

Abstract
Characterization of HIV Env glycoprotein with 28 glycosylation sites is the essential step of structure-based vaccine design programs. A comprehensive LC-MS/MS peptide mapping analysis was applied to assess the primary sequence, glycosylation profiles, and glycosite occupancy of Env to ensure the adequate mimicking of the native immunogen. Another structural feature was reported, related to its cleaved subunits within the trimeric assembly. We bring attention to the importance of thorough inspection of the results generated by the informatics tools which are currently available for the biopharmaceutical characterization. The complexity of Env translates into a vast amount of data with occasional information gaps that could not possibly be filled by means of the automatic data analysis. A series of data validation steps was applied, followed by the illustrations on how the high-quality results may be misinterpreted. It was shown that the glycan sites can only be characterized to a certain limit, and that any claim of full structural characterization of this molecule beyond these limits should be treated with caution. Following the result verification, the percent glycan occupancy was reported for 25 N-glycan sites, including 3 critical antibody-recognition sites. The exact glycan profiles were provided for 20 individual sites, whereas only the glycosylation type could be deduced for 5 sites, dictated by their location within Env sequence. The distribution of the unprocessed high mannose–type glycans correlated with the expected "mannose patch." Experimental procedure optimization and a workflow for glycan characterization with a focus on stringent data testing are presented in the current study.
Keywords: HIV, Complex glycoprotein structural, Mass spec, Characterization, Vaccine
Graphical Abstract
Introduction
The vaccine development programs have been focused on the study of HIV Env surface glycoprotein (Env), which enables the virus to enter the host cells [1]. Native Env forms a trimeric complex consisting of three noncovalently associated heterodimers made of gp120 and gp41 subunits. In the presence of co-transfected furin,the peptide bond between these subunits is expected to be fully cleaved. Among various Env constructs designed as the prototypes of the native-like immunogens, the BG505.DS.SOSIP.664 is the subject of the current report. In this stabilized soluble construct, "BG505" refers to the strain of HIV-1, "SOS" defines the disulfide bond C501-C605 binding the subunits gp120 and gp41, and "IP.664" describes I559P mutation for conformational stability and crystallization studies [2–4]. During the product development and process scale-up, a panel of LC-MS/MS peptide mapping analyses was applied to monitor the quality of a series of Env products at various manufacturing stages.
Known as one of the most densely glycosylated protein with 28 glycosylation sites, Env is expected to present a certain challenge for the routine mass spectrometry applications, in terms of both sample preparation and data processing. A dense glycan layer of Env is one of its distinctive characteristics: the glycans make up to half of total glycoprotein molecular weight. This glycan shield functions as the protection of the underlying protein viral epitopes from recognition by antibodies [5]; however, many broadly neutralizing antibodies target this particular glycan shield—a structural feature which has recently been used as the HIV-1 vaccine development strategy. Env is known to contain two main glycan subpopulations: complex-type glycans and under-processed high-mannose glycans, particularly a "high-mannose patch" region sterically protected from the glycan-processing enzymes (the patch represents a region rich in high-mannose glycans that limits accessibility of ER and Golgi α-mannosidases) [5–8]. The Env recognition by the broadly neutralizing antibodies is related to formation of high mannose at the specific "critical" highly conserved glycosylation sites. To ensure the integrity of this epitope in the designed mutated immunogen, the type of glycosylation and site occupancy of these sites needed to be evaluated during the product development, along with confirmation of the primary sequence and overall Env glycosylation characterization. In this report, we demonstrated the development of the LC-MS/MS strategy to characterize these complex structural features and highlighted some inevitable analytical challenges of such complex glycoprotein analysis. Previous work on characterization of the Env glycosylation type or occupancy included various MS instrumentation and proteomics tools for data processing [7–14]. Following these technological advances, a routine characterization of various Env constructs ensued, with vast amount of data generated by the automatic data processing. We now present the essential step of thorough inspection of the final results: omitting this step may compromise the resulting data quality, regardless of the prior stages of the sample preparation, the optimization of the LC-MS/MS method, and the amount and the quality of the generated data. Close inspection of the automatically produced results for Env protein analysis relative to its raw data sets from various alternative sources prompted the need for the use of the strict acceptance criteria. The current LC-MS/MS peptide mapping technique was performed according to a conventional biopharmaceutical workflow for glycoprotein characterization using a complementary set of proteolytic digests and a combination of glycosidases. A variety of traditional and innovative approaches for glycosite occupancy evaluation exist, which involves conversion of formerly glycosylated Asn residue to Asp upon deglycosylation [8, 12, 13]. We applied straightforward calculations of the glycan site occupancy based on comparing the intensities of the deglycosylation-induced deamidated components to their endogenous modification(s) using the routine QC compatible characterization. The current work is focused on the processing aspect of the resulting data: it was illustrated how the automatically generated results need to be followed by rigorous data inspection and filtering. The existing limitations of charactering the HIV trimer using the modern mass spectrometry tools were demonstrated and justified.
Experimental
LC-MS Materials and Reagents
High purity LC-MS grade water and acetonitrile containing formic acid used for mobile phase preparation and ammonium bicarbonate reagent were purchased from J.T. Baker (Phillipsburg, NJ). RapiGest™ surfactant and [Glu1]-Fibrino-peptide B lock mass standard (GluFib) were purchased from Waters (Milford, MA). Formic acid, urea, and Zeba™ spin desalting column (7 K MWCO) were purchased from Pierce (Rockford, IL). Dithiothreitol (DTT) and iodoacetamide were purchased from ThermoFisher Life Technologies (Grand Island, NY) and Sigma-Aldrich (St. Louis, MO), respectively. Trypsin (modified sequencing grade), chymotrypsin, and PNGase F glycosidase and endoglycosidase (Endo) H were purchased from Promega (Madison, WI). Alpha 1–2,3 mannosidase was purchased from New England Biolabs (Ipswich, MA).
Sample Preparation for the LC-MS/MS Analyses
BG505SOSIP.664 HIV trimer samples were produced in CHO cell lines. The purified samples (0.5–3 mg/mL concentration) were buffer exchanged into 50 mM ammonium bicarbonate solution using Zeba™ spin desalting columns. For denaturation, RapiGest detergent was added to the sample to final concentration of 0.1%, and the sample was heated at 60 °C for 30 min. For disulfide bonds reduction, DTT was added to the sample along with RapiGest, to 20-mM final DTT concentration.
The reduced samples were mixed with a lyophilized protease (trypsin, chymotrypsin, LysC, or a combination thereof), to a final enzyme:protein ratio of 1:20 w/w, and the solution was incubated at 37 °C for 4 h. For deglycosylation, the mixture of glycosidases PNGaseF (5 u per 8 εg Env), Endo H (10 u per 1 εg Env), and alpha 1–2,3 mannosidase (15 u per 20 εg Env) were added to the samples, and the solution was incubated overnight at 37 °C. The digestion was quenched by acidifying the solution with 0.1% formic acid, followed by incubation at 70 °C for 15 min and spinning down the hydrolyzed RapiGest by-product. The supernatant was pipetted out, and 10 εLofthe resulting peptide digest mixture was ready for the LC-MS/MS analysis.
