

Abstract
Even before the perception or interaction with pathogens, plants rely on constitutively guardian molecules, often specific to tissue or stage, with further expression after contact with the pathogen. These guardians include small molecules as antimicrobial peptides (AMPs), generally cysteine-rich, functioning to prevent pathogen establishment. Some of these AMPs are shared among eukaryotes (eg, defensins and cyclotides), others are plant specific (eg, snakins), while some are specific to certain plant families (such as heveins). When compared with other organisms, plants tend to present a higher amount of AMP isoforms due to gene duplications or polyploidy, an occurrence possibly also associated with the sessile habit of plants, which prevents them from evading biotic and environmental stresses. Therefore, plants arise as a rich resource for new AMPs. As these molecules are difficult to retrieve from databases using simple sequence alignments, a description of their characteristics and in silico (bioinformatics) approaches used to retrieve them is provided, considering resources and databases available. The possibilities and applications based on tools versus database approaches are considerable and have been so far underestimated.
Keywords: Defensin, lipid transfer protein, hevein, cyclotide, snakin, knotin, macadamia β-barrelins, impatiens-like, puroindoline, thaumatin
Introduction
Proteins and peptides play different roles depending on their amino acids (aa) constitution, which may vary from tens to thousands residues.1 Peptides are conventionally understood as having less than 50 aa.2 Proteins, on the contrary, would be any molecule presenting higher amino acid content and both—proteins and peptides—present a plethora of variations in plants. Despite that, plant proteomes have been much more studied than peptidomes. It is well-known that the biochemical machinery necessary for the synthesis and metabolism of peptides is present in every living organism. From the variations of this machinery, a wide structural and functional diversity of peptides was generated, justifying the growing interest in their study.
In eukaryotes, peptides are prevalent in intercellular communication, performing as hormones, growth factors, and neuropeptides, but they are also present in the defense system.3 Besides plants and animals, several pathogenic microorganisms, peptides can serve as classical virulence factors, which disrupt the epithelial barrier, damage cells, and activate or modulate host immune responses. An example of this performance is represented by Candidalysin,4 a fungal cytolytic peptide toxin found in the pathogenic fungus Candida albicans that damages epithelial membranes, triggers a response signaling pathway, and activates epithelial immunity. There are also reports of defense-related fungal peptides. For example, the Copsin, a peptide-based fungal antibiotic recently identified in the fungus Coprinopsis cinerea5 kills bacteria by inhibiting their cell wall synthesis. Regarding bacterial peptides, certain species from the gastrointestinal microbial community can release low-molecular-weight peptides, able to trigger immune responses.6 There are additionally peptides that act like bacterial “hormones” that allow bacterial communities to organize multicellular behavior such as biofilm formation.7 Some peptides are known for their medical importance, as defensins that present antibacterial, antiviral, and antifungal activities. For example, human alpha- and beta-defensins present in the saliva may potentially impede virus replication, including SARS-CoV-2,8 besides other roles as protection against intestinal inflammation (colitis).9
Considering the roles of plant peptides, they can also be multifunctional, and have been classified into 2 main categories10 (Supplementary Figure S1): (1) Peptides with no bioactivity, primarily resulting from the degradation of proteins by proteolytic enzymes, aiming at their recycling, and (2) bioactive peptides, which are encrypted in the structure of the parent proteins and are released mainly by enzymatic processes.
The first group is innocuous regarding signaling, regulatory functions, and bioactivity. So far, it has been reported that some of them may play a significant role in nitrogen mobilization across cellular membranes.11 The second group of bioactive peptides has a substantial impact on the plant cell physiology. Some peptides of this group can act in the plant growth regulation (through cell-to-cell signaling), endurance against pathogens and pests by acting as toxins or elicitors, or even detoxification of heavy metals by ion-sequestration.
Comprising bioactive peptides, additional subcategorization has been proposed regarding their function. Tavormina et al12 divided them into 3 groups (Supplementary Figure S1) based on the type of precursor:
Derived from functional precursors: originated from a functional precursor protein;
Derived from nonfunctional precursors: originated from a longer precursor that has no known biological function (as a preprotein, proprotein, or preproprotein);
Not derived from a precursor protein: some sORFs (small Open Read Frames; usually <100 codons) are considered to represent a potential new source of functional peptides (known as “short peptides encoded by sORFs”).
A more intuitive classification of bioactive peptides was further proposed by Farrokhi et al10 according to their intracellular role (Supplementary Figure S1):
Phytohormone peptides: the characteristic feature of these peptides is the regulation of fundamental plant physiological processes. They can be classified into those with (1) signaling roles in non-defense functions or those with (2) signaling roles in plant defense. Concerning the first group (Supplementary Figure S1), the peptide CLE25 (CLAVATA3/EMBRYO-SURROUNDING REGION-RELATED 25) is one of the representatives. This peptide transmits water-deficiency signals through vascular tissues in Arabidopsis thaliana, affecting abscisic acid biosynthesis and stomatal control of transpiration in association with BAM (BARELY ANY MERISTEM) receptors in leaves.13 Another example is the PLS (POLARIS) peptide that acts during early embryogenesis but later activates auxin synthesis, also affecting cytokine synthesis and ethylene response.14 Regarding the second group, it includes peptides with signaling roles in plant defense, comprising at least 4 subgroups, including SYST (systemin) (Supplementary Figure S1). The SYST peptides were identified in Solanaceae members, like tomato and potato15 (acting on the signaling response to herbivory). The SYST leads to the production of a plant protease inhibitor that suppresses insect’s proteases.16 Stratmann17 suggested that in plants, SYSTs act to stimulate the jasmonic acid signaling cascade within vascular tissues to induce a systemic wound response.
Defense peptides or antimicrobial peptides (AMPs): to be fitted into this class, a plant peptide must fulfill some specific biochemical and genetic prerequisites. Regarding biochemical features, in vitro antimicrobial activity is required. Concerning the genetic condition, the gene encoding the peptide should be inducted in the presence of infectious agents.18 In practice, this last requirement is not ever fulfilled as some AMPs are tissue-specific and are considered as part of the plant innate immunity, while other isoforms of the same class appear induced after pathogen inoculation.19
Plant AMPs are the central focus of the present review, comprising information on their structural features (at genomic, gene, and protein levels), resources, and bioinformatic tools available, besides the proposition of an annotation routine. Their biotechnological potential is also highlighted in the generation of both transgenic plants resistant to pathogens, and new drugs or bioactive compounds.
Overall Features of Plant AMPs
Antimicrobial peptides are ubiquitous host defense weapons against microbial pathogens. The overall plant AMP characterization regards the following variables (Figure 1): electrical charge, hydrophilicity, secondary and 3-dimensional (3D) structures, and the abundance or spatial pattern of cysteine residues.20 These features are primarily related to their defensive role(s) as membrane-active antifungal, antibacterial, or antiviral peptides.
Figure 1.

Plant antimicrobial peptide features considering DNA sequence level, protein structure, and physicochemical properties. LTP indicates lipid transfer protein.
Regarding the nucleotide sequence, plant AMPs are hypervariable and this genetic variability is considered crucial to provide diversity and the ability to recognize different targets. For their charges, AMPs can be classified as cationic or anionic (Figure 1). Most plant AMPs have positive charges, which is a fundamental feature for the interaction with the membrane lipids of pathogens.21 Concerning hydrophilicity, AMPs are generally amphipathic, that is, they exhibit molecular conformation with both hydrophilic and hydrophobic domains.22 With respect to their 3D structure, AMPs can be either linear or cyclic (Figure 1). Some linear AMPs adopt an amphipathic α-helical conformation, whereas non-α-helical linear peptides generally show 1 or 2 predominant amino acids.23 In turn, cyclic AMPs, including cysteine-containing peptides, can be divided into 2 subgroups based on the presence of a single or multiple disulfide bonds. A peculiar feature of these peptides is a cationic and amphipathic character, what improves their functioning as membrane-permeabilizing agents.23
Considering the secondary structures, AMPs may exhibit α-helices, β-chains, β-pleated sheets, and loops (Figure 1). Wang24 classified plant AMPs into 4 families (α, β, αβ, and non-αβ), based on the protein classification of Murzin et al,25 with some modifications. Antimicrobial peptides of the “α” family present α-helical structures,1 whereas AMPs from the “β” family contains β-sheet structures usually stabilized by disulfide bonds.26,27 Some plant AMPs show an α-hairpinin motif formed by antiparallel α-helices that are stabilized by 2 disulfide bridges.28 Such AMPs present a higher resistance to enzymatic, chemical, or thermal degradation.29 Antimicrobial peptides from the “αβ” family having both “α” and “β” structures are also stabilized by disulfide bridges. An example of AMP presenting “αβ” structures are defensins, usually with a cysteine-stabilized αβ motif (CSαβ), an α-helix, and a triple-stranded antiparallel β sheet stabilized mostly by 4 disulfide bonds.30 Finally, AMPs that do not belong to the “αβ” group exhibit no clearly defined “α” or “β” structures.26
Plant AMPs are also classified into families considering protein sequence similarity, cysteine motifs, and distinctive patterns of disulfide bonds, which determine the folding of the tertiary structure.31 Therefore, plant AMPs are commonly grouped as thionins, defensins, heveins, knottins (linear and cyclic), lipid transfer proteins (LTP), snakins, and cyclotides.27,31 These AMP categories will be detailed in the next sections, together with other groups here considered (Impatien-like, Macadamia [β-barrelins], Puroindoline (PIN), and Thaumatin-like protein [TLP]) and the recently described α-hairpinin AMPs. The description includes comments on their structure, pattern for regular expression (REGEX) analysis (when available), functions, tissue-specificity, and scientific data availability.
Thionin
Thionins are composed by 45 to 48 amino acid residues with a molecular weight around 5 kDa, considering the mature peptide. They are synthesized with a signal peptide together with the mature thionin and the so-called acidic domain.32 To date, there is no experimental information available about possible functions of the acidic domain, even though it is clearly not dispensable as shown by the high conservation of the cysteine residues.33
The thionin superfamily comprises 2 distinct groups of plant peptides α/β-thionins and γ-thionins with distinguished structural features.34 The α/β thionins have homologous amino acid sequences and similar structures.35 Besides, they are rich in arginine, lysine, and cysteine.36 In turn, γ-thionins have a greater similarity with defensins, and some authors classify them within this group.37 However, compared with the defensins, they present a longer conserved amino acid sequence.31
Regarding the cysteine motif, it can be divided into 2 subgroups, one with 8 residues connected by 4 disulfide bonds called 8C and the other with 6 residues connected by 3 disulfide bonds called 6C.38 The general designation of thionins has been proposed as a family of homologous peptides that includes purothionins. The first plant thionin was isolated in 1942 from wheat flour and labeled as purothionin.39 Since then, homologues from various taxa have been also identified, like viscotoxins (Viscum album) and crambins (Crambe abyssinica).40 They have also been isolated from different plant tissues like seeds, leaves, and roots.41,42
Thionins have been tested for different elicitors: Gram-positive43,44 or Gram-negative bacteria,45,46 yeast,38,43 insect larvae,47 nematode,33 and inhibitory proteinase.48 Thionins are hydrophobic in nature, interact with hydrophobic residues, and lyse bacteria cell membrane. Their toxicity is due to an electrostatic interaction with the negatively charged membrane phospholipids, followed by either pore formation or a specific interaction with a membrane.38 It has been reported that they are able to inhibit other enzymes possibly through covalent attachment mediated by the formation of disulfide bonds, as previously observed for other thionin/enzyme combinations.48
Thionin representatives with known 3D structures determined by X-ray crystallography are crambin (PDB ID: 1CRN), α1- and β-purothionins (PDB ID: 2PHN and 1BHP), β-hordothionin (PDB ID: 1WUW), and viscotoxin-A3 (PDB ID: 1OKH). The first to be determined was the mixed form of crambin.35,49 It showed a distinct capital Γ shape with the N terminus forming the first strand in a β-sheet. The architecture of this sheet is additionally strengthened by 2 disulfide bonds.50 After a short stretch of extended conformation, there is a helix-turn-helix motif. In crambin, there is a single disulfide involved in stabilizing the helix-to-helix contacts. At the center of this motif, there is a crucial Arg10 that forms 5 hydrogen bonds to tie together the first strand, the first helix, and the C terminus.50
Defensin
The first plant defensins were isolated from wheat51 and barley grains,52 initially called γ-hordothionins. Due to some similarities in cysteine content and molecular weight, they were classified as γ-thionins. Later, the term “γ-thionin” was replaced by “defensin” based on the higher number of primary and tertiary structures of these proteins and also on their antifungal activities more related to insect and mammalian defensins than to plant thionins.53 Plant defensins belong to a diverse protein superfamily called cis-defensin54 and exhibit cationic charge, consisting of 45 to 54 aa with 2 to 4 disulfide bonds.53,55 Plant defensins share similar tertiary structures and typically exhibit a triple-stranded antiparallel β sheet, enveloped by an α-helix and confined by intramolecular disulfide bonds1 (Figure 2A). This motif is called cysteine-stabilized αβ (CSαβ).56 The CSαβ defensins were classified into 3 groups based on their sequence, structure, and functional similarity.
Figure 2.

Three-dimensional structure of plant antimicrobial peptide representatives. Yellow arrows correspond to β-sheets; α-helices are represented in purple and disulfide bonds in red. (A) Defensin NaD1 of Nicotiana alata (PDB ID: 1MR4). (B) Lipid transfer protein TaLTP1.1 isolated from wheat (PDB ID: 1GH1). (C) Hevein of Hevea brasiliensis (PDB ID: 1HEV). (D) Knottin Ep-AMP1 of Echinopsis pachanoi (PDB ID: 2MFS). (E) MiAMP1 peptide of Macadamia integrifolia (PDB ID: 1C01). (F) Snakin-1 of Solanum tuberosum (PDB ID: 5E5Q). (G) Thaumatin from Thaumatococcus daniellii (1RQW). (H) Zeamatin of Zea mays (1DU5, chain A). (I). Theoretical 3-dimensional model of Impatiens balsamina IbAMP1. (J) Helical hairpin structure of the novel antimicrobial peptide EcAMP1 from seeds of barnyard grass (Echinochloa crus-galli) (PDB ID: 2L2R). PDB indicates protein data bank.
Defensins are known for their antimicrobial activity at low micromolar concentrations against Gram-positive and Gram-negative bacteria,57 fungi,58 viruses, and protozoa.59 In addition, they present protein inhibition, insecticidal, and antiproliferative activity, acting as an ion-channel blocker, being also associated with the inhibition of pathogen protein synthesis.60 Instead, plant defensins act in the regulation of signal transduction pathways and induce inflammatory processes, in addition to wound healing, proliferation control, and chemotaxis.61
In general, plant defensins do not present high toxicity to human cells, having in vivo efficacy records, with relevant therapeutic potential, and can be applied in treatments associated with traditional medicine.62 Cools et al63 reported that a peptide derived from a plant defensin (HsAFP1) acted synergistically with caspofungin (an antimycotic) (in vivo and in vitro) against the formation of Candida albicans biofilm on polystyrene and catheter substrate, indicating that the HsAFP1 variant presented a strong antifungal potential in the proposed treatment.
Other biotechnological applications of defensins are described, as in the case of EcgDf1, which was isolated from a legume (Erythrina crista-galli), heterologously expressed in Escherichia coli and purified. EcgDf1 inhibited the growth of various plant and human pathogens (such as Candida albicans and Aspergillus niger and the plant pathogens Clavibacter michiganensis ssp. michiganensis, Penicillium expansum, Botrytis cinerea and Alternaria alternate).64 Due to these features, EcgDf1 is a candidate for the development of antimicrobial products for both agriculture and medicine.64
Lipid transfer protein
Non-specific lipid transfer proteins (ns-LTPs) were first isolated from potato tubers65 and are actually identified in diverse terrestrial plant species. They comprise a large gene family, are abundantly expressed in most tissues, but absent in most basal plant groups as chlorophyte and charophyte green algae.66 They generally include an N-terminal signal peptide that directs the protein to the apoplastic space.67 Some LTPs have a C-terminal sequence that allows their post-translational modification with a glycosylphosphatidylinositol molecule, facilitating the integration of LTP on the extracellular side of the plasma membrane.
The ns-LTPs are small proteins which were thus named because of their function of transferring lipids between the different membranes carrying lipids (non-specifically, the list includes phospholipids, fatty acids, their acylCoAs, or sterols). They consist of approximately 100 aa and are relatively larger in size than other AMPs, such as defensins.
Depending on their sizes, LTPs may be classified into 2 subfamilies: LTP1 and LTP2, with relative molecular weight of 9 and 7 kDa, respectively.68,69 The limited sequence conservation turned this classification inadequate. Thus, a modified and expanded classification system was proposed, presenting 5 main types (LTP1, LTP2, LTPc, LTPd, and LTPg) and 5 additional types with a smaller number of members (LTPe, LTPf, LTPh, LTPj, and LTPk).66 The new classification system is not based on molecular size but rather on (1) the position of a conserved intron, (2) the identity of the amino acid sequence, and (3) the spacing between the cysteine (Cys) residues (Supplementary Figure S2). Although this latter classification system is the most recent, the conventional classification of LTP1 and LTP2 types has been maintained by most working groups.
Lipid transfer protein nomenclature has been confusing and without consistent guidelines or standards. There are several examples where specific LTPs receive different names in different scientific articles. The lack of a robust terminology sometimes turns it quite difficult, extremely time-consuming, and frustrating to compare LTPs with different roles/functions.67 Therefore, an additional nomenclature was also proposed by Salminen et al,67 naming LTPs as follows: AtLTP1.3, OsLTP2.4, HvLTPc6, PpLTPd5, and TaLTPg7, with the first 2 letters indicating the plant species (eg, At = Arabidopsis thaliana, Pp = Physcomitrella patens); LTP1, LTP2, and LTPc indicating the type; while the last digit (here 3-7) regards the specific number given to each gene or protein within a given LTP type. For the sake of clarity, the authors recommend the inclusion of a point between the type specification (LTP1 and LTP2) and the gene number. For LTPc, LTPd, LTPg, and other types of LTP defined with a letter, the punctuation mark was not recommended. This latter classification system is currently recommended as it comprises several features of LTPs and is more robust than the previous classification systems.
Lipid transfer proteins are small cysteine-rich proteins, having 4 to 5 helices in their tertiary structure (Figure 2B), which is stabilized by several hydrogen bonds. Such a folding gives LTPs a hydrophobic cavity to bind the lipids through hydrophobic interactions. This structure is stabilized by 4 disulfide bridges formed by 8 conserved cysteines, similar to defensins, although bound by cysteines in different positions. The disulfide bridges promote LTP folding into a very compact structure, which is extremely stable at different temperatures and denaturing agents.70-72 These foldings provide a different specificity of lipid binding at the LTP binding site, where the LTP2 structure is relatively more flexible and present a lower lipid specificity when compared with LTP1.34
The first 3D structure of an LTP was established for TaLTP1.1 based on 2D and 3D data of 1H-NMR, purified from wheat (Triticum aestivum) seeds in aqueous solution.73,74 Currently, several 3D structures of LTPs have been determined, either by nuclear magnetic resonance (NMR) or X-ray crystallography; in their free, unbound form or in a complex with ligands.
Hevein
The heveins were first identified in 1960 in the rubber-tree (Hevea brasiliensis), but its sequence was determined later, whereas a similarity was detected to the chitin-binding domain of an agglutinin isolated from Urtica dioica (L.)75 with 8 cysteine residues forming a typical Cys motif.76 The primary structure of the hevein consists of 29 to 45 aa, positively charged, with abundant glycine (6) and cysteine (8-10) residues,76 and aromatic residues.31,77 The chitin-binding domain is a determinant component in the identification of hevein-like peptides whose binding site is represented by the amino acid sequence SXFGY/SXYGY, where X regards any amino acid.76,78 Most heveins have a coil-β1-β2-coil-β3 structure that occurs by variations with the secondary structural motif in the presence of turns in 2 long coils in the β3 chain.31 Antiparallel β chains form the central β sheet of the hevein motif with 2 long coils stabilized by disulfide bonds (Figure 2C).
