

Abstract
Pancreatic acinar cell carcinoma (PAC) is a rare disease with a poor prognosis. Treatment options for metastatic PAC are limited and often follow chemotherapeutic regimens for pancreatic ductal adenocarcinoma. Although recurrent genomic alterations, such as BRAF fusions and defects in genes involved in homologous recombination DNA repair, have been described in PAC, data on the clinical efficacy of molecularly guided, targeted treatment are scarce. Here we describe the case of a 27-yr-old patient with BRAFV600E-mutated PAC who was successfully treated with a combination of BRAF and MEK inhibitors. The patient presented to our clinic with abdominal pain and weight loss. Imaging showed extensive retroperitoneal disease as well as mediastinal lymphadenopathy. Because of elevated α-fetoprotein (AFP) levels and inconclusive histologic findings, a germ cell tumor was suspected; however, PEI chemotherapy was unsuccessful. A repeat biopsy yielded the diagnosis of PAC and treatment with FOLFIRINOX was initiated. Comprehensive molecular profiling within the MASTER (Molecularly Aided Stratification for Tumor Eradication Research) precision oncology program revealed a somatic BRAFV600E mutation and a germline PALB2 stop-gain mutation. Therapy was therefore switched to BRAF/MEK inhibition, resulting in almost complete remission and disease control for 12 mo and a remarkable improvement in the patient's general condition. These results indicate that BRAF alterations are a valid therapeutic target in PAC that should be routinely assessed in this patient population.
Keywords: neoplasm of the pancreas
INTRODUCTION
Pancreatic acinar cell carcinomas (PACs) constitute a rare but distinct and aggressive group of neoplasms that differ from pancreatic ductal adenocarcinomas (PDACs) and neuroendocrine tumors. They differentiate similar to pancreatic acinar cells and, hence, display abundant eosinophilic cytoplasm and enlarged irregular nuclei with prominent nucleoli and positive immunostaining for the enzymes trypsin, chymotrypsin, and lipase (Stelow et al. 2010). Clinical symptoms upon diagnosis are usually nonspecific; however, up to 16% of patients describe systemic manifestations due to liberation of lipase, such as panniculitis and polyarthralgia (Klimstra et al. 1992). PACs most commonly affect adults in the sixth and seventh decade of life, although a wide age span has been described (Schmidt et al. 2008; Wisnoski et al. 2008). Similarly to other pancreatic neoplasms, PACs are often diagnosed at advanced disease stages with metastases present in ∼50% of patients. Rarely, PAC-type neoplasms may originate outside the boundaries of the pancreas including in the retroperitoneum, liver, and the gastrointestinal tract. Distinguishing this uncommon presentation from metastatic PAC is mandatory for appropriate therapy and prognostification (Agaimy et al. 2011). Although the clinical course tends to be more favorable in comparison to PDACs, unresectable or metastatic disease is still associated with a poor prognosis and overall survival ranges between 18 and 47 mo (Al-Hader et al. 2017). Despite recent work on the molecular pathogenesis, these tumors are still poorly understood. Comparative genomic hybridization analysis of 57 PAC samples demonstrated considerable chromosomal instability—that is, recurrent losses of Chromosome 1p, 3p, 4q, 5q, 6q, 8p, 9p, 11q, 13q, 16q, and 18 as well as gains of 1q, 7, 8q, 12, 17q, and 20q (Hoorens et al. 1993; Bergmann et al. 2014). Immunohistochemistry revealed DCC reduction or loss, MYC amplification, and increased epidermal growth factor receptor (EGFR) expression in major subgroups of 57 tumor samples investigated (Bergmann et al. 2014). Sequencing analyses identified mutations in TP53, ARID1A, BRAF, SMAD4, BRCA1, BRCA2, CDKN2A, CTNNB1, RB1, MEN1, MYC, JAK1, APC, GNAS, and FAT (Furlan et al. 2014; Jiao et al. 2014; Al-Hader et al. 2017; Jakel et al. 2017; La Rosa et al. 2018) as well as enrichment of mutational signatures linked to tobacco exposition or defective DNA repair mechanisms in some cases (Jakel et al. 2017). Mismatch repair deficiency has been reported in up to 14% of cases (Al-Hader et al. 2017). No randomized controlled trials are available for chemotherapeutic and or radiotherapeutic treatment for locally advanced or metastatic PACs. Across different regimens, an overall response rate of 23% and a median progression-free survival (PFS) of 5.6 mo were reported (Al-Hader et al. 2017); however, platinum-based regimens appear to be more effective because of inherent defects in DNA repair enzymes (BRCA1, BRCA2, ATM, PALB2) (Al-Hader et al. 2017; Yoo et al. 2017). Because of the rarity of the disease, no trials investigating targeted therapy options for the aforementioned mutations are currently underway.