LC-MS/MS Setup
LC-MS/MS analyses were performed using an Acquity H-Class chromatography system with mass spectrometry detection on a SYNAPT G2 QTof, both from Waters (Milford, MA). The mobile phase consisted of the aqueous solution of 0.1% formic acid as solvent A, and 0.1% formic acid in acetonitrile as solvent B. Protein digests were separated on a UPLC Peptide BEH C18 column (300 Å, 1.7 εm, 2.1 mm × 50 mm) (Waters), with the column temperature set to 65 °C, at a 0.2 mL/min flow rate; gradient: 0 min–3%, 1 min–3%, 91 min–57%, 91.5 min–85%, 102 min–85%, 103 min–3%, 105 min–3%. The MS data were acquired in positive ion/resolution analyzer mode; the MS/MS data were a result of collisionally activated dissociation (CAD), performed via MSE technique that simultaneously and independently collects the spectra for the precursor and fragment ions of any species present in the gas cell. The MSE "high-energy" channel settings used to produce the MS/MS spectra were as follows: linear ramping the collisional energy from 30 to 45 V, 0.5-s scan time over the acquisition range 100–2000 m/z. Capillary was set to 3.0 kV, cone 35 V, source 120 °C, desolvation 350 °C, and desolvation gas 800 L/h. Lock mass correction was done by means of 20 εL/min infusion of 100 fmol/εL GluFib in water/acetonitrile 1:1 with 0.1% formic acid (785.837 m/z peak, 30-s intervals, 3 scans to average). BiopharmaLynx v. 1.3 was used as a routine biopharmaceutical tool for streamlined data processing, whereas MassLynx v. 4.1 was applied for manual data inspection and for peptide de novo sequencing using MaxEnt3 deconvolution algorithm (resolution 25,000, peptide isotope distribution, 1 ensemble member, 50 iterations per ensemble member). The data search included semidigested and miscleaved peptides, with 10 ppm mass accuracy of the precursor ions and 20 ppm for the fragment ions for initial data scan prior to further data filtering, with a set of 25 custom-made glycoforms introduced to the targeted search list.
Results and Discussion
Optimization of the Sample Preparation Focusing on Env Structural Features
Following the analytical practice for routine product quality assessment, the Env samples were a subject of peptide mapping for sequence validation as well as detailed characterization of the glycan profiles and glycan % site occupancy at each individual glycosite of gp120 and gp41 subunits. High level of Env glycosylation suggests the need for sample preparation optimization, such as complementary proteolytic digests and harsh deglycosylation conditions. The proposed digestion was designed to generate a range of meaningful peptide components of adequate size and population of Asn residues, which subsequently could be used for site occupancy study. A particular attention was paid to the characterization of the "critical" glycosites, which represent the epitopes known for antibody recognition in BG505SOSIP.664: N156, N160, and N322 [4, 14–16]. The N-terminal signal peptide was expected to be fully cleaved; the gp120 and gp41 subunits were expected to be dissociated at the multi-Arg junction of furin cleavage site.
Glycan occupancy study dictates the requirement for the efficient deglycosylation of the glycoprotein; optimization of deglycosylation also helps the investigation of the primary structure. The traditional deglycosylation method established for antibodies (5 enzymatic units of PNGase F per 8 εg of protein) was applied to the denatured and reduced Env, following proteolytic digestion. The extracted ion chromatograms (XIC) of the oxonium glycan ions (204 m/z and 366 m/z) were used to monitor the amount of residual glycosylation (Figure 1a, top). Using the standard deglycosylation procedure with PNGase F, a complete deglycosylation was not achieved, even by using 4 × excess amount of PNGase F and 14-h digestion (Figure 1a, middle trace). As the Env protein is known to contain a high percentage of high-mannose and hybrid glycan species, the deglycosylation was therefore approached by the mixture of glycosidases: PNGase F, Endo H, and alpha 1–2,3 mannosidase, which were expected to target the immature glycan moieties. The mannosidase assisted with cleaving the terminal mannosyl residues, "clearing the path" for the other two enzymes, whereas EndoH was applied to boost overall deglycosylation efficiency of the hybrid- and high mannose–type glycan moieties. No significant heterogeneity in the residual glycan profiles was observed: in addition to fully deglycosylated glycopeptides, there were only GlcNAc peptides detected (the result of EndoH treatment), which was verified by XIC of various glycan signature ions. This residual glycosylation was accounted for the subsequent deamidation calculations, as a part of the original glycopeptide. The glycosidase mixture ensured an efficient deglycosylation (Figure 1a, bottom trace), and was since applied throughout all the following experiments.
Figure 1.

(a) XIC of 204.1 ± 0.05 Da corresponding to characteristic glycan peak in the original tryptic digest (top); the digest treated with PNGase F for 10 h (middle), with remaining glycopeptides circled; the digest treated the mixture of PNGase F, Endo H, and α-1–2,3 mannosidase results (bottom). The peaks in the bottom trace correspond to non-glycan peaks, with negligible residual glycosylation. The XIC peak intensities are normalized. (b) Type of the proteolytic digestion customized for individual glycopeptides: combined [tryptic + chymotryptic] digest (top) and combined [LysC + chymotryptic] digest (bottom) applied for glycosylation characterization of the peptide containing critical N332 glycosite
For optimum separation and detection, and for the glycosylation occupancy calculations, the peptides resulting from the efficient proteolytic digestion must match the following criteria: (1) to be of the adequate size for the column retention and MS ionization, with optimum hydrophilicity for the RP-LC separation, (2) to have one glycosylation site per peptide, (3) to have minimum potential deamidation sites per peptide, (4) to possess enough charge to produce a good quality MS/MS spectrum. To satisfy these requirements, in silico digestion using various enzymes was performed to evaluate the peptide length and Asn distribution, focusing on critical glycosylation sites (the in silico algorithm is embedded in the processing software; however, we found it helpful to reproduce the results by manual labeling for clear visualization of potentially challenging sequence motifs, the glycosite and disulfide bond arrangement, etc.) Several commonly used proteases were selected as potential candidates for Env digestion: trypsin, chymotrypsin, and LysC, as well as the combined digestion of [trypsin + chymotrypsin] and [LysC + chymotrypsin]. Two potential problems appeared with streamlining this approach. First, in silico results were not expected to accurately reflect the actual protein cleavage due to potential overdigestion and missed cleavages, so that the digestion efficiency needed to be evaluated using the empirical data. The second problem was the lack of a universal set of proteases applicable for characterization of the entire Env: different types of a digest favored characterization of different glycosites. For example, for the glycopeptide containing the critical N332 glycosylation site (Figure 1b), the combined [LysC + chymotrypsin] digest produced cleaner MS/MS spectrum than the combined [trypsin + chymotrypsin]. However, most of the information for the rest of the glycosites originated from the combined [tryptic + chymotryptic] digest with occasional use of the chymotryptic components.
