

Abstract
In the heart, the serine carboxypeptidase cathepsin A (CatA) is distributed between lysosomes and the extracellular matrix (ECM). CatA-mediated degradation of extracellular peptides may contribute to ECM remodeling and left ventricular (LV) dysfunction. Here, we aimed to evaluate the effects of CatA overexpression on LV remodeling. A proteomic analysis of the secretome of adult mouse cardiac fibroblasts upon digestion by CatA identified the extracellular antioxidant enzyme superoxide dismutase (EC-SOD) as a novel substrate of CatA, which decreased EC-SOD abundance 5-fold. In vitro, both cardiomyocytes and cardiac fibroblasts expressed and secreted CatA protein, and only cardiac fibroblasts expressed and secreted EC-SOD protein. Cardiomyocyte-specific CatA overexpression and increased CatA activity in the LV of transgenic mice (CatA-TG) reduced EC-SOD protein levels by 43%. Loss of EC-SOD–mediated antioxidative activity resulted in significant accumulation of superoxide radicals (WT, 4.54 μmol/mg tissue/min; CatA-TG, 8.62 μmol/mg tissue/min), increased inflammation, myocyte hypertrophy (WT, 19.8 μm; CatA-TG, 21.9 μm), cellular apoptosis, and elevated mRNA expression of hypertrophy-related and profibrotic marker genes, without affecting intracellular detoxifying proteins. In CatA-TG mice, LV interstitial fibrosis formation was enhanced by 19%, and the type I/type III collagen ratio was shifted toward higher abundance of collagen I fibers. Cardiac remodeling in CatA-TG was accompanied by an increased LV weight/body weight ratio and LV end diastolic volume (WT, 50.8 μl; CatA-TG, 61.9 μl). In conclusion, CatA-mediated EC-SOD reduction in the heart contributes to increased oxidative stress, myocyte hypertrophy, ECM remodeling, and inflammation, implicating CatA as a potential therapeutic target to prevent ventricular remodeling.
Keywords: cathepsin A, carboxypeptidase, extracellular matrix protein, oxidative stress, extracellular superoxide dismutase, cardiac hypertrophy, cardiac remodeling, left ventricular dysfunction, heart disease, secretome, superoxide dismutase (SOD), carboxypeptidase, oxygen radicals, heart failure, fibrosis, EC-SOD
Cardiac function depends on structural and functional integrity of the extracellular matrix (ECM). ECM components are synthesized and secreted by cardiac fibroblasts (CFs) (1), and their proper composition and turnover is controlled by proteolysis (2–4). Degradation of ECM proteins occurs either within the cell after fusion of ECM-containing phagosomes with lysosomes or in the extracellular space by secreted proteolytic enzymes (4). Cathepsins are lysosomal proteases that target a broad range of intra- and extracellular proteins, like laminin, fibronectin, elastin, and fibrillar collagens (2, 3). Increased activation of cathepsins results in remodeling of subcellular organelles and the ECM and is associated with cardiac complications, including hypertrophic cardiomyopathy, diabetic cardiomyopathy, dilated cardiomyopathy, and myocardial infarction (2, 3). Cysteine protease cathepsins like cathepsins B, K, L, and S play pathophysiological roles in cardiac structural changes and progression of heart failure (2, 3). However, the role of the serine protease cathepsin A (CatA) during cardiac disease is unclear. CatA is widely distributed in mammalian tissues, with highest expression found in kidney, lung, endothelium, liver, placenta, and heart (5, 6). Besides its catalytic function as a protease, lysosomal CatA forms a protein complex with neuraminidase 1 and β-galactosidase, which prevents proteolysis of its binding partners, thereby regulating and stabilizing lysosomal activity and function (7). In humans, disruption of this protein complex by CatA deficiency or mutations in the gene coding for CatA results in the lysosomal storage disease galactosialidosis (8). CatA is also localized on the cell surface and in the extracellular space, where it has been suggested to be involved in ECM formation, possibly by degradation of extracellular peptides (5, 6, 9). In animal models of myocardial infarction, type 2 diabetes, and angiotensin II-stimulated hypertrophy, cardiac expression of CatA is upregulated, and pharmacological inhibition of CatA activity exhibited cardioprotective, antihypertrophic, and antifibrotic effects under these conditions (10–13), but its mechanistic role in cardiovascular disease is unknown.
