

Abstract
Objective
Up to 50% of patients with hypertrophic cardiomyopathy (HCM) show no disease-causing variants in genetic studies. TRIM63 has been suggested as a candidate gene for the development of cardiomyopathies, although evidence for a causative role in HCM is limited. We sought to investigate the relationship between rare variants in TRIM63 and the development of HCM.
Methods
TRIM63 was sequenced by next generation sequencing in 4867 index cases with a clinical diagnosis of HCM and in 3628 probands with other cardiomyopathies. Additionally, 3136 index cases with familial cardiovascular diseases other than cardiomyopathy (mainly channelopathies and aortic diseases) were used as controls.
Results
Sixteen index cases with rare homozygous or compound heterozygous variants in TRIM63 (15 HCM and one restrictive cardiomyopathy) were included. No homozygous or compound heterozygous were identified in the control population. Familial evaluation showed that only homozygous and compound heterozygous had signs of disease, whereas all heterozygous family members were healthy. The mean age at diagnosis was 35 years (range 15–69). Fifty per cent of patients had concentric left ventricular hypertrophy (LVH) and 45% were asymptomatic at the moment of the first examination. Significant degrees of late gadolinium enhancement were detected in 80% of affected individuals, and 20% of patients had left ventricular (LV) systolic dysfunction. Fifty per cent had non-sustained ventricular tachycardia. Twenty per cent of patients suffered an adverse cerebrovascular event (20%).
Conclusion
TRIM63 appears to be an uncommon cause of HCM inherited in an autosomal-recessive manner and associated with concentric LVH and a high rate of LV dysfunction.
Keywords: hypertrophic cardiomyopathy, familial cardiomyopathies, genetics
Introduction
Hypertrophic cardiomyopathy (HCM) is the most common inherited cardiac disorder, with an estimated prevalence of at least 1:500.1 The understanding of the genetic architecture of HCM has greatly improved since the advent of next generation sequencing, but approximately 50% of cases remain unresolved.2 3
TRIM63 encodes an E3 ubiquitin-protein ligase known as muscle-specific RING-finger protein 1 (MuRF1), which belongs to a family of proteins with RING zinc-finger motifs. It mainly localises to the Z-disk and M-line of the sarcomere, where it interacts with several sarcomeric proteins.4 5 MuRF1 participates in the regulation of sarcomeric protein degradation through a process known as ubiquitination and plays an important role in linking myofibril components with intermediate filaments and microtubules.6 TRIM63 expression is upregulated by atrophic stimuli,4 and TRIM63 knockout mice models exhibit an exaggerated hypertrophic response to stress and an increase in hypertrophy biomarkers.7 8
Genetic variants in TRIM63 were first suggested as a possible cause of HCM following the identification of two heterozygous missense variants and a heterozygous nonsense variant, p.(Gln247*), in patients but not in controls.8 However, subsequent data on p.(Gln247*) showed that this variant is relatively common in the European population, and the relationship between TRIM63 and the development of HCM has not been confirmed yet.9 10 Some authors have suggested that variants in TRIM63 might act as modifiers of cardiac hypertrophy in the presence of a sarcomere pathogenic variant.11
On the basis of its potential role in cardiovascular diseases, we sought to determine the role of TRIM63 as a candidate gene for the development of cardiomyopathies in a large cohort of patients referred for clinical genetic testing.
Methods
From March 2013 to January 2019, TRIM63 was sequenced as a candidate gene using next-generation sequencing in 4867 index cases with a clinical diagnosis of HCM and in 3628 probands with other cardiomyopathies referred to our centre for molecular genetic diagnosis. Additionally, 3136 index cases with familial cardiovascular diseases other than cardiomyopathies (mainly channelopathies and aortic diseases) were used as controls. The phenotypes had been established by the referring centres prior to the genetic study. Patient samples were mainly received from hospitals in Spain and the UK, and the predominant ethnicity was European (>90%). The frequencies of variants in the general population were determined by using the gnomAD database (http://gnomad.broadinstitute.org; V.2.1.1, December 2019). According to gnomAD, the constraint score (observed/expected number of variants) for missense and loss-of-function (LoF) variants in TRIM63 suggests that this gene is tolerant to the aforementioned genetic changes. However, the variants reported in gnomAD are mainly found in heterozygosity. To investigate the role of non-synonymous TRIM63 (NM_032588.3) variants in homozygosity or compound heterozygosity, probands harbouring rare variants (defined as those with minor allele frequency (MAF)<0.001) in homozygous or compound heterozygous state were analysed and invited to participate in segregation studies. Clinical and genetic familial cascade screening was performed when possible. In cases where a second pathogenic variant in a sarcomeric gene was identified, the latter variant was also included in family screening to determine the role of each of the identified variants in the familial phenotype. Finally, the clinical characteristics and outcomes of homozygous and compound heterozygous individuals (including probands and relatives) were assessed. Our study was conducted in accordance with the Declaration of Helsinki. Written consent was obtained from all participants.