Similarly, various proteases were tested during the deglycosylated digest analysis, because the trypsin alone resulted in the gap in sequence coverage around N322 site (QAHCNL332VSK). A target searching for a non-reduced dimer or partially deglycosylated species did not yield any results. However, a combination of harsh denaturation conditions, using urea instead of RapiGest detergent, longer incubation times, addition of chymotrypsin, and replacing the glass sample vials with plastic ones for digest loading, allowed to detect the veritable deamidated peptide QAHCD332VSK eluting at 2 min of the 90-min gradient. Being a relatively hydrophilic peptide, it bound poorly to the RP C18 column, resulting in low peak intensity, insufficient for the routine glycan occupancy study. Therefore, the alternative digests were tested to obtain longer peptides covering N332 glycosite, for which the precursor ion intensity and the fragmentation efficiency could be ranked in the order of tryptic digest < combined [LysC + chymotrypsin] < LysC digests. The good quality of the MS/MS spectra was essential for the peptide identity confirmation and assigning the deamidated and non-modified peptide analogs, necessary for glycan occupancy calculations.
Based on the results of the digestion optimization, it was decided to proceed with a set of orthogonal digests for all routine Env sample analyses. Tryptic, chymotryptic, LysC, combined [LysC + chymotrypsin], and combined [tryptic + chymotryptic] digests were applied in parallel for the primary structure investigation, as each of the digests helped elucidate various portions of the sequence, complementing each other to provide better sequence coverage. By using an overlay of two combined digests along with a LysC digest, 99% sequence coverage of Env was obtained.
Another example of the need for complementary digestion was assessment of the primary sequence, with LysC protease predicted to work best based on the structural feature of the C-terminus of gp120 subunit. When cleaved successfully by furin, Env precursor forms gp120 and gp41 subunits connected via disulfide bond. This characteristic Env cleavage is necessary for Env membrane fusion during infection of the cell. The furin recognition site of BG505SOSIP.664 construct was mutated to form polybasic (RRRRRR) linker at the C-terminus of gp120 subunit (Figure 2a) in order to improve Env cleavage efficiency. One important goal of the LC-MS/MS analysis, therefore, was to confirm the engineered design to ensure the efficient Env precursor cleavage, and identify possible uncleaved species. The exact furin cleavage site of gp120 was characterized by the complementary LysC, chymotryptic, or the mixture [LysC + chymotrypsin] digests. To capture possible fused gp120-gp41 fragment, the data were searched against the individual subunits as well as non-cleaved string of subunits. Based on peptide mapping evaluation using the LysC digest, the ratio of the N-terminal peptide of gp41 to the peptide product of the fused (gp120-gp41) precursor was about 4:1 (Figure 2a), suggesting that the major furin proteolytic cleavage was achieved. At the same time, the peptide mapping approach was never designed for the accurate estimation of the Env cleavage, due to the expected large difference in the ionization efficiencies of these peptides. However, this approximate ratio was still reported, since it was aligned with the previously reported results of the MS analysis of the intact protein and the subsequently developed RPLC-UV method for quantitative monitoring of the Env integrity. The MS/MS spectrum of the peptide resulting from the fused subunit structure displayed high percentage of the ion fragments and good mass accuracy (SI S1a, S2), allowing unambiguous assignment of this component.
Figure 2.

LysC digest enables detection of (a) partially uncleaved [gp120+gp41] Env species, (b) truncated furin site. Upon furin cleavage, out of 6 possible Arg, either one Arg or no Arg are left at the C-terminus of gp120 subunit
It was shown that upon furin cleavage, the C-terminus of gp120 is presented as a mixture of the components with 1 terminal Arg and 0 Arg residues only left from the original RRRRRR string (Figure 2b). The detailed structure characterization related to this cleavage site will be discussed in a separate manuscript in preparation. The MS/MS spectra were used to confirm the identity of all species found (SI 1b), in addition to the mass accuracy filters and the relevant retention time range (Figure 2b).
Characterization of the Individual Glycan Profiles
The established orthogonal digestion approach with multiple glycosidases was applied to generate glycopeptides for a specific profiling of the individual glycosites. A set of the complex, high-mannose, and hybrid structures was used in glycan search, providing high level of Env immature glycosylation. The automatically processed data were aligned with manual inspection of the glycopeptide MS/MS spectra as another level of results proofing; the XIC of the glycan oxonium ions were largely aligned with the software-generated results. To justify complementarity, the glycan profiles were checked for consistency among various digest approaches: the characterization of the glycosite N88 using chymotryptic digest (peptide component C8), combined [trypsin + chymotrypsin] digest (component CT11), or combined [LysC + chymotrypsin] digest (miscleaved component CK11–12) resulted in the similar profiles (Figure 3). Predominant G2 and Man5 species were detected in all three digests along with a low-level distribution of various complex glycoforms and a small percentage of non-glycosylated peptides. Such reproducibility offers flexibility of selecting the most relevant protease combination for glycoprofile reporting: whichever generates best quality spectra and is most suitable for a particular glycopeptide sequence.
Figure 3.

Reproducibility of the glycan profiling of glycosite N88 using various digests and peptide components: (a) chymotryptic digest, component C8, (b) combined [trypsin + chymotrypsin] digest, component CT11, (c) combined [LysC + chymotrypsin] digest, miscleaved CK11–12 component
Out of 28 possible glycosites of the gp120 and gp41 subunits, the exact glycosylation profile was reported for 20 (Figure 4); the other 8 glycosites were used as an illustration of the analytical challenges of the routine peptide mapping analysis later in the text. The majority of the glycosylation identified in the gp120 subunit belonged to the high-mannose type, with smaller percentage of the complex glycans, whereas the complex-type glycans dominated in the gp41 subunit. A negligible amount of the hybrid-type glycans was found and was not included in the chart. Other low amounts (< 5%) of glycans are reported to reflect full qualitative profile, particularly for the critical sites. Another G0F-2GlcNAc structure was identified at N182 glycosite only. N332 glycosite was found to be predominantly occupied with Man9 and minor distribution of other high-mannose species (Man5 to Man8): these high mannose-type glycans are a key epitope of the N332-dependent bNAbs [17, 18]. Compact quaternary structure of Env and high glycan density of gp120 protects this "intrinsic mannose patch" from trimming by ER or Golgi α-mannosidase, resulting in the immature high mannose–type glycosylation [5]. This patch serves a target for HIV-1 vaccine design. The gp41 subunit is less compact, with its Asn sites fully exposed to mannosidase, resulting in the efficient processing the high-mannose glycans into the complex type, which was demonstrated by distribution of glycan structures at N611, N618, and N637 sites: despite the low percentage glycan occupancy at N611 and N618, all three sites are populated with the complex-type glycans. The conclusion at this point was that the straightforward LC-MS/MS analysis with minimal procedure optimization confirmed (1) the engineered design of the BG505SOSIP.664 with full high-mannose glycosylation at the critical sites N156, N160, and N332 [14] and (2) nearly fully cleaved gp120 and gp41 subunits. Nevertheless, it is likely that none of the thoroughly designed peptide mapping techniques would guarantee full molecule characterization, because the Env intrinsic sequence features cause inevitable challenges in characterization, which are described below.
Figure 4.

