

Abstract
Background
Idiopathic pulmonary fibrosis (IPF) is a chronic and progressive lung disease which presents a grave prognosis for diagnosed patients. Nintedanib (a triple tyrosine kinase inhibitor) and pirfenidone (unclear mechanism of action) are the only approved therapies for IPF, but have limited efficacy. The pathogenic mechanisms of this disease are not fully elucidated; however, a role for mast cells (MCs) has been postulated.
Objectives
The aim of this work was to investigate a role for MCs in IPF and to understand whether nintedanib or pirfenidone could impact MC function.
Methods and results
MCs were significantly elevated in human IPF lung and negatively correlated with baseline lung function (FVC). Importantly, MCs were positively associated with the number of fibroblast foci, which has been linked to increased mortality. Furthermore, MCs were increased in the region immediately surrounding the fibroblast foci, and co-culture studies confirmed a role for MC–fibroblast crosstalk in fibrosis. Nintedanib but not pirfenidone inhibited recombinant stem cell factor (SCF)–induced MC survival. Further evaluation of nintedanib determined that it also inhibited human fibroblast-mediated MC survival. This was likely via a direct effect on ckit (SCF receptor) since nintedanib blocked SCF-stimulated ckit phosphorylation, as well as downstream effects on MC proliferation and cytokine release. In addition, nintedanib ablated the increase in lung MCs and impacted high tissue density frequency (HDFm) in a rat bleomycin model of lung fibrosis.
Conclusion
Nintedanib inhibits MC survival and activation and thus provides a novel additional mechanism by which this drug may exert anti-fibrotic effects in patients with IPF.
Keywords: idiopathic pulmonary fibrosis, hypersensitivity pneumonitis, innate immunity
Key messages.
What is the key question?
Do mast cells (MCs) correlate with idiopathic pulmonary fibrosis (IPF) disease activity, and can either nintedanib or pirfenidone modulate MC survival or activation?
What is the bottom line?
MCs are increased in IPF lung and negatively correlate with baseline lung function (FVC) and positively correlate with the number of fibroblast foci. Nintedanib, but not pirfenidone, can inhibit human MC survival and activation in vitro and MC recruitment in the rat bleomycin model.
Why read on?
Inhibition of MC survival or activation may be a novel additional mechanism by which nintedanib exerts anti-fibrotic effects in patients with IPF.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive and ultimately fatal lung disease with unknown aetiology and increasing incidence.1 While still incompletely understood, the pathogenesis of IPF is thought to involve recurrent injury to the alveolar epithelium inducing an aberrant wound healing response, characterised by activation and proliferation of fibroblasts and myofibroblasts.2 Formation of the fibroblastic foci, as well as the deposition of extracellular matrix (ECM) are downstream pathological features3 that result in an abnormal lung structure referred to as the characteristic ‘honeycombing’ morphology, which is associated with impaired lung function.4 The role of structural cells such as fibroblasts in the pathogenesis of fibrosis is well accepted; however, less understood is the contribution of immune cells which are also present in IPF lung.5–9 The mast cell (MC) is an innate immune cell well known for its involvement in allergy, asthma and in host defence against parasites.10 11 However, MCs are also a key cell responding to tissue injury and are found very early at the site of the wound.12 On activation, MCs can release an array of mediators, which have been shown to partake in all stages of the wound healing process, including inflammation, proliferation and re-modelling.13 Thus, given the dysregulated nature of repair in IPF, this cell could be a potential pathogenic link to this disease.
MCs have been postulated to be pro-fibrotic in IPF, although the data are conflicting and their role remains controversial. Several studies have shown that MCs and their mediators, such as histamine, are increased in lung tissue and bronchoalveolar lavage from patients with IPF.14–20 Mechanistically, reports have demonstrated that human lung fibroblast and MC crosstalk leads to increased MC activation and survival, as well as fibroblast proliferation.17 Others have suggested that the MC mediators tryptase, chymase and histamine induce both lung and skin fibroblast proliferation and collagen production,19 21–23 and Shimbori and colleagues recently demonstrated that stretch-induced degranulating MCs activate the pro-fibrotic cytokine TGF-β1.20 Andersson and colleagues provide evidence for a switch to the connective tissue MC subtype expressing both chymase and tryptase proteases (MCTC) in IPF lung,24 and this increase in MCTCs in IPF has been confirmed recently.20 In contrast, Cha and colleagues propose a protective role for lung MCs since patients with higher numbers of these cells had less decline in lung function.16
Stem cell factor (SCF) is a key survival factor for mast cells, acting through the tyrosine kinase receptor ckit (also known as CD117 or SCF receptor). Importantly, a phase II clinical trial with a ckit inhibitor Imatinib (Gleevec) had no significant effect on lung function decline (or mortality), although the effect on MCs was not determined in this study.25
Recently, two small molecule drugs, pirfenidone and nintedanib, were licensed for IPF following successful phase III clinical trials demonstrating a slowing in lung function decline.26–28 Pirfenidone is a small molecule whose precise target is unknown, that exerts effects on transforming growth factor (TGF)-β1 signalling and PDGF as well as acting as an antioxidant,29 whereas nintedanib is a broad tyrosine kinase inhibitor with known activity on platelet-derived growth factor (PDGF), fibroblast growth factor (FGF) and vascular endothelial growth factor (VEGF) receptors.30 Thus, it is plausible that nintedanib may also have activity against the ckit receptor, which is found on MCs. Notably, the effects of nintedanib or pirfenidone on MC survival and function have not yet been investigated.
In this study, we sought to confirm that MCs are increased in IPF versus non-diseased lung, and compared MC density in other fibrotic lung diseases such as chronic hypersensitivity pneumonitis (HP) and non-specific interstitial pneumonia (NSIP). We also assessed MCs for pro-fibrotic functions. Furthermore, we investigated whether nintedanib or pirfenidone could impact MC survival, and therefore whether this could be an additional novel mechanism explaining why these small molecules are efficacious in IPF.
Materials and methods
See online supplementary for a more detailed version of these methods.
thoraxjnl-2019-214000supp001.pdf (3.4MB, pdf)
MC quantification and phenotyping
Formalin-fixed paraffin-embedded (FFPE) sections from lung biopsy specimens from patients with IPF (n=17), chronic HP (n=9) and NSIP (n=9) and normal adjacent lung specimens taken from patients with lung cancer (normals; n=20) were obtained from Royal Papworth Hospital Research Tissue Bank. Patients gave written consent and the study was approved by NRES Committee East of England—Cambridge East via Royal Papworth Hospital Research Tissue Bank generic ethical approval.
Consecutive 3 µm sections of the FFPE samples were stained for H&E or dual immunohistochemical staining for chymase and tryptase (Ventana Discovery Ultra; Roche). Tissue area was calculated by subtraction of white space from the total lung section area. MCs were analysed per area of lung section and per area of tissue. Fibroblastic foci, established fibrosis and non-fibrotic areas of the IPF lung sections were annotated by a registered senior pathologist (online supplementary figure S1), and the MC populations quantified. Subsequently, the outer edge of the foci was extended by 500 µm and MCs in the surrounding area were quantified. See online supplementary methods for more details.