We present the case of a young man with extrapancreatic acinar cell carcinoma with retroperitoneal lymphadenopathy and high α-fetoprotein (AFP) serum levels, leading to initial misdiagnosis as metastatic germ cell tumor, who reached almost complete remission upon targeted therapy with BRAF/MEK inhibition based on a BRAFV600E driver mutation.
RESULTS
A 27-yr-old male patient presented to our outpatient department with abdominal pain, weight loss, nausea, and fatigue. Imaging workup showed massive retroperitoneal bulky disease and mediastinal lymphadenopathy. Tumor marker analysis revealed an elevated AFP (1358.9 IU/mL), whereas others, including beta human chorionic gonadotropin (β-HCG), soluble interleukin-2 receptor (sCD25), lactic acid dehydrogenase (LDH), CA19-9, and carcinoembryonic antigen (CEA), were within normal range. Initial core needle biopsy was inconclusive; hence, with respect to the high tumor burden, chemotherapy with cisplatin, etoposide, and ifosfamid (PEI) under the assumption of metastatic germ cell tumor was initiated. Short-term imaging follow-up and AFP levels showed progressive disease. Repeated biopsy findings (Fig. 1) were consistent with PAC, presumably of peripancreatic origin. Treatment with 5-fluoruracil, oxaliplatin, and irinotecan (FOLFIRINOX) was initiated in analogy to PDAC, leading to a decline in AFP levels after two cycles (1038 IU/mL) and stable disease on magnetic resonance imaging (MRI) scans. Because of adverse events (nausea [Common Terminology Criteria for Adverse Events (CTCAE) 3°], loss of appetite [CTCAE 2°], and weight loss [CTCAE 3°]), chemotherapy was deescalated to the FOLFIRI regimen for eight cycles, yielding formally stable disease with an AFP of 760 IU/mL.
Figure 1.

(A) Hematoxylin and eosin (H&E) staining and immunohistochemical stainings of (B) trypsin and (C) α-fetoprotein (AFP), confirming AFP-producing acinar cell carcinoma of pancreatic-type (magnification, 400×).
To identify additional treatment options, the patient was enrolled in the MASTER precision oncology program of NCT Heidelberg and the German Cancer Consortium (Horak et al. 2017). Whole-genome and transcriptome sequencing revealed a low mutational burden (14 nonsynonymous single-nucleotide variants [SNVs] and two insertions/deletions [indels] within coding regions) and no signs of microsatellite instability (MSIsensor score of 1.65), which was in line with immunohistochemical assessment. The genome-wide copy-number assessment revealed a number of chromosomal and subchromosomal gains and losses (see Fig. 2). Further analysis identified an activating mutation (NM_004333.4, rs113488022, c.T1799A, p.V600E, allele frequency 30%) in BRAF exon 15 and a germline heterozygous PALB2 stop-gain mutation (NM_024675, rs180177100, c.C1240T, p.R414X, allele frequency 47%) (Table 1), which is considered pathogenic according to the current American College of Medical Genetics (ACMG) criteria (Richards et al. 2015) and which has previously been linked to familial pancreatic and breast cancer (Slater et al. 2010).
Figure 2.

DNA copy number plot displaying chromosomal gains and losses in the tumor sample. Dark and light blue lines show total and allele-specific copy numbers, respectively. (TCN) Total copy number.
Table 1.
Variant table
| Gene | Chromosome | HGVS DNA reference | HGVS protein reference | Variant type | Predicted effect (substitution, deletion, etc.) | dbSNP/dbVar ID | Genotype (heterozygous/homozygous) | ClinVar ID | Parent of origin |
|---|---|---|---|---|---|---|---|---|---|
| BRAF | 7 | NM_004333: c.T1799A | NP_004324: p.V600E | SNV | Substitution | rs113488022 | Heterozygous (somatic) | VCV000013961 | — |
| PALB2 | 16 | NM_024675: c.C1240T | NP_078951: p.R414X | Stop-gain-SNV | Substitution | rs180177100 | Heterozygous (germline) | VCV000128117 | Unknown |
(HGVS) Human Genome Variation Society, (SNV) single-nucleotide variant.
The patient's family history revealed a case of breast cancer in a first-degree relative but was otherwise unsuspicious. The patient was therefore recommended to receive genetic counseling. Functional indicators of defective homologous recombination DNA repair were inconclusive with very low genomic rearrangement scores (homologous repair deficiency [HRD], 1; large scale transition [LST], 0; telomeric allelic imbalance [TAI], 3) and mutational signature 3 supported by 23% of all somatic SNVs (Alexandrov et al. 2013).