Patient involvement
Patients were not involved in the design of this study. The index cases actively participated in the recruitment of first-degree relatives. They were central to the dissemination of the baseline information and our hypothesis, which helped to motivate the involvement of their families. All index cases received information on the basis of genetic inheritance in order to adequately inform their relatives prior to contacting the research team. Once our study is published, we aim to disseminate the main results to all trial participants.
Genetic studies, variant filtering and variant classification
Coding exons and intronic boundaries of 219 genes related to inherited cardiovascular diseases and sudden cardiac death (online supplementary table 1) were captured using a custom probe library (SureSelect Target Enrichment Kit for Illumina paired-end multiplexed sequencing method; Agilent Technologies, Santa Clara, California, USA) and sequenced on the HiSeq 1500 platform (Illumina, San Diego, California, USA) following Illumina protocols. The read depth (number of times that a base was sequenced by independent reads) of every nucleotide of genes related to the referred phenotype (including TRIM63) was >30 (mean 250–400). Exons that did not fulfil this standard were complementary sequenced using the Sanger method. Only likely protein-altering variants (missense, in-frame insertions/deletions, frameshifts, nonsense and consensus splice-site variants) in the TRIM63 gene were analysed. Bioinformatic analysis was performed by means of a custom pipeline including software for variant calling, genotyping and annotation. To determine the pathogenicity of the identified variants, the American College of Medical Genetics and Genomics recommendations were used.12
heartjnl-2020-316913supp001.pdf (7.4MB, pdf)
Statistical analysis
Continuous variables were expressed as mean±SD, and comparison between groups was performed using the Student’s t test or the Mann-Whitney U test according to values distribution. Non-continuous variables were expressed as an integer number (per cent of total) and compared using the χ² test or Fisher’s exact test, as appropriate. A two-sided p<0.05 was considered to indicate statistical significance.
Analysis was performed using the R V.3.4.3 (The R Foundation for Statistical Computing Platform).
LOD score (Logarithms of Odds) calculation
We calculated two-point LOD score for informative families by using the PARAMLINK package for R software. The model was set for a recessive inheritance pattern, with θ=0, phenocopy rate=0.005 and a penetrance of 0.95 for homozygous carriers. An indeterminate status was assigned to family members with a confounding cardiac diagnosis, as well as to individuals younger than 40 years who did not meet clinical criteria for HCM and could develop the disease afterwards.
Results
A total of 20 (0.39%) index cases harbouring homozygous or compound heterozygous rare variants in TRIM63 were identified, all of them of European descent (non-Finnish). Nineteen had a clinical diagnosis of HCM and one was diagnosed with restrictive cardiomyopathy (RCM). No individuals with biallelic variants were identified in the disease controls. Likewise, in the gnomAD database, no homozygous individuals harbouring rare variants (MAF<0.001) are identified.
Fifteen of the homozygous/compound heterozygous patients with a clinical diagnosis of HCM and the patient with restrictive cardiomyopathy accepted to participate in this study. Five missense and three protein-truncating variants in TRIM63 (two frameshifts and one nonsense) were identified in these families. All the identified variants are present in the gnomAD database, with a low frequency (MAF<0.001) and only in heterozygosity, except for the p.(Leu37Val) and p.(Lys146Thrfs*24) variants, which are absent in this database. The identified variants, their frequency in gnomAD, the result of the bioinformatic predictors, their classification in ClinVar (whenever present) and their significance according to the ACMG recommendations can be found in online supplementary table 2. Only one HCM individual harbouring a homozygous truncating variant in TRIM63 also carried a pathogenic truncating variant in a sarcomeric gene (MYBPC3), whereas in the rest of the included individuals, no other variants associated with the phenotype were identified.