MC protease signature analysis in IPF lung
Additional lung tissue was obtained from patients with IPF (n=10) undergoing lung volume reduction surgery or lung transplantation at Temple University Lung Center as part of a separate study. Patients gave written consent and the study was approved by the institutional review board at Temple University Lung Center. Normal lung tissue samples (n=9) were obtained from the National Disease Research Interchange (NDRI), in accordance with local internal review board ethical approval. Microarray analysis was performed using Affymetrix HG-U133Plus2.0 microarrays. For more information, see online supplementary methods.
Cell culture and in vitro assays
Cord blood–derived MCs (CBMCs) were differentiated from CD133+ cord blood precursors (Lonza). These cells are predominantly of the MCT phenotype (approximately 70%), as determined using a similar staining protocol to that used to phenotype the lung tissue MCs. Conditioned media was prepared from CBMCs that had been cultured for 7 days. CBMC lysates were prepared by sonication and freeze thawing. See online supplementary methods for details.
CBMC survival
CBMCs were cultured with recombinant SCF (MedImmune, see online supplementary methods for details) in the presence or absence of nintedanib, pirfenidone (both Selleckchem.com) or DMSO vehicle (Sigma Aldrich). After 7 days, CBMCs were stained with a viability dye and anti-ckit mIgG1 APC (BD Pharmingen) and the number of viable ckit-positive MCs in a fixed volume was counted in a flow cytometer (LSRII Fortessa) and compared with control.
CBMC proliferation, CCL2 release and phosphorylation of ckit
CBMCs were stimulated with recombinant SCF (MedImmune) in the presence or absence of nintedanib (LC Laboratories), DMSO vehicle, anti-SCF pAb (AF-255-NA) or goat IgG isotype (both antibodies R&D Systems). Phosphorylation of ckit was measured using an electrochemiluminescence-based phospho-ckit assay (MesoScale Discovery) after a 10 min stimulation and CBMC proliferation was measured by 3H-thymidine incorporation after a 3-day stimulation. CCL2 was measured in culture supernatants by MSD (MesoScale Discovery). See online supplementary methods for details.
CBMC:NHLF co-culture and flow cytometry assessment of MC and fibroblast numbers
Normal human lung fibroblasts (NHLFs) and CBMCs were cultured alone or co-cultured at a 1:1 ratio in media containing anti-SCF, goat IgG isotype control (67 nM; both R&D Systems), nintedanib (30 nM; LC Laboratories), DMSO vehicle or media alone over a time course up to 21 days. At each time point, cells were stained with a viability dye and anti-ckit APC mIgG1 and the number of ckit-positive (MCs) and ckit-negative viable cells (NHLFs) in a fixed volume was counted in a flow cytometer (LSRII Fortessa). See online supplementary methods for more details.
Fibroblast stimulation
NHLFs were stimulated with recombinant human bFGF in the presence of nintedanib (LC Laboratories), pirfenidone (Selleckchem.com), or DMSO vehicle for 48 hours or for 48–72 hours with MC lysates, conditioned media or MCs at a ratio of 1:1. TGF-β1 (R&D Systems) was included as a positive control for α smooth muscle actin (αSMA) experiments. Proliferation was assessed by 3H-thymidine incorporation. αSMA expression was assessed by immunofluorescence following a 72-hour stimulation.
Animal studies
Rats (male CD, 6–8 weeks old) received 0.9 U/kg bleomycin sulphate (Bleo-Kyowa) or saline (1 mL/kg) intratracheally under isoflurane anaesthesia (study day 0). Nintedanib (LC Laboratories) was dosed at 60 mg/kg daily by oral gavage, from day 0 to day 13. Changes in MC number and fibrosis markers were assessed on day 14. Animal housing and procedures were approved by the local ethical committees (The Babraham Institute, UK) in accordance with institutional and the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines (see online supplementary methods for more details).
Sections were stained using H&E and Giemsa for MC quantification, HE alone or Masson’s trichrome (MTC) for collagen using standard protocols (Damsgaard; Leica Autostainer). HE and MTC scanned images were analysed by Biocellvia using their IPF assay based on assessment of parenchymal tissue density using a proprietary software program. The high tissue density frequency (HDFm) was calculated to specifically quantify fibrotic tissue in lung parenchyma. The proportion of collagen staining by MTC was measured using standard image analysis methods (see31 for validation of this method and online supplementary for more details).
Statistical analysis
Details of the sample size and statistical analysis performed can be found in the relevant figure legend. Two group comparisons were carried out by t-test for normally distributed data or Mann-Whitney U non-parametric test. Multiple group comparisons were assessed by one-way analysis of variance (ANOVA) with Sidak’s post hoc multiple comparisons for normally distributed data, or where assumptions of normality or homogeneity of variance were not valid, by Kruskal-Wallis with Dunn’s post hoc multiple comparisons test. Analysis of MCs in different regions of the lung within the same patient was assessed by repeated measures ANOVA with Geisser-Greenhouse correction (for assumption of sphericity) with Sidak’s multiple comparisons. Correlations were assessed by Pearson’s correlation or non-parametric Spearman’s correlation. All analyses and plots were generated in GraphPad Prism, and a p value of less than 0.05 was considered statistically significant.
Results
MCs are significantly elevated in IPF lung
We sought to determine the number, phenotype and localisation of MCs in the lungs of patients with IPF (n=17) and compared this with MCs in non-diseased lung taken from age-matched patients undergoing cancer resections (normals; n=20). We also examined whether MCs are elevated across other fibrotic lung conditions in samples taken from nine patients with chronic HP and nine patients with NSIP (table 1). We found that the number of MCs per area of lung were significantly increased in both IPF and HP lung samples. In contrast, MC numbers were not altered in NSIP lung samples when compared with normal (non-fibrotic) controls (figure 1A). Although the control and IPF groups are not matched according to gender, this is unlikely to be responsible for the observed difference in MC density; we observed no significant difference in MC density between males and females in healthy lung (online supplementary figure S2).
Table 1.
Demographic and spirometric data at baseline
| Controls (n=20) | IPF (n=17) | Chronic HP (n=9) | NSIP (n=9) | Control vs IPF | Control vs chronic HP | Control vs NSIP | |
| Age, years | 70 (63–73) (n=18) | 64 (57–71) | 60 (48–67) | 53 (48–65) | p=0.6313 | p=0.0576 | p=0.0254 |
| Gender, male/female | 8/12 | 14/3 | 4/5 | 2/7 | p=0.0176 | p>0.9999 | p=0.4311 |
| Smoking status, ever/never-smokers | 11/4 | 11/5 | 3/4 | 5/3 | p>0.9999 | p=0.3426 | p=0.657 |
| FVC, L | 3.2 (2.6–4.3) (n=18) | 2.6 (2.5–3.3) (n=16) | 1.9 (1.4–2.6) (n=5) | 2.0 (1.5–3.1) (n=6) | p=0.372 | p=0.0077 | p=0.0418 |
| FVC % predicted, % | 108.5 (103.5–118.3) | 77.0 (65.8–95.0) | 58.7 (41.0–70.8) | 77.0 (60.2–91.2) | p=0.0009 | p=0.0001 | p=0.0057 |
| DLCO, mL/min/mm Hg | 6.3 (5.5–7.4) (n=17) | 4.2 (3.4–5.4) (n=14) | 4.2 (2.1–4.2) (n=5) | 4.2 (3.3–4.6) (n=5) | p=0.0021 | p=0.0009 | p=0.0127 |
| DLCO % predicted, % | 80.0 (72.0–88.5) | 54.5 (45.5–62.0) | 55.0 (22.7–55.6) | 56.4 (43.6–57.2) | p=0.0002 | p=0.0003 | p=0.0039 |
| Steroid use at baseline yes/no | 3/11 | 1/14 | 3/3 | 3/3 | p=0.3295 | p=0.3027 | p=0.3027 |
Data expressed as median (IQR) unless specified. Age is recorded at time of diagnosis apart from controls whose age is captured at the time of baseline measurements. Groups compared by Kruskal-Wallis test with Dunn’s post hoc test for continuous variables, and by Fisher’s exact test for categorical variables.
