the edema formation. We also demonstrate that elevated LAP results in the accumulation of neutrophils and macrophages in the lungs associated with increased wet-to-dry weight ratios. This was later demonstrated by the macrophage and neutrophil depletion experiments, which prevent much of the edema formation in our model. Therefore, our data support our hypothesis that proinflammatory cells mediate pulmonary fibrosis development via increased cytokine secretion in HF. Interestingly, macrophage depletion, as compared to neutrophil depletion, did not seem to affect TNF-α levels implying that at least one other proinflammatory cytokine is involved in cardiogenic pulmonary fibrosis development.
Changes in our model resemble those of human pulmonary fibrosis, namely inflammatory cell infiltration, increased collagen content, and reduced lung volumes and compliance. We further propose the following mechanisms to be involved in pulmonary fibrosis formation due to chronic edema: When tissues, including the lung, become edematous, the interstitial matrix swells, thus increasing the space between adjacent cells and changing the environment surrounding the cells. Edema may, thus, affect both cell-cell and cell-matrix interactions. According to previous studies, these changes in cell-matrix or cell-cell interactions may result in fibrosis development [27]. Cellular contact with the extracellular matrix may also regulate the synthesis of cytokines that influence extracellular matrix production. As such, our data show that TNF-α was significantly elevated in BAL fluid 7 days following HF, indicating pro-inflammatory activities in our model of HF that is further substantiated by the neutrophil depletion experiment. Thus, our study shows that activated neutrophils have an important role in initiating the inflammatory processes following elevated LAP, probably by releasing TNF-α. These results suggest that cell interactions might play a major role in pulmonary fibrosis. Therefore, cross-talk between fibroblasts, neutrophils, and macrophages remains to be studied in our model of elevated LAP-induced pulmonary fibrosis.
In summary, we have developed a small animal model of chronic LAP elevation mimicking the left ventricular backward failure seen in chronic HF. The resulting chronic pulmonary edema was associated with pulmonary tissue changes, including increased hydroxyproline and collagen content indicating fibrosis formation. Furthermore, our studies show that activated neutrophils have an important role in initiating the inflammatory processes following elevated LAP, probably by releasing TNF-α. Consequently, our animal model is suitable for further studies focusing on the mechanisms of pulmonary fibrosis formation associated with chronic pulmonary edema.
Acknowledgements
The authors would like to thank Kelli Wallen for assisting in the manuscript submission.
Disclosure of conflict of interest
None.
References
- 1.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB American Heart Association Statistics Committee; Stroke Statistics Subcommittee. Heart disease and stroke statistics-2016 update: a report from the american heart association. Circulation. 2016;133:e38–360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 2.Ahmed MS, Oie E, Vinge LE, von Lueder TG, Attramadal T, Attramadal H. Induction of pulmonary connective tissue growth factor in heart failure is associated with pulmonary parenchymal and vascular remodeling. Cardiovasc Res. 2007;74:323–33. doi: 10.1016/j.cardiores.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 3.Dimopoulou I, Daganou M, Tsintzas OK, Tzelepis GE. Effects of severity of long-standing congestive heart failure on pulmonary function. Respir Med. 1998;92:1321–5. doi: 10.1016/s0954-6111(98)90136-6. [DOI] [PubMed] [Google Scholar]
- 4.Forfia PR, Vaidya A, Wiegers SE. Pulmonary heart disease: the heart-lung interaction and its impact on patient phenotypes. Pulm Circ. 2013;3:5–19. doi: 10.4103/2045-8932.109910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iversen KK, Kjaergaard J, Akkan D, Kober L, Torp-Pedersen C, Hassager C, Vestbo J, Kjoller E ECHOS-Lung Function Study Group. Chronic obstructive pulmonary disease in patients admitted with heart failure. J Intern Med. 2008;264:361–9. doi: 10.1111/j.1365-2796.2008.01975.x. [DOI] [PubMed] [Google Scholar]
- 6.Kee K, Naughton MT. Heart failure and the lung. Circ J. 2010;74:2507–16. doi: 10.1253/circj.cj-10-0869. [DOI] [PubMed] [Google Scholar]
- 7.Wasserman K, Zhang YY, Gitt A, Belardinelli R, Koike A, Lubarsky L, Agostoni PG. Lung function and exercise gas exchange in chronic heart failure. Circulation. 1997;96:2221–7. doi: 10.1161/01.cir.96.7.2221. [DOI] [PubMed] [Google Scholar]