Familial cosegregation
Twelve of the included families accepted further clinical and genetic screening as part of the evaluation. Three other families accepted to undergo only clinical evaluation, and in one homozygous index case, family study was not possible. Clinical and genetic data on all evaluated family members as well as the family pedigrees are available online (online supplementary table S3 and figure S1).
After familial evaluation, three more homozygous and one compound heterozygous individuals were identified. Two of these met current diagnostic criteria for HCM,13 whereas the remaining two were considered likely to be affected, manifesting early signs of the disease given the grossly abnormal ECG, even though the left ventricular (LV) wall thickness did not fulfil the classical criteria for HCM (ECGs available as online supplementary figure 2). None of the heterozygous or wild-type family members showed signs of the disease. Figure 1 shows the difference in LV maximal wall thickness in homozygous/compound heterozygous individuals, compared with their heterozygous or wild-type family members. After segregation analysis in the families, a combined LOD score of 4.25 was obtained, which is indicative of strong cosegregation (online supplementary table 4).
Figure 1.
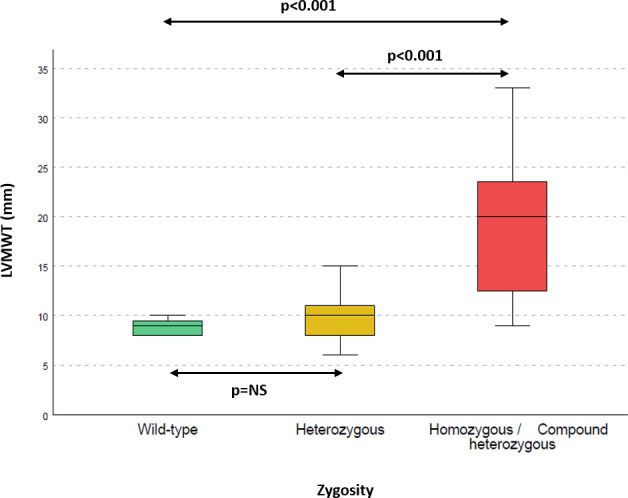
Left ventricular maximal wall thickness according to zygosity. Left ventricular maximal wall thickness is higher in homozygous/compound heterozygous patients than in their heterozygous (p<0.001) or wild-type (p<0.001) family members. No statistically significant differences are observed between wild-type and heterozygous individuals.
Clinical characteristics of homozygous and compound heterozygous individuals
The baseline characteristics of all homozygous and compound heterozygous individuals after family screening and their comparison with wild-type and heterozygous family members are shown in table 1.
Table 1.
Baseline characteristics of homozygous/compound heterozygous individuals
| Homozygous/compound heterozygous (n=20) | Heterozygous/ wild-type (n=44) | P value | |
| Age (range) | 33 (15–69) | 48 (12–80) | 0.002 |
| Hypertension (%) | 4/20 (20) | 7/44 (16) | 0.4 |
| HCM (%) | 19/20 (95) | 0/44 | <0.001 |
| ASH | 8/16 (50) | ||
| Concentric | 8/16 (50) | ||
| Restrictive cardiomyopathy (%) | 1/20 (5) | 0/44 | 0.15 |
| Reason for diagnosis (%) | |||
| Syncope | 2/20 (10%) | N/A | N/A |
| Angina | 2/20 (10%) | ||
| Dyspnoea | 3/20 (15%) | ||
| ECG disturbance | 5/20 (25%) | ||
| Palpitations | 3/20 (15%) | ||
| Family screening | 4/20 (20%) | ||
| Others | 1/20 (5%) | ||
| NYHA functional class I–II (%) | 19/20 (95) | N/A | N/A |
| ECG LVH (%) | 13/16 (81) | 0/44 | <0.001 |
| LVMWT (mm) | 19.2 (±5.6) | 9.8 (±1.9) | <0.001 |
| LVMWT>25 mm (%) | 3/20 (15) | 0/44 | <0.001 |
| PW (mm) | 13.6 (±3) | 8.8 (±2) | 0.01 |
| LVEDD (mm) | 49.4 (±7.5) | 45.4 (±4.8) | 0.02 |
| LVEF (%) | 60 (±12) | 63.4 (±3.7) | <0.001 |
| LV dysfunction (EF<50%) (%) |
4/20 (20) | 0/44 | 0.003 |
| LA diameter (mm) | 43 (±5) | 32 (±5) | 0.7 |
| LVOTO | 0/20 | N/A | N/A |
| CMR LGE (%) | 10/12 (83) | N/A | N/A |
| Atrial fibrillation (%) | 3/20 (15) | 0/40 | 0.012 |
| NSVT on Holter (%) | 9/18 (50) | N/A | N/A |
| ICD implant (%) | 7/20 (35) | N/A | N/A |
| Betablockers (%) | 12/20 (60) | N/A | N/A |
