

Abstract
Background
Lynch syndrome, the most common colorectal cancer (CRC) syndrome, is caused by germline mismatch repair (MMR) genes. Precise estimates of age-specific risks are crucial for sound counseling of individuals managing a genetic predisposition to cancer, but published risk estimates vary. The objective of this work is to provide gene-, sex-, and age-specific risk estimates of CRC for MMR mutation carriers that comprehensively reflect the best available data.
Methods
We conducted a meta-analysis to combine risk information from multiple studies on Lynch syndrome–associated CRC. We used a likelihood-based approach to integrate reported measures of CRC risk and deconvolved aggregated information to estimate gene- and sex-specific risk.
Results
Our comprehensive search identified 10 studies (8 on MLH1, 9 on MSH2, and 3 on MSH6). We estimated the cumulative risk of CRC by age and sex in heterozygous mutation carriers. At age 70 years, for male and female carriers, respectively, risks for MLH1 were 43.9% (95% confidence interval [CI] = 39.6% to 46.6%) and 37.3% (95% CI = 32.2% to 40.2%), for MSH2 were 53.9% (95% CI = 49.0% to 56.3%) and 38.6% (95% CI = 34.1% to 42.0%), and for MSH6 were 12.0% (95% CI = 2.4% to 24.6%) and 12.3% (95% CI = 3.5% to 23.2%).
Conclusions
Our results provide up-to-date and comprehensive age-specific CRC risk estimates for counseling and risk prediction tools. These will have a direct clinical impact by improving prevention and management strategies for both individuals who are MMR mutation carriers and those considering testing.
Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer (CRC) syndrome, accounts for approximately 3.0%–5.0% of all CRCs and is an autosomal dominant condition caused by germline pathogenic variants in mismatch repair (MMR) genes (1,2). Carriers of pathogenic variants in any of the MMR genes MLH1, MSH2, MSH6, PMS2, or EPCAM have an increased risk of developing several types of cancers, including colorectal, endometrial, stomach, small bowel, and biliary tract cancers (3). Lynch syndrome is generally identified following investigation of familial aggregation of multiple and/or early-onset cancers based on the Amsterdam II criteria, National Comprehensive Cancer Network guidelines, Bethesda guidelines (3–5) or more quantitative risk assessment (6). More recently, it is also being found incidentally through panel genetic testing and by microsatellite instability or immunohistochemistry testing of all CRCs. In addition, Hampel et al. (7) have recently called for sequencing of all CRC.
Carriers of pathogenic variants in MMR genes can benefit from reliable information about their cancer risk to better inform effective management and targeted surveillance strategies. Published estimates of penetrance (age-specific risk of cancer for carriers) vary. Studies typically provide different measures of CRC risk, including cumulative penetrance, relative risks, or standardized incidence ratios from family-based studies, and odds ratios from case-control studies.
The objective of this work is to combine results from published studies to provide more accurate age- and sex-specific penetrance estimates of MLH1, MSH2, and MSH6 on CRC for individuals with Lynch syndrome. Cumulative lifetime penetrance estimates of CRC range from 30.0% to 74.0% for MLH1 and MSH2 gene mutation carriers and from 10.0% to 22.0% for MSH6 mutation carriers (8). Variation in published estimates could arise from differences in study designs, selection criteria for molecular testing, and statistical adjustments for ascertainment (9). Without adjustment, estimated lifetime risk in studies of high-risk families can be higher than that estimated from population-based studies. In sensitivity analyses, studies have shown that different ascertainment schemes can lead to inconsistent risk estimates (10,11). To address these concerns, we explicitly considered properly adjusting for ascertainment as an inclusion criterion for our meta-analysis. Previous meta-analyses of CRC risk in individuals with Lynch syndrome were based on studies that report gene- and sex-specific cumulative penetrance estimates (12,13). This excludes additional published risk measures from studies that provide aggregated information across sex and genes. In our analysis, we did not make these exclusions, because they may miss important information and may lead to bias.
Methods
Literature Search
We performed 3 separate PubMed searches for MLH1, MSH2, and MSH6, with the following queries: MLH1 or colorectal: (“MutL Protein Homolog 1”[Mesh] OR “MLH1”[TIAB] OR “Lynch syndrome”[TIAB]) AND (”Risk”[Mesh] OR ”Risk”[TI] OR “Penetrance”[TIAB] OR “Hazard ratio”[TIAB]) AND (“Colorectal Neoplasms”[Mesh] OR “Colorectal Neoplasms, Hereditary Nonpolyposis”[Mesh] OR “colorectal cancer”[TIAB]); MSH2 or colorectal: (“MutS Homolog 2 Protein”[Mesh] OR “MSH2”[TIAB] OR “Lynch syndrome”[TIAB]) AND (“Risk”[Mesh] OR “Risk”[TI] OR “Penetrance”[TIAB] OR “Hazard ratio”[TIAB]) AND (“Colorectal Neoplasms”[Mesh] OR “Colorectal Neoplasms, Hereditary Nonpolyposis”[Mesh] OR “colorectal cancer”[TIAB]), MSH6 or colorectal: (“G-T mismatch-binding protein” [Supplementary Concept] OR “MSH6”[TIAB]) AND (”Risk”[Mesh] OR “Risk”[TI] OR “Penetrance”[TIAB] OR “Hazard ratio”[TIAB]) AND (“Colorectal Neoplasms”[Mesh] OR “Colorectal Neoplasms, Hereditary Nonpolyposis”[Mesh] OR “colorectal cancer”[TIAB]). We performed a similar search in EMBASE with the following query: (“MutL protein homolog 1”/exp OR “DNA mismatch repair protein MSH2”/exp OR “protein MutS”/exp OR MLH1: ab, ti OR MSH2: ab, ti OR MSH6: ab, ti OR Lynch: ab, ti) AND (“rectum tumor”/exp OR “colon tumor”/exp OR [(colon OR rectal OR rectum OR colorectal) NEAR/3 (cancer* OR neoplasm* OR carcinoma* OR tumor* OR tumour*)]:ab, ti)AND(“risk”/exp OR risk*:ab, ti OR penetrance: ab, ti OR “hazard ratio”:ab, ti).
References from relevant articles and previous meta-analyses were reviewed to identify additional studies not captured by the PubMed or EMBASE searches. In selecting articles from those found by the query, we required the following inclusion criteria: studies must report risk (and corresponding 95% confidence interval) of CRC for carriers of germline mutations in MLH1, MSH2, or MSH6; adjust for ascertainment if cohort is not population based or design is not case control; and include nonoverlapping participants with other studies (Figure 1). We excluded studies that focus on patients with polymorphisms and/or CRC as a secondary cancer. We chose not to include the PMS2 gene, though it is also involved in mismatch repair and associated with Lynch syndrome. In a PubMed literature search similar to that performed for our main analysis (for MLH1, MSH2, and MSH6), 3 studies reported the risk of CRC for PMS2 mutation carriers (14–16), and only 1 of these provided disaggregated data for PMS2 (15). PMS2 carriers generally have a later age of onset than their MLH1 or MSH2 counterparts, resulting in lower numbers of events for comparable observation years. Moreover, the low sensitivity of clinical criteria and less widespread diagnostic testing for identifying PMS2 carriers (17,18) make it challenging to extend our meta-analysis to PMS2 at the present time.