Glycan profiles for the glycosites of gp120 (N88–N4460) and gp41 (N611-N637) subunits
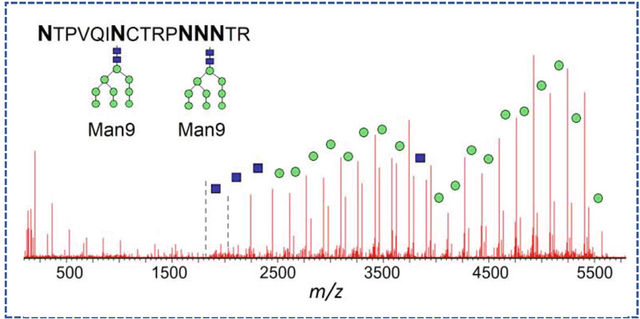
One of the typical challenges of the LC-MS/MS analysis occurs when the resulting peptide contains more than 1 glycosites, and this peptide cannot be broken down further into smaller peptides using conventional choice of proteases. For the analytical groups with the ultimate goal of routine product characterization under strictly controlled conditions, the LC-MS/MS techniques are limited with the choice of reproducible, highly specific enzymatic proteolysis techniques; hence, a combination of trypsin, LysC, and chymotrypsin is usually employed. For the resulting digestion products with multiple Asn sites, only the overall crude glycan population can be deduced, without glycan profiling per individual site. Figure 5 demonstrated how the glycan characterization of a peptide with 5 Asn residues was achieved, using the MS/MS spectrum of the product of the combined [LysC + chymotrypsin] digest, peptide NTPVQIN295CTRPNN301NTR (CT43 component of gp120 subunit). In this case, two glycosylation sites N295 and N301 are separated by the string of amino acids "CTRPN." No specific protease is capable to cleave in between these sites (not accounting for possible non-reproducible overdigested species or non-specific proteases). A total of 18 mannoses per resulting peptide were translated into the proposed structures of two Man9 moieties at each of its theoretically possible glycosites. As listed in Table 1, various combinations of the tentative high-mannose structures were identified belonging to this glycopeptide component in the original (non-deglycosylated) digest; however, it is impossible to assign the exact number of Man to either N295 or N301 glycosite. In this case, only the glycosylation type could be reported as opposed to the specific glycan profile, even under fully optimized conditions.
Figure 5.

The example of the glycan moiety characterization: peptide with 5 Asn (product of the combined [trypsin + chymotrypsin] digest, CT43 component in gp120 subunit, RT 20.4 min). The total of 18 mannoses translates into the proposed structures of two Man9 moieties on both N295 and N301 glycosites
Table 1.
The list of all components detected in the original and deglycosylated digests of CT43 component of gp120 subunit, containing 5 asn with 2 potential glycosites (NTPVQIN295CTRPNN301NTR). MS/MS spectra of the deamidated peptides confirmed the exact deamidation location and indicated 100% glycan occupancy at both N295 and N301 glycosites
| Tentative glycan composition | RT, min | Intensity, cts | Mass, Da | Mass error, ppm |
|---|---|---|---|---|
| Original digest | ||||
| Man8, Man8 | 20.6 | 88,655 | 5246.049 | −1.8 |
| Man9, Man8 | 20.5 | 77,947 | 5408.104 | −1.3 |
| Man7, Man8 | 20.7 | 58,239 | 5084.005 | −0.2 |
| Man7, Man7 | 20.8 | 42,674 | 4921.953 | 0.0 |
| Man7, Man6 | 21.0 | 23,529 | 4759.913 | 2.6 |
| Man5, Man6 | 21.1 | 17,258 | 4435.801 | 1.6 |
| Man7, Man7 | 20.5 | 16,770 | 4921.951 | −0.4 |
| Man7, Man6 | 20.5 | 16,684 | 4759.895 | −1.2 |
| Man6, Man6 | 21.1 | 14,624 | 4597.856 | 2.0 |
| Man7, Man8 | 20.5 | 11,911 | 5084.000 | −1.1 |
| Man6, Ma6 | 20.6 | 10,778 | 4597.851 | 0.7 |
| Man5, Man5 | 21.3 | 6099 | 4273.741 | −0.2 |
| Man5, Man6 | 20.6 | 5723 | 4435.787 | −1.6 |
| Man9, Man9 | 20.4 | 5542 | 5570.152 | −2.1 |
| Man5, Man6 | 20.7 | 4015 | 4435.792 | −0.7 |
| Man9, Mang | 22.5 | 1442 | 3705.534 | 1.1 |
| Deglycosylated digest | ||||
| Deamidation N(2): N295, N301 | 25.7 | 479,849 | 1842.866 | 0.8 |
| Deamidation N(3): N295, N300, N301 | 26.5 | 20,537 | 1843.850 | 0.9 |
The same problem occurred with sites N133 and N137 of the CT16 component LTPLCVTLQCTN133 VTNN137ITDDMRGELK, where two sites with possible glycosylation are separated by "VTN" residues. A mixture of the complex and the high mannose–type glycosylation was detected for these sites, without realistic prospect to assign the exact glycan profile to specific site. No further method optimization could lead to reporting these unassigned sites, as this deficiency is the intrinsic feature of heavily glycosylated Env and its sequence arrangement, as opposed to the lack of method development.
The 3 sites, for which neither glycan profile nor the glycan type was reported, were N398, N406, and N411: the smallest peptide which could be obtained by proteolytic digestion was ISN398TSVQGSN406STGSN411DSITLPCRIK, which still contained 3 glycan moieties, thus was poorly suitable for the reversed phase separation and the MS ionization. Claiming these sites as identified should prompt examination of the raw data and justification of the identified species (e.g., relevant chromatographic retention, mass accuracy of the precursor and fragment ions, MS/MS spectra quality, including the characteristic glycan fragments and the glycan moiety confirmation), to exclude potential false-positives.
The last Env site, for which only the glycan occupancy was determined, was N625 of gp41 subunit. Only the miscleaved SNRN618LSEIWDN625MTW component has been consistently produced by chymotryptic digestion or a combination of LysC with chymotrypsin, employing various denaturation/reduction conditions—still resistant to cleaving at W623 site. As shown later in the text, N625 site revealed the lowest level (8%) of the glycosylation site occupancy among all Env glycosites, and the resulting low-intensity glycopeptide spectra could not be used for the reliable confirmation of the species. For the large part, the glycans observed in this component were reported as residing at the neighboring N618. In summary, 8 glycosites out of theoretically possible 28 (predicted using the Asn-X-Ser/Thr, X≠Pro motif) were only reported in terms of the glycan type, and the exact glycan distribution was provided for 20 glycosites.
Glycosylation Site Occupancy and Correct Site Allocation
The glycosylation occupancy calculations were performed using the traditional approach by taking advantage of the Asn resides with the attached glycans being converted to Asp upon deglycosylation, and the two peptides different by 0.98 Da being sufficiently MS resolved. For routine analytical characterization, the deamidation and N-succinimide were included in the default list of PTM search. Asn deamidation produces isomers, which are difficult to differentiate by the conventional MS studies [19, 20]. Although the general PTM characterization is not a primary focus of this work, deamidation is directly related to the glycan occupancy study, so it was carefully investigated. The control set of samples held overnight at 37 °C without glycosidase mixture showed that virtually no deamidation was introduced during the sample preparation and a short-term storage (diagram in illustrated in supplementary fig. S3). The potential Asn glycosites, including the ones prone to rapid deamidation (NN, NG), as well as the Asn which were a part of a heavily glycosylated peptide, resulted in only marginal increase in deamidation (up to 2%). However, two critical steps were needed to be considered: it was essential (1) to pinpoint the exact deamidation location(s) within the peptide and (2) to distinguish among the deamidation resulting from the glycosidase treatment and the endogenous (process-related) deamidation. To address these tasks, the MS/MS spectra were inspected before and after deglycosylation, and the Asp/Asn site(s) were assigned using a set of strict filtering criteria. Potential difference in the ionization efficiencies of the deamidated and non-deamidated peptides was assumed to be minimal [21].