Although the presence of chitin has not been identified in plants, there are chitin-like structures present in proteins that exhibit a strong affinity to this polysaccharide isolated from different plant sources.79
The presence of 3 aromatic amino acids in the chitin-binding domain favors chitin binding by providing stability to the hydrophobic group C-H and the π electron system through van der Waals forces, as well as the hydrogen bonds between serine and N-acetylglucosamine (GlcNAc) present in the chitin structure.76,77 This domain is commonly found in chitinases of classes I to V, in addition to other plant antimicrobial proteins, such as lectins and PR-4 (Pathogenesis-Related protein 4) members.80,81 It may also occur in other proteins that bind to polysaccharide chitin,80 such as the antimicrobial proteins AC-AMP1 and AC-AMP2 of Amaranthus caudatus (Amaranthaceae) seeds which are homologous to hevein but lack the C-terminal glycosylated region.82 Plant chitinases (class I) have the hevein-like domains, called HLDs. Due to the similar structural epitopes between chitinases and heveins, they are responsible for the cross reactive syndrome (latex-fruit syndrome).83,84
Among the several classes of proteins mentioned, the proteins with a high degree of similarity to hevein are chitinases I and IV.76 Chitinases are known to play an essential role in plant defense against pathogens,85 also inhibiting in vitro fungal growth,86 especially when combined with β-1,3-glucanases.87 It also interferes with the growth of hyphae, resulting in abnormal ramification, delay, and swelling in their stretching.81 However, it has been shown that heveins have a higher inhibitory potential than chitinases and that their antifungal effect is not related only to the presence of chitinases88; Pn-AMP1 and Pn-AMP2 AMPs with hevein domains have potent antifungal activities against a broad spectrum of fungi, including those without chitin in their cell walls.88,89 Modes of action of chitinases usually include degradation and disruption of the fungal cell wall and plasma membrane due to its hydrolytic action, causing extravasation of plasma particles.21,89 Therefore, heveins have good antifungal activity, and only a few are active against bacteria, most of them with low activity.
Another role of hevein chitinases regards the antagonistic effect in triggering the aggregation of rubber particles in the latex extraction process in rubber trees. Unlike heveins, other chitinases inhibit rubber particle aggregation. However, its action in conjunction with other proteins (β-1,3-glucanase) increases the effect of β-1,3-glucanase on rubber particle aggregation.90 A study by Shi et al91 found that the interaction of the protein network related to the antipathogenic activity released by lutoids (lysosomal microvacuole in latex) is essential in closing laticiferous cells (cells that produce and store latex), not only providing a physical barrier, but a biochemical barrier used by laticiferous cells affected by pathogen invasion.
Knottins (cysteine-knot peptides)
Knottins are part of the cysteine-rich peptides (CRPs) superfamily, sharing the Cysteine-knot motif and therefore resembling other families as defensins, heveins, and cyclotides.92 Their structure was initially identified by crystallography of carboxypeptides isolated from potato, showing the Cysteine-knot motif with 39 aa and 6 cysteine residues.93 They are also called “Cysteine-knot peptides,” “inhibitor Cysteine-knot peptides,” or even “Cysteine-knot miniproteins” because their mature peptide presents less than 50 aa, forming 3 interconnected disulfide bonds in the Cysteine-knot motif, characterizing a particular scaffold.92 This conformation confers thermal stability at high temperatures. For example, the cysteine-stabilized β-sheet (CSB) motif derived from knottins presents stability at approximately 100°C with only 2 disulfide bonds.94 The knottins may have linear or cyclic conformation. However, both exhibit connectivity between the cysteines at positions 1-4C, 2-5C, and 3-6C, forming a ring at the last bridge92 (Figure 2D).
Knottins have different functions, such as signaling molecules,95 response against biotic and abiotic stresses,96 root growth,97 symbiotic interactions as well as antimicrobial activity against bacteria,98 fungi,99 virus,100 and insecticidal activity,101 among others. Knottins antimicrobial activity has been attributed to the action of functional components of the plasma membrane, leading to alterations of lipids, ion flux, and exposed charge.99 The accumulation of peptides on the surface of the membrane results in the weakening of the pathogen membrane,102 resulting in transient and toroidal perforations.99
Macadamia (β-barrelins)
In the course of a large-scale survey to identify novel AMPs from Australian plants,103,104 an AMP with no sequence homology was purified. Its complementary DNA (cDNA) was cloned from Macadamia integrifolia (Proteaceae) seeds, containing the complete peptide coding region. The peptide was named MiAMP1, being highly basic with an estimated isoelectric point (pI) of 10 and a mass of 8 kDa.
The MiAMP1 is 102 aa long, including a 26 aa signal peptide in the N-terminal region, bound to a 76 aa mature region with 6 cysteine residues.105 Its 3D structure was determined using NMR spectroscopy,104 revealing a unique conformation among plant AMPs, with 8 beta-strands arranged in 2 Greek key motifs, forming a Greek key beta-barrel (Figure 2E). Due to its particularities, MiAMP1 was classified as a new structural family of plant AMPs, and the name β-barrelins was proposed for this class.104 This structural fold resembles a superfamily of proteins called γ-crystallin-like characterized by the precursors βγ-crystallin.106 This family includes AMPs from other organisms, for example, WmKT, a toxin produced by the wild yeast Williopsis mraki.107
The MiAMP1 exhibited in vitro antimicrobial activity against various phytopathogenic fungi, oomycetes, and gram-positive bacteria103 with a concentration range of 0.2 to 2 μM generally required for a 50% growth inhibition (IC50). In addition, the transient expression of MiAMP1 in canola (Brassica napus) provided resistance against blackleg disease caused by the fungus Leptosphaeria maculans,108 turning MiAMP1 potentially useful for genetic engineering aiming at disease resistance in crop plants.
There are few scientific publications with Macadamia-like peptides, maybe because they prevail in primitive plant groups (eg, lycophytes, gymnosperms to early angiosperms as Amborella and Papaver), being apparently absent in derived angiosperms (eg, Asteridae, including Brassicaceae as Arabidopsis thaliana). On the contrary, they have been identified in some monocots (as Zantedeschia, Zea, and Sorghum).109 In fact, peptides similar to MiAMP1 appear to play a role in the defense against pathogens in gymnosperms, including species of economic importance (as Pinus and Picea) thus deserving attention for their biotechnological potential.109
Impatiens-like (Ib-AMPs)
Four closely related AMPs (Ib-AMP1, Ib-AMP2, Ib-AMP3, and Ib-AMP4) were isolated from seeds of Impatiens balsamina (Balsaminaceae) with antimicrobial activity against a variety of fungi and bacteria, and low toxicity to human cells in culture. These AMPs are the smallest isolated from plants to date, consisting of only 20 aa in length. The Ib-AMPs are highly basic and contain 4 cysteine residues that form 2 disulfide bonds. Interestingly, they have no significant homology with other AMPs available in public databases. Sequencing of cDNAs isolated from I. balsamina revealed that all 4 peptides are encoded within a single transcript. Concerning the predicted precursor of Ib-AMP protein, it consists of a pre-peptide followed by 6 mature peptide domains, each one of them flanked by propeptide domains ranging from 16 to 35 aa in length (Supplementary Figure S3). This primary structure with repeated domains of alternating basic peptides and acid propeptide domains has, to date, not been reported in other plant species.110
Patel et al111 conducted an experiment to purify Ib-AMP1 from seeds of Impatiens balsamina. After purification, this peptide had its secondary structure tested by Circular Dichroism (CD). The results revealed a peptide that may include a β-turn but do not show evidences for either helical or β-sheet structure over a range of temperature and pH. Structural information from 2D 1H-NMR was obtained in the form of proton-proton internuclear distances inferred from nuclear overhauser enhancements (NOEs) and dihedral angle restraints from spin-spin coupling constants, which were used for distance geometry calculations. Owing to the difficulty in obtaining the correct disulfide connectivity by chemical methods, the authors had built and performed 3 separate calculations: (1) a model with no disulfides; (2) another with predicted disulfide bonds; and (3) a model with alternative connectivity disulfide, as assigned from the Nuclear Overhauser Effect spectroscopy (NOESY) NMR spectra. As a result, 2 hydrophilic patches were observed at opposite ends and opposite sides of the models, whereas in between them a large hydrophobic patch was identified. However, the study did not conclude which of the 3 models would be the most likely representative of Ib-AMP1, reporting only that cysteines are necessary for maintaining the structure.
Based on the experiment performed by Patel et al,111 the present work built 3 different models: Model 1: without disulfide bonds, and the other 2 models with different disulfide connections—Model 2: NMR prediction by Patel et al111 6-Cys;16-Cys and 7-Cys;20-Cys, and Model 3: Disulfide Bond partner prediction by DiANNA 7-Cys;16-Cys and 6-Cys;20-Cys. Calculations have shown that although the peptide is small, the cysteines constrain part of it to adopt a well-defined main chain conformation. From residue 4 to 20 (except 11), the main chain is well-defined, whereas residues 1 to 3 in the N-terminal region present few restrictions and appear to be more flexible (Supplementary Figure S4). Analyzing the RMSD (root mean square deviation), we observed that all the models lost the initial conformation and, among them, Model 3 was the most stable. Models 1 and 2 showed a similar pattern (Supplementary Figure S5), as in the models of Patel et al,111 although Model 1 was the most flexible.
Little is known about Impatiens-like AMPs mode of action. Lee et al112 investigated the antifungal mechanism of Ib-AMP1 noting that when oxidized (bound by disulfide bridges), there occurs a 4-fold increase in antifungal activity against Aspergillus flavus and Candida albicans, as compared with reduced Ib-AMP1 (without disulfide bridges). Confocal microscopy analyses have shown that Ib-AMP1 can either bind to the cell surface or penetrate cell membranes, indicating an antifungal activity by inhibiting a distinct cellular process, rather than ion channel or membrane pore formation. Fan et al113 reported the Ib-AMP4 antimicrobial activity dependent of β-sheet configuration to enable insertion into the lipid membrane, thus killing the bacteria through a non-lytic mechanism.114
Current approaches aim to make changes in Ib-AMP to improve its antimicrobial activity. As an example, synthetic variants of Ib-AMP1 were fully active against yeasts and fungi, where the replacement of amino acid residues by arginine or tryptophan improved more than twice the antifungal activity.115 Another study involving AMP modification generated a synthetic peptide without the disulfide bridges (ie, a linear analog of Ib-AMP1), which showed an antimicrobial specificity 3.7 to 4.8 times higher than the wild-type Ib-AMP1.116
Puroindoline
Puroindolines are small basic proteins that contain a single domain rich in tryptophan. These proteins were isolated from wheat endosperm, have a molecular mass around 13 kDa, and a calculated isoelectric point higher than 10. At least 2 main isoforms (called PIN-a and PIN-b) are known, which are encoded by Pina-D1 and Pinb-D1 genes, respectively. These genes share 70.2% identical coding regions but exhibit only 53% identity in the 3′ untranslated region.117
Both PIN-a and PIN-b contain a structure with 10 conserved cysteine residues and a tertiary structure similar to LTPs, consisting of 4 α-helices separated by loops of varying lengths, with the tertiary structure joined by 5 disulfide bonds, 4 of which identical to ns-LTPs.117
The conformation of the 2 PIN isoforms was studied by infrared and Raman spectroscopy. Both PIN-a and PIN-b have similar secondary structures comprising approximately 30% helices, 30% β-sheets, and 40% non-ordered structures at pH 7. It has been proposed that the folding of both PINs is highly dependent on the pH of the medium. The reduction of the disulfide bridges results in a decrease of PINs solubility in water and to an increment of the β-sheet content by about 15% at the expense of the α-helix content.118 No high-resolution structure for any of the PIN isoforms is available, bringing challenges to understanding the function of their hydrophobic regions, with some evidence coming only from partially homolog peptides.117 However, Wilkinson et al119 proposed a theoretical model for several sequences of this AMP.
Puroindolines are proposed to be functional components of wheat grain hardness loci, control core texture, besides antifungal activity.120-123 Although the biological function of PINs is unknown, their involvement in lipid binding has been proposed. While LTPs bind to hydrophobic molecules in a large cavity, PINs interact only with lipid aggregates, that is, micelles or liposomes, through a single stretch of tryptophan residues. This stretch of tryptophan residues is especially significant in the main form, PIN-a (WRWWKWWK), while it is truncated in the smaller form, PIN-b (WPTWWK).124-126
Puroindolines form protein aggregates in the presence of membrane lipids, and the organization of such aggregates is controlled by the lipid structure. In the absence of lipids, these proteins may aggregate, but there is no accurate information on the relationship between aggregation and interaction with lipids. The antimicrobial activity of PINs is targeted to cell membranes. Charnet et al127 indicated that PIN is capable of forming ion channels in artificial and biological membranes that exhibit some selectivity over monovalent cations. The stress and Ca2+ ions modulate the formation and/or opening of channels. Puroindolines may also be membranotoxins, which may play a role in the plant defense mechanism against microbial pathogens.
Morris128 reported that the PIN-a and PIN-b act through similar but somewhat different modes, which may involve “membrane binding, membrane disruption and ion channel formation” or “intracellular nucleic acid binding and metabolic disruption.” Natural and synthetic mutants have allowed the identification of PINs as key elements for antimicrobial activity.
Snakin
Snakins are CRPs first identified in potato (Solanum tuberosum).129,130 Due to their sequence similarity to GASA (Gibberellic Acid Stimulated in Arabidopsis) proteins, the snakins were classified as members of the snakin/GASA family.131 The genes that encode these peptides have (1) a signal sequence of approximately 28 aa, (2) a variable region, and (3) a mature peptide of approximately 60 residues, with 12 highly conserved cysteine residues. These cysteine residues maintain the 3D structure of the peptide through disulfide bonds, besides providing stability to the molecule when the plant is under stress129,130,132,133 (Figure 2F; Supplementary Figure S6).
Snakins may be expressed in different parts of the plant, like stem, leaves, flowers, seeds, and roots,134-137 both constitutive or induced by biotic or abiotic stresses. In vitro activity was observed against a variety of fungi, bacteria, and nematodes, acting as a destabilizer of the plasma membrane.129,138,139 Moreover, they were reported as essential agents in biological processes such as cell division, elongation, cell growth, flowering, embryogenesis, and signaling pathways.140-143
Alpha-hairpinins
As reported by Nolde et al,144 alpha-hairpin emerged as a new AMP with unusual motif configuration. These peptides prevail in plants and their structure was resolved based on NMR data obtained from the EcAMP-1 peptide isolated from Barnyard grass seeds (Echinoa crus-galli).144 Some α-hairpinins comprise trypsin inhibitors with helical hairpin structure and this group was recently proposed as a new plant AMP family.145 Similar to other AMPs, the amino acid sequences of α-hairpinins are variable. They share the conserved cysteine motif (CX3CX1-15CX3C) that form a helix-loop-helix fold and may have 2 disulfide bridges C1-C4 and C2-C3.146 Its structural stability is maintained by forming hydrogen bonds, so that the side chains have a relatively stable spatial orientation.147
As reviewed by Slavokhotova et al,148 members of alpha-hairpin family have been described in both mono and dicot groups, including species as Echinochloa crus-galli and Zea mays (both Poaceae, monocot), Fagopyrum esculentum (Polygonaceae, eudicot), and Stellaria media (Caryophyllaceae, eudicot). Several transcripts with α-hairpinin motif exhibit similarities to snakin/GASA genes and are sometimes positioned within this family.
Although the α-hairpinins structure has been published, its mechanism of action is still not resolved (Figure 2J, PDB ID: 2L2R). However, studies indicate they present a potential DNA binding capacity.149
Cyclotide
The term cyclotide was created at the end of the past century to designate a family of plant peptides with approximately 30 aa in size and a structural motif called cyclic cysteine knot (CCK).150 This motif is composed by a head-to-tail cyclization that is stabilized by a knotted arrangement of disulfide bridges, with 6 conserved cysteines, connected as follows: C1-2, C3-6, C4-5.151 Cyclotides are generally divided into 2 subfamilies, Mӧbius and Bracelets, based on structural aspects. In addition to CCKs, 2 loops (between C1-2 and C4-5) have high similarity between both subfamilies, while the other 2 loops (between C2-3 and C3-4) exhibit some conservation within the subfamilies152,153 (Supplementary Figure S7).
To date, several cyclotides were identified in eudicot families such as Rubiaceae,154 Violaceae,155 Fabaceae,156 and Solanaceae,157 in addition to some monocots of Poaceae family.158 In general, cyclotides may act in defense against a range of agents like insects, helminths, or mollusks. In addition, they can also act as ecbolic (inducer of uterus contractions),154 antibacterial,159 anti-HIV,100 and anticancer factors.160 All these characteristics added to the stability conferred by the CCK motif turn these peptides into excellent candidates for drug development.161,162
Thaumatin-like protein
Thaumatins or TLPs belong to the PR-5 (Pathogen-related protein) family and received this name due to its first isolation from the fruit of Thaumatococcus daniellii (Maranthaceae) from West Africa.163 Thaumatin-like proteins are abundant in the plant kingdom,164 being found in angiosperms, gymnosperms, and bryophytes,163 being also identified in other organisms, including fungi,165,166 insects,167 and nematodes.168
Thaumatin-like proteins are known for their antifungal activity, either by permeating fungal membranes169 or by binding and hydrolyzing β-1,3-glucans.170,171 In addition, they may act by inhibiting fungal enzymes, such as xylanases,172 α-amylases, or trypsin.173 Besides, the expression of TLPs is regulated in response to some stress factors, such as drought,174 injuries,175 freezing,176 and infection by fungi177,178 viruses, and bacteria.179
As to the TLP structure, this protein presents characteristic thaumatin signature (PS00316): G-x-[GF]-x-C-x-T-[GA]-D-C-x(1,2)-[QG]-x(2,3)-C.180,181 Most of the TLPs have molecular mass ranging from 21 to 26 kDa,163 possessing 16 conserved cysteine residues (Supplementary Figure S8) involved in the formation of 8 disulfide bonds,182 which help in the stability of the molecule, allowing a correct folding even under extreme conditions of temperature and pH.183 Thaumatin-like proteins also contain a signal peptide at the N-terminal, which is responsible for targeting the mature protein to a particular secretory pathway.163 The tertiary structure presents 3 distinct domains, which are conserved and form the central cleft, responsible for the enzymatic activity of the protein, being located between domains I and II.184 This central cleft may be of an acidic, neutral, or basic nature depending on the binding of the different linkers/receptors. All plant TLPs with antifungal activity have an acidic cleft known as motif REDDD due to 5 highly conserved amino acid residues (arginine, glutamic acid, and 3 aspartic acid; Supplementary Figure S8), being very relevant for specific receptor binding and antifungal activity.169,185,186
Crystallized structures were determined for some plant TLPs, such as thaumatin187 (Figure 2G), zeamatin169 (Figure 2H), tobacco PR-5d185 and osmotin,186 the cherry allergen PruAv2,188 and banana allergen Ba-TLP,184 among other TLPs.
Some TLPs are known as small TLPs (sTLPs) due to the deletion of peptides in one of their domains, culminating in the absence of the typical central cleft. These sTLPs exhibit only 10-conserved cysteine residues, forming 5 disulfide bonds, resulting in a molecular weight of approximately 16 to 17 kDa. They have been described in monocots, conifers, and fungi, so far.163,189,190 Other TLPs exhibit an extracellular TLP domain and an intracellular kinase domain, being known as PR5K (PR5-like receptor kinases)191 and are present in both monocots and dicots. For example, Arabidopsis contains 3 PR5K genes, while rice has only 1.163
Bioinformatic Tools and Databases for Plant AMPs
Databases
With the rapid growth in the number of available sequences, it is unfeasible to handle such amount of data manually. Thus, AMP sequences (as well as their biological information) have been deposited in large general databases, such as UniProt and TrEMBL, which contain sequences of multiple origins.192,193 In this sense, the construction of databases that deal specifically with AMPs was an important step to organize the data.
During the past decade, several databases were built to support the deposition, consultation, and mining of AMPs. Thus, these databases can be classified into 2 groups: general and specific.194 The specific databases can be divided into 2 subgroups: those containing only 1 specific group (defensins or cyclotides) and those containing data from a supergroup of peptides (plant, animal, or cyclic peptides) (Supplementary Table 1). In general, both types of databases share some characteristics such as the way that the data are available or the tools to analyze AMPs.
The Collection of Antimicrobial Peptides (CAMPR3) is a database that comprises experimentally validated peptides, sequences experimentally deduced and still those with patent data, besides putative data based on similarity.195-197 The current version includes structures and signatures specific to families of prokaryotic and eukaryotic AMPs.197 The platform also includes some tools for AMP prediction.
The antimicrobial peptide database (APD)198 collects mature AMPs from natural sources, ranging from protozoa to bacteria, archaea, fungi, plants, and animals, including humans. AMPs encoded by genes that undergo post-translational modifications are also part of the scope, besides some peptides synthesized by multienzyme systems. The APD provides interactive interfaces for peptide research, prediction, and design, statistical data for a specific group, or for all peptides available in the database.