Figure 1.

Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) flow diagram of the literature review for our meta-analysis. EMBASE = Excerpta Medica dataBASE; NLP = natural language processing; CRC = colorectal cancer; MMR = mismatch repair.
Studies were first assessed based on title and abstract using a natural language processing algorithm (19). This algorithm uses a support vector machine, which learns a linear decision rule based on the bag-of-ngrams representation of each title and abstract. At least 2 reviewers independently examined the study abstracts, and those deemed relevant underwent full text review. For studies that remained relevant after full text review, we extracted the following information: first author’s last name, year of publication, study population, ascertainment method, number of events, number of carriers, gene type, and relevant risk estimates with corresponding confidence intervals.
Statistical Analysis
Common approaches for combining evidence across multiple studies include fixed effects models, which assume an underlying true effect size for all included studies, and random effects models, which allow for the true effect size to vary from study to study. Typically, these approaches cannot be used directly to combine heterogeneous measures of CRC risk that result from different study designs. Marabelli et al. (20) developed a likelihood-based method allowing meta-analytic integration of different types of cancer risk estimates (eg, penetrance, relative risk, standardized incidence ratio, and odds ratio). This method, however, does not address the challenge of combining studies that report gene-aggregated (a combination of 2 or more MMR genes) or sex-aggregated cancer risks, which are common in the Lynch syndrome literature. The deconvolution of aggregated risk information is crucial for personalized prevention, because male and/or MLH1 or MSH2 mutation carriers typically have higher risks of CRC than their female and/or MSH6 counterparts (10,21–25). In this work, we used a more general likelihood-based approach that allows the integration of aggregated cancer risks to provide accurate age-, gene-, and sex-specific penetrance of CRC for MMR mutations carriers. As a preliminary step, we used the Q2 and I2 values to explore between-study heterogeneity. A P value of less than .05 was considered representative of statistically significant heterogeneity. All tests were 2-sided and performed using the meta (26) package in R (version 3.3) (27). To investigate potential publication bias, we created funnel plots and used a 2-sided Egger (28) test to assess asymmetry. We then conducted our meta-analyses based on 2 complementary approaches. In the first approach we used the DerSimonian and Laird random effects model (29) (see details in Supplementary Material) to perform separate meta-analyses of cumulative risk by decade of age. We assumed the underlying penetrances are heterogeneous, with between-study variance captured by the Δ2 parameter in (29). The DerSimonian and Laird random effects model does not provide a way to handle aggregated estimates and does not lend itself to extrapolation of estimates to older ages, as required in genetic counseling and decision support tools.
To address these issues, in the second approach we used a likelihood-based approach to obtain penetrance estimates by yearly age. This approach extends the method of Marabelli et al. (20), which allows the meta-analytic integration of different risk measures into age-specific penetrance curves and is described in detail in the Supplementary Material. Briefly, we modeled the penetrance in mutation carriers as a probability distribution function characterized by 2 parameters. We specified the likelihood terms based on the study design and the risk estimates reported and estimated the parameters by maximizing the likelihood. Penetrance was assumed to follow a log-logistic distribution. The log-logistic distribution was chosen because, among the commonly used parametric distributions, it was the most similar to penetrance curves reported in the literature (21,30) and to the trend indicated by the meta-analytic results of the DerSimonian and Laird random effects model the first approach. Parameter estimates based on the log-logistic distribution are provided in the Supplementary Material. In addition, we conducted leave-1-study-out sensitivity analyses to better understand the sources of heterogeneity. We used the meta (26) and stats4 (27) packages in R to perform the DerSimonian and Laird random effects model analysis and the maximum likelihood estimation for the likelihood-based approach, respectively.
We extended the Marabelli et al. (20) method to incorporate studies that provide aggregated risk information. For studies that report sex-aggregated risk, we modeled the penetrance function as a weighted average of the male- and female-specific penetrance functions, which can be estimated separately as long as we have at least some studies that provide sex-specific risk. Weights correspond to the proportion of male or female carriers in the study. Similarly, for studies that report gene-aggregated risk, we modeled the penetrance as a weighted average based on the proportion of different carriers in the study. By allowing studies that report aggregated risk estimates to borrow information from those that report gene- or sex-specific risk estimates, this likelihood-based method combines both direct (gene- or sex-specific) and indirect (aggregated) evidence from the literature to provide comprehensive risk estimates of CRC.
Studies typically report risk estimates for carriers who are younger than 80. Penetrance estimates from 81 to 110 years of age were obtained by multiplying the risk of noncarriers at each age by the risk ratio comparing the risk of carriers with that of noncarriers at age 80 years (relative risk):
We obtained the risk of CRC for noncarriers from the Surveillance, Epidemiology, and End Results Program database (SEER) (31), which provides the combined risk of CRC for carriers and noncarriers. As mutations are sufficiently rare, we assume that the general population risk provided by the SEER database approximates the CRC risk for noncarriers (32).
Results
Overall, our searches resulted in 4759 abstracts as of March 8, 2019. Among the 4759 abstracts, 586 were deemed relevant by the natural language processing algorithm. After human review, 576 were excluded because of the following criteria: unclear or inappropriate ascertainment adjustment (n = 23), not relevant for MMR or CRC (n = 129), overlap with included studies (n = 16), reports penetrance modified by other risk factors (n = 10), second cancer (n = 50), missing full text (n = 7), nonpathogenicity (n = 2), polymorphisms (n = 74), and not relevant for penetrance (n = 265). For our final meta-analysis, we included 10 studies (Figure 1). Table 1 shows a synopsis of the included studies along with a description of the study design, ascertainment mechanism, and risk estimation methods. Studies vary in terms of population, ascertainment, and design. Among the studies, 1 reported aggregated risk for sex, 3 reported aggregated risk for the MMR genes, and 1 reported both sex- and gene-aggregated risk. Eight studies reported risk for MLH1 carriers, 9 reported risk for MSH2 carriers, and 3 reported risk for MSH6 carriers. To quantify the between-study variation, we performed tests of heterogeneity and calculated the corresponding I2 values. With 3 genes, 6 age intervals (age 30, 40, …, 80), and 2 sexes, a total of 36 tests were performed. For MLH1, the P values were less than .001 at age 40-70 years for both sexes. The corresponding I2 values ranged from 83.3% (68.5% to 91.1%) to 90.3% (83.2% to 94.4%) for males and from 78.0% (56.6% to 88.8%) to 86.6% (75.7% to 92.6%) for females. The P values at age 80 years for males and females, respectively, were .005 (I2 = 83.0% 56.5% to 93.3%) and .002 (I2 = 84.8% 62.0% to 93.9%). For MSH2 male carriers, the P values were less than .0001 at all age intervals with corresponding I2 values ranging from 89.2% (81.7% to 93.6%) to 94.1% (89.8% to 96.6%). For MSH2 female carriers, the P value was .04 (I2 = 35.1% 0.0% to 70.1%) at age 50 years and less than .0001 at age 60 years (I2 = 76.0% 54.0% to 87.5%) and 70 years (I2 = 77.4% 57.1% to 88.1%). For MSH6, the only statistically significant P value at the .05 level was that of female mutation carriers at age 70 years (P = .04, I2 = 68.4% 0.0% to 90.8%). Overall, there is evidence for heterogeneity in the risk estimates across the decades for MLH1 and MSH2 mutation carriers but less so for MSH6. Results from tests of asymmetry in the funnel plots suggest there is little evidence of publication bias. Details on publication bias assessment can be found in the Supplementary Material.