The importance of the exact deamidation location assignment is illustrated first. More than one deamidation site can be present in the peptides, despite the attempt to minimize such outcome by the design of proteolytic cleavage. As demonstrated in Figure 6, the miscleaved chymotryptic component C8–9 of the gp-41 subunit (SNRN618LSEIWDN625MTW) contained two potential glycosylation sites (N618 and N625) and three potential deamidation sites (N616, N618, and N625). XIC of 883.9 ± 0.5 Da mass (covering the mass of the doubly charged ion for either deamidated or non-deamidated peptide) revealed four peaks (Figure 6a). Inspection of the MS/MS spectra (Figure 6b) allowed the assignment of each peak as the non-modified peptide (56.5 min), deamidated at N618 (57.6 min), deamidated at N625 (58.0 min), and the peptide with both N618 and N625 being deamidated (59.2 min). For glycosylation occupancy calculations, only the relevant, glycosylation site–related Asp residues were accounted for, among other possible modifications. Analogous situation was observed with the peptide LIN195CN197TSACTQACPK (combined tryptic + chymotryptic digestion), which contains only one possible glycosylation site N197 and another possible deamidation site N195. In the deglycosylated digest, only the non-modified peptide and its deamidated analog were observed, for which the exact deamidation location (N197) was verified by means of the high-energy fragmentation, confirming its deglycosylation origin.
Figure 6.

Deamidation sites assigned by peptide sequencing, containing 2 potential deamidation sites: chymotryptic C8–9 component of gp41 subunit: (a) XIC of SNRN618LSEIWDN625MTW peptide-related components ([2+], 883.9 ± 0.5 Da) for selection of the relevant deamidation site for glycosite occupancy calculations; (b) the exact deamidation location is confirmed by the presence of 1214 m/z and 1215 m/z ion fragments in the corresponding N625- and D625-containing peptides
The next essential step was to evaluate the amount of the process-related (hereafter as "endogenous") deamidation. The workflow for the glycosylation % site occupancy calculations is illustrated in Scheme 1 using the peptide RLDVVQIN177EN179QGN182R with 3 Asp residues: N182 glycosite and two possible deamidation sites N177 and N179 (CT26–27 component of gp120 subunit, the product of the combined [trypsin + chymotrypsin] digest). The observables, including the intensity values, are listed in Table 2. To obtain the percentage of glycosylation occupancy, the intensity of the deglycosylation-related Asp-containing peptide was divided by the sum of the intensities of all its modified and non-modified components. Endogenous deamidation in the original non deglycosylated digest was taken into account, and its amount was subtracted from the amount of post-deglycosylation deamidation, yielding only the deglycosylation-related deamidation. XIC of the [2+] charge state of CT26–27-related components revealed a set of components containing N/D177 and N/D182. According to the MS/MS spectra of each chromatographic peak, N179 site was not deamidated, whereas each N177 and N182 had a low amount of endogenous deamidation prior to the glycosidases treatment. First step in Scheme 1 was the calculation of the endogenous deamidation of N182 glycosite in the non-deglycosylated digest. Following deglycosylation, the intensity of the N177/D182 XIC peak increased dramatically, and also, the new D177/DD182 component became apparent. Another low-intensity peak appeared at 27.5 min which was proved to be an artifact of Endo H treatment: the MS spectrum showed partial fragmentation of the labile GlcNAc group, contributing to the detection of a low amount of the N182-containing peptide. The next step according to the Scheme 1 was to adjust the intensity of the endogenous deamidation in the deglycosylated digest using the original digest data: only the portion of peak intensity related to the PNGase F treatment should be accounted for the D182-containing component eluting at 28.6 min. This corrected value was used for final calculations of the percent site occupancy, yielding 59% for N182 glycosite. Following this workflow, the calculations account for the endogenous deamidation and residual glycosylation, and ensure the use of the relevant deamidation site.
Scheme 1.

An example of a workflow for glycan occupancy calculations for N182 glycosite (RLDVVQIN177ENQGN182R, CT26–27 component of gp120 subunit, the missed-cleaved product of the combined [trypsin + chymotrypsin] digest). XIC of the CT26027 [2+] charge state displays several peptides containing N177, N182, and their deamidated analogs. The workflow accounts for the endogenous deamidation and residual glycosylation. The calculations are illustrated using the Table 2 values. The brackets denote the MS peak intensity (counts)
Table 2.
A set of data required for glycosite occupancy calculations, Including non-modified components, deamidated components in both original and deglycosylated digests, and residual glycosylation: an example using N182 glycosite (CT26–27 component of gp120 subunit, the result of the combined [Trypsin + Chymotrypsin] digest). N177 and N179 are other possible deamidation sites. The calculation details are provided in Scheme 1
| Modification | RT, min | Intensity, cts | Intensity, % | Mass, Da | Mass error, ppm | b/y-ions, % | Peptide |
|---|---|---|---|---|---|---|---|
| Original digest | |||||||
| All glycopeptides | 111,601 | 58.9 | RLDVVQIN177ENQGN182R | ||||
| Non-modified | 28.0 | 68,578 | 36.2 | 1653.854 | −0.6 | 77 | RLDVVQIN177ENQGN182R |
| Deamidation N182 | 28.8 | 4750 | 2.5 | 1654.838 | −0.3 | 15 | RLDVVQINENQGD182R |
| Deamidation N177 | 29.2 | 4632 | 2.4 | 1654.841 | 1.6 | 15 | RLDVVQID177ENQGNR |
| Deglycosylated digest | |||||||
| Non-modified | 28.1 | 67,536 | 36.8 | 1653.854 | −0.4 | 73 | RLDVVQIN177ENQGN182R |
| Deamidation N182 | 28.6 | 85,188 | 46.4 | 1654.840 | 0.6 | 88 | RLDVVQINENQGD182R |
| Glycosylation GIcNAc | 27.7 | 18,918 | 10.3 | 1856.934 | 0.2 | 19 | RLDVVQINENQGN182R |
| Deamidation N 177, N182 | 29.8 | 6109 | 3.3 | 1655.822 | −0.5 | 23 | RLDVVQID177ENQGD182R |
| Deamidation N177 | 29.3 | 5874 | 3.2 | 1654.839 | 0.5 | 23 | RLDVVQID177ENQGNR |
There were typical cases of a straightforward assignment of 100% glycan site occupancy, when the peptide showed no sign of the endogenous deamidation in the original digest and the resulting deglycosylated digest demonstrated 100% deamidation. Even for the peptides with multiple deamidation sites, when no specific glycan profile could be established, the glycan site occupancy could be calculated using the information from the MS/MS spectra. Table 1 lists two components in the deglycosylated digest, with both components being deamidated at potential glycosites N295 and N301. As no endogenous deamidation was identified in the original digest, both glycosites were assumed to be fully glycosylated. Similarly, focusing on critical glycosite N332, various proteolytic digests yielded peptides of different length, none of which demonstrated an endogenous deamidation. The only component associated with this glycosite was found to be 100% deamidated post-deglycosylation, and this critical site was presumed to be fully occupied.