DLCO, diffusing capacity for carbon monoxide; HP, hypersensitivity pneumonitis; IPF, idiopathic pulmonary fibrosis; n, number of available data; NSIP, non-specific interstitial pneumonia.
Figure 1.

Increased mast cells (MCs) in idiopathic pulmonary fibrosis (IPF) are associated with a higher number of fibroblastic foci. (A) The number of MCs/mm2 lung section is significantly higher in patients with IPF (n=17) compared with normals (n=20). HP, hypersensitivity pneumonitis (n=9); NSIP, non-specific interstitial pneumonia (n=9). (B) The proportion of lung section that is tissue (vs airspace) is higher in patients with IPF. (C) The number of MCs/mm2 tissue is higher in IPF than normal lung. (D) Representative image of lung. Arrows point to tryptase MCT (red), chymase (yellow) and dual positive MCTC cells (orange). (E) The number of MCs per area lung is positively correlated with the number of fibroblast foci per area lung. (F) There is a negative correlation between the number of MCs/area lung section and the baseline FVC. Closed circles, normals; open circles, IPF. (G) Distribution of MCs across IPF lung. MCs are no higher within the foci than other regions. (H) MCs are higher in the 500 µm region surrounding the foci. Details of the statistical tests used: (A) Kruskal-Wallis with Dunn’s multiple comparison, (B) Mann-Whitney U test, (C) unpaired t-test, (E) Pearson’s correlation, (F) Spearman’s correlation and (G and H) repeated measures ANOVA with Geisser-Greenhouse correction (for assumption of sphericity) with Sidak’s multiple comparisons. Median and IQR plotted for non-parametric data and mean and SD for parametric data.
Since the fibrotic process involves the deposition of extracellular matrix, leading to increased tissue mass, we measured the proportion of tissue mass area in the lung sections (by subtracting the air spaces) and found this to be enhanced in IPF lung compared with normal lung (figure 1B). Thus, we employed a more stringent analysis which calculated the MC number per area of tissue to confirm whether MC density was actually elevated, and not simply due to the increase in tissue mass in IPF lung. This analysis was possible due to the use of automated software to subtract the white airspaces. The elevation in MC number in IPF lung remained statistically significant (figure 1C). All further data are expressed as MCs per tissue area unless specified, to account for this increase in tissue area in fibrotic lung.
To determine whether increased MCs resulted in a switch in MC subtypes, staining for MC proteases was performed and chymase, tryptase (MCT) or double-positive (MCTC) MCs (figure 1D) were quantified according to both total area (online supplementary table S1) and tissue area (table 2). The number of chymase+ MCs and double-positive MCTCs were significantly increased in IPF lung compared with non-fibrotic controls, with a similar trend in tryptase+ MCTs (table 2).
Table 2.
MC subtypes in IPF vs non-fibrotic lungs (number of MCs/mm2 tissue area)
| Non-fibrotic lung (n=20); number of MCs/mm2 tissue area | IPF (n=17); number of MCs/mm2 tissue area | P value | |
| Chymase only | 3 (0–39) | 13 (4–53) | 0.0003 |
| Tryptase only (MCT) | 65.5 (1–271) | 122 (5–374) | 0.0894 |
| Chymase+ tryptase (MCTC) | 1 (0–64) | 3 (1–37) | 0.0284 |
Median and range shown. Groups compared by Mann-Whitney U test.
IPF, idiopathic pulmonary fibrosis; MC, mast cell.
MC density is associated with the number of fibroblastic foci and inversely correlates with lung function
Increased fibroblast foci in the lungs of patients with IPF has been shown to predict mortality.32 33 Therefore, we examined whether there was an association between MCs and the number of fibroblastic foci in our cohort. Strikingly, we observed a moderately strong and significant positive correlation between the numbers of MCs and fibroblast foci in patients with IPF (r2=0.3545, figure 1E). Furthermore, we also found that MC numbers per area of lung were strongly inversely correlated with lung function (% predicted FVC), that is, patients with lower FVC had increased numbers of lung MCs (rs=−0.4616, figure 1F). Interestingly, a negative trend between MCs per area of tissue and the % predicted FVC was also observed, but this did not reach statistical significance (online supplementary figure S3).
We next examined the localisation of MCs within IPF lung. Areas of apparent normal non-fibrotic parenchyma, fibroblastic foci and established fibrosis were annotated by a trained pathologist. MCs were detectable in all areas of IPF lung, but surprisingly were not increased within the foci themselves. Instead, numbers were significantly elevated within areas of established fibrosis compared with the foci (figure 1G). Knowing that MCs interact with fibroblasts17 34–36 and hypothesising that activated, degranulating MCs could impact local fibroblast foci, we quantified the number of MCs immediately surrounding the foci (ie, within a 500 µm periphery). Strikingly, we found that the MC density was significantly higher in the region surrounding the foci compared with the foci itself, with the highest numbers being identified in areas of established fibrosis (figure 1H).
Increased MC signature and SCF expression in IPF lung
A second cohort from normal (NDRI) and IPF (Temple) donors was obtained to perform microarray gene expression analysis. To confirm MC presence, we applied a MC-specific signature, composed of three MC protease genes: cpa3, TPSB2 and TPSAB1 (encoding carboxypeptidase 3, tryptase beta-2 and tryptase alpha/beta 1, respectively) to the analysis. This signature was markedly elevated in IPF compared with normal lung (figure 2A). Furthermore, we observed increased expression of SCF (the key MC survival factor) in IPF lung (figure 2B).
Figure 2.