The present study was designed to identify potential mechanisms of CatA-mediated ECM remodeling processes in the heart using state-of-the-art proteomic analysis of the secretome of adult mouse CFs upon digestion by CatA and supportive in vivo investigations in a transgenic mouse model with cardiomyocyte-specific overexpression of CatA (CatA-TG).
Results
CatA processes ECM proteins
To identify novel ECM-related candidate substrates of CatA, we performed a proteomic analysis of the secretome of adult mouse CFs, which produce and secrete ECM proteins. Proteins in the conditioned medium of CFs treated with and without human recombinant CatA (n = 4 each) were analyzed by LC–MS/MS after filtering using 3-kDa columns to recover only small cleavage products rather than intact proteins (14). Using Progenesis® LC–MS software (Nonlinear Dynamics), the ion intensities of all detected peptides in the <3-kDa fraction were compared between control and CatA-treated CFs (Fig. 1A). CatA digestion significantly affected abundance of protein degradation products of collagens (CO5A1 [p = 0.001], CO5A2 [p = 0.0001], CO3A1 [p = 0.002], CO1A1 [p = 0.003]) and other ECM proteins (i.e. PGS2 [Decorin; p = 0.005], LAMA4 (laminin subunit alpha 4, p = 0.028], PGBM [basement membrane-specific heparan sulfate proteoglycan core protein, also known as perlecan; p = 0.025], and FINC [fibronectin; p = 0.002]) compared with the control. The antioxidant enzyme extracellular superoxide dismutase (SODE or EC-SOD) was one of the most significantly affected extracellular proteins after incubation with CatA (p = 0.0001) (Fig. 1B and Table S1). The proteomics data are also available via ProteomeXchange with identifier PXD019895. EC-SOD is an essential antioxidant enzyme that is exclusively located in the ECM, catalyzing the dismutation of superoxide to hydrogen peroxide and oxygen (15). Three fragment peptides and possible cleavage sites were detected for EC-SOD under control conditions but were undetectable upon treatment with CatA (Fig. 1C). This finding added a novel and highly relevant aspect on the role of CatA in cardiac disease, because EC-SOD provides the only direct defense mechanism against superoxide radicals within the ECM (15), and CatA-mediated loss of antioxidant protection may facilitate ECM remodeling.
Figure 1.
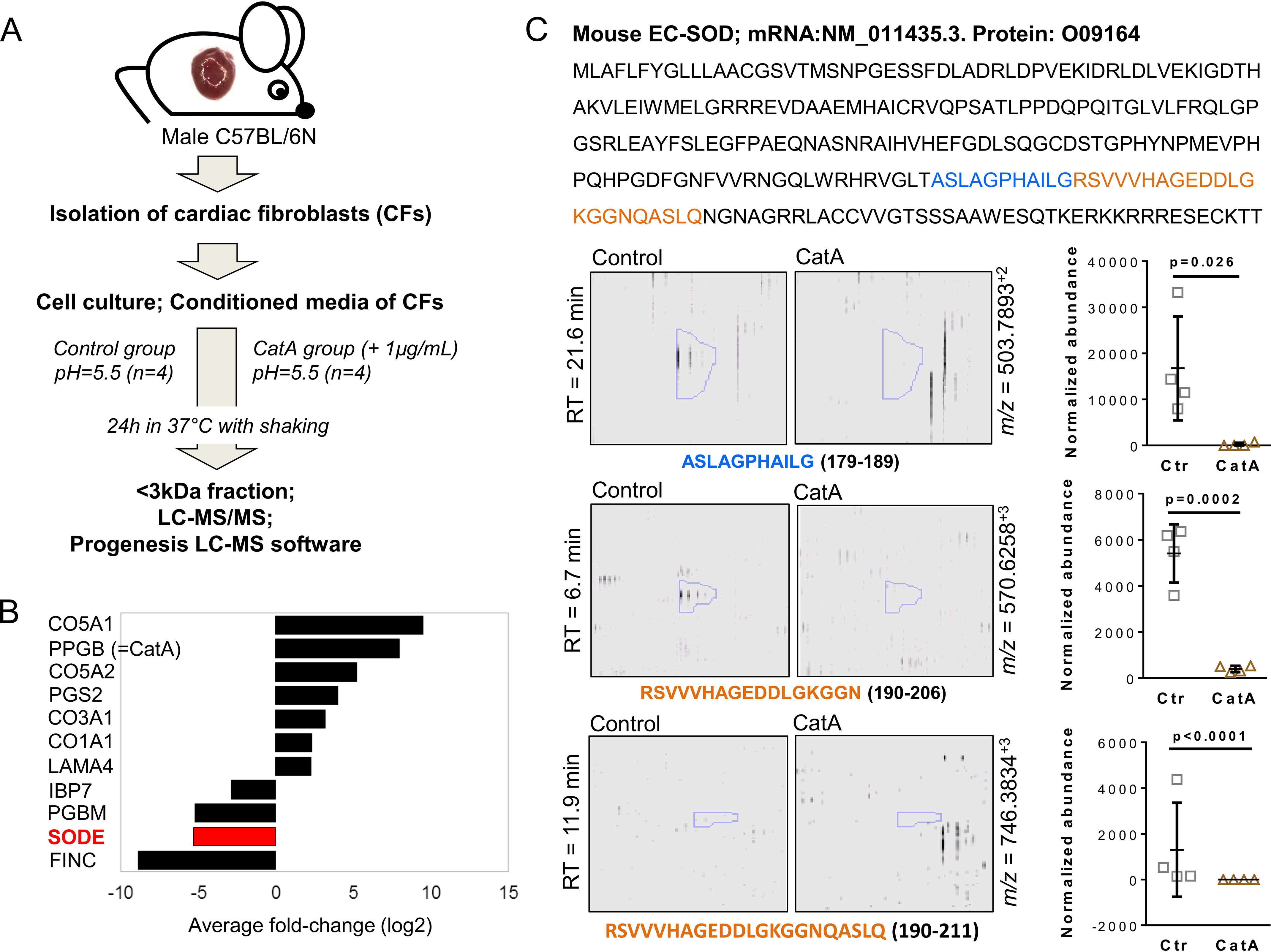