Table 1.
Summary of studies included in our meta-analysisa
| Study | Population | Ascertainment | Estimation | No. of events | No. of carriers | Gene(s) | Condition for unbiasedness |
|---|---|---|---|---|---|---|---|
| Aaltonen, 2007 (36) | Regional hospitals, Finland | FD relatives of CRC cases | Kaplan-Meier analysis where relatives were censored at ascertainment, emigration, or last contact with proband | 91 | 242 | MLH1, MSH2 | No additional familial aggregation other than MLH1/MSH2 |
| Bonadona, 2011 (24) | ERISCAM study France | Relatives of CRC cases identified from cancer genetics clinics and mutated for MMR genes | Genotype restricted likelihood conditioning on phenotypes of all relatives and genotype of proband | 768 | 1633 | MLH1, MSH2, MSH6 | No additional familial aggregation other than MLH1/MSH2/MSH6 |
| Borras, 2010 (33) | Genetic counseling clinic Spain | Relatives of CRC cases with MMR mutation | Modified segregation analysis conditioning on genotype and phenotype of proband and phenotype of all relatives | 28 | 180 | MLH1 | No additional familial aggregation other than MLH1 |
| Dowty, 2013 (35) | CCFR | FD and SD, or all relatives of cases with MMR mutation, for population- and clinic-based families, respectively | Modified segregation analysis conditioning on genotype and phenotype of proband and phenotypes of all relatives, for population and clinic-based families, respectively | 1112 | 2253 | MLH1, MSH2 | No additional familial aggregation other than MLH1/MSH2 |
| Dunlop, 1997 (38) | SNCR, Scotland | Relatives of early-onset CRC cases identified from population-based registries and mutated for MMR genes | Kaplan-Meier analysis excluding probands | 25 | 67 | MLH1, MSH2 | No effect from size-based sampling, or risks to patient carrier cases and relatives are no higher than carrier nonpatient cases |
| Kopciuk, 2009 (30) | Medical Genetics Clinic Canada | Multiple-case families with MMR mutation | Modified segregation analysis conditioning on phenotypes of all FDR | 101 | 145 | MSH2 | No additional familial aggregation other than MSH2 |
| Moller, 2017 (18) | Prospective multi center database by Europe Majorica group | Mutation carriers with increased risk of CRC identified by each center | Cumulative incidence rate excluding individuals with prior cancer | 711 | 1942 | MLH1, MSH2, MSH6 | None |
| Mukherjee, 2011 (11) | MECC, CHS | All participants, or carrier families with history of LS, identified from population study and cancer clinics, respectively | Modified segregation analysis conditioning on genotype and phenotype of proband or on genotype and phenotype of proband and phenotype of affected FD relatives | 74 | 88 | MSH2 | No additional familial aggregation other than MSH2 |
| Quehenberger, 2005 (37) | Dutch HNPCC family registry | Multiple-case families with MMR mutation | Modified segregation analysis conditioning on observed phenotypes and on event that at least 1 case in family was a carrier | 104 | 397 | MLH1, MSH2 | No additional familial aggregation other than MLH1/MSH2 |
| Stoffel, 2009 (10) | DFCI, U Michigan | Multiple-case families with MMR mutation | Modified segregation analysis conditioning on genotype and phenotype of proband and phenotype of all relatives | 99 | 307 | MLH1, MSH2, MSH6 | No additional familial aggregation other than MLH1/MSH2/MSH6 |
CRC = colorectal cancer; FD = first degree; SD = second degree; MMR = mismatch repair; ERISCAM = Estimation des Risques de Cancer chez les porteurs de mutation des gènes MMR; CCFR = Colon Cancer Family Registry; SNCR = Scottish National Cancer Registry; MECC = Molecular Epidemiology of Colorectal Cancer; CHS = Clalit Health Services; HNPCC = hereditary nonpolyposis colorectal cancer; DFCI = Dana-Farber Cancer Institute.
Next, we examined sources of heterogeneity from various aspects of study characteristics. This between-study heterogeneity could arise from differences in study design, mutation type, study population, and estimation strategy. Among the 10 included studies, Moller et al. (18) was the only study that conducted a prospective cohort analysis, whereas the rest focused on retrospective cohorts. Regarding mutation type, Borras et al. (33), Kopciuk et al. (30), and Mukherjee et al. (11) are studies that exclusively focused on founder mutations. All other studies included carriers of mixed mutation types, so it was not feasible to separate the effects of mutations from these studies at the present time. As a result, the findings from our meta-analysis represent the average risk among a group of carriers with a representative mix of mutations. Regarding study populations, it is likely that different populations may segregate different mutations. Though there are studies containing more than 1 subpopulation (18,34,35), they provide limited evidence of population-specific variation in penetrance. As shown in Table 1, each study used an analysis method that addressed an ascertainment mechanism in its design. Studies that were not population based (10,11,24,30,33,35–37) typically used estimation strategies that condition on information of the phenotype or genotype of included individuals to adjust for ascertainment.
Figure 2 shows the following: the means and 95% confidence intervals of the meta-analytic penetrances at each 10-year age interval that were estimated using the DerSimonian and Laird method, and the smoothed curves obtained from the likelihood-based approach that represent our final estimates by yearly age. The estimated cumulative penetrance by age 70 years from both approaches is displayed in Table 2 by sex and gene. Using the likelihood-based approach, the penetrances by age 70 years were estimated for males and females, respectively, to be 43.9% (95% CI = 39.6% to 46.6%) and 37.3% (95% CI = 32.2% to 40.2%) for MLH1 carriers, 53.9% (95% CI = 49.0% to 56.3%) and 38.6% (95% CI = 34.1% to 42.0%) for MSH2 carriers, and 12.0% (95% CI = 2.4% to 24.6%) and 12.3% (95% CI = 3.5% to 23.2%) for MSH6 carriers. In general, male carriers of MLH1 and MSH2 have a higher risk of developing CRC compared with their female counterparts. Estimates of MSH6 penetrance on CRC shows increased variability (wider confidence intervals) due to smaller sample sizes. Visual comparison of the confidence intervals within each 10-year age interval indicates overlap across studies for all 3 genes. Because all studies reported cumulative penetrance, we were able to include the same studies (8 on MLH1, 9 on MSH2, and 3 on MSH6) for both the DerSimonian and Laird and the likelihood-based approaches. In addition to Figure 2, Supplementary Figure 1 (available online) shows the study-specific penetrance estimates and 95% confidence intervals by decade of age.