Figure 7 presents the summary of the glycan type, glycosite occupancy, and primary sequence confirmation of the Env construct BG505SOSIP.664 (the exact glycan distribution per site was shown on Figure 4). Using a combination of the individual and combined proteolytic digests, 99% sequence of the glycoprotein was confirmed. The gaps in the sequence (DKKQK, CKDK, KY, and RR) were due to the enzyme cleavage yielding the peptides with less than 2 amino acid in length. No remnants of the signal peptide were identified, and the C-terminus of the gp120 subunit contained either 0 or 1 Arg instead of expected six.
Figure 7.

Sequence coverage and glycosylation mapping of Env with 25 characterized N-glycosites. Unconfirmed gaps in the sequence are highlighted. The glycan types are denoted as complex type only (boxed), high-mannose type (dotted underline), both complex and high-mannose type (solid underline). A negligible amount of hybrid-type N-glycans was also observed. Glycosylation % site occupancy is denoted. The exact glycan profiles are provided for all characterized glycosites except for N133, N137, N295, N301, and N625
The critical sites N156, N160, and N332 were fully characterized both in terms of glycosylation occupancy and the glycan distribution: full glycosylation at these sites was reported with predominant Man8–Man9 species detected, according to the proposed engineered structure [3, 22]. Overall, out of total 28 glycan sites of the BG505.DS.SOSIP.664 construct, percent glycan occupancy was reported for 25 sites (denoted in Figure 7), including 3 critical ones (N156, N160, and N332), which cover antibody recognition [4, 14–16]. For the other three glycosylation sites N398, N406, N41,1 no information was reported due to highly complex glycosylation at the glycopeptide, and 3 glycosites per resulting deglycosylated digested component. The glycan profiles were provided for the most glycosites except for N133, N137, N295, and N301 (two glycosites per digest component) and N625 (insufficient % glycosylation). The type of glycosylation was reported for the other 5 sites, and the other 3 sites were not characterized because of the amino acid composition in the Env sequence. In Figure 7, the glycosites with complex-type glycans only are marked in blue, green denotes the high-mannose type, and the sites at which both complex and high mannose–type glycans were identified are marked in purple, showing the visual difference between gp120 and gp41 glycosylation–type distribution. We believe that any missing information on the sequence coverage, site occupancy, or glycan profile is unlikely to be compensated by an alternative sample preparation or the LC-MS/MS setup in the setting of the routine confirmatory analytical assessment.
Data Inspection and Excluding False Component Assignment
Here, the importance of thorough inspection of the automatically processed data was illustrated. A highly efficient workflow designed for the glycoprotein data reporting by the state-of-the-art biopharmaceutical and proteomics analytical technologies may consider the Env samples as an ultimate test for improving the technological aspect of the data usability. Until such tools are implemented, we encourage a systematic manual analysis at the final stage of data reporting.
The data processing method contained the preliminary mass accuracy filters of 15 ppm for the precursor ion, 20 ppm for the ion fragments. For the confirmatory analysis of Env, these mass accuracy limits were further narrowed to 5 ppm for the precursor ion, 15 ppm for the fragment ions via the automatic data filtering. The preliminary mass accuracy range should remain wide enough not to discard potential misassigned outliers, which could subsequently be interpreted correctly in a manual fashion. Similarly, by manual data inspection, a set of other criteria was applied to filter the automatically processed results to ensure the legitimate component assignment. These criteria included the following: (1) matching the retention time to the peptide length and hydrophobicity; (2) the elution of glycosylated peptides as a relatively tight peak cluster; (3) the expected elution pattern for the deamidated peptides (including aspartyl and isoaspartyl residues), with their isotopic pattern being consistent with deamidation. MS/MS spectra of the glycosylated peptides had to contain specific glycan fragment ions characteristic of the glycosidic bond cleavage; the minimal 20% of b/y-ions was expected for the non-glycosylated peptides (the latter was narrowed down via software filtering tools). The combination of all these filtering criteria ensures an unambiguous sequence verification, free of false-positive assignments. Identification of the Env peptides was confirmed by the characteristic ion fragments generated by high-energy MSE fragmentation. For the MS peaks which failed to be assigned by the automatic data processing, a de novo manual assignment was performed using the characteristic ion fragment of the MS/MS spectra. Once the data is generated, the ultimate stage of reporting of legitimate species still required the user evaluation, to avoid the situations described below.
Despite setting tight mass accuracy limits, the glycosylated CT26–27 component of gp41 subunit was assigned as a glycopeptide CT70 of subunit gp120. The masses of CT26–27 and C70 peptide backbones are 1653.844 Da and 1288.706 Da, respectively, so that the mass difference of 365.138 Da between them is close to the mass of the combined sugars Hex + HexNAc (365.132 Da) given the current MS–resolving power. As a result, the CT70 component is wrongfully assigned by the automatic data processing, with an additional N-acetyl lactosamine (GalGlcNAc) in each glycopeptide structures (Table 3). Neither mass accuracy filters nor the retention time or the presence of the characteristic b/ y-ion fragments could prove this assignment wrong. Only the manual inspection of the MS/MS spectrum identified the true component as one of the CT26–27-related glycoforms G2F + Sia (Figure 8a): probing the glycan composition was deemed necessary for an unambiguous structure assignment.
Table 3.
Manual results testing and correction of the automatically processed data. 1CT26–27 component is misassigned due to coincidentally close mass of their glycosylated derivatives. Verification of glycan composition of CT26–27 glycopeptide is presented on Figure 8, demonstrating unambiguous assignment based on the manually confirmed structure. 2Automatically misassigned CK12–13 of gp41 in reality is the overdigested component CK27c4 with internal disulfide bond still intact. Figure 4 demonstrates the actual glycan moiety composition and the glycopeptide mass excluding the carbohydrate component. 3The glycosylation fragment peaks of C15 component are wrongfully assigned as C23n1 complete glycoform. 3The glycosylation fragment peaks of C15 component are wrongfully assigned as C23n1 complete glycoform
| RT, min | Intensity, cts | Mass, Da | Incorrect assignment |
Correct assignment |
||||
|---|---|---|---|---|---|---|---|---|
| Modification | Mass error, ppm | % b/y-ions | Modification | Mass error, ppm | % b/y-ions | |||
| 1. | CVSNITGULTR (CT70 of gp120) | RLDVVQINENQGNR (CT26–27 of gp120) | ||||||
| 26.9 | 18,587 | 3713.594 | G2F + GlcNAc + Gal + Sia | 2.6 | 0 | G2F + Sia | 1.2 | 8 |
| 26.4 | 14,850 | 2895.311 | G1F | 2.4 | 0 | G0F-GIcNAc | 0.7 | 4 |
| 26.0 | 12,859 | 3422.492 | G2F + GlcNAc + Gal | 1.0 | 0 | G2F | −0.5 | 0 |
| 27.3 | 6308 | 3713.595 | G2F + GlcNAc + Gal + Sia | 2.9 | 0 | G2F + Sia | 1.5 | 0 |
| 28.0 | 4303 | 3713.585 | G2F + GlcNAc + Gal + Sia | 0.2 | 0 | G2F + Sia | −1.2 | 0 |
| 2. | DNMTWLQW (CK12–13 of gp41) | RLiNCNTSACTQ (007c4 of gp120 with a disulfide bond) | ||||||
| 20.6 | 11,463 | 2699.073 | G1F | 6.2 | 0 | Man6 | 2.3 | 0 |
| 20.6 | 6052 | 2861.130 | G2F | 7.1 | 0 | Man7 | 3.4 | 0 |
| 20.4 | 4525 | 2699.080 | G1F | 8.7 | 0 | Man6 | 4.8 | 0 |
| 20.4 | 1724 | 2861.122 | G2F | 4.4 | 0 | Man7 | 0.8 | 8 |
| 3. | TGDUGDIRQAHCNVSKATW (C23n1 of gp120) | HLDVVQINENQGNRSNNSNKEY (C15 of gp120) | ||||||
| 26.8 | 7849 | 3628.630 | Glycosylation G0F | 6.0 | 0 | G0F-2GlcNAc | 2.0 | 0 |
| 26.8 | 6228 | 3482.563 | Glycosylation G0 | 3.7 | 0 | G0–2GlcNAc | −2.7 | 5 |
Figure 8.