Proteomics of CatA degradation products in conditioned media from CFs. A, proteomics workflow of degradation products in conditioned media from CFs after digestion with CatA. For isolation and culture of primary mouse CFs, primary mouse CFs were isolated from the hearts of male C57BL/6N mice (n = 4). For secretome analysis, 80% confluent CFs were cultured in serum-free medium for 3 days. The secreted proteins from the conditioned media were either incubated with human recombinant CatA (1 µg/ml) or without (control group) in assay buffer (pH5.5). Degradation products in the <3-kDa fractions were analyzed by LC–MS/MS, and significantly changed peptides (>2-fold intensity change, p < 0.05) were identified by Mascot (Matrix Science; version 2.3.01) in a no-enzyme search against the UniProt/SwissProt mouse database. B, proteins with differential abundance in the conditioned medium after filtering using 3-kDa columns to recover protein fragments, i.e. collagens (CO5A1, CO5A2, CO3A1, and CO1A1), decorin (PGS2), laminin subunit alpha 4 (LAMA4), basement membrane-specific heparan sulfate proteoglycan core protein (PGBM), fibronectin (FINC), and extracellular superoxide dismutase (SODE or EC-SOD). C, three peptides were detected for EC-SOD. Shown are the retention times (RT, y axis) versus the m/z (m/z, x axis) for the three EC-SOD peptides. Quantification based on normalized abundance is shown for each of the peptides (n = 4 per group). All values are presented as means ± S.D.
Differential expression pattern of CatA and EC-SOD in CMs and CFs
An initial gene expression analysis comparing isolated primary adult mouse CFs with cardiomyocytes (CMs) demonstrated mRNA transcription of CatA in both cell lines, whereas EC-SOD mRNA was only detectable in CFs (Fig. S1). These mRNA data were confirmed at the protein level by analyzing cultured neonatal rat CMs and rat CFs. Whereas CatA protein was expressed and secreted by both cell types (Fig. 2, A and B), the presence and secretion of EC-SOD protein was only evident in CFs (Fig. 2, C and D).
Figure 2.

Differential expression and secretion of CatA and EC-SOD by CMs and CFs. CMs and CFs were isolated from 4- to 6-day-old Sprague-Dawley rats of mixed sex. A, Western blot analysis demonstrating cellular protein expression of CatA in isolated neonatal CMs and in CFs. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as a loading control of cell lysates. B, Western blot analysis of secreted CatA protein comparing serum-free cell culture medium derived from CM and CF after 48 h. C, Western blot analysis of EC-SOD protein in isolated CMs and CFs. Collagen 1a2 (Col1a2) protein served as the fibroblast-specific control, and GAPDH served as the loading control of cell lysates. D, Western blot analysis of secreted EC-SOD protein comparing serum-free cell culture medium derived from CMs and CFs after 48 h.