Based on the genetic findings and in accordance with the recommendation by the molecular tumor board of NCT Heidelberg, combined BRAF and MEK inhibition with dabrafenib and trametinib was initiated. Follow-up imaging at 3 mo demonstrated almost complete remission with only residual abdominal lymph nodes and lung nodules (Fig. 3). Only pyrexia CTCAE 1° was observed as side effect. AFP levels had dropped to normal range. At the patient's 6-mo follow-up, the remaining lymph nodes further decreased in size and the initial lung lesions were no longer detectable. Hence, he underwent systematic abdominal lymphadenectomy with pancreatectomy and splenectomy with the aim of resecting all macroscopic tumor lesions. Histopathological workup revealed vital tumor within the head of the pancreas, surrounded by a dense chronic inflammatory wall, as well as vital tumor in 37 of 62 resected lymph nodes. Postoperatively, treatment with dabrafenib/trametinib had to be halted because of dysphagy and malabsorption, leading to a 2-mo treatment break. AFP levels showed a marked increase after discontinuation of treatment (249 IU/mL). Follow-up imaging unfortunately revealed progressive disease within the abdominal lymph nodes. After reinitiation of targeted therapy, the patient reported good general health; however, follow-up imaging showed progressive disease with increasing lymph nodes in both the abdominal and mediastinal compartments. BRAF/MEK inhibition was continued beyond progression because it was well-tolerated, and the young patient refused systemic chemotherapy because of the experienced side effects. After 12 mo of dabrafenib/trametinib the patient developed rapidly progressive disease with enlarging mediastinal and abdominal lymph nodes. He declined any further salvage treatment and succumbed to the disease 21 mo after diagnosis.
Figure 3.
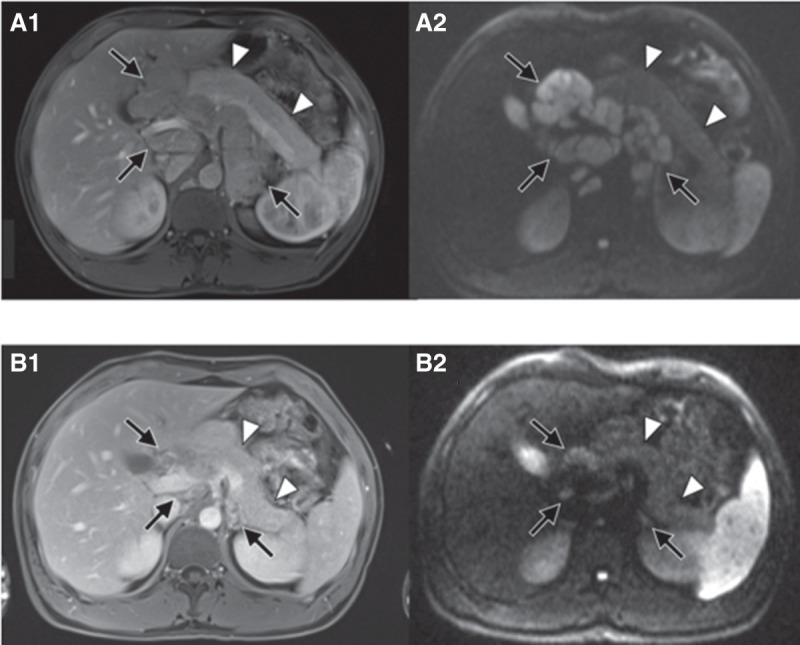
Almost complete remission at 3-mo follow-up after initiation of trametinib/dabrafenib. Baseline imaging: axial early venous phase MR image (A1) and axial diffusion weighted MR image with b-value of 800 sec/mm2 (A2) show extensive lymphadenopathy in the retroperitoneum and liver hilum (black arrows). There is no lesion in the pancreas (white arrowheads). Follow-up imaging: Axial early venous phase MR image (B1) and axial diffusion weighted MR image with b-value of 800 sec/mm2 (B2) show marked reduction of lymphadenopathy (black arrows). Still there is no lesion in the pancreas (white arrowheads).
DISCUSSION
Chemotherapeutic treatment is currently the mainstay of palliative therapy for PAC patients. Small cohort studies and case reports list a number of regimens with an overall response rate of 23% and a median PFS of 5.6 mo (Al-Hader et al. 2017; Yoo et al. 2017). Chemotherapeutic substances include gemcitabine, capecitabine, 5-fluoruracil, irinotecan, cisplatin/oxaliplatin, and taxanes (Lowery et al. 2011; Al-Hader et al. 2017; Yoo et al. 2017; Brunetti et al. 2018) and also targeted therapies such as erlotinib or panitumumab in KRAS wild-type tumors (Morales et al. 2013; Kruger et al. 2016). Regarding predictors of treatment response, defects in DNA repair genes such as BRCA1, BRCA2, and PALB2 have been linked to prolonged survival following platinum-based regimens in some patients (Al-Hader et al. 2017; Yoo et al. 2017). Interestingly, the patient reported here had a germline heterozygous stop-gain mutation in PALB2, which has previously been reported in the context of familial pancreatic and breast cancer (Slater et al. 2010). However, he did not benefit from cisplatin or oxaliplatin because he developed new lung lesions under the first cycle of PEI treatment and only demonstrated stable disease with oxaliplatin-containing FOLFIRINOX. Other molecular predictors of platinum response, such as the very low HRD, LST, and TAI scores (Waddell et al. 2015) and the rather low level of the mutational signature 3 (Telli et al. 2016), did not point to a major defect in homologous recombination DNA repair mechanisms and were therefore in line with the insensitivity to platinum-containing treatment regimens. The pathologic and clinical significance of the identified PALB2 mutation for the pathogenesis of the patient's disease therefore remains uncertain.