Figure 2.
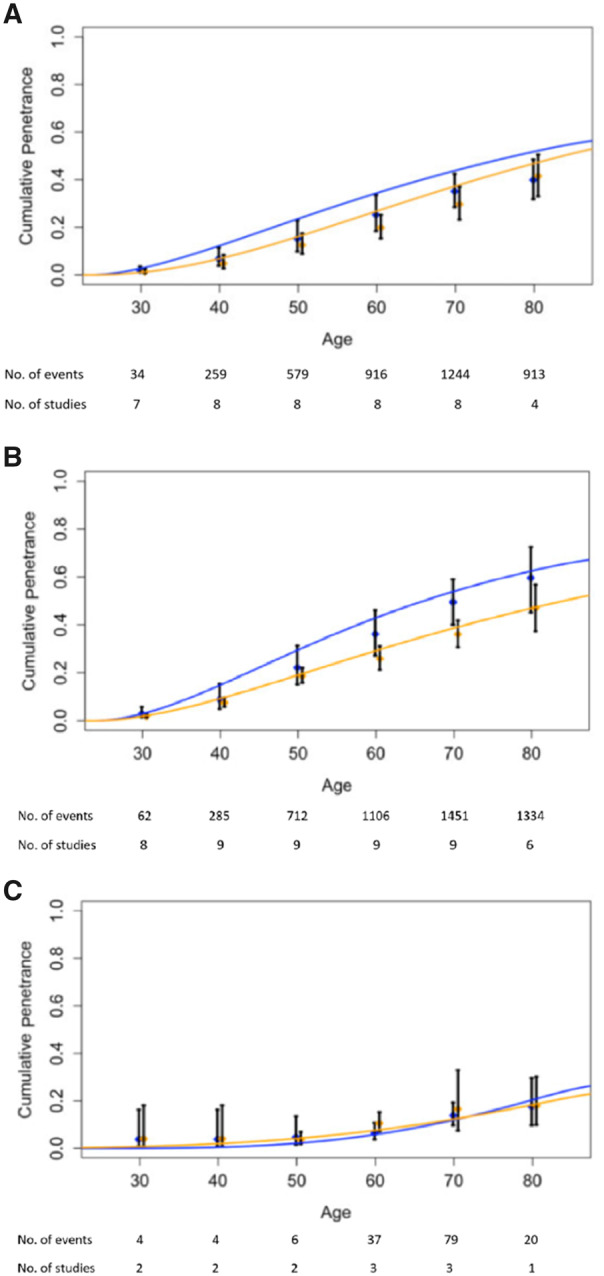
Age-specific colorectal cancer risk for mismatch repair gene mutation carriers. Panels A, B, and C correspond to MLH1, MSH2, and MSH6 mutation carriers, respectively. DerSimonian and Laird random effects model results: the age range is divided into 10-year intervals. Within each we show the meta-analytic estimate from the DerSimonian and Laird random effects model (thick vertical black bars). The height of vertical bars represents 95% confidence intervals. Likelihood-based approach results: Smooth blue and orange lines represent penetrance estimated from the likelihood-based approach by yearly age. Blue corresponds to male carriers, and orange corresponds to female carriers.
Table 2.
Estimated cumulative penetrance by age 70 years of CRC for MLH1, MSH2, and MSH6 mutation carriers by sex and screening statusa
| Sex | Gene | Method | Study population | Cum. penetrance (%) with 95% CI |
|---|---|---|---|---|
| Male | MLH1 | DerSimonian and Laird | All | 35.1 (28.5 to 42.4) |
| Unscreened | 36.5 (26.6 to 46.7) | |||
| Unspecified | 34.5 (22.6 to 48.7) | |||
| Likelihood-based | All | 43.9 (39.6 to 46.6) | ||
| Unscreened | 35.3 (29.4 to 40.0) | |||
| Unspecified | 49.7 (43.3 to 54.2) | |||
| MSH2 | DerSimonian and Laird | All | 50.0 (40.3 to 59.6) | |
| Unscreened | 51.8 (36.4 to 66.9) | |||
| Unspecified | 47.3 (35.7 to 59.1) | |||
| Likelihood-based | All | 53.9 (49.0 to 56.3) | ||
| Unscreened | 53.2 (47.1 to 57.4) | |||
| Unspecified | 57.0 (49.2 to 62.3) | |||
| MSH6 | DerSimonian and Laird | All | 13.8 (9.7 to 19.3) | |
| Unscreened | 14.0 (7.2 to 25.6) | |||
| Unspecified | 13.7 (9.0 to 20.3) | |||
| Likelihood-based | All | 12.0 (2.4 to 24.6) | ||
| Unscreened | 19.2 (5.1 to 32.8) | |||
| Unspecified | 13.2 (0.6 to 76.2) | |||
| Female | MLH1 | DerSimonian and Laird | All | 29.7 (23.2 to 37.1) |
| Unscreened | 31.8 (24.4 to 40.2) | |||
| Unspecified | 27.4 (15.2 to 44.2) | |||
| Likelihood-based | All | 37.3 (32.2 to 40.2) | ||
| Unscreened | 34.0 (27.1 to 39.4) | |||
| Unspecified | 36.7 (29.6, 42.4) | |||
| MSH2 | DerSimonian and Laird | All | 36.0 (30.6 to 41.8) | |
| Unscreened | 34.6 (26.9 to 43.2) | |||
| Unspecified | 37.5 (28.8 to 47.2) | |||
| Likelihood-based | All | 38.6 (34.1 to 42.0) | ||
| Unscreened | 37.3 (32.9 to 40.6) | |||
| Unspecified | 41.0 (34.4 to 46.3) | |||
| MSH6 | DerSimonian and Laird | All | 16.6 (7.4 to 32.9) | |
| Unscreened | 10.7 (4.9 to 21.9) | |||
| Unspecified | 22.3 (10.5 to 41.2) | |||
| Likelihood-based | All | 12.3 (3.5 to 23.2) | ||
| Unscreened | 5.3 (0.002 to 16.5) | |||
| Unspecified | 29.6 (2.5 to 79.5) |
CI = confidence interval; CRC = colorectal cancer.