Cardiomyocyte-specific CatA overexpression in mice results in posttranslational down-regulation of EC-SOD, increased oxidative stress, and enhanced inflammation in the left ventricle (LV)
To further understand the function of CatA in ECM remodeling in vivo, transgenic mice with a cardiomyocyte-specific postnatal overexpression of active human CatA were generated (CatA-TG) using the alpha myosin heavy chain promoter (Fig. S2). CatA-TG mice developed normally and showed no apparent abnormal phenotype. Quantitative real-time PCR, Western blot analysis, and immunohistological staining confirmed overexpression of human CatA in the myocardium of CatA-TG mice (Fig. 3, A–C).
Figure 3.
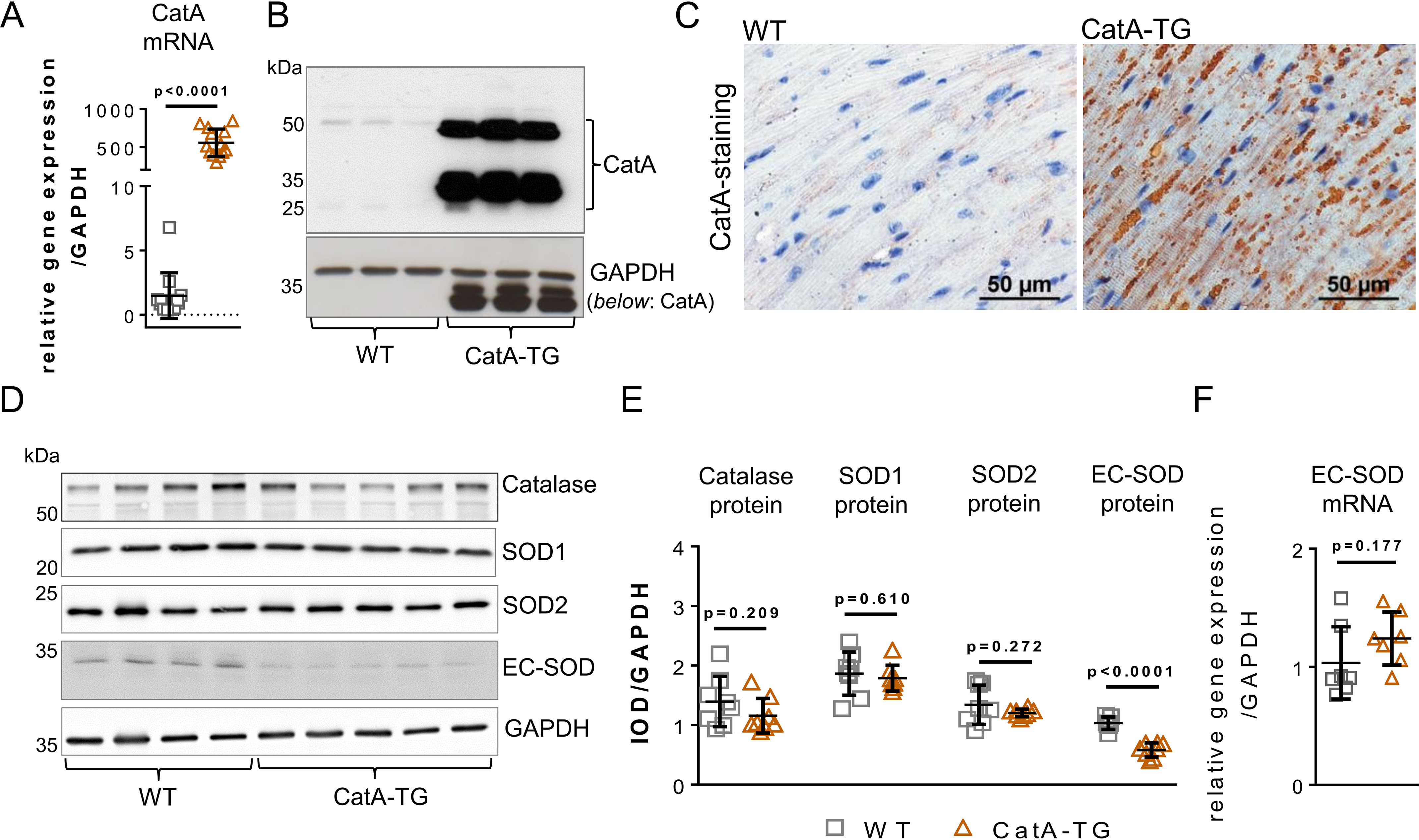
Cardiomyocyte-specific overexpression of CatA affects EC-SOD processing. A and B, left ventricular (LV) expression of human CatA mRNA (A) and protein (B) in male 6-month-old mice with cardiomyocyte-specific overexpression of human CatA (CatA-TG) and their WT littermates (WT). C, representative images of LV tissue immunohistologically stained for overexpressed human CatA. D–F, representative Western blotting (D) and quantification (E) of protein expression of catalase, superoxide dismutase 1 (SOD1), SOD2, and extracellular SOD (EC-SOD) in LV tissue of CatA-TG mice (n = 8) and their WT littermates (WT, n = 7–8) as well as mRNA expression of EC-SOD (n = 7 per group) (F). All values are presented as means ± S.D.