Until today, targeted treatment options in PAC are not well established. However, there are a number of reports of RAF alterations in PACs, especially SND1-BRAF and HERPUD1-BRAF fusions (Wang et al. 2018), and sensitivity of BRAF-fused PAC cell lines to MEK inhibitors has been demonstrated previously (Chmielecki et al. 2014). It has even been proposed that this BRAF-altered cohort of PACs form a unique patient cohort distinctive from other PAC patients characterized by frequent alterations in HR genes and clinical high sensitivity to platinum-based treatment (Chmielecki et al. 2014). In contrast to fusions, BRAF point mutations have been found only rarely (Chmielecki et al. 2014; Jiao et al. 2014) or not at all (Bergmann et al. 2014) in PACs. BRAF mutations occur in up to 15% across all cancers, predominantly as point mutations with substitution of glutamic acid (E) for valine (V) at position 600 (70%–90%) alongside other BRAF mutations, amplifications, and fusions (Turski et al. 2016). Combination therapy using BRAF and MEK inhibition has proven to be superior over monotherapy, both for reasons of efficacy and reduced side effects (Eroglu and Ribas 2016), and has led to approval—for example, BRAFV600E-mutated melanoma and lung cancer. Effectiveness of BRAF and/or MEK inhibition has also been observed for other activating mutations (Dahlman et al. 2012) and even BRAF fusions (Ross et al. 2016). A recent basket trial for BRAFV600E-mutated cancer patients demonstrated efficacy in 13 unique cancer types, including cholangiocarcinoma, sarcoma, glioma, neuroendocrine carcinoma, and salivary gland carcinoma (Subbiah et al. 2020). Schreck et al. report on two patients with high-grade glioma who were successfully treated with BRAF/MEK inhibition (Schreck et al. 2018). In BRAFV600E-mutated pancreatic cancer, within early basket trials, one patient with PDAC responded to vemurafenib (Hyman et al. 2015) and another PDAC patient showed prolonged survival under dabrafenib/trametinib combination treatment (Guan et al. 2018). In contrast to PDAC, to our knowledge, there have been no reports of BRAF-targeted treatments in PAC patients until today. We present a PAC patient carrying a BRAFV600E mutation who responded well to combined BRAF/MEK inhibitor treatment with an almost complete response. This allowed for extensive surgery aiming at complete resection of all visible tumor burden. However, the debulking surgery did not translate into long-term tumor control because postoperative complications delayed the reuptake of targeted therapy. After reinitiation, dabrafenib/trametinib did no longer halt disease progression, possibly because of the outgrowth of BRAFV600E-negative and/or -resistant clones.
The transient response to BRAF/MEK inhibitors in many patients remains a significant therapeutic challenge as our case also documents. Acquired mechanisms of BRAF resistance can be divided in upstream reactivation of the MAPK/ERK pathway through, for example, overexpression of the RAF isoforms ARAF and CRAF or activating RAS mutations and downstream activation through either BRAF overexpression and dimerization or activation of the PI3K/AKT pathway (Griffin et al. 2017). Although combination treatment with BRAF/MEK inhibition may slow down the development of BRAF inhibitor resistance because tumors cannot exploit the MEK pathway, most patients do eventually progress under combination treatment. Additionally, as demonstrated in our case, resection of all macroscopic tumor burden does not seem to prevent relapse and does not deter the development of drug resistance. Hence, current research focuses on other new possible combination therapies outside the MAP/ERK pathway (Griffin et al. 2017). Interestingly, intermittent dosing, both in vitro and in vivo models, seemed to delay the onset of BRAF inhibitor resistance (Griffin et al. 2017), with one case report describing an ongoing complete remission with intermittent vemurafenib dosing (Dooley et al. 2016). Also, in BRAFV600E-mutated colorectal cancer, in which feedback activation of EGFR forms a preexisting resistance mechanism to BRAF/MEK inhibition, a triple combination of cetuximab, encorafenib, and binimetinib has recently shown to successfully overcome this resistance leading to an impressive improvement in response and overall survival within the BEACON trial (Kopetz et al. 2019). Because of the rapid progression of disease and denial of further diagnostics, analysis of possible resistance mechanisms and their targetability was not possible in our case. Still, in this young patient with metastatic PAC, BRAF/MEK-targeted therapy allowed for prolonged disease control with minimal side effects.