The LAMP (Database Linking Antimicrobial Peptides) comprises natural and synthetic AMPs, which can be separated into 3 groups: experimentally validated, predicted, and patented. Their data were primarily collected from the scientific literature, including UniProt and other AMP-related databases.199
The Database of Antimicrobial Activity and Structure of Peptides (DBAASP)200 contains information about AMPs from different origins (synthetic or non-synthetic) and complexity levels (monomers and dimers) that were retrieved from PubMed using the following keywords: antimicrobial, antibacterial, antifungal, antiviral, antitumor, anticancer, and antiparasitic peptides. This database is manually curated and provides information about peptides that have specific targets validated experimentally. It also includes information on chemical structure, post-translational modifications, modifications in the N/C terminal amino acids, antimicrobial activities, cell target and experimental conditions in which a given activity was observed, besides information about the hemolytic and cytotoxic activities of the peptides.200 Due to the diversity of AMPs and the need to accommodate the most representative subclasses, several databases were established, focusing on specific types, sources, or features. There are several ways to classify AMPs, and they can range from biological sources such as bacterial AMPs (bacteriocins), plants, animals, and so on; biological activity: antibacterial, antiviral, antifungal, and insecticide; and based on molecular properties, pattern of covalent bonds, 3D structure and molecular targets.201,202
The “Defensins Knowledgebase” is a database with manual curation and focused exclusively on defensins. This database contains information about sequence, structure, and activity, with a web-based interface providing access to information and enabling text-based search. In addition, the site presents information on patents, grants, laboratories, researchers, clinical studies, and commercial entities.203,205
The CyBase is a database dedicated to the study of sequences and 3D structures of cyclized proteins and their synthetic variants, including tools for the analysis of mass spectral fingerprints of cyclic peptides, also assisting in the discovery of new circular proteins.205
The PhytAMP is a database designed to be solely dedicated to plant AMPs based on information collected from the UniProt database and from the scientific literature through PubMed.206
PlantPepDB is a database with manual curation of plant-derived peptides, mostly experimentally validated at the protein level. It includes data on the physical-chemical properties and tertiary structure of AMPs, also useful to identify their therapeutic potential. Different search options for simple and advanced compositing are provided for users to perform dynamic search and retrieve the desired data. Overall, PlantPepDB is the first database that comprises detailed analysis and comprehensive information on phyto-peptides from a wide functional range.207
Biological data banks (DBs) are organized collections of data of diverse nature that can be retrieved using different inputs. The management of this information is done through various software and hardware resources, whose retrieval and organization can be performed in a quick and efficient way.208 Considering biological data, information can be classified into (1) primary (sequences), (2) secondary (structure, expression, metabolic pathways, types of drugs, etc), and (3) specialized, for example, containing information on a species or on a class of protein.209 Within this third group, some references to AMPs can be mentioned, such as CAMPR3196 and APD198 that compile sequence data and structure retrieved from diverse sources, and also the Defensin knowledgebase203 and the CyBase205 which are dedicated to specific classes of peptides (defensins and cyclotides, respectively), in addition to PhytAMP,206 a specific database of plant AMPs (Supplementary Table 2).
Retrieving and annotating sequences from databases
The first step to infer the function of a given sequence (annotation) is to retrieve it in databases. For this purpose, 3 approaches have been used mostly: (1) local alignments, especially by using Basic Local Alignment Search Tool (BLAST)210 and FASTA211; by searching for specific patterns using (2) REGEX or (3) Hidden Markov Model (HMM).194
The first approach has been widely used, since most of the information is available in databases as sequences, together with tools to align them, whereas the BLAST is the primary tool for doing so.212 This tool splits the sequence into small pieces (words), comparing it with the database. However, this approach has a limitation. Small motifs may not be significantly aligned as they comprise small portions of the sequences that can be smaller than 20% of the total size.31,194 Due to the high variability of AMPs, only few highly conserved sequences can be identified using this type of inference. To reduce the effects of local alignment limitations, other strategies based on the search for specific patterns were introduced, such as REGEX213 (Supplementary Table 1) and HMM.214
The REGEX is a precise way of describing a pattern in a string where each REGEX position must be set, although ambiguous characters (or wildcards) can also be used. For example, if we want to find a match for both amino acid sequences CAIESSK and WAIESK, we can use the following expression: [CW]AIES{1,2}K, this expression would find a pattern starting with the letter “C” or “W,” followed by an “A,” an “I,” and an “E,” 1 or 2 “S,” and ending with a “K.”
The HMMs are well-known for their effectiveness in modeling the correlations between adjacent symbols, domains, or events, and they have been extensively used in various fields of biological analysis, including pairwise and multiple sequence alignment, base-calling, gene prediction, modeling DNA sequencing errors, protein secondary structure prediction, noncoding RNA (ncRNA) identification, protein and RNA structural alignments, acceleration of RNA folding and alignment, fast noncoding RNA annotation, and many others. Using HMM, a statistic profile is included in the model, which is calculated from a sequence alignment, and a score is determined site-to-site, with conserved and variable positions defined a priori.194,215
Predicting antimicrobial activity
The design of new AMPs led to the development of methods for the discovery of new peptides, thus allowing new experiments to be done by researchers. In this sense, the new challenge lies in the construction of new prediction models capable of discovering peptides with desired activities.
The APD DB has established a prediction interface based on some parameters defined by the entire set of peptides available in this database. These values are calculated from natural AMPs to consider features like length, net charge, hydrophobicity, amino acid composition, and so on. If we take as an example the net load, the AMPs deposited in the APD range from –12 to +30. This is the first parameter incorporated into the prediction algorithm. However, most AMPs have a net load ranging from –5 to +10, which then becomes the alternative prediction condition. Therefore, the same method is applied to the remaining parameters. The prediction in APD is performed in 3 main steps. First, the sequence parameters will be calculated and compared. If defined as an AMP, the peptide can then be classified into 3 groups: (1) rich in given amino acids, (2) stabilized by disulfide, and (3) linear bridges. Finally, sequence alignments will be conducted to find 5 peptides of higher similarity.198,216,217
The advent of machine learning (ML) methods has promoted new possibilities for drug discovery. In ML inferences, both a positive and a negative dataset are usually required to train the predictive models. The positive data, in this case, regard preferably experimentally validated AMPs that can be collected in databases, whereas negative data are randomly selected protein sequences that do not have AMP characteristics.197,218 Machine learning methods based on support vector machine (SVM), random forest (RF), and neural networks (NN) have been the most widely used. SVM is a specific type of supervised method of ML, aiming to classify data points by maximizing the margin between classes in a high-dimensional space.219,220 Random forest is a non-parametric tree-based approach that combines the ideas of adaptive neighbors with bagging for efficient adaptive data inference. Neural networks is an information processing paradigm inspired by how a biological nerve system process information. It is composed of highly interconnected processing elements (neurons or nodes) working together to solve specific problems.221-223
Evaluating proteomic data
Regarding the use of AMPs in peptide therapeutics, as an alternative to antimicrobial treatment, new efficient and specific antimicrobials are demanded. As aforementioned, AMPs are naturally occurring across all classes of life, presenting high active potential as therapeutic agents against various kinds of bacteria.224 The identification of novel AMPs in databases is primarily dependent on knowing about specific AMPs together with a sufficient sequence similarity.225 However, orthologs may be divergent in sequence, mainly because they are under strong positive selection for variation in many taxa,226 leading to remarkably lower similarity, even in closely related species. In this scenario, where alignment tools present limited use, 1 strategy to identify AMPs is related to proteomic approaches.
Proteins and peptides are biomolecules responsible for various biochemical events in living organisms, from formation and composition to regulation and functioning. Thus, understanding of the expression, function, and regulation of the proteins encoded by an organism is fundamental, leading to the so-called “Proteomic Era.” The term “proteome” was first used by Marc Wilkins in 1994 and it represents the set of proteins encoded by the genome of a biological system (cell, tissue, organ, biological fluid, or organism) at a specific time under certain conditions.227 Protein extraction, purification, and identification methods have significantly advanced our capacity to elucidate many biological questions using proteomic approaches.228,229 Due to the wide diversity of proteomic analysis, methods makes the choice of the correct approach dependent on the type of material and compounds to be analyzed.213,230 Two main tools are used to isolate proteins: (1) the 2-dimensional electrophoresis (2-DE) associated with mass spectrometry (MS) and (2) liquid chromatography associated with MS, each one with its own limitations.230-232 Obtaining native proteins is a challenge in proteomics or peptidomics, due to high protein complexity in samples, as the occurrence of post-translational modifications. Alternative strategies applied to extraction, purification, biochemical, and functional analyses of these molecules have been proposed, favoring access to structural and functional information of hard-to-reach proteins and peptides.233
Based on 2D gel, Al Akeel et al234 evaluated 14 spots obtained from seeds of Foeniculum vulgare (Apiaceae) aiming at proteomic analyses and isolation of small peptides. Extracted proteins were subjected to 3 kDa dialysis, and separation was carried out by DEAE-ion exchange chromatography while further proteins were identified by 2D gel electrophoresis. One of its spots showed high antibacterial activity against Pseudomonas aeruginosa, pointing to promising antibacterial effects, but requiring further research to authenticate the role of the anticipated proteins. For AMPs, 2DE is challenging due to the low concentration of the peptide molecules captured by this approach, their small sizes, and their ionic features (strongly cationic). In addition, the limited number of available specific databases and high variability turn their identification through proteolysis techniques and mass spectrometry, matrix-assisted laser desorption/ionization (MALDI-MS) difficult. In addition, the partial hydrophobicity characteristics and surface charges facilitate peptide molecular associations, making analysis difficult by any known proteomic approaches.232 In addition, peptides are most often cleaved from larger precursors by various releasing or processing enzymes.235 Furthermore, profiles generated do not represent integral proteome, as 2DE has limitations to detect proteins with low concentration, values of extreme molecular masses, pIs, and hydrophobic proteins, including those of membranes.236 Due to these limitations, multidimensional liquid chromatography–high-performance liquid chromatography (MDLC-HPLC) has been successfully employed as an alternative to 2D gels. Techniques and equipments for the newly developed separation and detection of proteins and peptides, such as nano-HPLC and multidimensional HPLC, have improved proteomics evaluation.237 Molecular mass values obtained are used in computational searches in which they are compared with in silico digestion results of proteins in databases. In silico approaches, usually by the action of trypsin as a proteolytic agent, may generate a set of unique peptides whose masses are determined by MS.238,239 These methodologies are widely adopted for large-scale identification of peptide from MS/MS spectra.240 Theoretical spectra are generated using fragmentation patterns known for specific series of amino acids. The first 2 widely used search engines in database searching were SEQUEST241 and MASCOT (Matrix Science, Boston, MA; www.matrixscience.com).242 They rank peptide matches based on a cross-correlation to match the hypothetical spectra to the experimental one.
MASCOT is widely used for peptidomics and proteomics analysis, including AMP identification in many organisms, or to evaluate the antibacterial efficacy of new AMPs. Evaluating new AMP against multidrug-resistant (MDR) Salmonella enterica, Tsai et al243 used 2D gel electrophoresis and liquid chromatography–electrospray ionization–quadrupole-time-of-flight tandem MS to determine the protein profiles. The protein identification was performed using the MASCOT with trypsin as cutting enzyme, whereas NCBI nr protein was set as a reference database. The methodology used in this study indicated that the novel AMP might serve as a potential candidate for drug development against MDR strains, confirming the usability of MASCOT. In a similar way, Umadevi et al244 described the AMP profile of black pepper (Piper nigrum L.) and their expression on Phytophthora infection using label-free quantitative proteomics strategy. For protein/peptide identification, MS/MS data were searched against the APD database245 using an in-house MASCOT server, established full tryptic peptides with a maximum of 3 missed cleavage sites and carbamidomethyl on cysteine, besides an oxidized methionine included as variable modifications. The APD database was used for AMP signature identification,245 together with PhytAMP206 and CAMPR3.197 To enrich the characterization parameters, isoelectric point, aliphatic index, and grand average of hydropathy were also used246 (GRAVY) (using ProtParam tool), besides the net charge from PhytAMP database. Based on label-free proteomics strategy, they established for the first time the black pepper peptidomics associated with the innate immunity against Phytophthora, evidencing the usability of proteomics/peptidomics data for AMP characterization in any taxa, including plant AMPs, aiming the exploitation of these peptides as next-generation molecules against pathogens.244
Other tools use database searching algorithms, such as X!TANDEM,247 Open mass spectrometry search algorithm (OMSSA),248 ProbID,249 RADARS,250 and so on. These search engines are based on database search but use different scoring schemes to determine the top hit for a peptide match. General information on database search engines, their algorithms, and scoring schemes were reviewed by Nesvizhskii et al.251 Despite its efficient ability to identify peptides, database searching presents several drawbacks, like false positive identifications due to overly noisy spectra and lower quality peptides score (related to the short size of peptides). So, the identification is strongly influenced by the amount of protein in the sample, the degree of post-translational modification, the quality of automatic searches, and the presence of the protein in the databases.252,253 In this scenario, the knowledge about the genome from a specific organism is important to allow the identification of the exact pattern of a given peptide. If an organism has no sequenced genome, it is not searchable using these methods.235,240 Once the sequences are obtained, bioinformatic tools can be used to predict peptides structure and estimate bioactive peptides.254
More recently, an interactive and free web software platform, MixProTool, was developed, aiming to process multigroup proteomics data sets. This tool is compiled in R (www.r-project.org), providing integrated data analysis workflow for quality control assessment, statistics, gene ontology enrichment, and other facilities. The MixProTool is compatible with identification and quantification outputs from other programs, such as MaxQuant and MASCOT, where results may be visualized as vector graphs and tables for further analysis, in contrast to existing softwares, such as GiaPronto.255 According to the authors, the web tool can be conveniently operated, even by users without bioinformatics expertise, and it is beneficial for mining the most relevant features among different samples.24
Antimicrobial peptide modeling
The central tenet of structural biology is that structure determines function. For proteins, it is often said the “function follows form” and “form defines function.” Therefore, to understand protein function in detail at the molecular level, it is mandatory to know its tertiary structure.256 Experimental techniques for determining structures, such as X-ray crystallography, NMR, electron paramagnetic resonance, and electron microscopy, require significant effort and investments.257
All methods mentioned have their own limitations, and the gap between the number of known proteins and the number of known structures is still substantial. Thus, there is a need for computational framework methods to predict protein structures based on the knowledge of the sequence.256 In addition, in recent years, there has been impressive progress in the development of algorithms for protein folding that may aid in the prediction of protein structures from amino acid sequence information.258
Historically, the prediction of a protein structure has been classified into 3 categories: comparative modeling, threading, and ab initio. The first 2 approaches construct protein models by aligning the query sequences with already solved model structures. If the models are absent in the Protein Data Bank (PDB), the models must be constructed from scratch, that is, by ab initio modeling, considered the most challenging way to predict protein structures.256
In the case of comparative modeling methods, when inserting a target sequence, the programs identify evolutionarily related models of solved structures based on their sequence or profile comparison, thus constructing structure models supported by these previously resolved models.259 This approach comprises 4 main steps: (1) fold assignment, which identifies similarity between the target and the structure of the solved model; (2) alignment of the target sequence to the model; (3) generation of a model based on alignment with the chosen template; and (4) analysis of errors considering the generated model.260
There are several servers and computer models that automate the comparative modeling process, with SWISS-MODEL and MODELER figuring as the most used.261,262 Although automation makes comparative modeling accessible to experts and beginners, some adjustments are still needed in most cases to maximize model accuracy, especially in the case of more complex proteins.262 Therefore, some caution must be taken regarding the generated models, considering the resolution and quality of the model used, as well as homology between the model and the protein of interest.
Threading modeling methods are based on the observation that known protein structures appear to comprise a limited set of stable folds, and those similarity elements are often found in evolutionarily distant or unrelated proteins. The most used servers based on this approach are MUSTER,263 SPARKS-X,264 RaptorX,259 ProSa-Web,265 and most notably the I-TASSER.266 In some cases, the incorporation of structural information to combine the sequence used in the search with possible models allows the detection of similarity in the fold, even in the absence of an explicit evolutionary relation.
The prediction of structures from known protein models is, at first sight, a more straightforward task than the prediction of protein structures from available sequences. Therefore, when no solved model is available, another approach is recommended, namely, the ab initio modeling. This method is intended to predict the structure only from the sequence information, without any direct assistance from previously known structures. The ab initio modeling aims to predict the best model, based on the minimum energy for a potential energy function by sampling the potential energy surface using various searchable information.267,268 Such approaches turn it challenging to produce high-resolution modeling, essential for determining the native protein folding and its biochemical interpretation. On the contrary, later resolved structures and comparisons with previously predicted proteins point to a higher successful modeling generated by ab initio methods than those generated by pure energy minimization methods, classical or even pure methods.256
Among the most used servers and programs for ab initio modeling, we highlight the ROSETTA,257 QUARK,269 and TOUCHSTONE II.267 The accuracy of the models calculated by many of these methods is evaluated by CAMEO (Continuous Automated Model EvaluatiOn)270 and by CASP (Critical Assessment of protein Structure Prediction).258 Probably the first reasonably accurate ab initio model was built in CASP4. Since then, sustained progress was achieved in ab initio prediction, but mainly for small proteins (120 residues or less). In CASP11, for the first time, a novel 256-residue protein with a sequence identity with known structures lower than 5% was constructed with high precision for sequences of this size.271
In CASP12, a significant improvement was reported in 4 areas: contact prediction, free modeling, template-based modeling, and estimating the accuracy of models. The authors report that this improvement is due to the accuracy of modeling and alignment methods, as well as increased data availability for both sequence and structure.258
Due to the number of AMPs deposited in the PDB (to date approximately 1099 structures), comparative modeling is the most used. However, when it comes to de novo peptide design, the most recommended choice would be ab initio272 or a hybrid approach that uses more than 1 modeling method.273
Molecular dynamics simulation
After the generation of a model, the AMP stability should be evaluated using molecular dynamics (MD). Molecular dynamics comprises the application of computational simulations that predict the changes in the positions and velocities of the constituent atoms of a system under given time and condition. This calculation is done through a classical approximation of empirical parameters, called “force field.”274 If, on one hand, this approximation makes the dynamics of a system containing thousands of atoms numerically accessible, it obviously limits the nature of the processes that can be observed during the simulations. No quantum effect is visualized in a MD simulation; just as no chemical bond is broken, no interactions occur between orbitals, resonance, polarization, or charge transfer effects.275 However, the molecules go beyond a static system. Thus, MD is a computational technique that can be used for predicting or refining structures, dynamics of molecular complexes, drug development, and action of molecular biological systems.276 Molecular dynamics simulation is widely used for protein research, aiming to extract information about the physical properties of individual proteins. The results of such simulations are then compared with experimental results. As these experiments are generally carried out in solvents, it is necessary to simulate molecular systems of protein in water. These simulations have a variety of applications, such as determining the folding of a structure to a native structure and analyzing the dynamic stability of this structure.277
The use of MD to simulate protein folding processes is one of the most challenging applications and should be relatively long (in the order of microseconds to milliseconds) to allow observing a single fold event. In addition, the force field used must correctly describe the relative energies of a wide variety of shapes, including unfolding and poorly folded shapes that may occur during the simulation.275 The considerable application potential led to the implementation of MD simulation in many software packages, including GROMACS,278-280 AMBER,281 NAMD,282 CHARMM,283 LAMMPS,284 and Desmond.285 In addition to the above mentioned, there are other simulation types available, such as the Monte Carlo Method, Stochastic Dynamics, and Brownian Dynamics.280
In the last decades, MD simulation has become a standard tool in theoretical studies of large biomolecular systems, including DNA or proteins, in environments with near realistic solvents. Indeed, simulations have proven valuable in deciphering functional mechanisms of proteins and other biomolecules, in uncovering the structural basis for disease, and in the design and optimization of small molecules, peptides, and proteins.286 Historically, the computational complexity of this type of computation has been extremely high, and much research has focused on algorithms to achieve unique simulations that are as long or as large as possible.278
Plant-pathogen interaction and molecular docking
The interplay between a given pathogen (eg, virus, bacteria, fungus) must be studied through a holistic approach. Host-pathogen relationships are very complex and occur at diverse conceivable levels, including the cellular/molecular level of both, pathogen and host, under given environmental conditions. A most approximate understanding of these interactions at every level is the ultimate goal of “systems biology” (SB). It comprises a holistic approach, integrating distinct disciplines, as biology, computer science, engineering, bioinformatics, physics, and others to predict how a given system behaves under given conditions and what is the role of its parts. Systems biology stands out because it is capable of correlating omics data for the understanding of plant-pathogen interaction. The construction of a plant-pathogen interaction network includes the reconstruction of metabolic pathways of these organisms, identification of the degree of pathogenicity, besides the expression of genes and proteins from both plant and pathogen. The networks can be classified into 5 types: (1) regulatory; (2) metabolic; (3) protein-protein interaction; (4) signaling and regulatory; and (5) signaling, regulatory, and metabolic.287 Each of these networks can be plotted according to computational approaches.