Among the 10 studies, 4 focused on individuals who were not screened or had not had prior surgery by censoring participants at the age of colonoscopy screening or prophylactic surgery (24,30,35,37). For the remainder of the studies, it was unclear whether screened individuals were included. Although screening and surgery were not part of the recruitment criteria, it is reasonable to assume that a number of participants from these 6 studies (10,11,18,33,36,38) may have undergone screening or surgery according to current screening recommendations (39). We divided the studies into 2 groups: studies that focused on unscreened populations (24,30,35,37) and studies that did not provide details on screening and therefore were assumed to be a mix of screened and unscreened populations (10,11,18,33,36,38). Figure 3 shows the cumulative penetrance of CRC for MLH1, MSH2, and MSH6 mutation carriers after stratifying studies by screening status. Estimated cumulative penetrance by age 70 years from both the DerSimonian and Laird and likelihood-based approaches is displayed in Table 2 by sex, gene, and screening status. For the 4 studies that included unscreened participants, the penetrance by age 70 years was estimated for males and females, respectively, to be 35.3% (95% CI = 29.4% to 40.0%) and 34.0% (95% CI = 27.1% to 39.4%) for MLH1 carriers, 53.2% (95% CI = 47.1% to 57.4%) and 37.3% (95% CI = 32.9% to 40.6%) for MSH2 carriers, and 19.2% (95% CI = 5.1% to 32.8%) and 5.3% (95% CI = 0.002% to 16.5%) for MSH6 carriers. For the 6 studies that potentially included both screened and unscreened participants (unspecified), the penetrance by age 70 years was estimated for males and females, respectively, to be 49.7% (95% CI = 43.3% to 54.2%) and 36.7% (95% CI = 29.6% to 42.4%) for MLH1 carriers, 57.0% (95% CI = 49.2% to 62.3%) and 41.0% (95% CI = 34.4% to 46.3%) for MSH2 carriers, and 13.2% (95% CI = 0.6% to 76.2%) and 29.6% (95% CI = 2.5% to 79.5%) for MSH6 carriers (Figure 4). Studies on unscreened populations report lower cumulative risk for MLH1 and female MSH6 mutation carriers compared with studies on both screened and unscreened populations. However, the converse is true for male MSH6 mutation carriers. Among the MSH6 studies that report CRC risk in both screened and unscreened populations, Stoffel et al. (10) made conservative ascertainment adjustments, which could lead to lower risk estimates. Although differences in CRC risk between the cohorts appear to be more pronounced for MSH6 mutation carriers, this could be attributed to the lack of studies in the unscreened group at age 80 years. Overall, there is considerable overlap in the 95% confidence intervals across all 3 genes and both sexes, indicating insufficient evidence to substantiate differences in CRC risk between unspecified (likely a mix of screened and unscreened) and unscreened populations. In addition to Figure 3, Supplementary Figure 2 (available online) shows the study-specific estimates and 95% confidence intervals by decade of age.
Figure 3.

Colorectal cancer risk stratified by studies on unscreened or no prior surgery population (top) or unspecified (ie, likely a mix of screened and unscreened populations) (bottom). Panels A, B, and C correspond to MLH1, MSH2, and MSH6 mutation carriers, respectively. DerSimonian and Laird random effects model results: The age range is divided into 10-year intervals. Within each we show the meta-analytic estimate from the DerSimonian and Laird random effects model (thick vertical black bars). The height of vertical bars represents 95% confidence intervals. Likelihood-based approach results: Smooth blue and orange lines represent penetrance estimated from the likelihood-based approach by yearly age. Blue corresponds to male carriers, and orange corresponds to female carriers.
Figure 4.
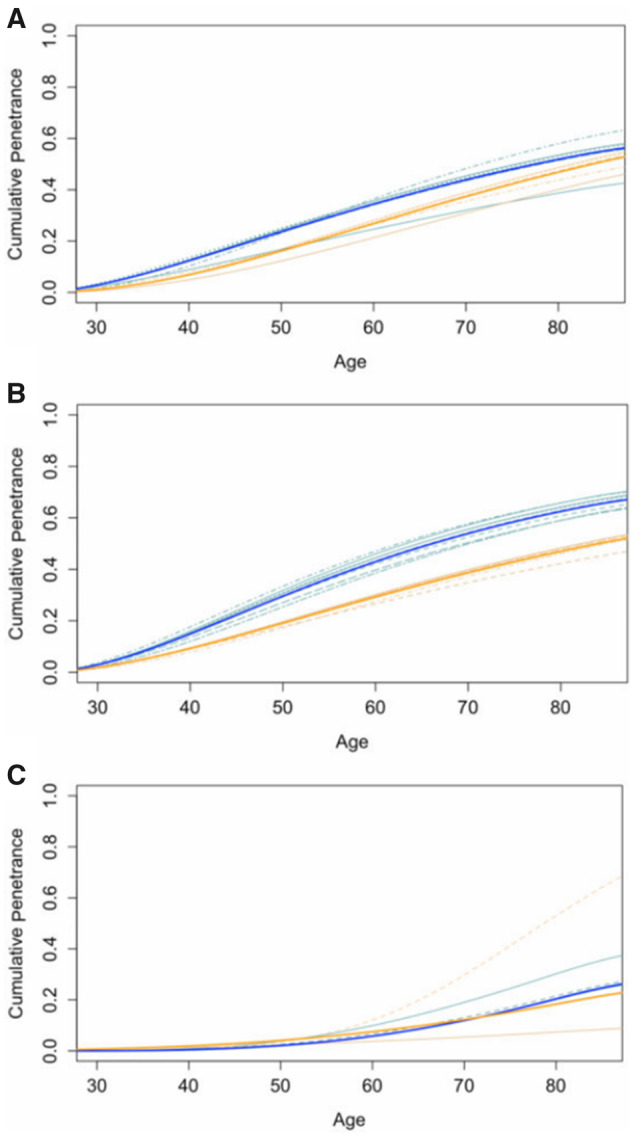
Leave-1-study-out sensitivity analysis for mutation carriers. Panels A, B, and C correspond to MLH1, MSH2, and MSH6 mutation carriers, respectively. Bold solid lines: Cumulative penetrance estimates of CRC based on our likelihood-based approach. Dashed lines: Cumulative penetrance estimates by yearly age of CRC from leave-1-study-out tests of sensitivity. Blue corresponds to male carriers, and orange corresponds to female carriers. Visually, small deviations of a dashed line from the solid line suggest our meta-analysis is robust to the removal of that study.
Next, we conducted sensitivity analysis by design or analysis strategy, study population, and mutation type. Mukherjee et al. (11) focused on founder mutations in MSH2 for individuals of Ashkenazi Jewish descent. Because previous evidence shows there is an increased risk of CRC in Ashkenazi Jews (40), we conducted our meta-analysis with and without this study. Removal of Mukherjee et al. had little effect on the combined penetrance estimates for MSH2 mutation carriers. Similarly, we conducted a systematic leave-1-study-out sensitivity analysis and concluded that the meta-analytic results of MLH1 and MSH2 mutation carriers are quite robust to leave-1-study-out sensitivity analysis (Figure 4). Estimated penetrance for female MSH6 mutation carriers is sensitive to the removal of studies by Bonadona et al. (24) and Moller et al. (18). Penetrance for male MSH6 mutation carriers is sensitive to the removal of Moller et al. (18). Because these 2 studies were weighted more heavily in the analysis because of their sample sizes, it is not surprising that removing one would affect the risk estimates. This variation in penetrance estimates for MSH6 carriers can be attributed to the smaller sample size (both in number of included studies and in number of mutation carriers) compared with their MLH1 or MSH2 counterparts. Moreover, because MSH6 mutation carriers tend to have a later age of onset, the risk information reported by studies was limited to age 50 years and older. Among the 3 studies that reported sex-specific risk for MSH6 mutation carriers, 2 studies indicated that female risks were associated with more variability than male risks (10,18), resulting in more variable maximum likelihood estimates for the female carriers. Overall, the meta-analytic risk estimates for MLH1 or MSH2 carriers were robust to the removal of studies, whereas the estimates for MSH6 were more easily affected because of the smaller number of available studies.