CONCLUSION
Here we present the case of a young patient with BRAFV600E-mutated PAC who achieved almost complete response upon initiation of combined BRAF/MEK inhibition.
This case underscores the value of comprehensive genomic assessment in patients with rare cancers whose treatment options are often limited. Hence, for PAC patients we recommend at least extended next-generation sequencing (NGS) panel sequencing for druggable targets including BRAF point mutations and fusions. BRAF/MEK inhibition is an efficient and well-tolerated treatment option in the BRAF-mutated subcohort of PAC patients.
METHODS
Microsatellite Instability Analysis
Genomic DNA was extracted from tissue samples after manual microdissection using the QIAGEN DNA Blood and Tissue Kit (QIAGEN) according to the manufacturer's recommendation. Measurements of DNA content were performed using a NanoDrop (Thermo Scientific). Microsatellite instability typing was carried out using the Bethesda marker panel (Boland et al. 1998) and CAT25 as described previously (Findeisen et al. 2005). Two or more MSI markers were scored as high-level MSI (MSI-H). MSISensor (Niu et al. 2014) was applied with a minimum required coverage of 15 reads in both tumor and control. The MSI score for a sample is the percentage of somatic, instable microsatellites, relative to the total number of microsatellites found in the control sample. According to the paper, a score of >3.5 implies microsatellite instability.
Whole-Genome Sequencing
Tissue samples were provided by the NCT Heidelberg Tissue Bank in accordance with its regulations and after approval by the Ethics Committee of Heidelberg University. DNA isolation from the tumor specimen and the blood sample was performed using the AllPrep DNA/RNA/Protein Mini Kit (QIAGEN), followed by quality control and quantification using a Qubit 2.0 Fluorometer (Life Technologies), a 2200 TapeStation system (Agilent), and a 2100 Bioanalyzer system (Agilent).
For genome sequencing on an Illumina HiSeq X instrument, 100 ng of genomic DNA were fragmented to an insert size of 450 base pairs (bp) with a Covaris LE220 or E220 device, and libraries were prepared using the TruSeq Nano Kit (Illumina). Paired-end sequencing was carried out according to the manufacturer's recommendations, yielding read lengths of 151 bp (HiSeq X). MSIsensor, HRD, LST, and TAI scores were calculated as previously described (Abkevich et al. 2012; Birkbak et al. 2012; Popova et al. 2012; Niu et al. 2014).
RNA Sequencing
RNA sequencing libraries were prepared using the TruSeq RNA Sample Preparation Kit v2 (Illumina). Briefly, mRNA was purified from 1 µg total RNA using oligo(dT) beads, poly(A)+ RNA was fragmented to 150 bp and converted into cDNA, and cDNA fragments were end-repaired, adenylated on the 3' end, adapter-ligated, and amplified with 12 cycles of polymerase chain reaction. The final libraries were validated using a Qubit 2.0 Fluorometer (Life Technologies) and a Bioanalyzer 2100 system (Agilent). Libraries were sequenced on an Illumina-patterned flowcell v2.5.
Mapping and Analysis of Whole-Genome Sequencing Data
Mapping and analysis of whole-genome sequencing data were performed as previously reported (Heining et al. 2018; Groschel et al. 2019). Sequencing coverages are provided in Supplemental Table 1.
ADDITONAL INFORMATION
Data Deposition and Access
Sequencing data were deposited in the European Genome-phenome Archive (https://www.ebi.ac.uk/ega/) under accession EGAS00001004282.
Ethics Statement
Tissue samples were provided by the NCT Heidelberg Tissue Bank after written informed consent was obtained under the NCT MASTER protocol (S-206/2011, approved by the Ethics Committee of Heidelberg University), which covers all aspects relevant to clinical cancer genome sequencing as well as further research and publication. This study was conducted in accordance with the Declaration of Helsinki.
Acknowledgments
The authors thank the NCT/DKFZ Sample Processing Laboratory, the DKFZ Genomics and Proteomics Core Facility, the DKFZ-Heidelberg Center for Personalized Oncology (DKFZ-HIPO) HIPO-021, the DKFZ Omics IT, and Data Management Core Facility for technical support and expertise.
Author Contributions
E.B., S.K., S.Z., S.F., P.M., D.J., A.A., G.M., and C.S. collected and interpreted patient data and were involved in clinical management. E.B., S.K., S.U., P.H., B.B., B.H., and M.F. were involved in genomic profiling and data analysis. S.F., H.G., E.S., and A.S. supervised data interpretation. All authors have approved the current version of the manuscript and its submission to Cold Spring Harbor Molecular Case Studies.