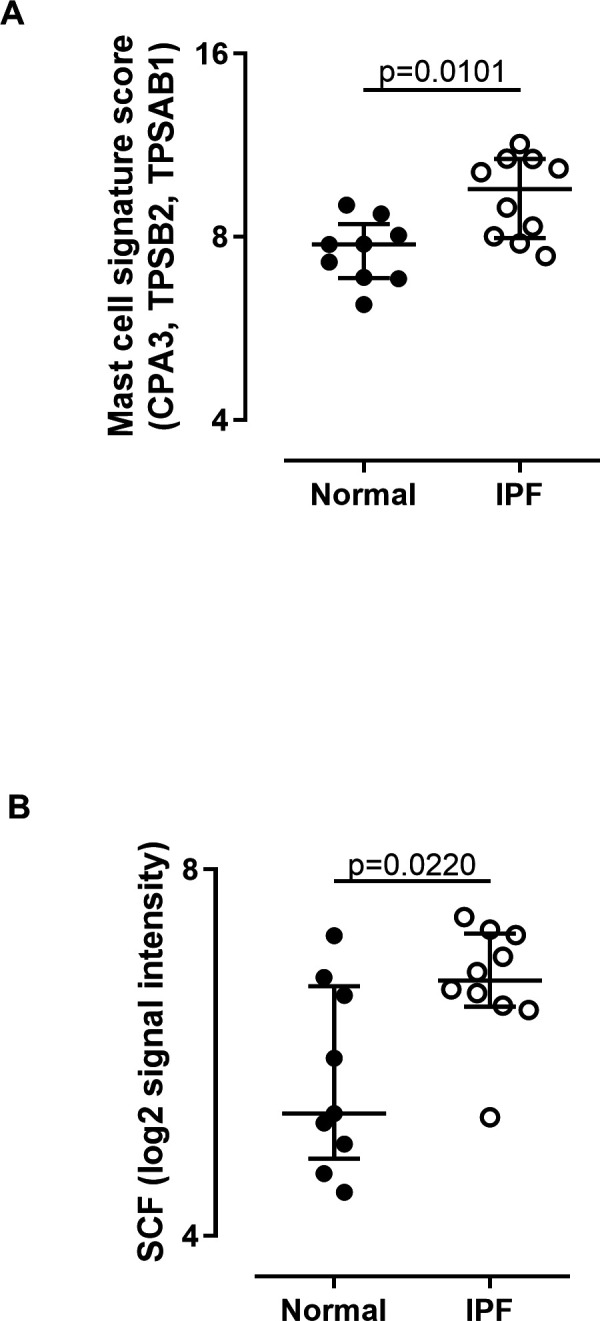
Increased mast cell signature and SCF expression in IPF lung. (A) Increased expression of mRNA encoding three MC protease genes and (B) SCF in lung of patients with IPF undergoing lung volume reduction surgery or lung transplantation (n=10) compared with normal lung tissue samples (n=9). CPA3, TPSB2 and TPSAB1 were among the strongest differentially expressed transcripts using a moderated t-test and Benjimini-Hochberg correction for multiple comparisons. These three genes comprise a MC signature. A signature score was calculated for each patient by summation of the fRMA normalised expression value reported by the microarray for each gene in the signature. The median and IQR for each group is plotted and groups were compared by Mann-Whitney U test. IPF, idiopathic pulmonary fibrosis; MC, mast cell; SCF, stem cell factor.
MCs induce fibroblast proliferation and activation
We sought to confirm and extend previous findings that an increased MC presence may drive the fibrotic pathology.19 21–23 37 When NHLFs were cultured in the presence of MC conditioned media (media cultured with MCs for 7 days) (figure 3A) or resting MC lysates (figure 3B), a significantly enhanced NHLF proliferation was observed. A similar effect was also seen when IPF fibroblasts were stimulated with MC conditioned media (online supplementary figure S4a) and when we substituted the conditioned media for recombinant MC mediators (tryptase, chymase, histamine, PDGF-AA; online supplementary figure S4b). Since these MC mediators induce fibroblast activation and subsequent differentiation into myofibroblasts,38 we performed MC–fibroblast co-culture experiments to investigate the effects of MCs on fibroblast activation. Co-culture for 72 hours led to significantly enhanced numbers of high intensity α smooth muscle actin (αSMA)–expressing cells; an increase was also seen following TGF-β stimulation (the positive control, representative images, figure 3C). Similarly, MC conditioned media induced more high intensity αSMA expressing fibroblasts; however, this did not reach statistical significance (figure 3D). Formation of αSMA fibres by fibroblasts is associated with activation of these cells into a myofibroblast phenotype and thus supports a pro-fibrotic role of MCs on fibroblasts.
Figure 3.

Mast cells (MCs) induce fibroblast proliferation and activation. MCs stimulate fibroblast proliferation, assessed by 3H-thymidine-incorporation assay. Normal human lung fibroblasts (NHLFs) were stimulated for 48 hours with (A) conditioned media obtained from MC culture and (B) MC lysates (2 different MC donors, multiple batches of lysate and conditioned media). Data are mean (SE) for at least three separate experiments, and the fold change compared with untreated cells is plotted. (C) MC conditioned media and co-culture with MCs stimulate αSMA expression in NHLFs. TGF-β1 stimulation was included as a positive control. Representative images at ×40 magnification for greater resolution. (D) A blinded quantification of high αSMA expressing cells (×20 magnification, 5 fields/well) in three separate experiments was carried out. Mean values for 3 separate experiments are plotted plus the overall median value and IQR for each condition. Groups were compared by Kruskal-Wallis with Dunn’s post hoc test. CBMC, cord blood–derived mast cell; CPM, counts per minute.
Nintedanib but not pirfenidone inhibits NHLF proliferation and MC survival
We next examined whether either of the two current therapies for IPF had an effect on MC survival or activation. Since nintendanib is a tyrosine kinase inhibitor, we hypothesised that it may induce effects downstream of the tyrosine kinase receptor ckit, the receptor for the MC survival factor SCF. Nintedanib significantly inhibited recombinant human bFGF-mediated fibroblast proliferation (IC50=526 nM; figure 4A), consistent with previous reports,30 confirming nintedanib’s expected activity. Moreover, nintedanib also markedly inhibited recombinant human SCF-mediated MC survival in vitro (IC50=13.7 nM; figure 4B). Notably, pirfenidone had no impact on either fibroblast proliferation or MC survival.
Figure 4.

Nintedanib but not pirfenidone inhibits normal human lung fibroblast (NHLF) proliferation and mast cell (MC) survival. (A) Nintedanib inhibited fibroblast proliferation induced by 100pM basic fibroblast growth factor (bFGF). (B) Nintedanib inhibited survival of MCs cultured with 4 nM rhSCF. ckit-positive MCs were quantified by flow cytometry after 7 days of treatment with nintedanib or pirfenidone. DMSO was used as a vehicle control. Data are presented as mean (SE) of three separate experiments. CPM, counts per minute.
Nintedanib inhibits fibroblast-mediated MC survival
Our data suggest that a significant crosstalk exists between MCs and fibroblasts and that this interaction may be required for a pathological role of MCs in IPF. Thus, we employed a MC–fibroblast co-culture system similar to that used by Wygrecka and colleagues,17 and quantified MC survival via flow cytometry (figure 5A). Here, we confirmed that NHLFs provided an essential survival benefit to MCs, whereas in the absence of NHLFs MCs died within 7–14 days (figure 5B). Importantly, nintedanib (30 nM) almost completely ablated this fibroblast-mediated MC survival (84% reduction day 14; figure 5C) and lower concentrations (10 nM) also inhibited survival (online supplementary figure S5). The reduction in survival was similar in magnitude to that observed in the presence of an anti-SCF antibody (82% at day 14; figure 5C). In our co-culture system, no fibroblast proliferation was observed (online supplementary figure S6). However, a potential fibroblast survival benefit was seen when cells were co-cultured with MCs. Nintedanib did not affect fibroblasts alone or the co-culture system. Collectively, these data suggest that nintedanib can inhibit fibroblast-mediated MC survival which is largely dependent on endogenous SCF.