- 8.Jasmin JF, Calderone A, Leung TK, Villeneuve L, Dupuis J. Lung structural remodeling and pulmonary hypertension after myocardial infarction: complete reversal with irbesartan. Cardiovasc Res. 2003;58:621–31. doi: 10.1016/s0008-6363(03)00290-6. [DOI] [PubMed] [Google Scholar]
- 9.Kapanci Y, Burgan S, Pietra GG, Conne B, Gabbiani G. Modulation of actin isoform expression in alveolar myofibroblasts (contractile interstitial cells) during pulmonary hypertension. Am J Pathol. 1990;136:881–9. [PMC free article] [PubMed] [Google Scholar]
- 10.Kingsbury MP, Huang W, Donnelly JL, Jackson E, Needham E, Turner MA, Sheridan DJ. Structural remodelling of lungs in chronic heart failure. Basic Res Cardiol. 2003;98:295–303. doi: 10.1007/s00395-003-0419-6. [DOI] [PubMed] [Google Scholar]
- 11.Drake RE, Doursout MF. Pulmonary edema and elevated left atrial pressure: four hours and beyond. News Physiol Sci. 2002;17:223–6. doi: 10.1152/nips.01399.2002. [DOI] [PubMed] [Google Scholar]
- 12.Davis KL, Laine GA, Geissler HJ, Mehlhorn U, Brennan M, Allen SJ. Effects of myocardial edema on the development of myocardial interstitial fibrosis. Microcirculation. 2000;7:269–80. [PubMed] [Google Scholar]
- 13.Witte CL, Witte MH, Dumont AE. Pathophysiology of chronic edema, lympedema and fibrosis. In: Staub NC, Taylor AE, editors. Edema. New York: Raven; 1984. pp. 521–542. [Google Scholar]
- 14.Tashiro J, Rubio GA, Limper AH, Williams K, Elliot SJ, Ninou I, Aidinis V, Tzouvelekis A, Glassberg MK. Exploring animal models that resemble idiopathic pulmonary fibrosis. Front Med (Lausanne) 2017;4:118. doi: 10.3389/fmed.2017.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun Y, Weber KT. Animal models of cardiac fibrosis. Methods Mol Med. 2005;117:273–90. doi: 10.1385/1-59259-940-0:273. [DOI] [PubMed] [Google Scholar]
- 16.Kuwano K, Hagimoto N, Hara N. Molecular mechanisms of pulmonary fibrosis and current treatment. Curr Mol Med. 2001;1:551–73. doi: 10.2174/1566524013363401. [DOI] [PubMed] [Google Scholar]
- 17.Dubaybo BA. Role of tumor necrosis factor-alpha in regulating fibrotic lung repair. Res Commun Mol Pathol Pharmacol. 1998;101:69–83. [PubMed] [Google Scholar]
- 18.Sime PJ, Marr RA, Gauldie D, Xing Z, Hewlett BR, Graham FL, Gauldie J. Transfer of tumor necrosis factor-alpha to rat lung induces severe pulmonary inflammation and patchy interstitial fibrogenesis with induction of transforming growth factor-beta1 and myofibroblasts. Am J Pathol. 1998;153:825–32. doi: 10.1016/s0002-9440(10)65624-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doursout MF, Wouters P, Kashimoto S, Hartley CJ, Rabinovitz R, Chelly JE. Measurement of cardiac function in conscious rats. Ultrasound Med Biol. 2001;27:195–202. doi: 10.1016/s0301-5629(00)00330-6. [DOI] [PubMed] [Google Scholar]
- 20.Pulli B, Ali M, Forghani R, Schob S, Hsieh KL, Wojtkiewicz G, Linnoila JJ, Chen JW. Measuring myeloperoxidase activity in biological samples. PLoS One. 2013;8:e67976. doi: 10.1371/journal.pone.0067976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bergman I, Loxley R. Lung tissue hydrolysates: studies of the optimum conditions for the spectrophotometric determination of hydroxyproline. Analyst. 1969;94:575–84. doi: 10.1039/an9699400575. [DOI] [PubMed] [Google Scholar]
- 22.Bick RJ, Wood DE, Poindexter B, McMillin JB, Karoly A, Wang D, Bunting R, McCann T, Law GJ, Buja LM. Cytokines increase neonatal cardiac myocyte calcium concentrations: the involvement of nitric oxide and cyclic nucleotides. J Interferon Cytokine Res. 1999;19:645–53. doi: 10.1089/107999099313794. [DOI] [PubMed] [Google Scholar]
- 23.Borzone G, Moreno R, Urrea R, Meneses M, Oyarzun M, Lisboa C. Bleomycin-induced chronic lung damage does not resemble human idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2001;163:1648–53. doi: 10.1164/ajrccm.163.7.2006132. [DOI] [PubMed] [Google Scholar]
- 24.Gehlbach BK, Geppert E. The pulmonary manifestations of left heart failure. Chest. 2004;125:669–82. doi: 10.1378/chest.125.2.669. [DOI] [PubMed] [Google Scholar]
- 25.Pedersen HD, Haggstrom J. Mitral valve prolapse in the dog: a model of mitral valve prolapse in man. Cardiovasc Res. 2000;47:234–43. doi: 10.1016/s0008-6363(00)00113-9. [DOI] [PubMed] [Google Scholar]
- 26.Gilhodes JC, Jule Y, Kreuz S, Stierstorfer B, Stiller D, Wollin L. Quantification of pulmonary fibrosis in a bleomycin mouse model using automated histological image analysis. PLoS One. 2017;12:e0170561. doi: 10.1371/journal.pone.0170561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chapman HA. Disorders of lung matrix remodeling. J Clin Invest. 2004;113:148–57. doi: 10.1172/JCI20729. [DOI] [PMC free article] [PubMed] [Google Scholar]