Detoxification of reactive oxygen species (ROS) is catalyzed by antioxidative enzymes, including catalase and superoxide dismutases (SODs). Besides the extracellular isoform EC-SOD, two intracellular isozymes of SODs exist, cytosolic Cu/Zn-SOD (SOD1) and mitochondrial Mn-SOD (SOD2) (15). Western blot analysis demonstrated that overexpression of CatA did not affect protein expression of catalase, SOD1, and SOD2, which are located within the cell (Fig. 3, D and E). Using an EC-SOD antibody that specifically targets the N-terminal region demonstrated a significant reduction of EC-SOD protein in the LV of CatA-TG compared with their WT littermate controls (Fig. 3, D and E). Interestingly, EC-SOD mRNA levels were unaltered (Fig. 3F), strongly suggesting a posttranslational regulation, i.e. consistent with proteolysis of EC-SOD.
To characterize downstream consequences of reduced EC-SOD and its loss of antioxidant protection (15), we determined the levels of superoxide radicals in LV tissue of CatA-TG and WT mice. Using electron spin resonance (ESR) spectroscopy measurements, CatA-TG mice demonstrated increased LV oxidative stress, as indicated by accumulation of superoxide radicals (Fig. 4A). Oxidative stress was associated with elevated gene expression of connective tissue growth factor (CTGF), an important redox-sensitive inducer of fibrosis (16) (Fig. 4B). Increased gene expression of tumor necrosis factor alpha (TNF-α), interleukin 6 (IL6), and interleukin 2 (IL2) as well as repressed transcription of interleukin 10 (IL10) demonstrated an enhanced inflammatory response in CatA-TG mice (Fig. 4C). Interleukin 1 beta (IL1b) was not regulated. Of note, mRNA levels of IL2 and IL10 were considerably lower than those with gene expression of TNF-α or IL6, and immunohistological stainings for infiltration of macrophages or neutrophils in the heart of CatA-TG and WT mice showed no significant inflammatory infiltration (Fig. S3). CatA-TG demonstrated a higher proportion of apoptotic cells, as detected by TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) staining (Fig. 4D), independent of a differential expression of pro-enzyme caspase 1 and caspase 3 protein (caspase 1 protein, WT, 1.00 ± 0.20; CatA-TG, 1.25 ± 0.61; p = 0.261; caspase 3 protein, WT, 1.01 ± 0.22; CatA-TG, 1.24 ± 0.31; p = 0.098) and in the absence of active caspase subunits (Fig. S4). LV protein expression of NAD(P)H oxidase 2 (Nox2/gp91phox), Nox4, and xanthine oxidase, representing major sources of cellular ROS (17), were unchanged between WT and CatA-TG mice (Nox2 protein, WT, 1.00 ± 0.20; CatA-TG, 1.06 ± 0.32; p = 0.611; Nox4 protein, WT, 1.00 ± 0.09; CatA-TG, 1.16 ± 0.35; p = 0.420; xanthine oxidase protein, WT, 1.00 ± 0.10; CatA-TG, 1.09 ± 0.27; p = 0.352; Fig. S5).
Figure 4.