| Oral anticoagulants (%) | 5/20 (25) | 0/44 | <0.001 |
| Events | |||
| Cardiovascular death | 0/20 | ||
| CVE | 4/20 | 1/44 | |
| Heart failure death/heart transplant | 0/20 | ||
| SCD | 0/20 | ||
ASH, asymmetric septal hypertrophy; CMR, cardiac magnetic resonance; CVE, cerebrovascular event; ECG, electrocardiogram; HCM, hypertrophic cardiomyopathy; ICD, implantable cardioverter defibrillator; LA, left atrium; LVEDD, left ventricular end-diastolic diameter; LVEF, left ventricular ejection fraction; LVH, left ventricular hypertrophy; LVMWT, left ventricular maximal wall thickness; LVOTO, left ventricular outflow tract obstruction; NSVT, non-sustained ventricular tachycardia; NYHA, New York Heart Association; PW, posterior wall; SCD, sudden cardiac death.
More detailed clinical, echocardiographic and electrocardiographic data on each of the homozygous/compound heterozygous are shown in online supplementary table 5. The most frequently identified variant in our cohort was p.(Gln247*), followed by the p.(Cys23Tyr) variant. The distribution of the identified variants throughout the different functional domains of the protein is shown in figure 2. HCM was the most frequent diagnosis (95%), with only one individual with biallelic variants in TRIM63 with a different phenotype (restrictive cardiomyopathy). The mean age at diagnosis was 35 years (range 15–69). Forty-five per cent were asymptomatic at the first examination, with abnormal ECG being the most frequent cause of diagnosis (25%). Fifty per cent of patients with available information had concentric left ventricular hypertrophy (LVH), and none had LV outflow tract obstruction. The mean maximal LV wall thickness was 19 mm (±5). At the moment of inclusion, no patient had extreme LV hypertrophy (ie, >30 mm); however, one patient with a maximal LV wall thickness of 17 mm showed significant wall thinning, with a previously reported septal wall thickness of 33 mm (without septal reduction therapies). Significant degrees of late gadolinium enhancement (LGE) (ie, transmural and involving more than one myocardial segment) were observed in almost 80% of homozygous/compound heterozygous and 20% of our population progressed to LV systolic dysfunction (defined as LV ejection fraction (LVEF) <50%). All patients with LV dysfunction showed LGE on MRI studies. Figure 3 shows two examples of patients’ cardiac MRIs. Fifty per cent of the patients had non-sustained ventricular tachycardia on Holter monitoring. One patient had hemodynamically stable sustained ventricular tachycardia, which prompted the implantation of an implantable cardioverter defibrillator for secondary prevention. Twenty per cent of the included individuals underwent a formal neurological examination, including muscular imaging techniques, and none of them showed evident neuromuscular alterations. Only one individual underwent muscle biopsy, which was normal. Creatine-kinase leves (CK) levels were elevated in 3 out of 11 evaluated patients. None of the evaluated cases had previous episodes of sudden cardiac death. Four patients (20%) had a previous cerebrovascular event, none of them disabling.
Figure 2.

Schematic representation of the protein’s domains. Location of the identified homozygous/compound heterozygous variants in TRIM63 at protein level.
Figure 3.