Also, further studies are required to contemplate the construction of evolutionary in silico models and the characterization of these molecular targets in vitro.288,289 Studies of protein-protein interactions to understand the regulatory process are essential290 and new computational methods are necessary for this purpose with more optimized algorithms, also to remove potential false positives. Thus, in-depth studies on the orientation of molecules and their linkages to the formation of a stable complex are of great importance for understanding plant-pathogen studies and also to develop new drugs.291
Molecular docking
The understanding of the regulatory principles by which protein receptors recognize, interact, and associate with molecular substrates or inhibitors is of paramount importance to generate new therapeutic strategies.292 In modern drug discovery, docking plays an important role in predicting the orientation of the binder when it is attached to a protein receptor or enzyme, using forms and electrostatic interactions, van der Walls, Coulombic, and hydrogen bond as parameters to quantify or predict a given interaction.293,294 Molecular docking aims at exploring the predominant mode(s) of binding of a molecule (protein or ligand) when it binds to a protein with a known 3D structure based on a scoring function that has 3 main functions: the first is to determine the binding mode and the binding site of a protein, the second is to predict the absolute binding affinity between protein and ligand (or other protein) in lead optimization, and the third is virtual screening, which can identify potential drug leads for a given protein target by searching a large ligand or protein in database.295
Protein-protein interactions are essential for cellular and immune function. In many cases, due to the absence of an experimentally determined structure of the complex, these interactions must be modeled to obtain an understanding about their structure and molecular basis.296 Few studies on plant-pathogen interactions include docking approaches and most studies focus on drug development for medical purposes. Drug research based on structure is a powerful technique for the rapid identification of small molecules against the 3D structure of available macromolecular targets, usually by X-ray crystallography, NMR structures, or homology models.
Due to abundant information on protein sequences and structures, the structural information on specific proteins and their interactions have become crucial for current pharmacological research.297 Even in the absence of knowledge about the binding site and limited backbone movements, a variety of algorithms have been developed for docking over the past 2 decades. Although the ZDOCK,296 the rDOCK,298 and the HEX299 have provided results with high coupling precision, the complexes provided are not very useful for designing inhibitors for protein interfaces due to constraints on rigid body docking.294 In this context, more flexible approaches have been developed which generally examine very limited conformations compared with rigid body methods. These docking methods predict that binding is more likely to occur in broad surface regions and then defines the sites in complex structures of high affinity.300 The best example is the HADDOCK software,297 which has been successful in solving a large number of precise models for protein-protein complexes. A good example of its use is the study of the complex formed between plectasin, a member of the innate immune system, and a precursor lipid of bacterial cell wall II. The study identified the residues involved in the binding site between the 2 proteins, providing valuable information for planning new antibiotics.301
However, the absolute energies associated with intermolecular interaction are not estimated with satisfactory accuracy by the current algorithms. Some significant issues as solvent effects, entropic effects, and receptor flexibility still need to be addressed. However, some methods, such as MOE-Dock,302 GOLD,303 Glide,304 FlexX,305 and Surflex306 which deal with lateral chain flexibility, have proven to be effective and adequate in most cases. Realistic interactions between small molecules and receptors still depend on experimental wet-lab validation.294,307
Despite the current difficulties, there is a growing interest in the mechanisms and prediction of small molecules such as peptides, as they bind to proteins in a highly selective and conserved manner, being promising as new medicinal and biological agents.308 While both “small molecule docking methods” and “custom protocols” can be used, short peptides are challenging targets because of their high torsional flexibility.307 Protein-peptide docking is generally more challenging than those related to other small molecules, and a variety of methods have been applied so far. However, few of these approaches have been published in a way that can be reproduced with ease.309-311 Although it is difficult to use peptide docking, a recent focus of basic and pharmacological research has used computational tools with modified peptides to predict the selective disruption of protein-protein interactions. These studies are based on the involvement of some critical amino acid residues that contribute most to the binding affinity of a given interaction, also called hot-spots.312,313
Despite the number of docking programs, existing algorithms still demand improvements. However, approaches are being developed to improve all issues related to punctuation, protein flexibility, interaction with plain water, among other issues.314 In this context, the CAPRI (Critical Assessment of Predicted Interactions) is a community that provides a quality assessment of different docking approaches. It started in 2001 and since then has aided the development and improvement of the methodologies applied for docking.315 An evaluation was carried out for CAPRI in 2016, resulting in an improvement in the integration of different modeling tools with docking procedures, as well as the use of more sophisticated evolutionary information to classify models. However, adequate modeling of conformational flexibility in interacting proteins remains an essential demand with a crucial need for improvement.314 Different docking programs are currently available,294 and new alternatives continue to appear. Some of these alternatives will disappear, just as others will become the top choices among field users.
Molecular docking technique is not often used for AMPs, due to its standard mechanism of action based on the classical association with the external membrane of the pathogen. Despite that, some AMPs have the ability to bind other proteins and/or enzymes, a feature still scarcely studied. In such cases, molecular docking can be useful. An example of success is the study performed by Melo et al,47 where they showed the specific binding of a trypsin to a cowpea (Vigna unguiculata) thionine, revealing that this interaction occurs in a canonical manner with Lys11, located in an extended exposed loop. Therefore, further application of docking may bring new evidences about the antimicrobial mechanisms revealing other molecular targets of interest.
It is clear that the combination of data bank information with bioinformatic tools (especially those allowing the identification of patterns, rather than sequence order) is able to revolutionize the identification of AMPs and prediction of their activity. The data may come from genomic, transcriptomic, or proteomic databases, or a combination of different information sources (eg, genomic and transcriptomics, transcriptomics and proteomics).
Supplementary Figure S9 brings a schematic flowchart describing the steps for mining, annotation, and structural/functional analysis of AMPs, in addition to some wet-lab analyses that can be integrated to assess/confirm candidate AMPs.
Similar bioinformatic approaches have been actually used to identify potential peptide candidates with anti-SARS-CoV-2 activity, especially those potentially able to interact with the spike protein and proteases involved in viral penetration.316,317
Concluding Remarks and Perspectives
As emphasized, plant AMPs show greater diversity and abundance, when compared with other kingdoms. It can be speculated that plants shelter many yet undescribed AMP classes, given their vast abundance and isoform diversity.
The genomic and peptidic structure of AMPs can be variable, with few key residues conserved, which turns their identification, classification, and comparison challenging even in the omics age. Nevertheless, advances in the generation of new bioinformatics tools and specialized databases have led to new and more efficient approaches for both the identification of primary sequences and molecular modeling, besides the analysis of the stability of the generated models.
Despite the large availability of omics data and bioinformatics tools, most new plant peptides have been discovered by wet-lab approaches regarding single candidates. High throughput in silico methods have the potential to transform this scenario, revealing many new candidates, including some new or “non-canonical” peptides. It may be also speculated that a myriad of new peptides may exist considering even smaller peptides, still less considered and more difficult to identify. Finally, in silico approaches shall in future studies be mandatory to define the design of wet-lab studies, turning the identification more efficient and requiring reasonably less time to track, identify, and confirm new candidate AMPs.
Considering the actual pandemic scenario of COVID-19, plant AMPs may be regarded as an important source of antiviral drug candidates, especially considering that some AMP categories present not only antiviral effects but also a wide spectrum antimicrobial activity, act as anti-inflammatory, and also induce the immune response.
Supplemental Material
Supplemental material, Figure_S2_xyz356052dbb1c09 for Plant Antimicrobial Peptides: State of the Art, In Silico Prediction and Perspectives in the Omics Era by Carlos André dos Santos Silva, Luisa Zupin, Marx Oliveira-Lima, Lívia Maria Batista Vilela, João Pacifico Bezerra-Neto, José Ribamar Ferreira-Neto, José Diogo Cavalcanti Ferreira, Roberta Lane de Oliveira-Silva, Carolline de Jesús Pires, Flavia Figueira Aburjaile, Marianne Firmino de Oliveira, Ederson Akio Kido, Sergio Crovella and Ana Maria Benko-Iseppon in Bioinformatics and Biology Insights
Supplemental material, Figure_S3_xyz356055a6a5139 for Plant Antimicrobial Peptides: State of the Art, In Silico Prediction and Perspectives in the Omics Era by Carlos André dos Santos Silva, Luisa Zupin, Marx Oliveira-Lima, Lívia Maria Batista Vilela, João Pacifico Bezerra-Neto, José Ribamar Ferreira-Neto, José Diogo Cavalcanti Ferreira, Roberta Lane de Oliveira-Silva, Carolline de Jesús Pires, Flavia Figueira Aburjaile, Marianne Firmino de Oliveira, Ederson Akio Kido, Sergio Crovella and Ana Maria Benko-Iseppon in Bioinformatics and Biology Insights
Supplemental material, Figure_S4_xyz35605158f10b9 for Plant Antimicrobial Peptides: State of the Art, In Silico Prediction and Perspectives in the Omics Era by Carlos André dos Santos Silva, Luisa Zupin, Marx Oliveira-Lima, Lívia Maria Batista Vilela, João Pacifico Bezerra-Neto, José Ribamar Ferreira-Neto, José Diogo Cavalcanti Ferreira, Roberta Lane de Oliveira-Silva, Carolline de Jesús Pires, Flavia Figueira Aburjaile, Marianne Firmino de Oliveira, Ederson Akio Kido, Sergio Crovella and Ana Maria Benko-Iseppon in Bioinformatics and Biology Insights
Supplemental material, Figure_S5_xyz35605fb2523e9 for Plant Antimicrobial Peptides: State of the Art, In Silico Prediction and Perspectives in the Omics Era by Carlos André dos Santos Silva, Luisa Zupin, Marx Oliveira-Lima, Lívia Maria Batista Vilela, João Pacifico Bezerra-Neto, José Ribamar Ferreira-Neto, José Diogo Cavalcanti Ferreira, Roberta Lane de Oliveira-Silva, Carolline de Jesús Pires, Flavia Figueira Aburjaile, Marianne Firmino de Oliveira, Ederson Akio Kido, Sergio Crovella and Ana Maria Benko-Iseppon in Bioinformatics and Biology Insights
Supplemental material, Figure_S6_xyz35605b863c136 for Plant Antimicrobial Peptides: State of the Art, In Silico Prediction and Perspectives in the Omics Era by Carlos André dos Santos Silva, Luisa Zupin, Marx Oliveira-Lima, Lívia Maria Batista Vilela, João Pacifico Bezerra-Neto, José Ribamar Ferreira-Neto, José Diogo Cavalcanti Ferreira, Roberta Lane de Oliveira-Silva, Carolline de Jesús Pires, Flavia Figueira Aburjaile, Marianne Firmino de Oliveira, Ederson Akio Kido, Sergio Crovella and Ana Maria Benko-Iseppon in Bioinformatics and Biology Insights
Supplemental material, Figure_S7_xyz3560531a00dc6 for Plant Antimicrobial Peptides: State of the Art, In Silico Prediction and Perspectives in the Omics Era by Carlos André dos Santos Silva, Luisa Zupin, Marx Oliveira-Lima, Lívia Maria Batista Vilela, João Pacifico Bezerra-Neto, José Ribamar Ferreira-Neto, José Diogo Cavalcanti Ferreira, Roberta Lane de Oliveira-Silva, Carolline de Jesús Pires, Flavia Figueira Aburjaile, Marianne Firmino de Oliveira, Ederson Akio Kido, Sergio Crovella and Ana Maria Benko-Iseppon in Bioinformatics and Biology Insights
Supplemental material, Figure_S8_xyz35605c5b2d379 for Plant Antimicrobial Peptides: State of the Art, In Silico Prediction and Perspectives in the Omics Era by Carlos André dos Santos Silva, Luisa Zupin, Marx Oliveira-Lima, Lívia Maria Batista Vilela, João Pacifico Bezerra-Neto, José Ribamar Ferreira-Neto, José Diogo Cavalcanti Ferreira, Roberta Lane de Oliveira-Silva, Carolline de Jesús Pires, Flavia Figueira Aburjaile, Marianne Firmino de Oliveira, Ederson Akio Kido, Sergio Crovella and Ana Maria Benko-Iseppon in Bioinformatics and Biology Insights
Supplemental material, Figure_S9_xyz3560525a5abea for Plant Antimicrobial Peptides: State of the Art, In Silico Prediction and Perspectives in the Omics Era by Carlos André dos Santos Silva, Luisa Zupin, Marx Oliveira-Lima, Lívia Maria Batista Vilela, João Pacifico Bezerra-Neto, José Ribamar Ferreira-Neto, José Diogo Cavalcanti Ferreira, Roberta Lane de Oliveira-Silva, Carolline de Jesús Pires, Flavia Figueira Aburjaile, Marianne Firmino de Oliveira, Ederson Akio Kido, Sergio Crovella and Ana Maria Benko-Iseppon in Bioinformatics and Biology Insights
Supplemental material, Supplementary_Table_1_AND_2_Final_13-07-20_xyz43729463bfb8d for Plant Antimicrobial Peptides: State of the Art, In Silico Prediction and Perspectives in the Omics Era by Carlos André dos Santos Silva, Luisa Zupin, Marx Oliveira-Lima, Lívia Maria Batista Vilela, João Pacifico Bezerra-Neto, José Ribamar Ferreira-Neto, José Diogo Cavalcanti Ferreira, Roberta Lane de Oliveira-Silva, Carolline de Jesús Pires, Flavia Figueira Aburjaile, Marianne Firmino de Oliveira, Ederson Akio Kido, Sergio Crovella and Ana Maria Benko-Iseppon in Bioinformatics and Biology Insights
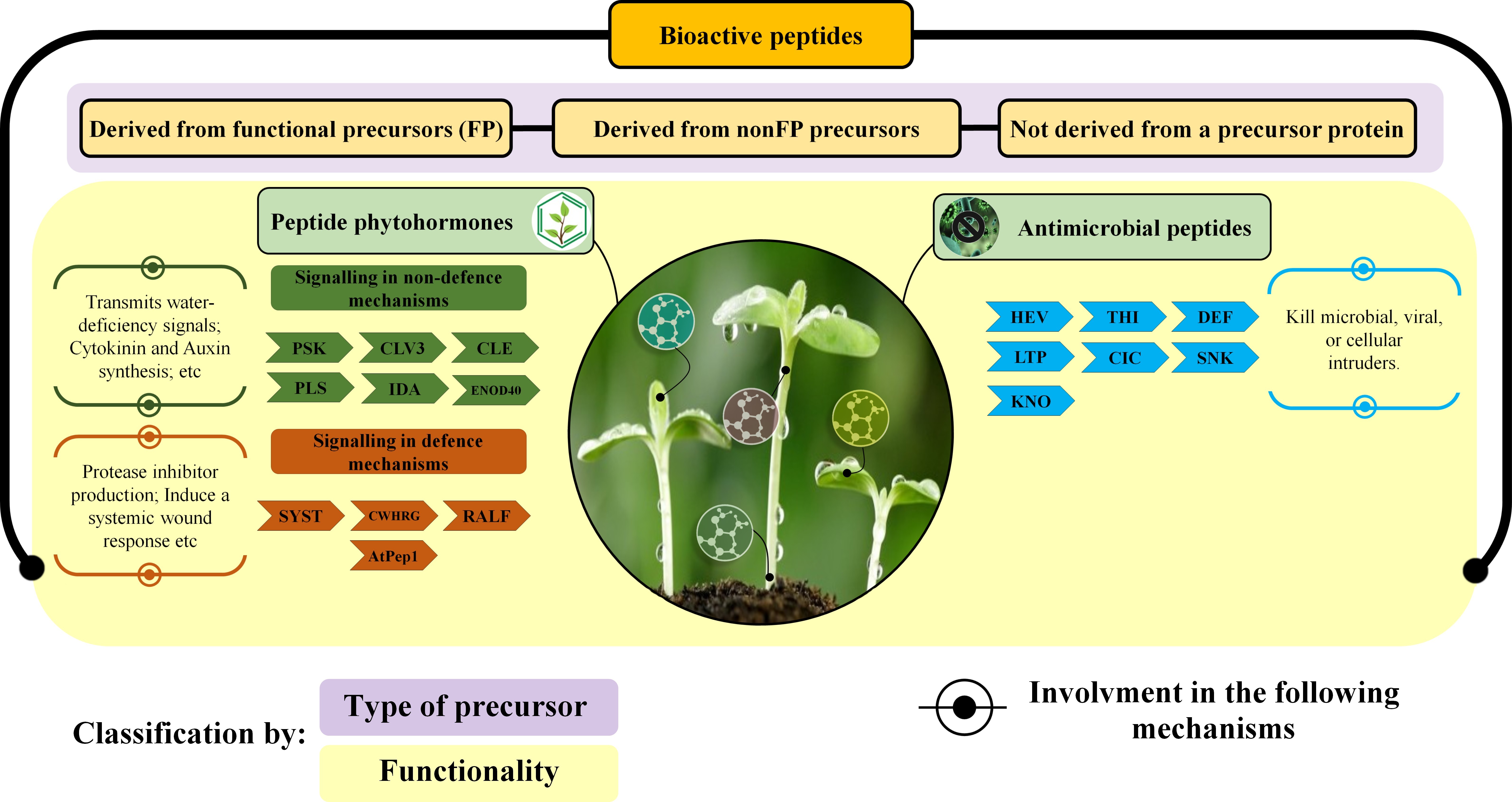{kind=link}
Supplemental material, Sup_Figure_1_13-07-20_Corrected_Version_xyz43729c25f998f for Plant Antimicrobial Peptides: State of the Art, In Silico Prediction and Perspectives in the Omics Era by Carlos André dos Santos Silva, Luisa Zupin, Marx Oliveira-Lima, Lívia Maria Batista Vilela, João Pacifico Bezerra-Neto, José Ribamar Ferreira-Neto, José Diogo Cavalcanti Ferreira, Roberta Lane de Oliveira-Silva, Carolline de Jesús Pires, Flavia Figueira Aburjaile, Marianne Firmino de Oliveira, Ederson Akio Kido, Sergio Crovella and Ana Maria Benko-Iseppon in Bioinformatics and Biology Insights
Acknowledgments
The authors are very grateful to CAPES (Coordination for the Improvement of Higher Education Personnel, BioComputational Program), CNPq (Brazilian National Council for Scientific and Technological Development), and FACEPE (Fundação de Amparo à Ciência e Tecnologia de Pernambuco) for fellowships.
The project is supported by the Interreg Italia- Slovenia, ISE-EMH 07/2019 and RC 03/20 from IRCCS Burlo Garofolo/Italian Ministry of Health
Footnotes
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors are very grateful to CAPES (Coordination for the Improvement of Higher Education Personnel, BioComputational Program), CNPq (Brazilian National Council for Scientific and Technological Development), and FACEPE (Fundação de Amparo à Ciência e Tecnologia de Pernambuco) for valuable financial support. The project is also supported by the Interreg Italia- Slovenia, ISE-EMH 07/2019 and RC 03/20 from IRCCS Burlo Garofolo/Italian Ministry of Health.
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: CASS performed the literature review and whore the manuscript. LZ, MOL, LMBV, JPBN, JRFN, JDCF, RLOS, CJP, FFA, and MFO wrote specific chapters, EAK and SC critically revised the text and included relevant suggestions. AMBI conceived the review, wrote the introduction and concluding remarks, besides critically revising the manuscript. All authors have read the manuscript and agree to its content.
ORCID iDs: Luisa Zupin  https://orcid.org/0000-0001-5886-9129
https://orcid.org/0000-0001-5886-9129
Roberta Lane de Oliveira-Silva
https://orcid.org/0000-0003-1876-0094
Flavia Figueira Aburjaile
https://orcid.org/0000-0002-1067-1882
Supplemental Material: Supplemental material for this article is available online.