Funding
This work was supported by grant H021 from the Heidelberg Center for Personalized Oncology (DKFZ-HIPO).
Competing Interest Statement
The authors have declared no competing interest.
Referees
Davide Melisi
Anonymous
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Abkevich V, Timms KM, Hennessy BT, Potter J, Carey MS, Meyer LA, Smith-McCune K, Broaddus R, Lu KH, Chen J, et al. 2012. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer 107: 1776–1782. 10.1038/bjc.2012.451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agaimy A, Kaiser A, Becker K, Brasen JH, Wunsch PH, Adsay NV, Kloppel G. 2011. Pancreatic-type acinar cell carcinoma of the liver: a clinicopathologic study of four patients. Mod Pathol 24: 1620–1626. 10.1038/modpathol.2011.127 [DOI] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. 2013. Signatures of mutational processes in human cancer. Nature 500: 415–421. 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hader A, Al-Rohil RN, Han H, Von Hoff D. 2017. Pancreatic acinar cell carcinoma: a review on molecular profiling of patient tumors. World J Gastroenterol 23: 7945–7951. 10.3748/wjg.v23.i45.7945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann F, Aulmann S, Sipos B, Kloor M, von Heydebreck A, Schweipert J, Harjung A, Mayer P, Hartwig W, Moldenhauer G, et al. 2014. Acinar cell carcinomas of the pancreas: a molecular analysis in a series of 57 cases. Virchows Arch 465: 661–672. 10.1007/s00428-014-1657-8 [DOI] [PubMed] [Google Scholar]
- Birkbak NJ, Wang ZC, Kim JY, Eklund AC, Li Q, Tian R, Bowman-Colin C, Li Y, Greene-Colozzi A, Iglehart JD, et al. 2012. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov 2: 366–375. 10.1158/2159-8290.CD-11-0206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, et al. 1998. A National Cancer Institute Workshop on Microsatellite Instability for Cancer Detection and Familial Predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 58: 5248–5257. [PubMed] [Google Scholar]
- Brunetti O, Aprile G, Marchetti P, Vasile E, Casadei Gardini A, Scartozzi M, Barni S, Delfanti S, De Vita F, Di Costanzo F, et al. 2018. Systemic chemotherapy for advanced rare pancreatic histotype tumors: a retrospective multicenter analysis. Pancreas 47: 759–771. 10.1097/MPA.0000000000001063 [DOI] [PubMed] [Google Scholar]
- Chmielecki J, Hutchinson KE, Frampton GM, Chalmers ZR, Johnson A, Shi C, Elvin J, Ali SM, Ross JS, Basturk O, et al. 2014. Comprehensive genomic profiling of pancreatic acinar cell carcinomas identifies recurrent RAF fusions and frequent inactivation of DNA repair genes. Cancer Discov 4: 1398–1405. 10.1158/2159-8290.CD-14-0617 [DOI] [PubMed] [Google Scholar]
- Dahlman KB, Xia J, Hutchinson K, Ng C, Hucks D, Jia P, Atefi M, Su Z, Branch S, Lyle PL, et al. 2012. BRAFL597 mutations in melanoma are associated with sensitivity to MEK inhibitors. Cancer Discov 2: 791–797. 10.1158/2159-8290.CD-12-0097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley AJ, Gupta A, Middleton MR. 2016. Ongoing response in BRAF V600E-mutant melanoma after cessation of intermittent vemurafenib therapy: a case report. Target Oncol 11: 557–563. 10.1007/s11523-015-0410-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu Z, Ribas A. 2016. Combination therapy with BRAF and MEK inhibitors for melanoma: latest evidence and place in therapy. Ther Adv Med Oncol 8: 48–56. 10.1177/1758834015616934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findeisen P, Kloor M, Merx S, Sutter C, Woerner SM, Dostmann N, Benner A, Dondog B, Pawlita M, Dippold W, et al. 2005. T25 repeat in the 3′ untranslated region of the CASP2 gene: a sensitive and specific marker for microsatellite instability in colorectal cancer. Cancer Res 65: 8072–8078. 10.1158/0008-5472.CAN-04-4146 [DOI] [PubMed] [Google Scholar]
- Furlan D, Sahnane N, Bernasconi B, Frattini M, Tibiletti MG, Molinari F, Marando A, Zhang L, Vanoli A, Casnedi S, et al. 2014. APC alterations are frequently involved in the pathogenesis of acinar cell carcinoma of the pancreas, mainly through gene loss and promoter hypermethylation. Virchows Arch 464: 553–564. 10.1007/s00428-014-1562-1 [DOI] [PubMed] [Google Scholar]