Cardiac MRI. Panels A and B show patient II.3 from family 6897. Severe concentric LV hypertrophy (20 mm IVS) and severe transmural LGE were observed. Panels C and D show patient II.2 from family 2523, who developed LV systolic dysfunction and dilatation. Severe transmural LGE is observed in the apical segments and near- transmural LGE in the interventricular septum. Hypertrabeculation of the lateroapical segments was found. LGE, late gadolinium enhancement; LV, left ventricular.
Discussion
Our study demonstrates a relationship between homozygous and compound heterozygous rare variants in TRIM63 and the development of HCM. Autosomal-recessive (AR) inheritance has been considered the least common form of inheritance in inherited cardiac muscle diseases and is mainly associated with metabolic or complex multi-system disorders.13 14 More than 11 600 probands with different cardiovascular inherited disorders were sequenced, and rare homozygous and compound heterozygous variants in TRIM63 were identified only in subjects with HCM (and one case with RCM), whereas they were absent in our control population. Likewise, homozygous individuals harbouring rare variants in TRIM63 are absent in the gnomAD database. Moreover, familial evaluation confirmed these findings, as heterozygous carriers were healthy and only homozygous and compound heterozygous individuals showed manifestations of the disease. Even though two family members (homozygous) did not fulfil the classical criteria for HCM, we classified them as possibly affected on the basis of an abnormal ECG (online supplementary figure 2). It has been described that ECG changes in the absence of diagnostic morphological abnormalities (in the context of a familial disease) may represent an early or mild form of HCM.13
Sarcomeric proteins undergo constant turnover in order to adapt to the physiological needs as well as to repair damaged proteins.4 Central to this turnover is the ubiquitin proteasome system (UPS). E3 ubiquitin ligases direct the addition of ubiquitin to specific target proteins, marking them for degradation.15 Substrates targeted by MuRF1 include several members of the contractile apparatus, such as myosin heavy chain, troponins, tropomyosin and titin.6 16 17 Online supplementary figure 3 shows a schematic representation of the role of ubiquitin ligases in the UPS. Due to its interactions with sarcomeric proteins, TRIM63, encoding MuRF1, has been proposed as a candidate gene for the development of cardiomyopathies. However, most descriptions focused on heterozygous carriers or isolated homozygous individuals without familial cosegregation studies,8 18 and its true relevance as a cardiomyopathy-causing gene has not yet been elucidated.
One previous study19 reported a family in which a male harbouring the p.(Gln247*) variant in TRIM63 in homozygosity and the heterozygous missense TRIM54 variant p.(Asp106Asn) had HCM and skeletal myopathy. His siblings heterozygous for the TRIM63 variant were asymptomatic and healthy. One of his sisters was also a homozygous for the p.(Gln247*) variant, but not a carrier of the TRIM54 variant, and was diagnosed with LV hypertrophy thought to be secondary to hypertension. This group reported that the presence of digenic mutations (TRIM63 and TRIM54) would be necessary for the development of the phenotype and that the homozygous TRIM63 variant would deserve further investigation in order to establish its contribution to the development of LV hypertrophy.
The presence of concentric hypertrophy is not a frequent presentation in sarcomeric HCM and may be more common in metabolic and infiltrative disorders.13 However, 50% of the patients with HCM included in our study showed this distribution. The finding of concentric HCM together with extensive LGE distribution prompted the search for an infiltrative disease in three of our patients (15%), and was ruled out in all of them. The clinical suspicion of infiltrative disorder together with the presence of extensive LGE and LV systolic dysfunction in our population matches the description of the cardiac phenotype in the proband reported by Olivé et al.19
End-stage HCM (defined as EF <50%) has been identified in up to 10% of individuals with HCM in modern cohorts, and it has been suggested that the progression to this phenotype may be lower in individuals with sarcomeric HCM when compared with phenocopies.1 20–22 Nevertheless, in our study, 20% of individuals showed progression to LV dysfunction. Moreover, in one family, there was also a history of heart transplant due to end-stage HCM in an individual with unknown genotype. Animal studies have shown an increase in myocardial fibrosis in mice harbouring mutations in TRIM63, and myocardial fibrosis has been shown to be a key determinant of LV adverse remodelling in other cardiomyopathies.23–25
Interestingly, the previously described variant p.(Gln247*) was the most frequently identified variant in our cohort. Functional studies have shown that this variant reduces the localisation of the protein to the Z-disk and impairs the ubiquitination of MuRF1 substrates in cardiac myocytes.8 RT-PCR analysis suggested that the transcripts undergo nonsense-mediated decay (a mechanism responsible for haploinsufficiency); moreover, transient expression of wild-type MuRF1 rescued the phenotype in adult human myocytes.19 Sixty per cent of the identified missense variants in our cohort are located in the RING domain, a highly conserved region (see figure 2 and online supplementary figure 4) which mediates ubiquitination events.26 Other groups8 have shown that missense variants located in the RING domain can significantly decrease the enzyme’s autoubiquitination function and the ubiquitination of the substrates, as well as producing an activation in the mTOR pathway signalling (important for the development of ventricular hypertrophy). Furthermore, three out of five (60%) of all the missense variants identified involve the substitution of cysteine (a neutral polar side chain amino acid) for tyrosine (an aromatic hydrophobic side chain amino acid), which causes very significant changes in the physicochemical properties and may alter the structure and function of the protein.