References
- 1. Whisstock JC, Lesk AM. Prediction of protein function from protein sequence and structure. Q Rev Biophys. 2003;36:307-340. doi: 10.1017/S0033583503003901. [DOI] [PubMed] [Google Scholar]
- 2. Marmiroli N, Maestri E. Plant peptides in defense and signaling. Peptides. 2014;56:30-44. doi: 10.1016/j.peptides.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 3. Germain H, Chevalier E, Matton DP. Plant bioactive peptides: an expanding class of signaling molecules. Can J Bot. 2006;84:1-19. doi: 10.1139/b05-162. [DOI] [Google Scholar]
- 4. Moyes DL, Wilson D, Richardson JP, et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature. 2016;532:64-68. doi: 10.1038/nature17625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Essig A, Hofmann D, Münch D, et al. Copsin, a novel peptide-based fungal antibiotic interfering with the peptidoglycan synthesis. J Biol Chem. 2014;289:34953-34964. doi: 10.1074/jbc.M114.599878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Singh V, Singh K, Amdekar S, et al. Innate and specific gut-associated immunity and microbial interference. FEMS Immunol Med Microbiol. 2009;55:6-12. doi: 10.1111/j.1574-695X.2008.00497.x. [DOI] [PubMed] [Google Scholar]
- 7. Flaherty RA, Freed SD, Lee SW. The wide world of ribosomally encoded bacterial peptides. PLoS Pathog. 2014;10:e1004221. doi: 10.1371/journal.ppat.1004221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baghizadeh Fini M. Oral saliva and COVID-19. Oral Oncol. 2020;108:104821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Koeninger L, Armbruster NS, Brinch KS, et al. Human β-defensin 2 mediated immune modulation as treatment for experimental colitis. Front Immunol. 2020;11:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Farrokhi N, Whitelegge JP, Brusslan JA. Plant peptides and peptidomics. Plant Biotechnol J. 2008;6:105-134. doi: 10.1111/j.1467-7652.2007.00315.x. [DOI] [PubMed] [Google Scholar]
- 11. Higgins CF, Payne JW. Plant peptides. In: Boulter D, Parthier B, eds. Nucleic Acids and Proteins in Plants I. Berlin, Germany: Springer; 1982:438-458. doi: 10.1007/978-3-642-68237-7_13. [DOI] [Google Scholar]
- 12. Tavormina P, De Coninck B, Nikonorova N, De Smet I, Cammue BP. The plant peptidome: an expanding repertoire of structural features and biological functions. Plant Cell. 2015;27:2095-2118. doi: 10.1105/tpc.15.00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Takahashi F, Suzuki T, Osakabe Y, et al. A small peptide modulates stomatal control via abscisic acid in long-distance signalling. Nature. 2018;556:235-238. doi: 10.1038/s41586-018-0009-2. [DOI] [PubMed] [Google Scholar]
- 14. Liu J, Mehdi S, Topping J, Friml J, Lindsey K. Interaction of PLS and PIN and hormonal crosstalk in Arabidopsis root development. Front Plant Sci. 2013;4:75. doi: 10.3389/fpls.2013.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pearce G, Bhattacharya R, Chen Y-C. Peptide signals for plant defense display a more universal role. Plant Signal Behav. 2008;3:1091-1092. doi: 10.4161/psb.3.12.6907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ryan CA. Protease inhibitors in plants: genes for improving defenses against insects and pathogens. Annu Rev Phytopathol. 1990;28:425-449. doi: 10.1146/annurev.py.28.090190.002233. [DOI] [Google Scholar]
- 17. Stratmann J. Long distance run in the wound response—jasmonic acid is pulling ahead. Trends Plant Sci. 2003;8:247-250. doi: 10.1016/S1360-1385(03)00106-7. [DOI] [PubMed] [Google Scholar]
- 18. García-Olmedo F, Molina A, Alamillo JM, Rodríguez-Palenzuéla P. Plant defense peptides. Pept Sci. 1998;47:479-491. [DOI] [PubMed] [Google Scholar]
- 19. Benko-Iseppon AM, Lins Galdino S, Calsa T, Jr, et al. Overview on plant antimicrobial peptides. Curr Protein Pept Sci. 2010;11:181-188. doi: 10.2174/138920310791112075. [DOI] [PubMed] [Google Scholar]
- 20. Benko-Iseppon AM, Crovella S. Ethnobotanical bioprospection of candidates for potential antimicrobial drugs from Brazilian plants: state of art and perspectives. Curr Protein Pept Sci. 2010;11:189-194. [DOI] [PubMed] [Google Scholar]
- 21. Kaas Q, Westermann J-C, Craik DJ. Conopeptide characterization and classifications: an analysis using ConoServer. Toxicon. 2010;55:1491-1509. doi: 10.1016/j.toxicon.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 22. Yu J, Kan Y, Rapp M, et al. Adaptive hydrophobic and hydrophilic interactions of mussel foot proteins with organic thin films. Proc Natl Acad Sci USA. 2013;110:15680-15685. doi: 10.1073/pnas.1315015110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zanetti M. Cathelicidins, multifunctional peptides of the innate immunity. J Leukoc Biol. 2004;75:39-48. doi: 10.1189/jlb.0403147. [DOI] [PubMed] [Google Scholar]
- 24. Wang G, ed. Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies. Wallingford, UK: CABI; 2010. doi: 10.1079/9781845936570.0000. [DOI] [Google Scholar]
- 25. Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536-540. doi: 10.1016/S0022-2836(05)80134-2. [DOI] [PubMed] [Google Scholar]
- 26. Broekaert WF, Cammue BPA, De Bolle MFC, et al. Antimicrobial peptides from plants. Crit Rev Plant Sci. 1997;16:297-323. [Google Scholar]
- 27. Wang CK, Wacklin HP, Craik DJ. Cyclotides insert into lipid bilayers to form membrane pores and destabilize the membrane through hydrophobic and phosphoethanolamine-specific interactions. J Biol Chem. 2012;287:43884-43898. doi: 10.1074/jbc.M112.421198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Terras FR, Schoofs HM, De Bolle MF, et al. Analysis of two novel classes of plant antifungal proteins from radish (Raphanus sativus L.) seeds. J Biol Chem. 1992;267:15301-15309. [PubMed] [Google Scholar]
- 29. Vriens K, Cammue B, Thevissen K. Antifungal plant defensins: mechanisms of action and production. Molecules. 2014;19:12280-12303. doi: 10.3390/molecules190812280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bruix M, González C, Santoro J, et al. 1 H-NMR studies on the structure of a new thionin from barley endosperm: structure of a new thionin. Biopolymers. 1995;36:751-763. doi: 10.1002/bip.360360608. [DOI] [PubMed] [Google Scholar]
- 31. Tam J, Wang S, Wong K, Tan W. Antimicrobial peptides from plants. Pharmaceuticals. 2015;8:711-757. doi: 10.3390/ph8040711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bohlmann H, Apel K, Garcia-Olmedo F. Thionins. Plant Mol Biol Rep. 1994;12:S75. [Google Scholar]
- 33. Almaghrabi B, Ali MA, Zahoor A, Shah KH, Bohlmann H. Arabidopsis thionin-like genes are involved in resistance against the beet-cyst nematode (Heterodera schachtii). Plant Physiol Biochem. 2019;140:55-67. [DOI] [PubMed] [Google Scholar]
- 34. Goyal RK, Mattoo AK. Plant antimicrobial peptides. In: Epand RM, ed. Host Defense Peptides and Their Potential as Therapeutic Agents. Cham, Switzerland: Springer International Publishing; 2016:111-136. doi: 10.1007/978-3-319-32949-9_5. [DOI] [Google Scholar]
- 35. Stec B. Plant thionins—the structural perspective. Cell Mol Life Sci. 2006;63:1370-1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nawrot R, Barylski J, Nowicki G, Broniarczyk J, Buchwald W, Goździcka-Józefiak A. Plant antimicrobial peptides. Folia Microbiol (Praha). 2014;59:181-196. doi: 10.1007/s12223-013-0280-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. De Coninck B, De Smet I. Plant peptides—taking them to the next level. J Exp Bot. 2016;67:4791-4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yamuna S, Kumar V, Subramaniam D, Kumaravel K. Antimicrobial peptides from plants and their mode of action. Am Int J Res Sci Technol Eng Math. 2019;105:265-269. [Google Scholar]
- 39. Balls AK, Harris TH. The inhibitory effect of a protamine from wheat flour on the fermentation of wheat mashes. Cereal Chem. 1944;21:74-79. [Google Scholar]
- 40. Garcia-Olmedo F, Carmona MJ, Lopez-Fando JJ, et al. Characterization and analysis of thionin genes. In: Boller T, Meins F, eds. Genes Involved in Plant Defense. Vienna, Austria: Springer; 1992:283-302. [Google Scholar]
- 41. Gausing K. Thionin genes specifically expressed in barley leaves. Planta. 1987;171:241-246. [DOI] [PubMed] [Google Scholar]
- 42. Iqbal A, Khan RS, Shehryar K, et al. Antimicrobial peptides as effective tools for enhanced disease resistance in plants. Plant Cell Tissue Organ Cult. 2019;139:1-15. [Google Scholar]
- 43. Franco OL, Murad AM, Leite JR, Mendes PAM, Prates MV, Bloch C. Identification of a cowpea γ-thionin with bactericidal activity. FEBS J. 2006;273:3489-3497. [DOI] [PubMed] [Google Scholar]
- 44. Vasilchenko AS, Smirnov AN, Zavriev SK, Grishin EV, Vasilchenko AV, Rogozhin EA. Novel thionins from black seed (Nigella sativa L.) demonstrate antimicrobial activity. Int J Pept Res Ther. 2017;23:171-180. [Google Scholar]
- 45. Vila-Perelló M, Sánchez-Vallet A, García-Olmedo F, Molina A, Andreu D. Synthetic and structural studies on Pyrularia pubera thionin: a single-residue mutation enhances activity against Gram-negative bacteria. FEBS Lett. 2003;536:215-219. [DOI] [PubMed] [Google Scholar]
- 46. Choi Y, Choi YD, Lee JS. Antimicrobial activity of γ-thionin-like soybean SE60 in E. coli and tobacco plants. Biochem Biophys Res Commun. 2008;375:230-234. [DOI] [PubMed] [Google Scholar]
- 47. Melo FR, Rigden DJ, Franco OL, et al. Inhibition of trypsin by cowpea thionin: characterization, molecular modeling, and docking. Proteins. 2002;48:311-319. [DOI] [PubMed] [Google Scholar]
- 48. Kramer KJ, Klassen LW, Jones BL, Speirs RD, Kammer AE. Toxicity of purothionin and its homologues to the tobacco hornworm, Manduca sexta (L.) (lepidoptera:sphingidae). Toxicol Appl Pharmacol. 1979;48:179-183. [DOI] [PubMed] [Google Scholar]
- 49. Wada K, Ozaki Y, Matsubara H, Yoshizumi H. Studies on purothionin by chemical modifications. J Biochem (Tokyo). 1982;91:257-263. [DOI] [PubMed] [Google Scholar]
- 50. Stec B, Zhou R, Teeter MM. Full-matrix refinement of the protein crambin at 0.83 Å and 130 K. Acta Crystallogr D Biol Crystallogr. 1995;51:663-681. [DOI] [PubMed] [Google Scholar]
- 51. Colilla FJ, Rocher A, Mendez E. γ-purothionins: amino acid sequence of two polypeptides of a new family of thionins from wheat endosperm. FEBS Lett. 1990;270:191-194. [DOI] [PubMed] [Google Scholar]
- 52. Mendez E, Moreno A, Colilla F, et al. Primary structure and inhibition of protein synthesis in eukaryotic cell-free system of a novel thionin, gamma-hordothionin, from barley endosperm. Eur J Biochem. 1990;194:533-539. doi: 10.1111/j.1432-1033.1990.tb15649.x. [DOI] [PubMed] [Google Scholar]
- 53. Broekaert WF, Terras FRG, Cammue BPA, Osborn RW. Plant defensins: novel antimicrobial peptides as components of the host defense system. Plant Physiol. 1995;108:1353-1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Parisi K, Shafee TMA, Quimbar P, van der Weerden NL, Bleackley MR, Anderson MA. The evolution, function and mechanisms of action for plant defensins. Semin Cell Dev Biol. 2019;88:107-118. doi: 10.1016/j.semcdb.2018.02.004. [DOI] [PubMed] [Google Scholar]
- 55. Pelegrini PB, Franco OL. Plant γ-thionins: novel insights on the mechanism of action of a multi-functional class of defense proteins. Int J Biochem Cell Biol. 2005;37:2239-2253. doi: 10.1016/j.biocel.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 56. Zhao B-C, Lin H-C, Yang D, Ye X, Li Z-G. Disulfide bridges in defensins. Curr Top Med Chem. 2015;16:206-219. doi: 10.2174/1568026615666150701115911. [DOI] [PubMed] [Google Scholar]
- 57. Kraszewska J, Beckett MC, James TC, Bond U. Comparative analysis of the antimicrobial activities of plant defensin-like and ultrashort peptides against food-spoiling bacteria. Appl Environ Microbiol. 2016;82:4288-4298. doi: 10.1128/AEM.00558-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Oddepally R, Guruprasad L. Isolation, purification, and characterization of a stable defensin-like antifungal peptide from Trigonella foenum-graecum (fenugreek) seeds. Biochemistry (Moscow). 2015;80:332-342. doi: 10.1134/S0006297915030086. [DOI] [PubMed] [Google Scholar]
- 59. Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol. 2005;3:238-250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 60. Carvalho Ade O, Gomes VM. Plant defensins—prospects for the biological functions and biotechnological properties. Peptides. 2009;30:1007-1020. doi: 10.1016/j.peptides.2009.01.018. [DOI] [PubMed] [Google Scholar]
- 61. Shi J. Defensins and Paneth cells in inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:1284-1292. doi: 10.1002/ibd.20197. [DOI] [PubMed] [Google Scholar]
- 62. Sher Khan R, Iqbal A, Malak R, et al. Plant defensins: types, mechanism of action and prospects of genetic engineering for enhanced disease resistance in plants. 3 Biotech. 2019;9:192. doi: 10.1007/s13205-019-1725-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cools TL, Struyfs C, Drijfhout JW, et al. A linear 19-mer plant defensin-derived peptide acts synergistically with caspofungin against Candida albicans biofilms. Front Microbiol. 2017;8:2051. doi: 10.3389/fmicb.2017.02051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rodríguez-Decuadro S, Dans PD, Borba MA, Benko-Iseppon AM, Cecchetto G. Gene isolation and structural characterization of a legume tree defensin with a broad spectrum of antimicrobial activity. Planta. 2019;250:1757-1772. doi: 10.1007/s00425-019-03260-w. [DOI] [PubMed] [Google Scholar]
- 65. Mazliak P, Douady D, Demandre C, Kader JC. Recent Advances in the Chemistry and Biochemistry of Plant Lipids (ed Gailliard T, Mercer EJ.). New York, NY: Academic Press; 1975. [Google Scholar]
- 66. Edstam MM, Viitanen L, Salminen TA, Edqvist J. Evolutionary history of the non-specific lipid transfer proteins. Mol Plant. 2011;4:947-964. doi: 10.1093/mp/ssr019. [DOI] [PubMed] [Google Scholar]
- 67. Salminen TA, Blomqvist K, Edqvist J. Lipid transfer proteins: classification, nomenclature, structure, and function. Planta. 2016;244:971-997. doi: 10.1007/s00425-016-2585-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kader JC. Lipid-transfer proteins in plants. Annu Rev Plant Physiol Plant Mol Biol. 1996;47:627-654. [DOI] [PubMed] [Google Scholar]
- 69. Castro MS, Gerhardt IR, Orrù S, Pucci P, Bloch C. Purification and characterization of a small (7.3 kDa) putative lipid transfer protein from maize seeds. J Chromatogr B. 2003;794:109-114. doi: 10.1016/S1570-0232(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 70. Lindorff-Larsen K, Winther JR. Surprisingly high stability of barley lipid transfer protein, LTP1, towards denaturant, heat and proteases. FEBS Lett. 2001;488:145-148. doi: 10.1016/S0014-5793(00)02424-8. [DOI] [PubMed] [Google Scholar]
- 71. Berecz B, Mills ENC, Tamás L, Láng F, Shewry PR, Mackie AR. Structural stability and surface activity of sunflower 2S albumins and nonspecific lipid transfer protein. J Agric Food Chem. 2010;58:6490-6497. doi: 10.1021/jf100554d. [DOI] [PubMed] [Google Scholar]
- 72. Edstam MM, Edqvist J. Involvement of GPI-anchored lipid transfer proteins in the development of seed coats and pollen in Arabidopsis thaliana. Physiol Plant. 2014;152:32-42. doi: 10.1111/ppl.12156. [DOI] [PubMed] [Google Scholar]
- 73. Simorre JP, Caille A, Marion D, Marion D, Ptak M. Two- and three-dimensional proton NMR studies of a wheat phospholipid transfer protein: sequential resonance assignments and secondary structure. Biochemistry. 1991;30:11600-11608. doi: 10.1021/bi00113a016. [DOI] [PubMed] [Google Scholar]
- 74. Gincel E, Simorre J-P, Caille A, Marion D, Ptak M, Vovelle F. Three-dimensional structure in solution of a wheat lipid-transfer protein from multidimensional 1H-NMR data. A new folding for lipid carriers. Eur J Biochem. 1994;226:413-422. doi: 10.1111/j.1432-1033.1994.tb20066.x. [DOI] [PubMed] [Google Scholar]
- 75. Peumans WJ, De Ley M, Broekaert WF. An unusual lectin from stinging nettle (Urtica dioica) rhizomes. FEBS Lett. 1984;177:99-103. doi: 10.1016/0014-5793(84)80989-8. [DOI] [Google Scholar]
- 76. Slavokhotova AA, Shelenkov AA, Andreev YA, Odintsova TI. Hevein-like antimicrobial peptides of plants. Biochemistry (Moscow). 2017;82:1659-1674. doi: 10.1134/S0006297917130065. [DOI] [PubMed] [Google Scholar]
- 77. Asensio JL, Cañada FJ, Siebert HC, et al. Structural basis for chitin recognition by defense proteins: GlcNAc residues are bound in a multivalent fashion by extended binding sites in hevein domains. Chem Biol. 2000;7:529-543. doi: 10.1016/S1074-5521(00)00136-8. [DOI] [PubMed] [Google Scholar]
- 78. Wong KH, Tan WL, Serra A, et al. Ginkgotides: proline-rich hevein-like peptides from gymnosperm Ginkgo biloba. Front Plant Sci. 2016;7:1639. doi: 10.3389/fpls.2016.01639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Raikhel NV, Lee HI, Broekaert WF. Structure and function of chitin-binding proteins. Annu Rev Plant Physiol Plant Mol Biol. 1993;44:591-615. doi: 10.1146/annurev.pp.44.060193.003111. [DOI] [Google Scholar]
- 80. Beintema JJ. Structural features of plant chitinases and chitin-binding proteins. FEBS Lett. 1994;350:159-163. doi: 10.1016/0014-5793(94)00753-5. [DOI] [PubMed] [Google Scholar]
- 81. Rogozhin EA, Slezina MP, Slavokhotova AA, et al. A novel antifungal peptide from leaves of the weed Stellaria media L. Biochimie. 2015;116:125-132. doi: 10.1016/j.biochi.2015.07.014. [DOI] [PubMed] [Google Scholar]
- 82. Broekaert WF, Marien W, Terras FRG, et al. Antimicrobial peptides from Amaranthus caudatus seeds with sequence homology to the cysteine/glycine-rich domain of chitin-binding proteins. Biochemistry. 1992;31:4308-4314. doi: 10.1021/bi00132a023. [DOI] [PubMed] [Google Scholar]
- 83. Volpicella M, Leoni C, Fanizza I, Placido A, Pastorello EA, Ceci LR. Overview of plant chitinases identified as food allergens. J Agric Food Chem. 2014;62:5734-5742. doi: 10.1021/jf5007962. [DOI] [PubMed] [Google Scholar]
- 84. Wagner S, Breiteneder H. The latex-fruit syndrome. Biochem Soc Trans. 2002;30:935-940. doi: 10.1042/bst0300935. [DOI] [PubMed] [Google Scholar]
- 85. Iseli B, Boller T, Neuhaus JM. The N-terminal cysteine-rich domain of tobacco class I chitinase is essential for chitin binding but not for catalytic or antifungal activity. Plant Physiol. 1993;103:221-226. doi: 10.1104/pp.103.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Broekaert WF, Van Parijs J, Leyns F, Joos H, Peumans WJ. A chitin-binding lectin from stinging nettle rhizomes with antifungal properties. Science. 1989;245:1100-1102. doi: 10.1126/science.245.4922.1100. [DOI] [PubMed] [Google Scholar]
- 87. Leah R, Tommerup H, Svendsen I, Mundy J. Biochemical and molecular characterization of three barley seed proteins with antifungal properties. J Biol Chem. 1991;266:1564-1573. [PubMed] [Google Scholar]
- 88. Van Parijs J, Broekaert WF, Goldstein IJ, Peumans WJ. Hevein: an antifungalprotein from rubber-tree (Hevea brasiliensis) latex. Planta. 1991;183:258-264. doi: 10.1007/BF00197797. [DOI] [PubMed] [Google Scholar]
- 89. Koo JC, Lee SY, Chun HJ, et al. Two hevein homologs isolated from the seed of Pharbitis nil L. exhibit potent antifungal activity. Biochim Biophys Acta. 1998;1382:80-90. doi: 10.1016/S0167-4838(97)00148-9. [DOI] [PubMed] [Google Scholar]