Discussion
We performed a systematic review of the risk of CRC in mutation carriers of MLH1, MSH2, and MSH6 and combined evidence from 10 studies to provide age-, gene-, and sex-specific risk estimates. These comprehensively reflect the best available data. We conclude that the lifetime cumulative penetrance to age 70 years of CRC for males and female carriers, respectively, is 43.9% (95% CI = 39.6% to 46.6%) and 37.3% (95% CI = 32.2% to 40.2%) for MLH1 carriers, 53.9% (95% CI = 49.0% to 56.3%) and 38.6% (95% CI = 34.1% to 42.0%) for MSH2 carriers, and 12.0% (95% CI = 2.4% to 24.6%) and 12.3% (95% CI = 3.5% to 23.2%) for MSH6 carriers. The smaller number of MSH6 mutation carriers in our analysis led to less certain estimates for that gene, especially at younger ages. Interestingly, more recent studies tend to have narrower confidence intervals, suggesting increased precision in their penetrance estimates. Although more conservative ascertainment adjustment mechanisms in recent studies are at play, it is difficult to establish whether those may affect the study estimates or the confidence intervals. The narrower confidence intervals may be attributed to carrier sample size, because recent studies including Bonadona et al. (24), Dowty et al. (35), and Moller et al. (18) have the 3 largest carrier sample sizes among the included studies.
The differences in the penetrance estimates between the DerSimonian and Laird random effects model and our likelihood-based approach could be attributed to the parametric assumption of the likelihood-based approach. Overall, because the majority of the likelihood-based estimates fall within the meta-analytic 95% confidence interval of the random effects model, we conclude that our findings are likely to be robust to the choice of statistical approach.
To the best of our knowledge, this meta-analysis is the first to provide age-, gene-, and sex-specific penetrance estimates of MLH1, MSH2, and MSH6 mutations for CRC. A previous meta-analysis by Jenkins et al. (13) focused on combining evidence from 4 articles that report gene- and sex-specific penetrance for MLH1 and MSH2 mutation carriers to provide short-term (5 years) CRC risk. Although there is some overlap in included studies, the risk estimates provided by our meta-analysis are age specific, are based on several more studies, and include MSH6 mutation carriers.
A strength of the likelihood-based approach used here lies in its ability to deconvolve aggregated risks, allowing us to use all of the information available in the literature and provide more comprehensive penetrance estimates. Of note, our meta-analysis included only studies that made adjustments for ascertainment if the participants were recruited through high-risk families, so reported risk estimates were less likely to be biased upward. At the same time, many studies were excluded as a result. Our method can be applied in the future to address other Lynch syndrome genes and cancers, such as PMS2, EPCAM, endometrial cancer, and more generally to other gene and cancer combinations with no restriction on the mutation type as long as there are enough studies. A potential limitation of this approach is the use of a parametric distribution to model the penetrance; this assumption, although difficult to check, can be relaxed with richer data. For example, a leave-1-study-out sensitivity analysis can be used to assess the parametric modeling choice. Currently, our meta-analysis included only articles that reported cumulative penetrance. Extensions of our deconvolution method could potentially be designed to include studies that report other risk measures (eg, odds ratio, hazard ratio, etc). Regarding systematic sources of study heterogeneity, our meta-analysis included studies of mixed mutation types and populations. Although ideally one would desire to assess mutation- or population-specific variation in penetrance, the present information is insufficient, and it is not feasible to separate these effects. Overall, the meta-analytic results for MLH1 and MSH2 mutation carriers are robust according to the sensitivity analysis and show little evidence of publication bias. On the other hand, the same cannot be said for MSH6 mutation carriers due to the small number of studies.
It is well known that colonoscopic surveillance serves as an effective prevention strategy for individuals managing their CRC risk (41). Our results show that cancer penetrance estimated from populations that are a mix of unscreened and screened individuals is similar to that estimated from unscreened populations for MSH2 mutation carriers. However, the former is higher for MLH1 and female MSH6 mutation carriers. This may be because individuals with a family history of CRC are more likely to undergo screening. Thus, the remaining individuals who are unscreened in these studies may have a lower risk of cancer. Moreover, mutation carriers from clinics or population-based registries were referred for enhanced surveillance with colonoscopy, so cancers detected by colonoscopies may increase the cumulative lifetime risk in populations that are a mix of unscreened and screened individuals. Although results indicate otherwise for male MSH6 carriers, there is substantial overlap in confidence intervals across all ages, suggesting a lack of evidence to support differences in penetrance between the 2 groups. It is challenging to compare study results stratified by screening, because the majority of the studies did not fully clarify whether surveillance was part of the patient selection criteria. More refined data would be needed to extend our analysis to incorporate colonoscopic surveillance as a modifier of CRC risk along with other environmental factors previously shown to affect cancer risk, such as aspirin use (42), smoking (43,44), and body mass index (45).
MMRpro is a genetic counseling and clinical decision support tool that estimates the probability of carrying MMR mutations and of developing CRC for mutation carriers. It relies on meta-analytic penetrance estimates (12). Chen et al. assume the penetrance for MLH1 and MSH2 carriers are the same and that of MSH6 male and female carriers are the same, whereas our meta-analysis contains more studies to substantiate the estimation of gene- and sex-specific risk (Figure 5). In comparison, our results show higher lifetime penetrance estimates for MSH2 and female MLH1 carriers, lower estimates for female MSH6 carriers, and similar estimates for male MLH1 and MSH6 carriers compared with those of Chen et al. (Figure 5). Of the 5 studies included in the meta-analysis by Chen et al. (12), we included 2 in our current analysis (37,38). We excluded 1 because of overlap in study participants (21), 1 because of lack of ascertainment adjustment (46), and another because it does not provide colorectal-specific risks (47).
Figure 5.