Figure 5.

Mast cell (MC) survival in co-culture with fibroblasts is mediated largely through stem cell factor (SCF) and is inhibited by nintedanib. MCs were cultured alone or with normal human lung fibroblasts (NHLFs) in media lacking SCF or IL-6 growth factors for 21 days. (A) At each time point, cells were stained with a viability dye and anti-ckit APC mIgG1 and ckit-positive MCs quantified by flow cytometry. There is a survival benefit to MCs co-cultured with NHLFs, and this is inhibited in the presence of 67 nM anti-SCF. Inclusion of 30nM nintedanib in the co-culture has a similar effect. Data plotted as mean and SD of 3 separate experiments (2 MC donors). (B) Experimental time course plotted. (C) Day 14 data plotted as bar chart with mean and SD and showing percentage inhibition relative to isotype/vehicle control. Groups were compared by ANOVA with Sidak’s post hoc test for multiple comparisons. CBMC, cord blood–derived mast cell; FSC-SSC, forward scatter versus side scatter; LD, live dead viabilty dye.
Nintedanib significantly impacts KIT receptor signalling/phosphorylation
We next questioned whether inhibition of ckit receptor activation could be the mechanism by which nintedanib impacts MC survival. Thus, we examined the effect of nintedanib on rhSCF-mediated ckit phosphorylation on MCs. Nintedanib fully inhibited phosphorylation of ckit, which was comparable with the effect observed with anti-SCF, with an IC50 in the nanomolar range, and suggesting that inhibition of ckit activation is occurring at biologically relevant concentrations (IC50: 2.5 nM vs 13.3 nM for anti-SCF and nintedanib, respectively; figure 6A). Nintedanib also significantly inhibited rhSCF-mediated MC proliferation, a downstream effect of ckit receptor activation (IC50: 1.2 nM vs 14.8 nM for anti-SCF and nintedanib, respectively; figure 6B), as well as release of the cytokine CCL2 (IC50: 2.6 nM vs 12.2 nM for anti-SCF and nintedanib, respectively; figure 6C) which has been associated with MC activation post SCF or IgE stimulation.39 Thus, nintedanib is a potent inhibitor of ckit activation.
Figure 6.

Nintedanib inhibits ckit activation. (A) Nintedanib inhibits phosphorylation of ckit induced by 10 min stimulation with 4 nM rhSCF. IC50: 2.5 nM vs 13.3 nM for anti-SCF and nintedanib, respectively. (B) Nintedanib inhibits mast cell (MC) proliferation induced by stimulation of MCs with 4 nM rhSCF for 3 days. (C) Nintedanib inhibits CCL2 release induced by stimulation of MCs with 4 nM rhSCF for 3 days. Data are plotted as mean (SE) of 3 separate experiments. DMSO was used as a vehicle control and anti-SCF pAb was included as a positive control. CBMC, cord blood–derived mast cell; SCF, stem cell factor.
Nintedanib reduces MC recruitment in rat bleomycin model
Finally, we employed a bleomycin model to investigate the effect of nintedanib on MCs in vivo. Rats were selected since, like humans, MCs are present in the rat lung parenchyma, whereas lung MCs in mice are sparse and localised predominantly adjacent to upper airways.40 Rats were dosed orally with 60 mg/kg nintedanib or water (vehicle) daily following a single intratracheal installation of bleomycin (or saline control) (figure 7A) and MC density and fibrotic read-outs were assessed at day 14. Bleomycin induced a marked increase (p=0.05) in lung MCs (figure 7B and C), which was ablated with nintedanib treatment in bleomycin dosed rats (figure 7C). Furthermore, we observed a reduction in the levels of histamine in lung homogenates, although this did not reach statistical significance (online supplementary figure S7). Nintendanib also had an impact on the fibrosis endpoints in this model; HDFm, a measure which specifically quantifies fibrotic tissue in lung parenchyma, was significantly decreased with nintedanib treatment compared with bleomycin vehicle control (figure 7D, E), and there was a similar effect on mean tissue density (Dm) (online supplementary figure S8). In addition, we observed a trend towards decreased collagen by MTC with nintedanib treatment (figure 7F). The variability in the effects of nintedanib on fibrotic endpoints was likely due to the patchy nature of fibrosis induced by bleomycin since nintedanib had no significant effect on the levels of lung hydroxyproline, or collagen and fibronectin mRNA expression (online supplementary figure S7).
Figure 7.

Nintedanib inhibits lung mast cells (MCs) in the rat bleomycin model. (A) Outline of study design. (B) Rat lungs were stained with Giemsa to visualise the MCs. Representative sections from bleomycin-treated animals treated with nintedanib or vehicle are shown. (C) The number of MCs per area lung was increased with bleomycin and was inhibited by nintedanib. (D) Representative images of annotation of tissue density mapping in which high parenchymal tissue densities specifically located in pulmonary foci have been pseudocoloured in brown. (E) There is a significant increase in the HDFm in the lung parenchyma with bleomycin, and this is inhibited by nintedanib treatment (H&E staining, 5 sections per animal; Sal/vehicle n=6, Bleo/vehicle n=7, Bleo/nintedanib n=9, Sal/nintedanib n=5). (F) There was also a trend towards a decrease of collagen content with nintedanib treatment, although this was not statistically significant (Masson’s trichrome staining, 5 sections per animal) Kruskal-Wallis H=6.571, p=0.0869. Median and IQR are plotted and groups were compared by Kruskal-Wallis with Dunn’s multiple comparisons test.
Discussion
In this study, we sought to investigate a role for MCs in the pathogenesis of IPF and to determine whether nintedanib and pirfenidone, the only approved therapies for IPF, had an impact on MC function.