- 90. Wang X, Shi M, Wang D, et al. Comparative proteomics of primary and secondary lutoids reveals that chitinase and glucanase play a crucial combined role in rubber particle aggregation in Hevea brasiliensis. J Proteome Res. 2013;12:5146-5159. doi: 10.1021/pr400378c. [DOI] [PubMed] [Google Scholar]
- 91. Shi M, Li Y, Deng S, et al. The formation and accumulation of protein-networks by physical interactions in the rapid occlusion of laticifer cells in rubber tree undergoing successive mechanical wounding. BMC Plant Biol. 2019;19:8. doi: 10.1186/s12870-018-1617-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Molesini B, Treggiari D, Dalbeni A, Minuz P, Pandolfini T. Plant cystine-knot peptides: pharmacological perspectives: plant cystine-knot proteins in pharmacology. Br J Clin Pharmacol. 2017;83:63-70. doi: 10.1111/bcp.12932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Rees DC, Lipscomb WN. Refined crystal structure of the potato inhibitor complex of carboxypeptidase A at 2.5 Å resolution. J Mol Biol. 1982;160:475-498. doi: 10.1016/0022-2836(82)90309-6. [DOI] [PubMed] [Google Scholar]
- 94. Chiche L, Heitz A, Gelly J-C, et al. Squash inhibitors: from structural motifs to macrocyclic knottins. Curr Protein Pept Sci. 2004;5:341-349. doi: 10.2174/1389203043379477. [DOI] [PubMed] [Google Scholar]
- 95. Murphy E, Smith S, De Smet I. Small signaling peptides in Arabidopsis development: how cells communicate over a short distance. Plant Cell. 2012;24:3198-3217. doi: 10.1105/tpc.112.099010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Li G, Asiegbu FO. Use of Scots pine seedling roots as an experimental model to investigate gene expression during interaction with the conifer pathogen Heterobasidion annosum (P-type). J Plant Res. 2004;117:155-162. doi: 10.1007/s10265-003-0140-4. [DOI] [PubMed] [Google Scholar]
- 97. Iyer S, Acharya KR. Tying the knot: the cystine signature and molecular-recognition processes of the vascular endothelial growth factor family of angiogenic cytokines. FEBS J. 2011;278:4304-4322. doi: 10.1111/j.1742-4658.2011.08350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Aboye TL, Strömstedt AA, Gunasekera S, et al. A cactus-derived toxin-like cystine knot peptide with selective antimicrobial activity. ChemBioChem. 2015;16:1068-1077. doi: 10.1002/cbic.201402704. [DOI] [PubMed] [Google Scholar]
- 99. Göransson U, Burman R, Gunasekera S, Strömstedt AA, Rosengren KJ. Circular proteins from plants and fungi. J Biol Chem. 2012;287:27001-27006. doi: 10.1074/jbc.R111.300129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Gustafson KR, Sowder RC, Henderson LE, et al. Circulins A B. Novel human immunodeficiency virus (HIV)-inhibitory macrocyclic peptides from the tropical tree Chassalia parvifolia. J Am Chem Soc. 1994;116:9337-9338. doi: 10.1021/ja00099a064. [DOI] [Google Scholar]
- 101. Jennings CV, Rosengren KJ, Daly NL, et al. Isolation, solution structure, and insecticidal activity of kalata B2, a circular protein with a twist: do Möbius strips exist in nature? Biochemistry. 2005;44:851-860. doi: 10.1021/bi047837h. [DOI] [PubMed] [Google Scholar]
- 102. Burman R, Strömstedt AA, Malmsten M, Göransson U. Cyclotide–membrane interactions: defining factors of membrane binding, depletion and disruption. Biochim Biophys Acta. 2011;1808:2665-2673. doi: 10.1016/j.bbamem.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 103. Marcus JP, Goulter KC, Green JL, Harrison SJ, Manners JM. Purification, characterisation and cDNA cloning of an antimicrobial peptide from Macadamia integrifolia. Eur J Biochem. 1997;244:743-749. doi: 10.1111/j.1432-1033.1997.00743.x. [DOI] [PubMed] [Google Scholar]
- 104. McManus AM, Nielsen KJ, Marcus JP, et al. MiAMP1, a novel protein from Macadamia integrifolia adopts a Greek key β-barrel fold unique amongst plant antimicrobial proteins. J Mol Biol. 1999;293:629-638. doi: 10.1006/jmbi.1999.3163. [DOI] [PubMed] [Google Scholar]
- 105. Finkina EI, Melnikova DN, Bogdanov IV, Ovchinnikova TV. Peptides of the innate immune system of plants. Part II. Biosynthesis, biological functions, and possible practical applications. Russ J Bioorganic Chem. 2019;45:55-65. doi: 10.1134/S1068162019020043. [DOI] [Google Scholar]
- 106. Ohno A, Tate S, Seeram SS, et al. NMR structure of the Streptomyces metalloproteinase inhibitor, SMPI, isolated from Streptomyces nigrescens TK-23: another example of an ancestral βγ-crystallin precursor structure. J Mol Biol. 1998;282:421-433. doi: 10.1006/jmbi.1998.2022. [DOI] [PubMed] [Google Scholar]
- 107. Antuch W, Güntert P, Wüthrich K. Ancestral beta gamma-crystallin precursor structure in a yeast killer toxin. Nat Struct Biol. 1996;3:662-665. [DOI] [PubMed] [Google Scholar]
- 108. Kazan K, Rusu A, Marcus JP, Goulter KC, Manners JM. Enhanced quantitative resistance to Leptosphaeria maculans conferred by expression of a novel antimicrobial peptide in canola (Brassica napus L.). Mol Breed. 2002;10:63-70. doi: 10.1023/A:1020354809737. [DOI] [Google Scholar]
- 109. Manners JM. Primitive defence: the MiAMP1 antimicrobial peptide family. Plant Mol Biol Rep. 2009;27:237-242. doi: 10.1007/s11105-008-0083-y. [DOI] [Google Scholar]
- 110. Tailor RH, Acland DP, Attenborough S, et al. A novel family of small cysteine-rich antimicrobial peptides from seed of Impatiens balsamina is derived from a single precursor protein. J Biol Chem. 1997;272:24480-24487. doi: 10.1074/jbc.272.39.24480. [DOI] [PubMed] [Google Scholar]
- 111. Patel SU, Osborn R, Rees S, Thornton JM. Structural studies of Impatiens balsamina antimicrobial protein (Ib-AMP1). Biochemistry. 1998;37:983-990. doi: 10.1021/bi971747d. [DOI] [PubMed] [Google Scholar]
- 112. Lee DG, Shin SY, Kim D-H, et al. Antifungal mechanism of a cysteine-rich antimicrobial peptide, Ib-AMP1, from Impatiens balsamina against Candida albicans. Biotechnol Lett. 1999;21:1047-1050. doi: 10.1023/A:1005636610512. [DOI] [Google Scholar]
- 113. Fan X, Xu W, Han J, Jiang X, Wink M, Wu G. Antimicrobial peptide hybrid fluorescent protein based sensor array discriminate ten most frequent clinic isolates. Biochim Biophys Acta Gen Subj. 2019;1863:1158-1166. doi: 10.1016/j.bbagen.2019.04.010. [DOI] [PubMed] [Google Scholar]
- 114. Fan X, Korytowski A, Makky A, Tanaka M, Wink M. Ib-AMP4 insertion causes surface rearrangement in the phospholipid bilayer of biomembranes: implications from quartz-crystal microbalance with dissipation. Biochim Biophys Acta Biomembr. 2018;1860:617-623. doi: 10.1016/j.bbamem.2017.10.025. [DOI] [PubMed] [Google Scholar]
- 115. Thevissen K, François IE, Sijtsma L, et al. Antifungal activity of synthetic peptides derived from Impatiens balsamina antimicrobial peptides Ib-AMP1 and Ib-AMP4. Peptides. 2005;26:1113-1119. doi: 10.1016/j.peptides.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 116. Wang P, Bang J-K, Kim HJ, Kim J-K, Kim Y, Shin SY. Antimicrobial specificity and mechanism of action of disulfide-removed linear analogs of the plant-derived Cys-rich antimicrobial peptide Ib-AMP1. Peptides. 2009;30:2144-2149. doi: 10.1016/j.peptides.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 117. Gautier M-F, Aleman M-E, Guirao A, Marion D, Joudrier P. Triticum aestivum puroindolines, two basic cystine-rich seed proteins: cDNA sequence analysis and developmental gene expression. Plant Mol Biol. 1994;25:43-57: doi: 10.1007/BF00024197. [DOI] [PubMed] [Google Scholar]
- 118. Le Bihan T, Blochet J-É, Désormeaux A, Marion D, Pézolet M. Determination of the secondary structure and conformation of puroindolines by infrared and Raman spectroscopy. Biochemistry. 1996;35:12712-12722. doi: 10.1021/bi960869n. [DOI] [PubMed] [Google Scholar]
- 119. Wilkinson M, King R, Grimaldi R. Sequence diversity and identification of novel puroindoline and grain softness protein alleles in Elymus, Agropyron and related species. Diversity. 2018;10:114. doi: 10.3390/d10040114. [DOI] [Google Scholar]
- 120. Giroux MJ, Sripo T, Gerhardt S, Sherwood J. Puroindolines: their role in grain hardness and plant defence. Biotechnol Genet Eng Rev. 2003;20:277-290. doi: 10.1080/02648725.2003.10648047. [DOI] [PubMed] [Google Scholar]
- 121. Bhave M, Morris CF. Molecular genetics of puroindolines and related genes: allelic diversity in wheat and other grasses. Plant Mol Biol. 2008;66:205-219. doi: 10.1007/s11103-007-9263-7. [DOI] [PubMed] [Google Scholar]
- 122. Dhatwalia VK, Sati OP, Tripathi MK, Kumar A. Isolation, characterization and antimicrobial activity at diverse dilution of wheat puroindoline protein. World J Agric Sci. 2009;5:297-300. [Google Scholar]
- 123. Zhang J, Martin JM, Balint-Kurti P, Huang L, Giroux MJ. The wheat puroindoline genes confer fungal resistance in transgenic corn: the puroindolines confer corn SLB resistance. J Phytopathol. 2011;159:188-190. doi: 10.1111/j.1439-0434.2010.01744.x. [DOI] [Google Scholar]
- 124. Douliez J-P, Michon T, Elmorjani K, Marion D. Mini review: structure, biological and technological functions of lipid transfer proteins and indolines, the major lipid binding proteins from cereal Kernels. J Cereal Sci. 2000;32:1-20. doi: 10.1006/jcrs.2000.0315. [DOI] [Google Scholar]
- 125. Morris CF. Puroindolines: the molecular genetic basis of wheat grain hardness. Plant Mol Biol. 2002;48:633-647. doi: 10.1023/A:1014837431178. [DOI] [PubMed] [Google Scholar]
- 126. Marion D, Bakan B, Elmorjani K. Plant lipid binding proteins: properties and applications. Biotechnol Adv. 2007;25:195-197. doi: 10.1016/j.biotechadv.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 127. Charnet P, Molle G, Marion D, Rousset M, Lullien-Pellerin V. Puroindolines form ion channels in biological membranes. Biophys J. 2003;84:2416-2426. doi: 10.1016/S0006-3495(03)75046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Morris CF. The antimicrobial properties of the puroindolines, a review. World J Microbiol Biotechnol. 2019;35:86. doi: 10.1007/s11274-019-2655-4. [DOI] [PubMed] [Google Scholar]
- 129. Segura A, Moreno M, Madueño F, Molina A, García-Olmedo F. Snakin-1, a peptide from potato that is active against plant pathogens. Mol Plant Microbe Interact. 1999;12:16-23. doi: 10.1094/MPMI.1999.12.1.16. [DOI] [PubMed] [Google Scholar]
- 130. Berrocal-Lobo M, Segura A, Moreno M, Lopez G, Garcia-Olmedo F, Molina A. Snakin-2, an antimicrobial peptide from potato whose gene is locally induced by wounding and responds to pathogen infection. Plant Physiol. 2002;128:951-961. doi: 10.1104/pp.010685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Oliveira-Lima M, Benko-Iseppon A, Neto J, et al. Snakin: structure, roles and applications of a plant antimicrobial peptide. Curr Protein Pept Sci. 2017;18:368-374. doi: 10.2174/1389203717666160619183140. [DOI] [PubMed] [Google Scholar]
- 132. Mao Z, Zheng J, Wang Y, et al. The new CaSn gene belonging to the snakin family induces resistance against root-knot nematode infection in pepper. Phytoparasitica. 2011;39:151-164. doi: 10.1007/s12600-011-0149-5. [DOI] [Google Scholar]
- 133. Yeung H, Squire CJ, Yosaatmadja Y, et al. Radiation damage and racemic protein crystallography reveal the unique structure of the GASA/snakin protein superfamily. Angew Chem Int Ed. 2016;55:7930-7933. doi: 10.1002/anie.201602719. [DOI] [PubMed] [Google Scholar]
- 134. Zhang S, Yang C, Peng J, Sun S, Wang X. GASA5, a regulator of flowering time and stem growth in Arabidopsis thaliana. Plant Mol Biol. 2009;69:745-759. doi: 10.1007/s11103-009-9452-7. [DOI] [PubMed] [Google Scholar]
- 135. Almasia NI, Narhirñak V, Hopp HE, Vazquez-Rovere C. Isolation and characterization of the tissue and development-specific potato snakin-1 promoter inducible by temperature and wounding. Electron J Biotechnol. 2010;13:8-9. doi: 10.2225/vol13-issue5-fulltext-12. [DOI] [Google Scholar]
- 136. Zimmermann R, Sakai H, Hochholdinger F. The gibberellic acid stimulated-like gene family in maize and its role in lateral root development. Plant Physiol. 2010;152:356-365. doi: 10.1104/pp.109.149054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Guzmán-Rodríguez JJ, Ibarra-Laclette E, Herrera-Estrella L, et al. Analysis of expressed sequence tags (ESTs) from avocado seed (Persea americana var. drymifolia) reveals abundant expression of the gene encoding the antimicrobial peptide snakin. Plant Physiol Biochem. 2013;70:318-324. doi: 10.1016/j.plaphy.2013.05.045. [DOI] [PubMed] [Google Scholar]
- 138. Faccio P, Vazquez-Rovere C, Hopp E, et al. Increased tolerance to wheat powdery mildew by heterologous constitutive expression of the Solanum chacoense snakin-1 gene. Czech J Genet Plant Breed. 2011;47:S135-S141. doi: 10.17221/3268-CJGPB. [DOI] [Google Scholar]
- 139. Herbel V, Schäfer H, Wink M. Recombinant production of snakin-2 (an antimicrobial peptide from tomato) in E. coli and analysis of its bioactivity. Molecules. 2015;20:14889-14901. doi: 10.3390/molecules200814889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Kotilainen M. GEG participates in the regulation of cell and organ shape during corolla and carpel development in Gerbera hybrida. Plant Cell. 1999;11:1093-1104. doi: 10.1105/tpc.11.6.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Furukawa T, Sakaguchi N, Shimada H. Two OsGASR genes, rice GAST homologue genes that are abundant in proliferating tissues, show different expression patterns in developing panicles. Genes Genet Syst. 2006;81:171-180. doi: 10.1266/ggs.81.171. [DOI] [PubMed] [Google Scholar]
- 142. Roxrud I, Lid SE, Fletcher JC, Schmidt EDL, Opsahl-Sorteberg H-G. GASA4, one of the 14-member Arabidopsis GASA family of small polypeptides, regulates flowering and seed development. Plant Cell Physiol. 2007;48:471-483. doi: 10.1093/pcp/pcm016. [DOI] [PubMed] [Google Scholar]
- 143. Lucau-Danila A, Laborde L, Legrand S, et al. Identification of novel genes potentially involved in somatic embryogenesis in chicory (Cichorium intybus L.). BMC Plant Biol. 2010;10:122. doi: 10.1186/1471-2229-10-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Nolde SB, Vassilevski AA, Rogozhin EA, et al. Disulfide-stabilized helical hairpin structure and activity of a novel antifungal peptide EcAMP1 from seeds of barnyard grass (Echinochloa crus-galli). J Biol Chem. 2011;286:25145-25153. doi: 10.1074/jbc.M110.200378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Oparin PB, Mineev KS, Dunaevsky YE, et al. Buckwheat trypsin inhibitor with helical hairpin structure belongs to a new family of plant defence peptides. Biochem J. 2012;446:69-77. doi: 10.1042/BJ20120548. [DOI] [PubMed] [Google Scholar]
- 146. Slavokhotova AA, Rogozhin EA, Musolyamov AK, et al. Novel antifungal α-hairpinin peptide from Stellaria media seeds: structure, biosynthesis, gene structure and evolution. Plant Mol Biol. 2014;84:189-202. doi: 10.1007/s11103-013-0127-z. [DOI] [PubMed] [Google Scholar]
- 147. Wang X, Zhu G, Liang W, et al. Design, synthesis and docking of linear and hairpin-like alpha helix mimetics based on alkoxylated oligobenzamide. ChemistrySelect. 2019;4:6651-6655. doi: 10.1002/slct.201900171. [DOI] [Google Scholar]
- 148. Slavokhotova AA, Shelenkov AA, Korostyleva TV, et al. Defense peptide repertoire of Stellaria media predicted by high throughput next generation sequencing. Biochimie. 2017;135:15-27. doi: 10.1016/j.biochi.2016.12.017. [DOI] [PubMed] [Google Scholar]
- 149. Sousa D, Porto W, Silva M, da Silva T, Franco O. Influence of cysteine and tryptophan substitution on DNA-binding activity on maize α-hairpinin antimicrobial peptide. Molecules. 2016;21:1062. doi: 10.3390/molecules21081062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Craik DJ, Daly NL, Bond T, Waine C. Plant cyclotides: a unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif. J Mol Biol. 1999;294:1327-1336. doi: 10.1006/jmbi.1999.3383. [DOI] [PubMed] [Google Scholar]
- 151. Lima S, Benko-Iseppon A, Neto J, et al. Plants defense-related cyclic peptides: diversity, structure and applications. Curr Protein Pept Sci. 2017;18:375-390. doi: 10.2174/1389203717666160724194119. [DOI] [PubMed] [Google Scholar]
- 152. Weidmann J, Craik DJ. Discovery, structure, function, and applications of cyclotides: circular proteins from plants. J Exp Bot. 2016;67:4801-4812. doi: 10.1093/jxb/erw210. [DOI] [PubMed] [Google Scholar]
- 153. Park S, Yoo K-O, Marcussen T, et al. Cyclotide evolution: insights from the analyses of their precursor sequences, structures and distribution in violets (Viola). Front Plant Sci. 2017;8:2058. doi: 10.3389/fpls.2017.02058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Grain L. Isolation of oxytocic peptides from Oldenlandia affinis by solvent extraction of tetraphenylborate complexes and chromatography on sephadex LH-20. Lloydia. 1973;36:207-208. [PubMed] [Google Scholar]
- 155. Claeson P, Göransson U, Johansson S, Luijendijk T, Bohlin L. Fractionation protocol for the isolation of polypeptides from plant biomass. J Nat Prod. 1998;61:77-81. doi: 10.1021/np970342r. [DOI] [PubMed] [Google Scholar]
- 156. Poth AG, Colgrave ML, Philip R, et al. Discovery of cyclotides in the Fabaceae plant family provides new insights into the cyclization, evolution, and distribution of circular proteins. ACS Chem Biol. 2011;6:345-355. doi: 10.1021/cb100388j. [DOI] [PubMed] [Google Scholar]
- 157. Poth AG, Mylne JS, Grassl J, et al. Cyclotides associate with leaf vasculature and are the products of a novel precursor in petunia (Solanaceae). J Biol Chem. 2012;287:27033-27046. doi: 10.1074/jbc.M112.370841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Nguyen GKT, Zhang S, Nguyen NTK, et al. Discovery and characterization of novel cyclotides originated from chimeric precursors consisting of albumin-1 chain a and cyclotide domains in the Fabaceae family. J Biol Chem. 2011;286:24275-24287. doi: 10.1074/jbc.M111.229922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Pranting M, Loov C, Burman R, Goransson U, Andersson DI. The cyclotide cycloviolacin O2 from Viola odorata has potent bactericidal activity against Gram-negative bacteria. J Antimicrob Chemother. 2010;65:1964-1971. doi: 10.1093/jac/dkq220. [DOI] [PubMed] [Google Scholar]
- 160. Lindholm P, Göransson U, Johansson S, et al. Cyclotides: a novel type of cytotoxic agents. Mol Cancer Ther. 2002;1:365-369. [PubMed] [Google Scholar]
- 161. Craik DJ, Clark RJ, Daly NL. Potential therapeutic applications of the cyclotides and related cystine knot mini-proteins. Expert Opin Investig Drugs. 2007;16:595-604. doi: 10.1517/13543784.16.5.595. [DOI] [PubMed] [Google Scholar]
- 162. Northfield SE, Wang CK, Schroeder CI, et al. Disulfide-rich macrocyclic peptides as templates in drug design. Eur J Med Chem. 2014;77:248-257. doi: 10.1016/j.ejmech.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 163. Liu J-J, Sturrock R, Ekramoddoullah AKM. The superfamily of thaumatin-like proteins: its origin, evolution, and expression towards biological function. Plant Cell Rep. 2010;29:419-436. doi: 10.1007/s00299-010-0826-8. [DOI] [PubMed] [Google Scholar]
- 164. de Jesus-Pires C, Ferreira-Neto JRC, Pacifico Bezerra-Neto J, et al. Plant thaumatin-like proteins: function, evolution and biotechnological applications. Curr Protein Pept Sci. 2020;21:36-51. doi: 10.2174/1389203720666190318164905. [DOI] [PubMed] [Google Scholar]