Verification of glycan composition using MS/MS sequencing of the glycopeptide is necessary to justify the presence of components listed in Table 2. (a) CT26–27 (G2F + Sia) peptide of gp120 subunit (RT 26.9 min) overwrites incorrect automatic assignment of CT70; (b) another gp120 subunit peptide, CK27c4 (Man7), which contains a disulfide bond, overwrites incorrect assignment of CK12–13. Additional illustration of the MS and MS/MS channels are provided in supplementary Fig. S4
Similarly, due to the close values of their masses, the chymotryptic component C15 of gp120 was incorrectly reported as a C23n1 glycoform. The mass difference between these peptide backbones is 406.17 Da, which failed to be differentiated from the mass of 2 GlcNAc (406.16 Da) units. Using mass accuracy criterion alone was not sufficient for identity confirmation; however, the high-energy fragmentation clearly indicated the correct C15 glycopeptide, which contained less common structure G0F-2GlcNAc, rarely a part of a targeted MS search. Such glycoform was not detected in any of other glycosites and is not presented in the overall chart due to its negligible amount; however, its presence affects the overall quality of the glycosylation report.
Another example of the need for the analyst evaluation of the results listed in Table 3 is the case with the mass accuracy limits are extended to 10 ppm. These brackets can be a result of either intentional search for the large, heavily glycosylated species, often resulting in a higher mass error, or a consequence of not taking an advantage of available calibration tools. The range of the mass errors (4.4–8.7 ppm) is slightly higher for the wrong species than for the correctly assigned ones (0.8–4.8 ppm), although it is still within a commonly used acceptance limits in the confirmatory analysis. Upon manual inspection, automatically assigned CK12–13 of subunit gp41 was found to be the overdigested component CK27c4 of gp120 (RLINCNTSACTQ) containing a disulfide bond. This could be an artifact of the sample preparation, yet this example showed how the wrong assignment interferes with reporting of the legitimate components. Figure 8b displays the actual glycan moiety composition with the glycopeptide mass excluding the carbohydrate component. Unless the disulfide bond mapping is included in the automatic data processing workflow, the legitimate component would be either non-assigned or assigned incorrectly. Even though including disulfide bonds in the search is not always a time-saving step, it is recommended to browse through the MS/MS spectra of the top-intensity unassigned species, to perform de novo (glyco)peptide assignment to understand the reason behind their missing identity.
Conclusions
A complementary set of the individual and combined proteolytic digests was applied for the peptide mapping LC-MS/MS analysis of the HIV-1 Env glycoprotein construct BG505.DS.SOSIP.664. After optimization of the sample preparation, which included deglycosylation and denaturation conditions, 99% sequence of Env glycoprotein was confirmed. Minor amount of the uncleaved single chain was detected, confirming the furin cleavage site region at the C-terminus of gp120 subunit. Only 1 Arg or no Arg was detected in the cleavage site of gp120 C-terminus.
For the glycosylation site occupancy study, efficient deglycosylation of the tightly folded trimer glycoprotein was achieved using a set of 3 glycosidases. Deglycosylated and original samples were analyzed in parallel, each glycopeptide was paired up with its deglycosylated analog, the deamidated peptides were identified and assigned as (1) endogenous deamidation (present in the original sample), or (2) result of the deglycosylation; % of deamidated peptides associated with deglycosylation reported relative to the sum of all peptide modifications. Out of total theoretically possible 28 glycan sites of BG505.DS.SOSIP.664, % glycan occupancy was reported for 25 sites, including 3 critical sites (N156, N160, and N332), which are responsible for antibody recognition.
The specific glycan profiles were reported for 20 individual sites; the glycosylation type reported for the other 5 sites: high mannose–type glycans dominated in gp120 subunit, whereas the gp41 subunit was populated with complex glycans. The identified high mannose–type glycans correlated with the expected "intrinsic mannose patch," a target for vaccine design, as the key antibody epitopes.
The designed and optimized peptide mapping approach demonstrated little room for an improvement, which was not due to the lack of the method optimization, but rather defined by structural features of such complex protein as Env. Any alternative data supplying information for all 28 glycan sites should be carefully verified as a potential source of false assignment following the suggested identity confirmation guidelines. Using examples of the peptide and glycan assignment, we demonstrated the need for delivery of the high-quality data that should be confirmed using the set of filters, various criteria, and user judgment.
Supplementary Material
Acknowledgements
The authors would like to acknowledge Kuang-Chuan Cheng, Nicole A. Schneck, Joe Horwitz, and Christopher Barry from the Vaccine Production Program for scientific discussions and reviews.
This work was supported by the intramural research program of the Vaccine Research Center, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s13361-019-02225-3) contains supplementary material, which is available to authorized users.