We showed that total MCs were significantly increased in IPF compared with normal lung, which is consistent with studies by others including Cha and colleagues.14 16 17 20 The mismatch in gender between the control and IPF groups was not responsible for this difference; there was no difference in MC density according to gender. Importantly, our novel finding of a significant positive correlation between the number of MCs versus fibroblast foci in patients with IPF supports an association between MC density and IPF mortality since increased foci are reported predictors of mortality.32 33 Of relevance, MC numbers inversely correlated with FVC, a clinical measure of lung function, which may suggest a higher MC density presence in more severe disease. Despite the small sample size (n=20/group, similar to those cohorts used in other publications16 24), these significant correlations suggest our findings are worthy of validation in additional cohorts to determine a direct association of MC density and mortality. Interestingly, increases in MC number were not only associated with IPF as we also showed that MCs were increased in chronic HP (but not NSIP), and fibroblastic foci are a hallmark of the usual interstitial pneumonia pattern of fibrosis that is characteristic of IPF and occasionally found in chronic HP. Moreover, an association between MCs and IPF was strengthened by our findings in a separate cohort where we observed increased MC protease signature (CPA3, TPSAB1 and TPSB2) as well as SCF gene expression, one of the key factors involved in MC survival, proliferation and activation.41 42
Enhanced fibroblast number, activation and myofibroblast differentiation are detected in IPF lung. These cells are capable of laying down ECM and causing lung stiffening.38 Our observation that MC density was highest in the region immediately surrounding the fibroblast foci, specifically in areas of established fibrosis, corroborates the hypothesis that the MC–fibroblast interaction exists in IPF. Furthermore, we confirmed previous evidence that MC–fibroblast crosstalk is pro-fibrotic, by demonstrating that MCs induced fibroblast proliferation and increased αSMA fibre formation.17 21 23 An additional pro-fibrotic role for MCs has recently been suggested by Shimbori and colleagues, who demonstrated that the fibrotic lung ECM regulates MC function and that degranulating MCs induce TGF-β1 activation.20 Our observation that the chymase+ MCs and the dual positive MCTCs were significantly increased in IPF compared with non-fibrotic controls implies that the MC pro-fibrotic activity may be related to these subtypes, although we saw a trend across all MC subtypes. This hypothesis is consistent with Andersson and colleagues who proposed that MC subtypes are switched towards the double positive MCTC phenotype in IPF24 and with Shimbori and colleagues who recently observed an increase in MCTCs in IPF lung,20 although it should be noted that we observed fewer MCTCs compared with Andersson and colleagues, which could be due to the analysis techniques used. Collectively, our data provide evidence that MCs are increased in IPF lungs, possibly due to increased levels of SCF in the lung, and thus may play a pro-fibrotic role in the pathogenesis of the disease.
Since MCs express the receptor tyrosine kinase ckit (the receptor for SCF), we investigated whether nintedanib, a broad-spectrum tyrosine kinase inhibitor, impacted MC survival or function and also compared its mechanism of action on MCs with pirfenidone. Interestingly, nintedanib but not pirfenidone reduced SCF-mediated MC survival, at concentrations below the reported cmax for nintedanib (ie, 30 nM; the maximum plasma concentration achieved at steady state with 150 mg twice daily dosing is 70 nM),43 indicating these effects are achievable with physiologically relevant concentrations of nintedanib. Decreased MC survival was also observed at lower concentrations (online supplementary figure S5). Of note, the CBMCs used in these assays did not proliferate in co-culture with fibroblasts, which is in contrast to the effect observed with human lung MCs in co-culture,17 44 and may reflect differences between CBMCs and human lung MCs. Importantly, we demonstrated that the effects of nintedanib were mediated via the ckit receptor and provided evidence that nintedanib attenuated SCF-mediated ckit phosphorylation, which reduced subsequent MC proliferation and CCL2 release from these cells. Since the adhesion molecule cell adhesion molecule 1 (CADM1) has been shown to interact with ckit and promote MC survival,44 45 it is plausible that nintedanib’s effect on ckit could disrupt this interaction. One potential limitation of this work is that due to the impact of nintedanib on MC survival, we have not demonstrated a direct effect of nintedanib on MC-mediated fibroblast proliferation or activation. Nevertheless, to our knowledge, this is the first data demonstrating that nintedanib can inhibit ckit activation and reduce human MC survival and function.
Since access to lung tissue in patients before and after nintedanib treatment is not currently feasible, we availed of a rat bleomycin model to study the effects of this drug on MC survival or recruitment in vivo. Notably, nintedanib prevented the bleomycin-induced increase in lung MCs, confirming its activity on MC recruitment, although whether this is due to a direct effect of nintedanib on MC ckit or indirectly via effects on other cells is unclear. However, the effect of nintedanib on fibrosis was variable; we noted a significant reduction in high density tissue (HDFm) by quantitative image analysis, however little impact was seen on whole lung hydroxyproline levels. The regional image analysis method is perhaps more sensitive to detect subtle alterations in the parenchyma and less affected by the patchy nature of fibrosis or the contribution of airway collagen.31 The bleomycin model has limitations for modelling the development of fibrosis in human disease and a longer time point (ie, >2 weeks) may have been required to see greater anti-fibrotic effects with nintedanib, and determine whether nintedanib can deplete resident MCs, which are long lived.46 There are conflicting data on the effect of MC depletion in rodent bleomycin models, with MCs appearing pro-fibrotic in some studies and protective in others.19 47–49 Bradding and Pejler suggest this may relate to the short duration of injury exhibited in the rodent models versus the more chronic injury observed in human IPF.50 Nevertheless, our findings are largely consistent with studies showing nintedanib reduces fibrosis in rodent models,30 51 but here we importantly expand this drug’s mechanism of action to impacting MC numbers in vivo. Future studies using cultured human lung tissue from healthy individuals or patients with IPF, such as precision cut lung slices, could examine the effects of nintedanib treatment on human lung MCs in a more physiologically relevant multicellular assay system. This would also enable us to determine if other disease-associated factors could compensate for the ablated SCF signalling and also inhibit other potential cellular sources of SCF.
In summary, we have presented novel data suggesting that nintedanib inhibits MC survival both in vitro and in vivo via ckit; however, the efficacy of nintedanib in IPF likely resides in the drug’s ability to modulate multiple fibrotic pathways. For nintedanib, these positive effects include an impact on the receptors for VEGF, FGF and PDGF.30 Furthermore, nintedanib can act on a number of other tyrosine kinases, including Src, Flt3, Lyn and Lck,52 suggesting that the impact on MC function could also be mediated via other tyrosine kinases as shown for Lyn53 in addition to the novel effects shown here on ckit. Interestingly, Imatinib (Gleevec), an inhibitor of ckit, PDGFR and c-abl, was not efficacious in an IPF clinical study; however, the effects on MC density and function in this study are unknown.25 Pirfenidone is known to have antioxidant effects as well as blockade of TGF-β1 signalling and inhibitory effects on PDGF, but the precise receptor and anti-fibrotic mechanism of action is unknown.29 The lack of effect of pirfenidone on MCs in our studies may differentiate these therapies in IPF. Future work should be directed to investigate how nintedanib modulates MC functions in patients with IPF to determine whether there is a patient population that may gain additional benefits from treatment with nintedanib. Understanding the anti-fibrotic mechanisms of action of nintedanib could also enable the discovery of better novel therapies that more specifically target pro-fibrotic mechanisms and minimise the side effects associated with this therapy.43 Furthermore, it is plausible that nintedanib could be efficacious in other respiratory diseases where MC density is elevated, such as asthma and COPD,54 and further investigation is required to determine if nintedanib could ameliorate day-to-day airway dysfunction or the development of fixed airflow obstruction.
In conclusion, our data suggest that the ability of nintedanib to impair MC survival and activation may be a novel and additional mechanism by which nintedanib exerts its anti-fibrotic effects in patients with IPF.
Acknowledgments
We thank Mahmuda Khutan and Jingya Wang for statistical advice, and Dominic Corkill for in vivo study design expertise. We thank Biocellvia for their image analysis services and Oracle Bio for their histological staining services. We thank the Laboratory of Gisli Jenkins (University of Nottingham) for the IPF fibroblasts and Gerard Criner for the Temple Study samples. We also thank Alison Humbles and Donna Finch for their editorial input into the manuscript.