- 165. Grenier J, Potvin C, Asselin A. Some fungi express beta-1,3-glucanases similar to thaumatin-like proteins. Mycologia. 2000;92:841-848. doi: 10.2307/3761579. [DOI] [Google Scholar]
- 166. Sakamoto Y. Lentinula edodes tlg1 encodes a thaumatin-like protein that is involved in lentinan degradation and fruiting body senescence. Plant Physiol. 2006;141:793-801. doi: 10.1104/pp.106.076679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Brandazza A, Angeli S, Tegoni M, Cambillau C, Pelosi P. Plant stress proteins of the thaumatin-like family discovered in animals. FEBS Lett. 2004;572:3-7. doi: 10.1016/j.febslet.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 168. Kitajima S, Sato F. Plant pathogenesis-related proteins: molecular mechanisms of gene expression and protein function. J Biochem. 1999;125:1-8. doi: 10.1093/oxfordjournals.jbchem.a022244. [DOI] [PubMed] [Google Scholar]
- 169. Batalia MA, Monzingo AF, Ernst S, Roberts W, Robertus JD. The crystal structure of the antifungal protein zeamatin, a member of the thaumatin-like, PR-5 protein family. Nat Struct Biol. 1996;3:19-22. doi: 10.1038/nsb0196-19. [DOI] [PubMed] [Google Scholar]
- 170. Trudel J, Grenier J, Potvin C, Asselin A. Several thaumatin-like proteins bind to β-1,3-glucans. Plant Physiol. 1998;118:1431-1438. doi: 10.1104/pp.118.4.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Grenier J, Potvin C, Trudel J, Asselin A. Some thaumatin-like proteins hydrolyse polymeric beta-1,3-glucans. Plant J. 1999;19:473-480. doi: 10.1046/j.1365-313X.1999.00551.x. [DOI] [PubMed] [Google Scholar]
- 172. Fierens E, Rombouts S, Gebruers K, et al. TLXI, a novel type of xylanase inhibitor from wheat (Triticum aestivum) belonging to the thaumatin family. Biochem J. 2007;403:583-591. doi: 10.1042/BJ20061291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Schimoler-O’Rourke R, Richardson M, Selitrennikoff CP. Zeamatin inhibits trypsin and alpha-amylase activities. Appl Environ Microbiol. 2001;67:2365-2366. doi: 10.1128/AEM.67.5.2365-2366.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Jung YC, Lee HJ, Yum SS, et al. Drought-inducible—but ABA-independent—thaumatin-like protein from carrot (Daucus carota L.). Plant Cell Rep. 2005;24:366-373. doi: 10.1007/s00299-005-0944-x. [DOI] [PubMed] [Google Scholar]
- 175. Ruperti B, Cattivelli L, Pagni S, Ramina A. Ethylene-responsive genes are differentially regulated during abscission, organ senescence and wounding in peach (Prunus persica). J Exp Bot. 2002;53:429-437. doi: 10.1093/jexbot/53.368.429. [DOI] [PubMed] [Google Scholar]
- 176. Hon WC, Griffith M, Mlynarz A, Kwok YC, Yang DSC. Antifreeze proteins in winter rye are similar to pathogenesis-related proteins. Plant Physiol. 1995;109:879-889. doi: 10.1104/pp.109.3.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Kumar KRR, Kirti PB. Differential gene expression in Arachis diogoi upon interaction with peanut late leaf spot pathogen, Phaeoisariopsis personata and characterization of a pathogen induced cyclophilin. Plant Mol Biol. 2011;75:497-513. doi: 10.1007/s11103-011-9747-3. [DOI] [PubMed] [Google Scholar]
- 178. Rudd JJ, Kanyuka K, Hassani-Pak K, et al. Transcriptome and metabolite profiling of the infection cycle of Zymoseptoria tritici on wheat reveals a biphasic interaction with plant immunity involving differential pathogen chromosomal contributions and a variation on the hemibiotrophic lifestyle definition. Plant Physiol. 2015;167:1158-1185. doi: 10.1104/pp.114.255927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Breiteneder H, Radauer C. A classification of plant food allergens. J Allergy Clin Immunol. 2004;113:821-830. doi: 10.1016/j.jaci.2004.01.779. [DOI] [PubMed] [Google Scholar]
- 180. Jami SK, Swathi Anuradha T, Guruprasad L, Kirti PB. Molecular, biochemical and structural characterization of osmotin-like protein from black nightshade (Solanum nigrum). J Plant Physiol. 2007;164:238-252. doi: 10.1016/j.jplph.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 181. Tachi H, Fukuda-Yamada K, Kojima T, Shiraiwa M, Takahara H. Molecular characterization of a novel soybean gene encoding a neutral PR-5 protein induced by high-salt stress. Plant Physiol Biochem. 2009;47:73-79. doi: 10.1016/j.plaphy.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 182. Breiteneder H. Thaumatin-like proteins—a new family of pollen and fruit allergens. Allergy. 2004;59:479-481. doi: 10.1046/j.1398-9995.2003.00421.x. [DOI] [PubMed] [Google Scholar]
- 183. Fierens E, Gebruers K, Voet ARD, De Maeyer M, Courtin CM, Delcour JA. Biochemical and structural characterization of TLXI, the Triticum aestivum L. J Enzyme Inhib Med Chem. 2009;24:646-654. doi: 10.1080/14756360802321831. [DOI] [PubMed] [Google Scholar]
- 184. Leone P, Menu-Bouaouiche L, Peumans WJ, et al. Resolution of the structure of the allergenic and antifungal banana fruit thaumatin-like protein at 1.7-Å. Biochimie. 2006;88:45-52. doi: 10.1016/j.biochi.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 185. Koiwa H, Kato H, Nakatsu T, Oda J, Yamada Y, Sato F. Crystal structure of tobacco PR-5d protein at 1.8 Å resolution reveals a conserved acidic cleft structure in antifungal thaumatin-like proteins. J Mol Biol. 1999;286:1137-1145. doi: 10.1006/jmbi.1998.2540. [DOI] [PubMed] [Google Scholar]
- 186. Min K, Ha SC, Hasegawa PM, Bressan RA, Yun D-J, Kim KK. Crystal structure of osmotin, a plant antifungal protein. Proteins. 2003;54:170-173. doi: 10.1002/prot.10571. [DOI] [PubMed] [Google Scholar]
- 187. Ogata CM, Gordon PF, de Vos AM, Kim S-H. Crystal structure of a sweet tasting protein thaumatin I, at 1·65 Å resolution. J Mol Biol. 1992;228:893-908. doi: 10.1016/0022-2836(92)90873-I. [DOI] [PubMed] [Google Scholar]
- 188. Dall’Antonia Y, Pavkov T, Fuchs H, Breiteneder H, Keller W. Crystallization and preliminary structure determination of the plant food allergen Pru av 2. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2005;61:186-188. doi: 10.1107/S1744309104033822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Osherov N, Mathew J, Romans A, May GS. Identification of conidial-enriched transcripts in Aspergillus nidulans using suppression subtractive hybridization. Fungal Genet Biol. 2002;37:197-204. doi: 10.1016/S1087-1845(02)00502-9. [DOI] [PubMed] [Google Scholar]
- 190. Greenstein S, Shadkchan Y, Jadoun J, Sharon C, Markovich S, Osherov N. Analysis of the Aspergillus nidulans thaumatin-like cetA gene and evidence for transcriptional repression of pyr4 expression in the cetA-disrupted strain. Fungal Genet Biol. 2006;43:42-53. doi: 10.1016/j.fgb.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 191. Wang X, Zafian P, Choudhary M, Lawton M. The PR5K receptor protein kinase from Arabidopsis thaliana is structurally related to a family of plant defense proteins. Proc Natl Acad Sci USA. 1996;93:2598-2602. doi: 10.1073/pnas.93.6.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Apweiler R. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2004;32:115-119. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Torrent M, Nogues MV, Boix E. Discovering new in silico tools for antimicrobial peptide prediction. Curr Drug Targets. 2012;13:1148-1157. doi: 10.2174/138945012802002311. [DOI] [PubMed] [Google Scholar]
- 194. Porto WF, Pires AS, Franco OL. Computational tools for exploring sequence databases as a resource for antimicrobial peptides. Biotechnol Adv. 2017;35:337-349. doi: 10.1016/j.biotechadv.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 195. Thomas S, Karnik S, Barai RS, Jayaraman VK, Idicula-Thomas S. CAMP: a useful resource for research on antimicrobial peptides. Nucleic Acids Res. 2010;38:D774-D780. doi: 10.1093/nar/gkp1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Waghu FH, Gopi L, Barai RS, Ramteke P, Nizami B, Idicula-Thomas S. CAMP: collection of sequences and structures of antimicrobial peptides. Nucleic Acids Res. 2014;42:D1154-D1158. doi: 10.1093/nar/gkt1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Waghu FH, Barai RS, Gurung P, Idicula-Thomas S. CAMPR3: a database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res. 2016;44:D1094-D1097. doi: 10.1093/nar/gkv1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Wang G, Li X, Wang Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016;44:D1087-D1093. doi: 10.1093/nar/gkv1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Zhao X, Wu H, Lu H, Li G, Huang Q. LAMP: a database linking antimicrobial peptides. PLoS ONE. 2013;8:e66557. doi: 10.1371/journal.pone.0066557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Gogoladze G, Grigolava M, Vishnepolsky B, et al. DBAASP: database of antimicrobial activity and structure of peptides. FEMS Microbiol Lett. 2014;357:63-68. doi: 10.1111/1574-6968.12489. [DOI] [PubMed] [Google Scholar]
- 201. Jorge P, Lourenço A, Pereira MO. New trends in peptide-based anti-biofilm strategies: a review of recent achievements and bioinformatic approaches. Biofouling. 2012;28:1033-1061. doi: 10.1080/08927014.2012.728210. [DOI] [PubMed] [Google Scholar]
- 202. Lee HT, Lee CC, Yang JR, Lai JZC, Chang KY. A large-scale structural classification of antimicrobial peptides. Biomed Res Int. 2015;2015:475062. doi: 10.1155/2015/475062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Seebah S, Suresh A, Zhuo S, et al. Defensins knowledgebase: a manually curated database and information source focused on the defensins family of antimicrobial peptides. Nucleic Acids Res. 2007;35:D265-D268. doi: 10.1093/nar/gkl866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Liu S, Fan L, Sun J, Lao X, Zheng H. Computational resources and tools for antimicrobial peptides. J Pept Sci. 2017;23:4-12. doi: 10.1002/psc.2947. [DOI] [PubMed] [Google Scholar]
- 205. Mulvenna JP. CyBase: a database of cyclic protein sequence and structure. Nucleic Acids Res. 2006;34:D192-D194. doi: 10.1093/nar/gkj005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Hammami R, Ben Hamida J, Vergoten G, Fliss I. PhytAMP: a database dedicated to antimicrobial plant peptides. Nucleic Acids Res. 2009;37:D963-D968. doi: 10.1093/nar/gkn655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Das D, Jaiswal M, Khan FN, Ahamad S, Kumar S. PlantPepDB: a manually curated plant peptide database. Sci Rep. 2020;10:2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Eltabakh MY, Ouzzani M, Aref WG. bdbms—a database management system for biological data. https://arxiv.org/pdf/cs/0612127.pdf. Published 2006.
- 209. Rahman M, Shaheen T, Rahman M, Iqbal MA, Zafar Y. Bioinformatics: a way forward to explore “plant omics.” In: Abdurakhmonov IY. ed. Bioinformatics: Updated Features and Applications. London, England: IntechOpen; 2016. doi: 10.5772/64043. [DOI] [Google Scholar]
- 210. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403-410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 211. Pearson WR. Rapid and sensitive sequence comparison with FASTP and FASTA. Methods Enzymol. 1990;183:63-98. doi: 10.1016/0076-6879(90)83007-V. [DOI] [PubMed] [Google Scholar]
- 212. Polyanovsky VO, Roytberg MA, Tumanyan VG. Comparative analysis of the quality of a global algorithm and a local algorithm for alignment of two sequences. Algorithms Mol Biol. 2011;6:25. doi: 10.1186/1748-7188-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Thompson K. Programming techniques: regular expression search algorithm. Commun ACM. 1968;11:419-422. doi: 10.1145/363347.363387. [DOI] [Google Scholar]
- 214. Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755-763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 215. Yoon B-J. Hidden Markov models and their applications in biological sequence analysis. Curr Genomics. 2009;10:402-415. doi: 10.2174/138920209789177575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Zhou P, Yang C, Ren Y, Wang C, Tian F. What are the ideal properties for functional food peptides with antihypertensive effect? A computational peptidology approach. Food Chem. 2013;141:2967-2973. doi: 10.1016/j.foodchem.2013.05.140. [DOI] [PubMed] [Google Scholar]
- 217. Zhou P, Huang J, eds. Computational Peptidology. New York, NY: Springer; 2015. doi: 10.1007/978-1-4939-2285-7. [DOI] [Google Scholar]
- 218. Niarchou A, Alexandridou A, Athanasiadis E, Spyrou G. C-PAmP: large scale analysis and database construction containing high scoring computationally predicted antimicrobial peptides for all the available plant species. PLoS ONE. 2013;8:e79728. doi: 10.1371/journal.pone.0079728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Islam SMA, Sajed T, Kearney CM, et al. PredSTP: a highly accurate SVM based model to predict sequential cystine stabilized peptides. BMC Bioinformatics. 2015;16:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Islam SMA, Kearney CM, Baker EJ. Assigning biological function using hidden signatures in cystine-stabilized peptide sequences. Sci Rep. 2018;8:9049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Lin Y, Jeon Y. Random forests and adaptive nearest neighbors. J Am Stat Assoc. 2006;101:578-590. doi: 10.1198/016214505000001230. [DOI] [Google Scholar]
- 222. Karasuyama M, Takeuchi I. Multiple incremental decremental learning of support vector machines. IEEE Trans Neural Netw. 2010;21:1048-1059. doi: 10.1109/TNN.2010.2048039. [DOI] [PubMed] [Google Scholar]
- 223. Ding S, Li H, Su C, Yu J, Jin F. Evolutionary artificial neural networks: a review. Artif Intell Rev. 2013;39:251-260. doi: 10.1007/s10462-011-9270-6. [DOI] [Google Scholar]
- 224. Dobslaff K, Kreisig T, Berthold N, Hoffmann R, Zuchner T. Novel peptide–protein assay for identification of antimicrobial peptides by fluorescence quenching. Anal Bioanal Chem. 2012;403:2725-2731. doi: 10.1007/s00216-012-6050-3. [DOI] [PubMed] [Google Scholar]
- 225. Fedders H, Leippe M. A reverse search for antimicrobial peptides in Ciona intestinalis: identification of a gene family expressed in hemocytes and evaluation of activity. Dev Comp Immunol. 2008;32:286-298. doi: 10.1016/j.dci.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 226. Tennessen JA. Positive selection drives a correlation between non-synonymous/synonymous divergence and functional divergence. Bioinformatics. 2008;24:1421-1425. doi: 10.1093/bioinformatics/btn205. [DOI] [PubMed] [Google Scholar]
- 227. Wilkins MR, Sanchez J-C, Gooley AA, et al. Progress with proteome projects: why all proteins expressed by a genome should be identified and how to do it. Biotechnol Genet Eng Rev. 1996;13:19-50. doi: 10.1080/02648725.1996.10647923. [DOI] [PubMed] [Google Scholar]
- 228. Wasinger VC, Corthals GL. Proteomic tools for biomedicine. J Chromatogr B. 2002;771:33-48. doi: 10.1016/S1570-0232(02)00125-3. [DOI] [PubMed] [Google Scholar]
- 229. Sheoran IS, Ross ARS, Olson DJH, Sawhney VK. Compatibility of plant protein extraction methods with mass spectrometry for proteome analysis. Plant Sci. 2009;176:99-104. doi: 10.1016/j.plantsci.2008.09.015. [DOI] [Google Scholar]
- 230. Muroi M, Osada H. Proteomic profiling for target identification of biologically active small molecules using 2D DIGE. In: Ziegler S, Waldmann H, eds. Systems Chemical Biology. New York, NY: Springer; 2019:127-139. doi: 10.1007/978-1-4939-8891-4_7. [DOI] [PubMed] [Google Scholar]
- 231. Cho WCS. Proteomics technologies and challenges. Genomics Proteomics Bioinformatics. 2007;5:77-85. doi: 10.1016/S1672-0229(07)60018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Baracat-Pereira MC, de Oliveira Barbosa M, Magalhães Júnior MJ, et al. Separomics applied to the proteomics and peptidomics of low-abundance proteins: choice of methods and challenges—a review. Genet Mol Biol. 2012;35:283-291. doi: 10.1590/S1415-47572012000200009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Kołodziejek I, van der Hoorn RA. Mining the active proteome in plant science and biotechnology. Curr Opin Biotechnol. 2010;21:225-233. doi: 10.1016/j.copbio.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 234. Al Akeel R, Mateen A, Syed R, Alyousef A, Shaik M. Screening, purification and characterization of anionic antimicrobial proteins from Foeniculum vulgare. Molecules. 2017;22:602. doi: 10.3390/molecules22040602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Menschaert G, Vandekerckhove TTM, Baggerman G, Schoofs L, Luyten W, Criekinge WV. Peptidomics coming of age: a review of contributions from a bioinformatics angle. J Proteome Res. 2010;9:2051-2061. doi: 10.1021/pr900929m. [DOI] [PubMed] [Google Scholar]
- 236. Nägele E, Vollmer M, Hörth P, Vad C. 2D-LC/MS techniques for the identification of proteins in highly complex mixtures. Expert Rev Proteomics. 2004;1:37-46. doi: 10.1586/14789450.1.1.37. [DOI] [PubMed] [Google Scholar]
- 237. Mitulovic G, Mechtler K. HPLC techniques for proteomics analysis—a short overview of latest developments. Brief Funct Genomic Proteomic. 2006;5:249-260. doi: 10.1093/bfgp/ell034. [DOI] [PubMed] [Google Scholar]
- 238. Blueggel M, Chamrad D, Meyer H. Bioinformatics in proteomics. Curr Pharm Biotechnol. 2004;5:79-88. doi: 10.2174/1389201043489648. [DOI] [PubMed] [Google Scholar]
- 239. McHugh L, Arthur JW. Computational methods for protein identification from mass spectrometry data. PLoS Comput Biol. 2008;4:e12. doi: 10.1371/journal.pcbi.0040012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Hughes C, Ma B, Lajoie GA. De novo sequencing methods in proteomics. In: Hubbard SJ, Jones AR, eds. Proteome Bioinformatics. Totowa, NJ: Humana Press; 2010:105-121. doi: 10.1007/978-1-60761-444-9_8. [DOI] [PubMed] [Google Scholar]
- 241. Eng JK, Fischer B, Grossmann J, Maccoss MJ. A fast SEQUEST cross correlation algorithm. J Proteome Res. 2008;7:4598-4602. doi: 10.1021/pr800420s. [DOI] [PubMed] [Google Scholar]
- 242. Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551-3567. [DOI] [PubMed] [Google Scholar]
- 243. Tsai WC, Zhuang ZJ, Lin CY, Chen WJ. Novel antimicrobial peptides with promising activity against multidrug resistant Salmonella enterica serovar Choleraesuis and its stress response mechanism. J Appl Microbiol. 2016;121:952-965. doi: 10.1111/jam.13203. [DOI] [PubMed] [Google Scholar]
- 244. Umadevi P, Soumya M, George JK, Anandaraj M. Proteomics assisted profiling of antimicrobial peptide signatures from black pepper (Piper nigrum L.). Physiol Mol Biol Plants. 2018;24:379-387. doi: 10.1007/s12298-018-0524-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Wang Z. APD: the antimicrobial peptide database. Nucleic Acids Res. 2004;32:590D-592D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Gasteiger E, Hoogland C, Gattiker A, et al. Protein identification and analysis tools on the ExPASy server. In: Walker JM, ed. The Proteomics Protocols Handbook. Totowa, NJ: Humana Press; 2005:571-607. doi: 10.1385/1-59259-890-0. [DOI] [Google Scholar]
- 247. Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466-1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 248. Geer LY, Markey SP, Kowalak JA, et al. Open mass spectrometry search algorithm. J Proteome Res. 2004;3:958-964. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 249. Zhang N, Aebersold R, Schwikowski B. ProbID: a probabilistic algorithm to identify peptides through sequence database searching using tandem mass spectral data. Proteomics. 2002;2:1406-1412. [DOI] [PubMed] [Google Scholar]