Although our findings will affect a small subset of patients with HCM, they are important in clinical practice because they have a relevant impact on the genetic counselling of these families, as all the available evidence suggests that TRIM63 is a cause of HCM with AR inheritance. Moreover, the clinical profile of these patients (moderate to severe hypertrophy, high incidence of ventricular arrhythmias, extensive fibrosis and frequent LV systolic dysfunction) suggests that individuals homozygous and compound heterozygous for variants in TRIM63 are at high risk of adverse events. The absence of arrhythmic events in our cohort may be due to a lack of long-term follow-up of the identified carriers.
Study limitations
The fact that no other TRIM protein family members have been studied in our cohort made it impossible to study whether digenic variants could explain the development of clinical myopathy or elevated CK observed in some patients.
Conclusion
TRIM63 is a cause of HCM with an AR inheritance pattern, which may be associated with concentric LVH and a high rate of LV dysfunction. It accounts for approximately 0.4% of HCM cases, although its importance in sporadic cases could be higher. If these findings are validated by other groups, it is possible that TRIM63 is the most frequent cause of HCM with a recessive inheritance pattern.
Key messages.
What is already known on this subject?
Up to 50% of patients with hypertrophic cardiomyopathy (HCM) show no disease-causing variants in genetic studies. TRIM63 has been suggested as a candidate gene for the development of cardiomyopathies, although evidence for a causative role in HCM is limited.
What might this study add?
This study adds to the genetic architecture of HCM by showing that TRIM63 is a cause of a non-classical form of HCM, with an autosomal-recessive inheritance, presenting as concentric HCM with a high rate of left ventricular (LV) dysfunction.
How might this impact on clinical practice?
Our findings have a relevant impact on the genetic counselling of these families, as all the available evidence suggests that TRIM63 is a cause of HCM only with autosomal-recessive inheritance. Moreover, the clinical profile of these patients (moderate to severe hypertrophy, high incidence of ventricular arrhythmias, extensive fibrosis and frequent LV systolic dysfunction) suggests that homozygous and compound heterozygous individuals for variants in TRIM63 are at high risk of adverse events.
Acknowledgments
The authors would like to thank Health in Code's laboratory, bioinformatics and administrative teams for their everyday effort. We thank Mr Radek Suchac and Mrs Alicia Pallas for their support in English editing.
Footnotes
Twitter: @JoelSMG
Collaborators: GENESCOPIC Research group: Health in Code: Xusto Fernández; Elena Veira; David de Uña; Pablo Iglesias; Noël Brögger; Germán Fernández; Juan Pablo Trujillo-Quintero Hospital Universitario Puerta de Hierro: Marta Cobo-Marcos; María A. Restrepo-Córdoba; Esther González-LópezSaint Bartholomew’s Hospital: Luis R. Lopes Hospital Universitario de Getafe: Jesús Saavedra Hospital Clínico Universitario de Valladolid: María Sandín-Fuentes; Iria Duro-Aguado Hospital General Universitario de Alicante: María García Álvarez.