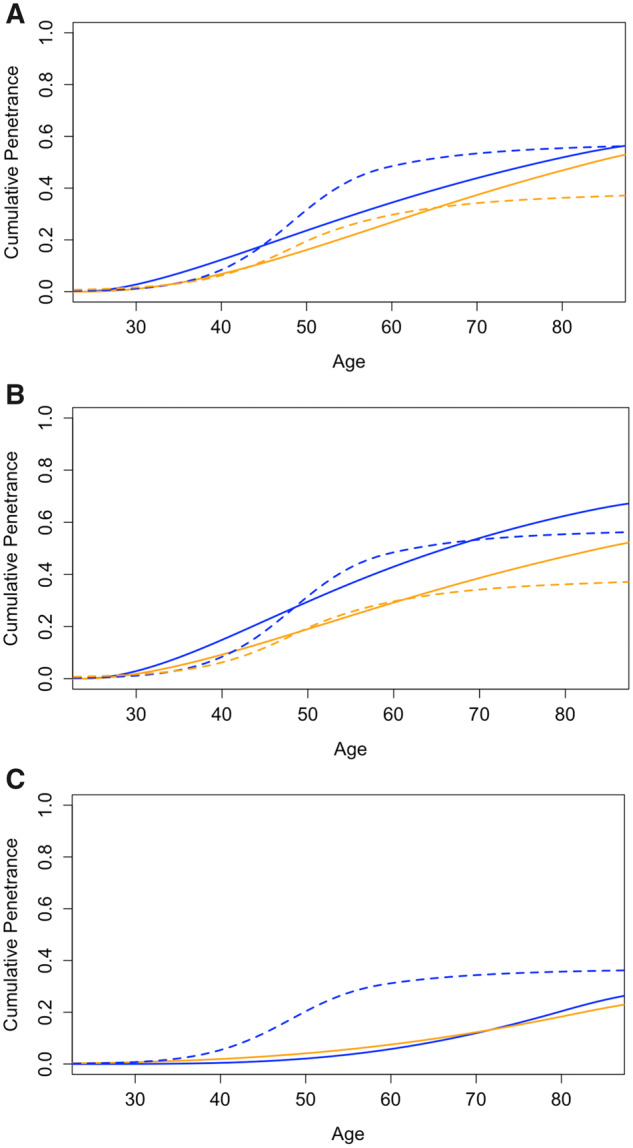
Cumulative penetrance estimates of colorectal cancer from current meta-analysis and MMRpro. Panels A, B, and C correspond to MLH1, MSH2, and MSH6 mutation carriers, respectively. Estimates from current meta-analysis and MMRpro are denoted by solid and dotted lines, respectively. Blue corresponds to male carriers, and orange corresponds to female carriers.
In conclusion, our analysis provides a principled empirical assessment of the risk of Lynch syndrome–associated CRC by combining evidence from relevant studies. For individuals with Lynch syndrome, the risk of cancer is dependent on sex and type of MMR mutation, with male MLH1 or MSH2 mutation carrier risk at age 70 years approximately 4 times higher than that of his female MSH6 counterpart. Risk estimates from our meta-analysis will be incorporated into the 2019 version of the risk prediction tool MMRpro (12), and the clinical decision support tool ASK2ME (All Syndrome Known to Man Evaluator) (48) to improve risk prediction and management strategies for individuals who have mutations in MLH1, MSH2, and MSH6. Our results can support the development of effective prevention strategies and personalized counseling.
Funding
This work was supported by the National Cancer Institute at the National Institutes of Health (5T32CA009337-32 to C.W. and 4P30CA006516-51 to G.P.) and by the Koch Institute/Dana-Farber/Harvard Cancer Center Bridge Project (Footbridge to C.W., K.S.H., G.P., and D.B.).
Notes
Role of the funders: The funders had no role in the design of the study; the collection, analysis, and interpretation of the data; the writing of the manuscript; and the decision to submit the manuscript for publication.
Disclosures: Dr Hughes receives Honoraria from Myriad Genetics Veritas Genetics, Advisory Board for Beacon (An RFID Biopsy Marker) and is a founder of and has a financial interest in Cancer Risk Apps, LLC. Dr Hughes’s interests were reviewed and are managed by Massachusetts General Hospital and Partners Health Care in accordance with their conflict of interest policies. Dr Parmigiani is a member of the Scientific Advisory Board and has a financial interest in Cancer Risk Apps LLC (CRA). CRA commercializes software for management of patients at high risk of cancer. He is a member of the Scientific Advisory Board of Konica-Minolta who owns Ambry genetics. Ms Wang, Dr Braun, and Dr Yan Wang declare that they have no conflict of interest.
Acknowledgements: The authors would like to thank Dr Kush Pathak and Dr Kanhua Yin for their assistance with the literature review process. We wish to thank Dr Swati Biswas and members of the BayesMendel lab for their valuable feedback during the preparation of this manuscript.
Supplementary Material
References
- 1. Rustgi AK. The genetics of hereditary colon cancer. Genes Dev. 2007;21(20):2525-2538. [DOI] [PubMed] [Google Scholar]
- 2. Jass JR. Hereditary non-polyposis colorectal cancer: the rise and fall of a confusing term. World J Gastroenterol. 2006;12(31):4943-4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96(4):261-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Provenzale D, Gupta S, Ahnen DJ, et al. Genetic/familial high-risk assessment: colorectal version 1.2016, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2016;14(8):1010-1030. [DOI] [PubMed] [Google Scholar]
- 5. Vasen H, Watson P, Mecklin J, et al. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116(6):1453-1456. [DOI] [PubMed] [Google Scholar]
- 6. Fay K,, Gregory I, Giovanni P. . Prediction Models for Lynch Syndrome in Hereditary Colorectal Cancer: Genetic Basis and Clinical Implications. Cham, Switzerland: Springer International Publishing; 2018:281-303. [Google Scholar]
- 7. Hampel H, Pearlman R, Beightol M, et al. Assessment of tumor sequencing as a replacement for Lynch syndrome screening and current molecular tests for patients with colorectal cancer. JAMA Oncol. 2018;4(6):806-813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Giardiello FM, Allen JI, Axilbund JE, et al. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-Society Task Force on colorectal cancer. Gastrointestinal Endosc. 2014;80(2):197-220. [DOI] [PubMed] [Google Scholar]
- 9. Kraft P, Thomas DC.. Bias and efficiency in family-based gene-characterization studies: conditional, prospective, retrospective, and joint likelihoods. Am J Hum Genet. 2000;66(3):1119-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stoffel E, Mukherjee B, Raymond VM, et al. Calculation of risk of colorectal and endometrial cancer among patients with Lynch syndrome. Gastroenterology. 2009;137(5):1621-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mukherjee B, Rennert G, Ahn J, et al. High risk of colorectal and endome- trial cancer in Ashkenazi families with the MSH2 A636P founder mutation. Gastroenterology. 2011;140(7):1919-1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen S, Wang W, Lee S, et al. Prediction of germline mutations and cancer risk in the Lynch syndrome. JAMA. 2006;296(12):1479-1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jenkins MA, Dowty JG, Ait Ouakrim D, et al. Short-term risk of colorectal cancer in individuals with Lynch syndrome: a meta-analysis. J Clin Oncol. 2015;33(4):326-331. [DOI] [PubMed] [Google Scholar]
- 14. Win AK, Young JP, Lindor NM, et al. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. J Clin Oncol. 2012;30(9):958-964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sanne W, Brohet RM, Tops Carli M, et al. Lynch syndrome caused by germline PMS2 mutations: delineating the cancer risk. J Clin Oncol. 2015;33(4):319-325. [DOI] [PubMed] [Google Scholar]