- 250. Field HI, Fenyö D, Beavis RC. RADARS, a bioinformatics solution that automates proteome mass spectral analysis, optimises protein identification, and archives data in a relational database. Proteomics. 2002;2:36-47. [PubMed] [Google Scholar]
- 251. Nesvizhskii AI, Vitek O, Aebersold R. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat Methods. 2007;4:787-797. doi: 10.1038/nmeth1088. [DOI] [PubMed] [Google Scholar]
- 252. Gazzana G, Borlak J. Improved method for proteome mapping of the liver by 2-DE MALDI-TOF MS. J Proteome Res. 2007;6:3143-3151. doi: 10.1021/pr070097p. [DOI] [PubMed] [Google Scholar]
- 253. Pestana-Calsa MC, Ribeiro IL, Calsa T., Jr. Bioinformatics-coupled molecular approaches for unravelling potential antimicrobial peptides coding genes in Brazilian native and crop plant species. Curr Protein Pept Sci. 2010;11:199-209. doi: 10.2174/138920310791112138. [DOI] [PubMed] [Google Scholar]
- 254. Tejano LA, Peralta JP, Yap EES, Panjaitan FCA, Chang Y-W. Prediction of bioactive peptides from Chlorella sorokiniana proteins using proteomic techniques in combination with bioinformatics analyses. Int J Mol Sci. 2019;20:1786. doi: 10.3390/ijms20071786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Weiner AK, Sidoli S, Diskin SJ, Garcia BA. Graphical interpretation and analysis of proteins and their ontologies (GiaPronto): a one-click graph visualization software for proteomics data sets. Mol Cell Proteomics. 2018;17:1426-1431. doi: 10.1074/mcp.TIR117.000438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Jothi A. Principles, challenges and advances in ab initio protein structure prediction. Protein Pept Lett. 2012;19:1194-1204. doi: 10.2174/092986612803217015. [DOI] [PubMed] [Google Scholar]
- 257. Kaufmann KW, Lemmon GH, DeLuca SL, Sheehan JH, Meiler J. Practically useful: what the ROSETTA protein modeling suite can do for you. Biochemistry. 2010;49:2987-2998. doi: 10.1021/bi902153g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Moult J, Fidelis K, Kryshtafovych A, Schwede T, Tramontano A. Critical assessment of methods of protein structure prediction (CASP)-round XII. Proteins. 2018;86:7-15. doi: 10.1002/prot.25415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Källberg M, Wang H, Wang S, et al. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. 2012;7:1511-1522. doi: 10.1038/nprot.2012.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Martí-Renom MA, Stuart AC, Fiser A, Sánchez R, Melo F, Šali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291-325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 261. Biasini M, Bienert S, Waterhouse A, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252-W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Webb B, Sali A. Comparative protein structure modeling using MODELLER: comparative protein structure modeling using modeller. In: Bateman A, Pearson WR, Stein LD, Stormo GD, Yates JR. eds. Current Protocols in Bioinformatics. Hoboken, NJ: John Wiley & Sons, Inc.; 2016:5.6.1-5.6.37. doi: 10.1002/cpbi.3. [DOI] [Google Scholar]
- 263. Wu S, Zhang Y. MUSTER: improving protein sequence profile-profile alignments by using multiple sources of structure information. Proteins. 2008;72:547-556. doi: 10.1002/prot.21945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Yang Y, Faraggi E, Zhao H, Zhou Y. Improving protein fold recognition and template-based modeling by employing probabilistic-based matching between predicted one-dimensional structural properties of query and corresponding native properties of templates. Bioinformatics. 2011;27:2076-2082. doi: 10.1093/bioinformatics/btr350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407-W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Zhang Y, Kolinski A, Skolnick J. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys J. 2003;85:1145-1164. doi: 10.1016/S0006-3495(03)74551-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Wu S, Skolnick J, Zhang Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 2007;5:17. doi: 10.1186/1741-7007-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Zhang W, Yang J, He B, et al. Integration of QUARK and I-TASSER for ab initio protein structure prediction in CASP11: ab initio structure prediction in CASP11. Proteins. 2016;84:76-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Haas J, Barbato A, Behringer D, et al. Continuous Automated Model EvaluatiOn (CAMEO) complementing the critical assessment of structure prediction in CASP12. Proteins. 2018;86:387-398. doi: 10.1002/prot.25431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Moult J, Fidelis K, Kryshtafovych A, Schwede T, Tramontano A. Critical assessment of methods of protein structure prediction: progress and new directions in round XI: progress in CASP XI. Proteins. 2016;84:4-14. doi: 10.1002/prot.25064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Porto WF, Irazazabal L, Alves ESF, et al. In silico optimization of a guava antimicrobial peptide enables combinatorial exploration for peptide design. Nat Commun. 2018;9:1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Song Y, DiMaio F, Wang RYR, et al. High-resolution comparative modeling with RosettaCM. Structure. 2013;21:1735-1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Robustelli P, Piana S, Shaw DE. Developing a molecular dynamics force field for both folded and disordered protein states. Proc Natl Acad Sci USA. 2018;115:E4758-E4766. doi: 10.1073/pnas.1800690115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Freddolino PL, Harrison CB, Liu Y, Schulten K. Challenges in protein-folding simulations. Nat Phys. 2010;6:751-758. doi: 10.1038/nphys1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nat Struct Biol. 2002;9:646-652. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- 277. Mitsutake A, Takano H. Relaxation mode analysis for molecular dynamics simulations of proteins. Biophys Rev. 2018;10:375-389. doi: 10.1007/s12551-018-0406-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435-447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 279. Pronk S, Páll S, Schulz R, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845-854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Abraham MJ, Murtola T, Schulz R, et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19-25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 281. Case DA, Cheatham TE, 3rd, Darden T, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668-1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781-1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187-217. doi: 10.1002/jcc.540040211. [DOI] [Google Scholar]
- 284. Plimpton S. Fast parallel algorithms for short-range molecular dynamics. J Comput Phys. 1995;117:1-19. doi: 10.1006/jcph.1995.1039. [DOI] [Google Scholar]
- 285. Bowers KJ, Chow DE, Xu H, et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. Paper presented at: ACM/IEEE SC 2006 Conference (SC’06); November 11-17, 2006:43; Tampa, FL. doi: 10.1109/SC.2006.54. [DOI] [Google Scholar]
- 286. Hollingsworth SA, Dror RO. Molecular dynamics simulation for all. Neuron. 2018;99:1129-1143. doi: 10.1016/j.neuron.2018.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Meylan S, Andrews IW, Collins JJ. Targeting antibiotic tolerance, pathogen by pathogen. Cell. 2018;172:1228-1238. doi: 10.1016/j.cell.2018.01.037. [DOI] [PubMed] [Google Scholar]
- 288. Hay SI, Rao PC, Dolecek C, et al. Measuring and mapping the global burden of antimicrobial resistance. BMC Med. 2018;16:78. doi: 10.1186/s12916-018-1073-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Neu HC, Gootz TD. Antimicrobial chemotherapy. In: Baron S, ed. Medical Microbiology. 4th ed. Galveston, TX: University of Texas Medical Branch at Galveston; 1996:1-10. http://www.ncbi.nlm.nih.gov/books/NBK7986/ [PubMed] [Google Scholar]
- 290. Hong W, Zeng J, Xie J. Antibiotic drugs targeting bacterial RNAs. Acta Pharm Sin B. 2014;4:258-265. doi: 10.1016/j.apsb.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Cheng G, Dai M, Ahmed S, Hao H, Wang X, Yuan Z. Antimicrobial drugs in fighting against antimicrobial resistance. Front Microbiol. 2016;7:470. doi: 10.3389/fmicb.2016.00470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Sousa SF, Fernandes PA, Ramos MJ. Protein-ligand docking: current status and future challenges. Proteins. 2006;65:15-26. [DOI] [PubMed] [Google Scholar]
- 293. London N, Raveh B, Schueler-Furman O. Peptide docking and structure-based characterization of peptide binding: from knowledge to know-how. Curr Opin Struct Biol. 2013;23:894-902. [DOI] [PubMed] [Google Scholar]
- 294. Pagadala NS, Syed K, Tuszynski J. Software for molecular docking: a review. Biophys Rev. 2017;9:91-102. doi: 10.1007/s12551-016-0247-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Sun J, Deng Z, Yan A. Bacterial multidrug efflux pumps: mechanisms, physiology and pharmacological exploitations. Biochem Biophys Res Commun. 2014;453:254-267. doi: 10.1016/j.bbrc.2014.05.090. [DOI] [PubMed] [Google Scholar]
- 296. Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics. 2014;30:1771-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. van Zundert GCP, Rodrigues JPGLM, Trellet M, et al. The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J Mol Biol. 2016;428:720-725. doi: 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 298. Ruiz-Carmona S, Alvarez-Garcia D, Foloppe N, et al. rDock: a fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLoS Comput Biol. 2014;10:e1003571. doi: 10.1371/journal.pcbi.1003571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Ghoorah AW, Devignes M-D, Smaïl-Tabbone M, Ritchie DW. Protein docking using case-based reasoning. Proteins. 2013;81:2150-2158. doi: 10.1002/prot.24433. [DOI] [PubMed] [Google Scholar]
- 300. Andrusier N, Mashiach E, Nussinov R, Wolfson HJ. Principles of flexible protein-protein docking. Proteins. 2008;73:271-289. doi: 10.1002/prot.22170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Schneider T, Kruse T, Wimmer R, et al. Plectasin, a fungal defensin, targets the bacterial cell wall precursor lipid II. Science. 2010;328:1168-1172. doi: 10.1126/science.1185723. [DOI] [PubMed] [Google Scholar]
- 302. Corbeil CR, Williams CI, Labute P. Variability in docking success rates due to dataset preparation. J Comput Aided Mol Des. 2012;26:775-786. doi: 10.1007/s10822-012-9570-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Bio. 1997;267:727-748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 304. Friesner RA, Murphy RB, Repasky MP, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J Med Chem. 2006;49:6177-6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 305. Sousa SF, Fernandes PA, Ramos MJ. Protein-ligand docking: current status and future challenges. Proteins. 2006;65:15-26. [DOI] [PubMed] [Google Scholar]
- 306. Spitzer R, Jain AN. Surflex-dock: docking benchmarks and real-world application. J Comput Aided Mol Des. 2012;26:687-699. doi: 10.1007/s10822-011-9533-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Rentzsch R, Renard BY. Docking small peptides remains a great challenge: an assessment using AutoDock Vina. Brief Bioinform. 2015;16:1045-1056. doi: 10.1093/bib/bbv008. [DOI] [PubMed] [Google Scholar]
- 308. Audie J, Swanson J. Advances in the prediction of protein-peptide binding affinities: implications for peptide-based drug discovery: protein-peptide binding affinities. Chem Biol Drug Des. 2013;81:50-60. doi: 10.1111/cbdd.12076. [DOI] [PubMed] [Google Scholar]
- 309. Audie J, Swanson J. Recent work in the development and application of protein–peptide docking. Future Med Chem. 2012;4:1619-1644. doi: 10.4155/fmc.12.99. [DOI] [PubMed] [Google Scholar]
- 310. London N, Raveh B, Schueler-Furman O. Peptide docking and structure-based characterization of peptide binding: from knowledge to know-how. Curr Opin Struct Biol. 2013;23:894-902. [DOI] [PubMed] [Google Scholar]
- 311. Sousa SF, Ribeiro AJM, Coimbra JTS, et al. Protein-ligand docking in the new millennium—a retrospective of 10 years in the field. Curr Med Chem. 2013;20:2296-2314. doi: 10.2174/0929867311320180002. [DOI] [PubMed] [Google Scholar]
- 312. Wilson A. Inhibition of protein–protein interactions using designed molecules. Chem Soc Rev. 2009;38:3289-3300. doi: 10.1039/b807197g. [DOI] [PubMed] [Google Scholar]
- 313. Unal EB, Gursoy A, Erman B. VitAL: Viterbi algorithm for de novo peptide design. PLoS ONE. 2010;5:e10926. doi: 10.1371/journal.pone.0010926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Lensink MF, Velankar S, Wodak SJ. Modeling protein-protein and protein-peptide complexes: CAPRI 6th edition. Proteins. 2017;85:359-377. doi: 10.1002/prot.25215. [DOI] [PubMed] [Google Scholar]
- 315. Lensink MF, Wodak SJ. Docking, scoring, and affinity prediction in CAPRI. Proteins. 2013;81:2082-2095. doi: 10.1002/prot.24428. [DOI] [PubMed] [Google Scholar]
- 316. Barh D, Tiwari S, Silva Andrade B, et al. Potential chimeric peptides to block the SARS-CoV-2 spike receptor-binding domain. F1000Research. 2020;9:576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Pant S, Singh M, Ravichandiran V, Murty US, Srivastava HK. Peptide-like and small-molecule inhibitors against Covid-19 [published online ahead of print May 6, 2020]. J Biomol Struct Dyn. doi: 10.1080/07391102.2020.1757510. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, Figure_S2_xyz356052dbb1c09 for Plant Antimicrobial Peptides: State of the Art, In Silico Prediction and Perspectives in the Omics Era by Carlos André dos Santos Silva, Luisa Zupin, Marx Oliveira-Lima, Lívia Maria Batista Vilela, João Pacifico Bezerra-Neto, José Ribamar Ferreira-Neto, José Diogo Cavalcanti Ferreira, Roberta Lane de Oliveira-Silva, Carolline de Jesús Pires, Flavia Figueira Aburjaile, Marianne Firmino de Oliveira, Ederson Akio Kido, Sergio Crovella and Ana Maria Benko-Iseppon in Bioinformatics and Biology Insights
Supplemental material, Figure_S3_xyz356055a6a5139 for Plant Antimicrobial Peptides: State of the Art, In Silico Prediction and Perspectives in the Omics Era by Carlos André dos Santos Silva, Luisa Zupin, Marx Oliveira-Lima, Lívia Maria Batista Vilela, João Pacifico Bezerra-Neto, José Ribamar Ferreira-Neto, José Diogo Cavalcanti Ferreira, Roberta Lane de Oliveira-Silva, Carolline de Jesús Pires, Flavia Figueira Aburjaile, Marianne Firmino de Oliveira, Ederson Akio Kido, Sergio Crovella and Ana Maria Benko-Iseppon in Bioinformatics and Biology Insights
Supplemental material, Figure_S4_xyz35605158f10b9 for Plant Antimicrobial Peptides: State of the Art, In Silico Prediction and Perspectives in the Omics Era by Carlos André dos Santos Silva, Luisa Zupin, Marx Oliveira-Lima, Lívia Maria Batista Vilela, João Pacifico Bezerra-Neto, José Ribamar Ferreira-Neto, José Diogo Cavalcanti Ferreira, Roberta Lane de Oliveira-Silva, Carolline de Jesús Pires, Flavia Figueira Aburjaile, Marianne Firmino de Oliveira, Ederson Akio Kido, Sergio Crovella and Ana Maria Benko-Iseppon in Bioinformatics and Biology Insights
Supplemental material, Figure_S5_xyz35605fb2523e9 for Plant Antimicrobial Peptides: State of the Art, In Silico Prediction and Perspectives in the Omics Era by Carlos André dos Santos Silva, Luisa Zupin, Marx Oliveira-Lima, Lívia Maria Batista Vilela, João Pacifico Bezerra-Neto, José Ribamar Ferreira-Neto, José Diogo Cavalcanti Ferreira, Roberta Lane de Oliveira-Silva, Carolline de Jesús Pires, Flavia Figueira Aburjaile, Marianne Firmino de Oliveira, Ederson Akio Kido, Sergio Crovella and Ana Maria Benko-Iseppon in Bioinformatics and Biology Insights
Supplemental material, Figure_S6_xyz35605b863c136 for Plant Antimicrobial Peptides: State of the Art, In Silico Prediction and Perspectives in the Omics Era by Carlos André dos Santos Silva, Luisa Zupin, Marx Oliveira-Lima, Lívia Maria Batista Vilela, João Pacifico Bezerra-Neto, José Ribamar Ferreira-Neto, José Diogo Cavalcanti Ferreira, Roberta Lane de Oliveira-Silva, Carolline de Jesús Pires, Flavia Figueira Aburjaile, Marianne Firmino de Oliveira, Ederson Akio Kido, Sergio Crovella and Ana Maria Benko-Iseppon in Bioinformatics and Biology Insights
Supplemental material, Figure_S7_xyz3560531a00dc6 for Plant Antimicrobial Peptides: State of the Art, In Silico Prediction and Perspectives in the Omics Era by Carlos André dos Santos Silva, Luisa Zupin, Marx Oliveira-Lima, Lívia Maria Batista Vilela, João Pacifico Bezerra-Neto, José Ribamar Ferreira-Neto, José Diogo Cavalcanti Ferreira, Roberta Lane de Oliveira-Silva, Carolline de Jesús Pires, Flavia Figueira Aburjaile, Marianne Firmino de Oliveira, Ederson Akio Kido, Sergio Crovella and Ana Maria Benko-Iseppon in Bioinformatics and Biology Insights
Supplemental material, Figure_S8_xyz35605c5b2d379 for Plant Antimicrobial Peptides: State of the Art, In Silico Prediction and Perspectives in the Omics Era by Carlos André dos Santos Silva, Luisa Zupin, Marx Oliveira-Lima, Lívia Maria Batista Vilela, João Pacifico Bezerra-Neto, José Ribamar Ferreira-Neto, José Diogo Cavalcanti Ferreira, Roberta Lane de Oliveira-Silva, Carolline de Jesús Pires, Flavia Figueira Aburjaile, Marianne Firmino de Oliveira, Ederson Akio Kido, Sergio Crovella and Ana Maria Benko-Iseppon in Bioinformatics and Biology Insights
Supplemental material, Figure_S9_xyz3560525a5abea for Plant Antimicrobial Peptides: State of the Art, In Silico Prediction and Perspectives in the Omics Era by Carlos André dos Santos Silva, Luisa Zupin, Marx Oliveira-Lima, Lívia Maria Batista Vilela, João Pacifico Bezerra-Neto, José Ribamar Ferreira-Neto, José Diogo Cavalcanti Ferreira, Roberta Lane de Oliveira-Silva, Carolline de Jesús Pires, Flavia Figueira Aburjaile, Marianne Firmino de Oliveira, Ederson Akio Kido, Sergio Crovella and Ana Maria Benko-Iseppon in Bioinformatics and Biology Insights
Supplemental material, Supplementary_Table_1_AND_2_Final_13-07-20_xyz43729463bfb8d for Plant Antimicrobial Peptides: State of the Art, In Silico Prediction and Perspectives in the Omics Era by Carlos André dos Santos Silva, Luisa Zupin, Marx Oliveira-Lima, Lívia Maria Batista Vilela, João Pacifico Bezerra-Neto, José Ribamar Ferreira-Neto, José Diogo Cavalcanti Ferreira, Roberta Lane de Oliveira-Silva, Carolline de Jesús Pires, Flavia Figueira Aburjaile, Marianne Firmino de Oliveira, Ederson Akio Kido, Sergio Crovella and Ana Maria Benko-Iseppon in Bioinformatics and Biology Insights
Supplemental material, Sup_Figure_1_13-07-20_Corrected_Version_xyz43729c25f998f for Plant Antimicrobial Peptides: State of the Art, In Silico Prediction and Perspectives in the Omics Era by Carlos André dos Santos Silva, Luisa Zupin, Marx Oliveira-Lima, Lívia Maria Batista Vilela, João Pacifico Bezerra-Neto, José Ribamar Ferreira-Neto, José Diogo Cavalcanti Ferreira, Roberta Lane de Oliveira-Silva, Carolline de Jesús Pires, Flavia Figueira Aburjaile, Marianne Firmino de Oliveira, Ederson Akio Kido, Sergio Crovella and Ana Maria Benko-Iseppon in Bioinformatics and Biology Insights