Contributors: JS-M is the principal investigator, wrote the proposal, coordinated the study, supervised the data and prepared the article. JPO had a central role in statistical analysis and critical revision. JP-D, FD, CD-L, MA, SR, AP-C, MMC, MR, MLPP, VC-P, FMS, CH, PEG-G, MVM, IC-R, MC, DG-G, AL, BG-F, FD-F and RS helped in data acquisition and coordinated the family studies. IG-D, LS and PS did a critical review on all the bioinformatic aspects of the identified genetic variants. MO critically revised the manuscript and led the neuromuscular investigations. PG-P, MO-G and PME made substantial contributions to the design of the work and added important intellectual content to the final manuscript. LM conceived the initial idea and acted as co-coordinator. JS-M and LM act as guarantors.
Funding: St. Bartholomew's Hospital (Drs P M Elliot and M Akhtar) and Hospital Universitario Puerta de Hierro Majadahonda (Drs P Garcia-Pavia and F Domínguez) are members of the European Reference Network on Rare and Complex Diseases of the Heart (Guard-Heart; http://guard-heart.ernnet.eu).
Competing interests: JS-M, JPO, IG-D, IJC-R, MNC, DG-G, AL, LS and MO-G are employees of Health in Code SL. LM is a stakeholder and CEO of Health in Code SL.
Patient and public involvement: Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: All data relevant to the study are included in the article or uploaded as supplementary information.
Contributor Information
on behalf of GENESCOPIC Research Group:
Xusto Fernández, Elena Veira, David de Uña, Pablo Iglesias, Noël Brögger, Marta Cobo-Marcos, María A. Restrepo-Córdoba, María Sandín-Fuentes, María García Álvarez, Esther González-López, Luis R Lopes, Jesús Saavedra, María Sandín-Fuentes, Iria Duro-Aguado, Germán Fernández, and Juan Pablo Trujillo-Quintero
References
- 1. Maron BJ. Clinical course and management of hypertrophic cardiomyopathy. N Engl J Med 2018;379:655–68. 10.1056/NEJMra1710575 [DOI] [PubMed] [Google Scholar]
- 2. Van Driest SL, Ellsworth EG, Ommen SR, et al. Prevalence and spectrum of thin filament mutations in an outpatient referral population with hypertrophic cardiomyopathy. Circulation 2003;108:445–51. 10.1161/01.CIR.0000080896.52003.DF [DOI] [PubMed] [Google Scholar]
- 3. Richard P, Charron P, Carrier L, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003;107:2227–32. 10.1161/01.CIR.0000066323.15244.54 [DOI] [PubMed] [Google Scholar]
- 4. Willis MS, Bevilacqua A, Pulinilkunnil T, et al. The role of ubiquitin ligases in cardiac disease. J Mol Cell Cardiol 2014;71:43–53. 10.1016/j.yjmcc.2013.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mearini G, Gedicke C, Schlossarek S, et al. Atrogin-1 and MuRF1 regulate cardiac MyBP-C levels via different mechanisms. Cardiovasc Res 2010;85:357–66. 10.1093/cvr/cvp348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gregorio CC, Perry CN, McElhinny AS. Functional properties of the titin/connectin-associated proteins, the muscle-specific RING finger proteins (MURFs), in striated muscle. J Muscle Res Cell Motil 2005;26:389–400. 10.1007/s10974-005-9021-x [DOI] [PubMed] [Google Scholar]
- 7. Willis MS, Ike C, Li L, et al. Muscle ring finger 1, but not muscle ring finger 2, regulates cardiac hypertrophy in vivo. Circ Res 2007;100:456–9. 10.1161/01.RES.0000259559.48597.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen SN, Czernuszewicz G, Tan Y, et al. Human molecular genetic and functional studies identify TRIM63, encoding muscle RING finger protein 1, as a novel gene for human hypertrophic cardiomyopathy. Circ Res 2012;111:907–19. 10.1161/CIRCRESAHA.112.270207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Płoski R, Pollak A, Müller S, et al. Does p.Q247X in TRIM63 cause human hypertrophic cardiomyopathy? Circ Res 2014;114:2–5. 10.1161/CIRCRESAHA.114.302662 [DOI] [PubMed] [Google Scholar]