Footnotes
Contributors: CO-S designed and performed the research, analysed the data and wrote the manuscript; EM, RD, ELM, LB, DR, HP, AC, AC, SK and GG-G performed the research; EM provided critical input to histological assessments; HP and DR provided critical input to study cohort design; MK conducted bioinformatic analyses; AL co-ordinated histopathological assessments; LM, RM and DC planned the project and conceived the experiments. All authors read and approved the final manuscript.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: At the time of this study, all authors apart from DR, HP and SK were employees of MedImmune (part of the AstraZeneca group). DC is now an employee of Galapagos. CO-S, LM, AL, SK, GG-G and ELM are current AstraZeneca employees and have stock/stock options in AstraZeneca, and CO-S and LM are developing drugs for IPF.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: Data are available on reasonable request. Additional gene expression data are available on reasonable request.
References
- 1. Raghu G, Rochwerg B, Zhang Y, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med 2015;192:e3–19. 10.1164/rccm.201506-1063ST [DOI] [PubMed] [Google Scholar]
- 2. Selman M, Pardo A. Idiopathic pulmonary fibrosis: an epithelial/fibroblastic cross-talk disorder. Respir Res 2002;3:3. 10.1186/rr175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Clarke DL, Carruthers AM, Mustelin T, et al. Matrix regulation of idiopathic pulmonary fibrosis: the role of enzymes. Fibrogenesis Tissue Repair 2013;6:20. 10.1186/1755-1536-6-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Daccord C, Maher TM. Recent advances in understanding idiopathic pulmonary fibrosis. F1000Res 2016;5. 10.12688/f1000research.8209.1. [Epub ahead of print: 31 May 2016]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marchal-Sommé J, Uzunhan Y, Marchand-Adam S, et al. Dendritic cells accumulate in human fibrotic interstitial lung disease. Am J Respir Crit Care Med 2007;176:1007–14. 10.1164/rccm.200609-1347OC [DOI] [PubMed] [Google Scholar]
- 6. Marchal-Sommé J, Uzunhan Y, Marchand-Adam S, et al. Cutting edge: nonproliferating mature immune cells form a novel type of organized lymphoid structure in idiopathic pulmonary fibrosis. J Immunol 2006;176:5735–9. 10.4049/jimmunol.176.10.5735 [DOI] [PubMed] [Google Scholar]
- 7. Xue J, Kass DJ, Bon J, et al. Plasma B lymphocyte stimulator and B cell differentiation in idiopathic pulmonary fibrosis patients. J Immunol 2013;191:2089–95. 10.4049/jimmunol.1203476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luzina IG, Todd NW, Iacono AT, et al. Roles of T lymphocytes in pulmonary fibrosis. J Leukoc Biol 2008;83:237–44. 10.1189/jlb.0707504 [DOI] [PubMed] [Google Scholar]
- 9. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature 2013;496:445–55. 10.1038/nature12034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abraham SN, St John AL. Mast cell-orchestrated immunity to pathogens. Nat Rev Immunol 2010;10:440–52. 10.1038/nri2782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Erjefält JS. Mast cells in human airways: the culprit? Eur Respir Rev 2014;23:299–307. 10.1183/09059180.00005014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Trautmann A, Toksoy A, Engelhardt E, et al. Mast cell involvement in normal human skin wound healing: expression of monocyte chemoattractant protein-1 is correlated with recruitment of mast cells which synthesize interleukin-4 in vivo. J Pathol 2000;190:100–6. [DOI] [PubMed] [Google Scholar]
- 13. Wulff BC, Wilgus TA. Mast cell activity in the healing wound: more than meets the eye? Exp Dermatol 2013;22:507–10. 10.1111/exd.12169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hunt LW, Colby TV, Weiler DA, et al. Immunofluorescent staining for mast cells in idiopathic pulmonary fibrosis: quantification and evidence for extracellular release of mast cell tryptase. Mayo Clin Proc 1992;67:941–8. 10.1016/S0025-6196(12)60924-0 [DOI] [PubMed] [Google Scholar]
- 15. Inoue Y, King TE, Tinkle SS, et al. Human mast cell basic fibroblast growth factor in pulmonary fibrotic disorders. Am J Pathol 1996;149:2037–54. [PMC free article] [PubMed] [Google Scholar]
- 16. Cha S-I, Chang CS, Kim EK, et al. Lung mast cell density defines a subpopulation of patients with idiopathic pulmonary fibrosis. Histopathology 2012;61:98–106. 10.1111/j.1365-2559.2012.04197.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wygrecka M, Dahal BK, Kosanovic D, et al. Mast cells and fibroblasts work in concert to aggravate pulmonary fibrosis: role of transmembrane SCF and the PAR-2/PKC-α/Raf-1/p44/42 signaling pathway. Am J Pathol 2013;182:2094–108. 10.1016/j.ajpath.2013.02.013 [DOI] [PubMed] [Google Scholar]
- 18. Rankin JA, Kaliner M, Reynolds HY. Histamine levels in bronchoalveolar lavage from patients with asthma, sarcoidosis, and idiopathic pulmonary fibrosis. J Allergy Clin Immunol 1987;79:371–7. 10.1016/0091-6749(87)90158-8 [DOI] [PubMed] [Google Scholar]
- 19. Veerappan A, O'Connor NJ, Brazin J, et al. Mast cells: a pivotal role in pulmonary fibrosis. DNA Cell Biol 2013;32:206–18. 10.1089/dna.2013.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shimbori C, Upagupta C, Bellaye P-S, et al. Mechanical stress-induced mast cell degranulation activates TGF-β1 signalling pathway in pulmonary fibrosis. Thorax 2019;74:455–65. 10.1136/thoraxjnl-2018-211516 [DOI] [PubMed] [Google Scholar]
- 21. Akers IA, Parsons M, Hill MR, et al. Mast cell tryptase stimulates human lung fibroblast proliferation via protease-activated receptor-2. Am J Physiol Lung Cell Mol Physiol 2000;278:L193–201. 10.1152/ajplung.2000.278.1.L193 [DOI] [PubMed] [Google Scholar]
- 22. Dong X, Zhang C, Ma S, et al. Mast cell chymase in keloid induces profibrotic response via transforming growth factor-β1/Smad activation in keloid fibroblasts. Int J Clin Exp Pathol 2014;7:3596–607. [PMC free article] [PubMed] [Google Scholar]
- 23. Garbuzenko E, Nagler A, Pickholtz D, et al. Human mast cells stimulate fibroblast proliferation, collagen synthesis and lattice contraction: a direct role for mast cells in skin fibrosis. Clin Exp Allergy 2002;32:237–46. 10.1046/j.1365-2222.2002.01293.x [DOI] [PubMed] [Google Scholar]
- 24. Andersson CK, Andersson-Sjöland A, Mori M, et al. Activated MCTC mast cells infiltrate diseased lung areas in cystic fibrosis and idiopathic pulmonary fibrosis. Respir Res 2011;12:139. 10.1186/1465-9921-12-139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Daniels CE, Lasky JA, Limper AH, et al. Imatinib treatment for idiopathic pulmonary fibrosis: randomized placebo-controlled trial results. Am J Respir Crit Care Med 2010;181:604–10. 10.1164/rccm.200906-0964OC [DOI] [PubMed] [Google Scholar]