- Griffin M, Scotto D, Josephs DH, Mele S, Crescioli S, Bax HJ, Pellizzari G, Wynne MD, Nakamura M, Hoffmann RM, et al. 2017. BRAF inhibitors: resistance and the promise of combination treatments for melanoma. Oncotarget 8: 78174–78192. 10.18632/oncotarget.19836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groschel S, Hubschmann D, Raimondi F, Horak P, Warsow G, Frohlich M, Klink B, Gieldon L, Hutter B, Kleinheinz K, et al. 2019. Defective homologous recombination DNA repair as therapeutic target in advanced chordoma. Nat Commun 10: 1635 10.1038/s41467-019-09633-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan MB, Bender RJ, Pishvaian MJ, Halverson DC, Tuli R, Klempner SJ, Wainberg ZA, Singhi AD, Petricoin E, Hendifar AE. 2018. Molecular and clinical characterization of BRAF mutations in pancreatic ductal adenocarcinomas (PDACs). J Clin Oncol 36: 214 10.1200/JCO.2018.36.4_suppl.214 [DOI] [Google Scholar]
- Heining C, Horak P, Uhrig S, Codo PL, Klink B, Hutter B, Frohlich M, Bonekamp D, Richter D, Steiger K, et al. 2018. NRG1 fusions in KRAS wild-type pancreatic cancer. Cancer Discov 8: 1087–1095. 10.1158/2159-8290.CD-18-0036 [DOI] [PubMed] [Google Scholar]
- Hoorens A, Lemoine NR, McLellan E, Morohoshi T, Kamisawa T, Heitz PU, Stamm B, Ruschoff J, Wiedenmann B, Kloppel G. 1993. Pancreatic acinar cell carcinoma. An analysis of cell lineage markers, p53 expression, and Ki-ras mutation. Am J Pathol 143: 685–698. [PMC free article] [PubMed] [Google Scholar]
- Horak P, Klink B, Heining C, Groschel S, Hutter B, Frohlich M, Uhrig S, Hubschmann D, Schlesner M, Eils R, et al. 2017. Precision oncology based on omics data: the NCT Heidelberg experience. Int J Cancer 141: 877–886. 10.1002/ijc.30828 [DOI] [PubMed] [Google Scholar]
- Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, Wolf J, Raje NS, Diamond EL, Hollebecque A, et al. 2015. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 373: 726–736. 10.1056/NEJMoa1502309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakel C, Bergmann F, Toth R, Assenov Y, van der Duin D, Strobel O, Hank T, Kloppel G, Dorrell C, Grompe M, et al. 2017. Genome-wide genetic and epigenetic analyses of pancreatic acinar cell carcinomas reveal aberrations in genome stability. Nat Commun 8: 1323 10.1038/s41467-017-01118-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Yonescu R, Offerhaus GJ, Klimstra DS, Maitra A, Eshleman JR, Herman JG, Poh W, Pelosof L, Wolfgang CL, et al. 2014. Whole-exome sequencing of pancreatic neoplasms with acinar differentiation. J Pathol 232: 428–435. 10.1002/path.4310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimstra DS, Heffess CS, Oertel JE, Rosai J. 1992. Acinar cell carcinoma of the pancreas. A clinicopathologic study of 28 cases. Am J Surg Pathol 16: 815–837. 10.1097/00000478-199209000-00001 [DOI] [PubMed] [Google Scholar]
- Kopetz S, Grothey A, Yaeger R, Van Cutsem E, Desai J, Yoshino T, Wasan H, Ciardiello F, Loupakis F, Hong YS, et al. 2019. Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N Engl J Med 381: 1632–1643. 10.1056/NEJMoa1908075 [DOI] [PubMed] [Google Scholar]
- Kruger S, Haas M, Burger PJ, Ormanns S, Modest DP, Westphalen CB, Kleespies A, Angele MK, Hartwig W, Bruns CJ, et al. 2016. Acinar cell carcinoma of the pancreas: a rare disease with different diagnostic and therapeutic implications than ductal adenocarcinoma. J Cancer Res Clin Oncol 142: 2585–2591. 10.1007/s00432-016-2264-7 [DOI] [PubMed] [Google Scholar]
- La Rosa S, Bernasconi B, Vanoli A, Sciarra A, Notohara K, Albarello L, Casnedi S, Billo P, Zhang L, Tibiletti MG, et al. 2018. c-MYC amplification and c-myc protein expression in pancreatic acinar cell carcinomas. New insights into the molecular signature of these rare cancers. Virchows Arch 473: 435–441. 10.1007/s00428-018-2366-5 [DOI] [PubMed] [Google Scholar]