References
- 1.Sattentau QJ: Envelope glycoprotein trimers as HIV-1 vaccine immunogens. Vaccines (Basel). 28,497–512 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanders RW, Moore JP: Native-like Env trimers as a platform for HIV-1 vaccine design. Immunol. Rev. 275(1), 161–182 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Georgiev IS, Joyce MG, Yang Y, Sastry M, Zhang B, Baxa U, Chen RE, Druz A, Lees CR, Narpala S, Schön A, Van Galen J, Chuang GY, Gorman J, Harned A, Pancera M, Stewart-Jones GB, Cheng C, Freire E, McDermott AB, Mascola JR, Kwong PD: Single-chain soluble BG505.SOSIP gp140 trimers as structural and antigenic mimics of mature closed HIV-1 Env. J. Virol. 89, 5318–5329 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doores KJ: The HIV glycan shield as a target for broadly neutralizing antibodies. FEBS J. 282, 4679–4691 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crispin M, Doores KJ: Targeting host-derived glycans on enveloped viruses for antibody-based vaccine design. Curr. Opin. Virol. 11,63–69 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stewart-Jones GB, Soto C, Lemmin T, Chuang GY, Druz A, Kong R, Thomas PV, Wagh K, Zhou T, Behrens AJ, Bylund T, Choi CW, Davison JR, Georgiev IS, Joyce MG, Kwon YD, Pancera M, Taft J, Yang Y, Zhang B, Shivatare SS, Shivatare VS, Lee CC, Wu CY, Bewley CA, Burton DR, Koff WC, Connors M, Crispin M, Baxa U, Korber BT, Wong CH, Mascola JR, Kwong PD: Trimeric HIV-1-Env structures define glycan shields from clades A, B, and G. Cell. 165,813–826 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Behrens AJ, Vasiljevic S, Pritchard LK, Harvey DJ, Andev RS, Krumm SA, Struwe WB, Cupo A, Kumar A, Zitzmann N, Seabright GE, Kramer HB, Spencer DI, Royle L, Lee JH, Klasse PJ, Burton DR, Wilson IA, Ward AB, Sanders RW, Moore JP, Doores KJ, Crispin M: Composition and antigenic effects of individual glycan sites of a trimeric HIV-1 envelope glycoprotein. Cell Rep. 14, 2695–2706 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao L, Diedrich JK, Kulp DW, Pauthner M, He L, Park SR, Sok D, Su CY, Delahunty CM, Menis S, Andrabi R, Guenaga J, Georgeson E, Kubitz M, Adachi Y, Burton DR, Schief WR, Yates JR, Paulson JC: Global site-specific N-glycosylation analysis of HIV envelope glycoprotein. Nat. Commun. 8,1–13 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Go EP, Hewawasam G, Liao HX, Chen H, Ping LH, Anderson JA, Hua DC, Haynes BF, Desaire H: Characterization of glycosylation profiles of HIV-1 transmitted/founder envelopes by mass spectrometry. J. Virol. 85, 8270–8284 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu X, Borchers C, Bienstock RJ, Tomer KB: Mass spectrometric characterization of the glycosylation pattern of HIV-gp120 expressed in CHO cells. Biochemistry. 39,11194–11204 (2000) [DOI] [PubMed] [Google Scholar]
- 11.Xu Y, Bailey UM, Schulz BL: Automated measurement of site-specific N-glycosylation occupancy with SWATH-MS. Proteomics. 15, 2177–2186 (2015) [DOI] [PubMed] [Google Scholar]
- 12.Zhu Z, Go EP, Desaire H: Absolute quantitation of glycosylation site occupancy using isotopically labeled standards and LC-MS. J. Am. Soc. Mass Spectrom. 25,1012–1017 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moh ES, Lin CH, Thaysen-Andersen M, Packer NH: Site-specific N-glycosylation of recombinant pentameric and hexameric human IgM. J. Am. Soc. Mass Spectrom. 27,1143–1155 (2016) [DOI] [PubMed] [Google Scholar]
- 14.Kwong PD, Mascola JR: Human antibodies that neutralize HIV-1: identification, structures, and B cell ontogenies. Immunity. 37, 412–4125 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwong PD, Doyle ML, Casper DJ, Cicala C, Leavitt SA, Majeed S, Steenbeke TD, Venturi M, Chaiken I, Fung M, Katinger H, Parren PW, Robinson J, Van Ryk D, Wang L, Burton DR, Freire E, Wyatt R, Sodroski J, Hendrickson WA, Arthos J: HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature. 420,678–682 (2002) [DOI] [PubMed] [Google Scholar]
- 16.Sok D, Doores KJ, Briney B, Le KM, Sa KL, Ramos A, Kulp DW, Julien JP, Menis S, Wickramasinghe L, Seaman MS, Schief WR, Wilson IA, Poignard P, Burton DR: Promiscuous glycan site recognition by antibodies to the high-mannose patch of gp120 broadens neutralization of HIV. Sci. Transl. Med. 6, 1–26 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pritchard LK, Vasiljevic S, Ozorowski G, Seabright GE, Cupo A, Ringe R, Kim HJ, Sanders RW, Doores KJ, Burton DR, Wilson IA, Ward AB, Moore JP, Crispin M: Structural constraints determine the glycosylation of HIV-1 envelope trimers. Cell Rep. 11, 1604–1613 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Binley JM, Sanders RW, Master A, Cayanan CS, Wiley CL, Schiffner L, Travis B, Kuhmann S, Burton DR, Hu SL, Olson WC, Moore JP: Enhancing the proteolytic maturation of human immunodeficiency virus type 1 envelope glycoproteins. J. Virol. 76, 2606–2616 (2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cournoyer JJ, Pittman JL, Ivleva VB, Fallows E, Waskell L, Costello CE, O’Connor PB: Deamidation: differentiation of aspartyl from isoaspartyl products in peptides by electron capture dissociation. Protein Sci. 14,452–463 (2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang H, Zubarev RA: Mass spectrometric analysis of asparagine deamidation and aspartate isomerization in polypeptides. Electrophoresis. 11, 1764–1772 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stavenhagen K, Hinneburg H, Thaysen-Andersen M, Hartmann L, Varón Silva D, Fuchser J, Kaspar S, Rapp E, Seeberger PH, Kolarich D: Quantitative mapping of glycoprotein micro-heterogeneity and macro-heterogeneity: an evaluation of mass spectrometry signal strengths using synthetic peptides and glycopeptides. J. Mass Spectrom. 48,627–639 (2013) [DOI] [PubMed] [Google Scholar]
- 22.Joyce MG, Georgiev IS, Yang Y, Druz A, Geng H, Chuang GY, Kwon YD, Pancera M, Rawi R, Sastry M, Stewart-Jones GBE, Zheng A, Zhou T, Choe M, Van Galen JG, Chen RE, Lees CR, Narpala S, Chambers M, Tsybovsky Y, Baxa U, McDermott AB, Mascola JR, Kwong PD: Soluble prefusion closed DS-SOSIP.664-Env trimers of diverse HIV-1 strains. Cell Rep. 21, 2992–3002 (2017) [DOI] [PubMed] [Google Scholar]
- 23.Behrens AJ, Kumar A, Medina-Ramirez M, Cupo A, Marshall K, Cruz Portillo VM, Harvey DJ, Ozorowski G, Zitzmann N, Wilson IA, Ward AB, Struwe WB, Moore JP, Sanders RW, Crispin M: Integrity of glycosylation processing of a glycan-depleted trimeric HIV-1 immunogen targeting key B-cell lineages. J. Proteome Res. 17,987–999 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Go EP, Ding H, Zhang S, Ringe RP, Nicely N, Steinbock RT, Golabek M, Alin J, Alam SM, Cupo A, Haynes BF, Kappes JC, Moore JP, Sodroski JG, Desaire H: Glycosylation benchmark profile for HIV-1 envelope glycoprotein production basedonelevenEnvtrimers.J.Virol.91,1–19 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Panico M, Bouché L, Binet D, O’Connor MJ, Rahman D, Pang PC, Canis K, North SJ, Desrosiers RC, Chertova E, Keele BF, Bess JW Jr., Lifson JD, Haslam SM, Dell A, Morris HR: Mapping the complete glycoproteome of virion-derived HIV-1 gp120 provides insights into broadly neutralizing antibody binding. Sci. Rep. 6(32956), 1–17 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.