- 26. Richeldi L, Cottin V, du Bois RM, et al. Nintedanib in patients with idiopathic pulmonary fibrosis: combined evidence from the TOMORROW and INPULSIS(®) trials. Respir Med 2016;113:74–9. 10.1016/j.rmed.2016.02.001 [DOI] [PubMed] [Google Scholar]
- 27. King TE, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2083–92. 10.1056/NEJMoa1402582 [DOI] [PubMed] [Google Scholar]
- 28. Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011;377:1760–9. 10.1016/S0140-6736(11)60405-4 [DOI] [PubMed] [Google Scholar]
- 29. Hilberg O, Simonsen U, du Bois R, et al. Pirfenidone: significant treatment effects in idiopathic pulmonary fibrosis. Clin Respir J 2012;6:131–43. 10.1111/j.1752-699X.2012.00302.x [DOI] [PubMed] [Google Scholar]
- 30. Wollin L, Wex E, Pautsch A, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J 2015;45:1434–45. 10.1183/09031936.00174914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gilhodes J-C, Julé Y, Kreuz S, et al. Quantification of pulmonary fibrosis in a bleomycin mouse model using automated histological image analysis. PLoS One 2017;12:e0170561. 10.1371/journal.pone.0170561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. King TE, Schwarz MI, Brown K, et al. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med 2001;164:1025–32. 10.1164/ajrccm.164.6.2001056 [DOI] [PubMed] [Google Scholar]
- 33. Enomoto N, Suda T, Kato M, et al. Quantitative analysis of fibroblastic foci in usual interstitial pneumonia. Chest 2006;130:22–9. 10.1378/chest.130.1.22 [DOI] [PubMed] [Google Scholar]
- 34. Rubinchik E, Levi-Schaffer F. Mast cells and fibroblasts: two interacting cells. Int J Clin Lab Res 1994;24:139–42. 10.1007/BF02592443 [DOI] [PubMed] [Google Scholar]
- 35. Gruber BL, Kew RR, Jelaska A, et al. Human mast cells activate fibroblasts: tryptase is a fibrogenic factor stimulating collagen messenger ribonucleic acid synthesis and fibroblast chemotaxis. J Immunol 1997;158:2310–7. [PubMed] [Google Scholar]
- 36. Artuc M, Steckelings UM, Henz BM. Mast cell–fibroblast interactions: human mast cells as source and inducers of fibroblast and epithelial growth factors. J Invest Dermatol 2002;118:391–5. 10.1046/j.0022-202x.2001.01705.x [DOI] [PubMed] [Google Scholar]
- 37. Gailit J, Marchese MJ, Kew RR, et al. The differentiation and function of myofibroblasts is regulated by mast cell mediators. J Invest Dermatol 2001;117:1113–9. 10.1046/j.1523-1747.2001.15211.x [DOI] [PubMed] [Google Scholar]
- 38. Moore MW, Herzog EL. Regulation and relevance of myofibroblast responses in idiopathic pulmonary fibrosis. Curr Pathobiol Rep 2013;1:199–208. 10.1007/s40139-013-0017-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oliveira SH, Lukacs NW. Stem cell factor and igE-stimulated murine mast cells produce chemokines (CCL2, CCL17, CCL22) and express chemokine receptors. Inflamm Res 2001;50:168–74. 10.1007/s000110050741 [DOI] [PubMed] [Google Scholar]
- 40. Miller HRP, Pemberton AD. Tissue-specific expression of mast cell granule serine proteinases and their role in inflammation in the lung and gut. Immunology 2002;105:375–90. 10.1046/j.1365-2567.2002.01375.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Costa JJ, Demetri GD, Harrist TJ, et al. Recombinant human stem cell factor (kit ligand) promotes human mast cell and melanocyte hyperplasia and functional activation in vivo. J Exp Med 1996;183:2681–6. 10.1084/jem.183.6.2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reber L, Da Silva CA, Frossard N. Stem cell factor and its receptor c-Kit as targets for inflammatory diseases. Eur J Pharmacol 2006;533:327–40. 10.1016/j.ejphar.2005.12.067 [DOI] [PubMed] [Google Scholar]
- 43. Ogura T, Taniguchi H, Azuma A, et al. Safety and pharmacokinetics of nintedanib and pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J 2015;45:1382–92. 10.1183/09031936.00198013 [DOI] [PubMed] [Google Scholar]
- 44. Moiseeva EP, Roach KM, Leyland ML, et al. CADM1 is a key receptor mediating human mast cell adhesion to human lung fibroblasts and airway smooth muscle cells. PLoS One 2013;8:e61579. 10.1371/journal.pone.0061579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hollins F, Kaur D, Yang W, et al. Human airway smooth muscle promotes human lung mast cell survival, proliferation, and constitutive activation: cooperative roles for CADM1, stem cell factor, and IL-6. J Immunol 2008;181:2772–80. 10.4049/jimmunol.181.4.2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kiernan JA. Production and life span of cutaneous mast cells in young rats. J Anat 1979;128:225–38. [PMC free article] [PubMed] [Google Scholar]
- 47. Reber LL, Daubeuf F, Pejler G, et al. Mast cells contribute to bleomycin-induced lung inflammation and injury in mice through a chymase/mast cell protease 4-dependent mechanism. J Immunol 2014;192:1847–54. 10.4049/jimmunol.1300875 [DOI] [PubMed] [Google Scholar]
- 48. Okazaki T, Hirota S, Xu ZD, et al. Increase of mast cells in the liver and lung may be associated with but not a cause of fibrosis: demonstration using mast cell-deficient Ws/Ws rats. Lab Invest 1998;78:1431–8. [PubMed] [Google Scholar]
- 49. Mori H, Kawada K, Zhang P, et al. Bleomycin-induced pulmonary fibrosis in genetically mast cell-deficient WBB6F1-W/Wv mice and mechanism of the suppressive effect of tranilast, an antiallergic drug inhibiting mediator release from mast cells, on fibrosis. Int Arch Allergy Appl Immunol 1991;95:195–201. 10.1159/000235429 [DOI] [PubMed] [Google Scholar]
- 50. Bradding P, Pejler G. The controversial role of mast cells in fibrosis. Immunol Rev 2018;282:198–231. 10.1111/imr.12626 [DOI] [PubMed] [Google Scholar]
- 51. Wollin L, Maillet I, Quesniaux V, et al. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J Pharmacol Exp Ther 2014;349:209–20. 10.1124/jpet.113.208223 [DOI] [PubMed] [Google Scholar]
- 52. Hilberg F, Roth GJ, Krssak M, et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res 2008;68:4774–82. 10.1158/0008-5472.CAN-07-6307 [DOI] [PubMed] [Google Scholar]
- 53. Gilfillan AM, Rivera J. The tyrosine kinase network regulating mast cell activation. Immunol Rev 2009;228:149–69. 10.1111/j.1600-065X.2008.00742.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cruse G, Bradding P. Mast cells in airway diseases and interstitial lung disease. Eur J Pharmacol 2016;778:125–38. 10.1016/j.ejphar.2015.04.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
thoraxjnl-2019-214000supp001.pdf (3.4MB, pdf)