- 10. Walsh R, Buchan R, Wilk A, et al. Defining the genetic architecture of hypertrophic cardiomyopathy: re-evaluating the role of non-sarcomeric genes. Eur Heart J 2017;38:ehw603–8. 10.1093/eurheartj/ehw603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Su M, Wang J, Kang L, et al. Rare variants in genes encoding MuRF1 and MuRF2 are modifiers of hypertrophic cardiomyopathy. Int J Mol Sci 2014;15:9302–13. 10.3390/ijms15069302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet Med 2015;17:405–23. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13., Elliott PM, Anastasakis A, et al. , Authors/Task Force members . 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of cardiology (ESC). Eur Heart J 2014;35:2733–79. 10.1093/eurheartj/ehu284 [DOI] [PubMed] [Google Scholar]
- 14. Rapezzi C, Arbustini E, Caforio ALP, et al. Diagnostic work-up in cardiomyopathies: bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working group on myocardial and pericardial diseases. Eur Heart J 2013;34:1448–58. 10.1093/eurheartj/ehs397 [DOI] [PubMed] [Google Scholar]
- 15. Willis MS, Schisler JC, Portbury AL, et al. Build it up-Tear it down: protein quality control in the cardiac sarcomere. Cardiovasc Res 2009;81:439–48. 10.1093/cvr/cvn289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McElhinny AS, Kakinuma K, Sorimachi H, et al. Muscle-specific RING finger-1 interacts with titin to regulate sarcomeric M-line and thick filament structure and may have nuclear functions via its interaction with glucocorticoid modulatory element binding protein-1. J Cell Biol 2002;157:125–36. 10.1083/jcb.200108089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Henderson CA, Gomez CG, Novak SM, et al. Overview of the muscle cytoskeleton. Compr Physiol 2017;7:891–944. 10.1002/cphy.c160033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jokela M, Baumann P, Huovinen S, et al. Homozygous nonsense mutation p.Q274X in Trim63 (MuRF1) in a patient with mild skeletal myopathy and cardiac hypertrophy. J Neuromuscul Dis 2019;6:143–6. 10.3233/JND-180350 [DOI] [PubMed] [Google Scholar]
- 19. Olivé M, Abdul-Hussein S, Oldfors A, et al. New cardiac and skeletal protein aggregate myopathy associated with combined MuRF1 and MuRF3 mutations. Hum Mol Genet 2015;24:3638–50. 10.1093/hmg/ddv108 [DOI] [PubMed] [Google Scholar]
- 20. Biagini E, Coccolo F, Ferlito M, et al. Dilated-hypokinetic evolution of hypertrophic cardiomyopathy: prevalence, incidence, risk factors, and prognostic implications in pediatric and adult patients. J Am Coll Cardiol 2005;46:1543–50. 10.1016/j.jacc.2005.04.062 [DOI] [PubMed] [Google Scholar]
- 21. Harris KM, Spirito P, Maron MS, et al. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation 2006;114:216–25. 10.1161/CIRCULATIONAHA.105.583500 [DOI] [PubMed] [Google Scholar]
- 22. Rosmini S, Biagini E, O'Mahony C, et al. Relationship between aetiology and left ventricular systolic dysfunction in hypertrophic cardiomyopathy. Heart 2017;103:300–6. 10.1136/heartjnl-2016-310138 [DOI] [PubMed] [Google Scholar]
- 23. Masci PG, Schuurman R, Andrea B, et al. Myocardial fibrosis as a key determinant of left ventricular remodeling in idiopathic dilated cardiomyopathy: a contrast-enhanced cardiovascular magnetic study. Circ Cardiovasc Imaging 2013;6:790–9. 10.1161/CIRCIMAGING.113.000438 [DOI] [PubMed] [Google Scholar]
- 24. Liu T, Song D, Dong J, et al. Current understanding of the pathophysiology of myocardial fibrosis and its quantitative assessment in heart failure. Front Physiol 2017;8:1–13. 10.3389/fphys.2017.00238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Karaahmet T, Gürel E, Tigen K, et al. The effect of myocardial fibrosis on left ventricular torsion and twist in patients with non-ischemic dilated cardiomyopathy. Cardiol J 2013;20:276–86. 10.5603/CJ.2013.0073 [DOI] [PubMed] [Google Scholar]
- 26. Ozato K, Shin D-M, Chang T-H, et al. TRIM family proteins and their emerging roles in innate immunity. Nat Rev Immunol 2008;8:849–60. 10.1038/nri2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
heartjnl-2020-316913supp001.pdf (7.4MB, pdf)