- 16. Guindalini RSC, Win AK, Gulden C, et al. Mutation spectrum and risk of colorectal cancer in African American families with Lynch syndrome. Gastroenterology. 2015;149(6):1446-1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sjursen W, Haukanes BI, Grindedal Eli M, et al. Current clinical criteria for Lynch syndrome are not sensitive enough to identify MSH6 mutation carriers. J Med Genet. 2010;47(9):579-585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Møller P, Seppälä T, Bernstein I, et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective Lynch syndrome database. Gut. 2017;66(3):464-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bao Y, Deng Z, Wang Y, et al. Using machine learning and natural language processing to review and classify the medical literature on cancer susceptibility genes. J Clin Oncol. 2019;1(3):1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marabelli M, Cheng S-C, Parmigiani G.. Penetrance of ATM gene mutations in breast cancer: a meta-analysis of different measures of risk. Genet Epidemiol. 2016;40(5):425-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jenkins M, Baglietto L, Dowty J, et al. Cancer risks for mismatch repair gene mutation carriers: a population-based early onset case-family study. Clin Gastroenterol Hepatol. 2006;4(4):489-498. [DOI] [PubMed] [Google Scholar]
- 22. Barrow E, Robinson L, Alduaij W, et al. Cumulative lifetime incidence of extracolonic cancers in Lynch syndrome: a report of 121 families with proven mutations. Clin Genet. 2009;75(2):141-149. [DOI] [PubMed] [Google Scholar]
- 23. Choi Y-H, Cotterchio M, McKeown-Eyssen G, et al. Penetrance of colorectal cancer among MLH1/MSH2 carriers participating in the colorectal cancer familial registry in Ontario. Hered Cancer Clin Pract. 2009;7(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bonadona V, Bonaïti B, Olschwang S, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA. 2011;305(22):2304-2310. [DOI] [PubMed] [Google Scholar]
- 25. Vasen HFA, Stormorken A, Menko FH, et al. MSH2 mutation carriers are at higher risk of cancer than MLH1 mutation carriers: a study of hereditary nonpolyposis colorectal cancer families. J Clin Oncol. 2001;19(20):4074-4080. [DOI] [PubMed] [Google Scholar]
- 26.Schwarzer G, Schwarzer MG. Package ‘meta’. The R Foundation for Statistical Computing. 2012:9. http://cran.r-project.org/web/packages/meta/index.html. [Google Scholar]
- 27.R Core Team. R: A Language and Environment for Statistical Computing Vienna, Austria: R Foundation for Statistical Computing; 2017.
- 28. Egger M, Smith GD, Schneider M, Minder C.. Bias in meta-analysis detected by a simple. BMJ. 1997;315(7109):629-634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. DerSimonian R, Laird N.. Meta-analysis in clinical trials. Control Clin Trials. 1986;7(3):177-188. [DOI] [PubMed] [Google Scholar]
- 30. Kopciuk KA, Choi Y-H, Parkhomenko E, et al. Penetrance of HNPCC-related cancers in a retrolective cohort of 12 large Newfoundland families carrying a MSH2 founder mutation: an evaluation using modified segregation models. Hered Cancer Clin Pract. 2009;7(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Surveillance, Epidemiology, and End Results (SEER) Program. SEER*Stat Database: Incidence - SEER Research Data, 9 Registries, 1975. –2017. https://seer.cancer.gov/data/. Accessed March 20, 2017.
- 32. Chapelle A. The incidence of Lynch syndrome. Fam Cancer. 2005;4(3):233-237. [DOI] [PubMed] [Google Scholar]
- 33. Borras E, Pineda M, Blanco I, et al. MLH1 founder mutations with moderate penetrance in Spanish Lynch syndrome families. Cancer Res. 2010;70(19):7379. [DOI] [PubMed] [Google Scholar]
- 34. Laura B, Lindor Noralane M, Dowty James G, et al. Risks of Lynch syndrome cancers for MSH6 mutation carriers. J Natl Cancer Inst. 2010;102(3):193-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dowty JG, Win Aung K, Buchanan Daniel D, et al. Cancer risks for MLH1 and MSH2 mutation carriers human mutation. Hum Mutat. 2013;34(22):490-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aaltonen L, Johns L, Jarvinen H, Mecklin J-P, Houlston R.. Explaining the familial colorectal cancer risk associated with mismatch repair (MMR)-deficient and MMR-stable tumors. Clin Cancer Res. 2007;13(1):356-361. [DOI] [PubMed] [Google Scholar]
- 37. Quehenberger F, Vasen HFA, Van Houwelingen HC.. Risk of colorectal and endometrial cancer for carriers of mutations of the hMLH1 and hMSH2 gene: correction for ascertainment. J Med Genet. 2005;42(6):491-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dunlop MG, Farrington SM, Carothers AD.. Cancer risk associated with germline DNA mismatch repair gene mutations. Hum Mol Genet. 1997;6(1):105-110. [DOI] [PubMed] [Google Scholar]
- 39. Vasen HF, Blanco I, Aktan-Collan K, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut. 2013;62(6):812–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Locker Gershon Y, Lynch Henry T.. Genetic factors and colorectal cancer in Ashkenazi Jews. Fam Cancer. 2004;3(3-4):215-221. [DOI] [PubMed] [Google Scholar]
- 41. Järvinen HJ, Mecklin J-P, Sistonen P.. Screening reduces colorectal cancer rate in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 1995;108(5):1405-1411. [DOI] [PubMed] [Google Scholar]
- 42. John B, Anne-Marie G, Finlay M, et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet. 2011;378(9809):2081-2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Watson P, Ashwathnarayan R, Lynch HT, Roy HK.. Tobacco use and increased colorectal cancer risk in patients with hereditary nonpolyposis colorectal cancer (Lynch syndrome). Arch Intern Med. 2004;164(22):2429-2431. [DOI] [PubMed] [Google Scholar]
- 44. Pande M, Lynch PM, Hopper JL, et al. Smoking and colorectal cancer in Lynch syndrome: results from the Colon Cancer Family Registry and the University of Texas MD Anderson Cancer Center. Clin Cancer Res. 2010;16(4):1331-1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Win AK, Dowty JG, English DR, et al. Body mass index in early adulthood and colorectal cancer risk for carriers and non-carriers of germline mutations in DNA mismatch repair genes. Br J Cancer. 2011;105(1):162-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hampel H, Stephens J, Pukkala E, et al. Cancer risk in hereditary nonpolyposis colorectal cancer syndrome: later age of onset. Gastroenterology. 2005;129(2):415-421. [DOI] [PubMed] [Google Scholar]
- 47. Buttin BM, Powell MA, Mutch DG, et al. Penetrance and expressivity of MSH6 germline mutations in seven kindreds not ascertained by family history. Am J Hum Genet. 2004;74(6):1262-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Braun D, Yang J, Griffin M, Parmigiani G, Hughes KS.. A clinical decision support tool to predict cancer risk for commonly tested cancer-related germline mutations. J Genet Couns. 2018;27(5):1187–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.