- Lowery MA, Klimstra DS, Shia J, Yu KH, Allen PJ, Brennan MF, O'Reilly EM. 2011. Acinar cell carcinoma of the pancreas: new genetic and treatment insights into a rare malignancy. Oncologist 16: 1714–1720. 10.1634/theoncologist.2011-0231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales M, Cabrera MA, Maeso MD, Ferrer-López N. 2013. Use of panitumumab in the treatment of acinar cell carcinoma of the pancreas: a case report. Oncol Lett 5: 969–971. 10.3892/ol.2012.1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, Wendl MC, Ding L. 2014. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics 30: 1015–1016. 10.1093/bioinformatics/btt755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popova T, Manie E, Rieunier G, Caux-Moncoutier V, Tirapo C, Dubois T, Delattre O, Sigal-Zafrani B, Bollet M, Longy M, et al. 2012. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res 72: 5454–5462. 10.1158/0008-5472.CAN-12-1470 [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross JS, Wang K, Chmielecki J, Gay L, Johnson A, Chudnovsky J, Yelensky R, Lipson D, Ali SM, Elvin JA, et al. 2016. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int J Cancer 138: 881–890. 10.1002/ijc.29825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt CM, Matos JM, Bentrem DJ, Talamonti MS, Lillemoe KD, Bilimoria KY. 2008. Acinar cell carcinoma of the pancreas in the United States: prognostic factors and comparison to ductal adenocarcinoma. J Gastrointest Surg 12: 2078–2086. 10.1007/s11605-008-0705-6 [DOI] [PubMed] [Google Scholar]
- Schreck KC, Guajardo A, Lin DDM, Eberhart CG, Grossman SA. 2018. Concurrent BRAF/MEK inhibitors in BRAF V600-mutant high-grade primary brain tumors. J Natl Compr Canc Netw 16: 343–347. 10.6004/jnccn.2017.7052 [DOI] [PubMed] [Google Scholar]
- Slater EP, Langer P, Niemczyk E, Strauch K, Butler J, Habbe N, Neoptolemos JP, Greenhalf W, Bartsch DK. 2010. PALB2 mutations in European familial pancreatic cancer families. Clin Genet 78: 490–494. 10.1111/j.1399-0004.2010.01425.x [DOI] [PubMed] [Google Scholar]
- Stelow EB, Shaco-Levy R, Bao F, Garcia J, Klimstra DS. 2010. Pancreatic acinar cell carcinomas with prominent ductal differentiation: mixed acinar ductal carcinoma and mixed acinar endocrine ductal carcinoma. Am J Surg Pathol 34: 510–518. 10.1097/PAS.0b013e3181cfcac7 [DOI] [PubMed] [Google Scholar]
- Subbiah V, Puzanov I, Blay JY, Chau I, Lockhart AC, Raje NS, Wolf J, Baselga J, Meric-Bernstam F, Roszik J, et al. 2020. Pan-cancer efficacy of vemurafenib in BRAFV600-mutant non-melanoma cancers. Cancer Discov 10: 657–663. 10.1158/2159-8290.CD-19-1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, Szallasi Z, Barry WT, Winer EP, Tung NM, et al. 2016. Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin Cancer Res 22: 3764–3773. 10.1158/1078-0432.CCR-15-2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turski ML, Vidwans SJ, Janku F, Garrido-Laguna I, Munoz J, Schwab R, Subbiah V, Rodon J, Kurzrock R. 2016. Genomically driven tumors and actionability across histologies: BRAF-mutant cancers as a paradigm. Mol Cancer Ther 15: 533–547. 10.1158/1535-7163.MCT-15-0643 [DOI] [PubMed] [Google Scholar]
- Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al. 2015. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518: 495–501. 10.1038/nature14169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Basturk O, Wang J, Benayed R, Middha S, Zehir A, Linkov I, Rao M, Aryeequaye R, Cao L, et al. 2018. A FISH assay efficiently screens for BRAF gene rearrangements in pancreatic acinar-type neoplasms. Mod Pathol 31: 132–140. 10.1038/modpathol.2017.106 [DOI] [PubMed] [Google Scholar]
- Wisnoski NC, Townsend CM Jr, Nealon WH, Freeman JL, Riall TS. 2008. 672 patients with acinar cell carcinoma of the pancreas: a population-based comparison to pancreatic adenocarcinoma. Surgery 144: 141–148. 10.1016/j.surg.2008.03.006 [DOI] [PubMed] [Google Scholar]
- Yoo C, Kim BJ, Kim KP, Lee JL, Kim TW, Ryoo BY, Chang HM. 2017. Efficacy of chemotherapy in patients with unresectable or metastatic pancreatic acinar cell carcinoma: potentially improved efficacy with oxaliplatin-containing regimen. Cancer Res Treat 49: 759–765. 10.4143/crt.2016.371 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data were deposited in the European Genome-phenome Archive (https://www.ebi.ac.uk/ega/) under accession EGAS00001004282.