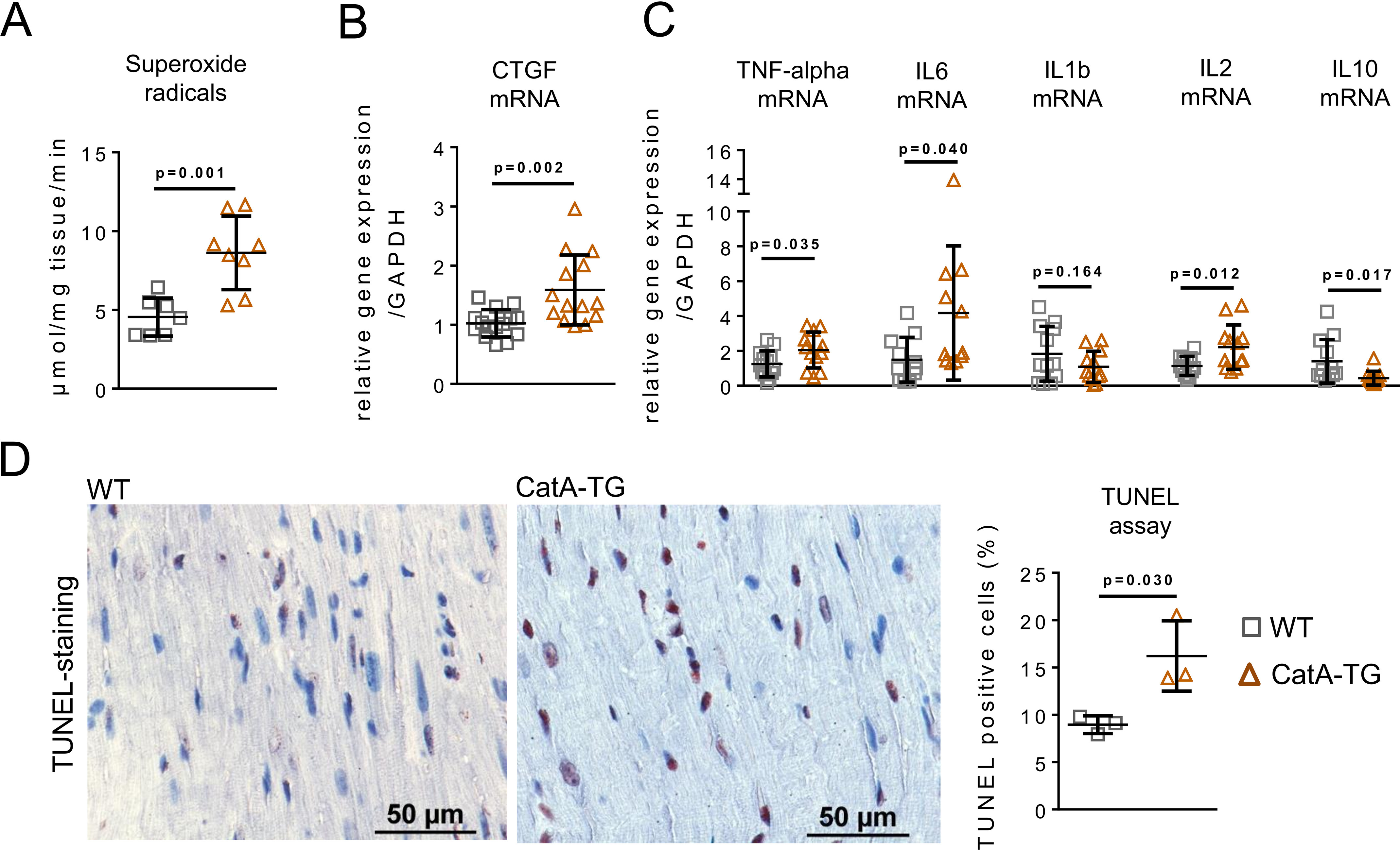
Impact of cardiomyocyte-specific CatA overexpression on cellular oxidative stress and inflammation. A and B, analysis of superoxide radical production (n = 7–8 per group) in LV tissue of WT and CatA-TG (A) as well as mRNA expression of redox-sensitive connective tissue growth factor (CTGF) (n = 14 per group) (B). C, real-time PCR quantification of proinflammatory marker genes for tumor necrosis factor alpha (TNF-α [WT, n = 13; CatA, n = 12]), interleukin 6 (IL6; n = 11 per group), interleukin 1 beta (IL1b; n = 12 per group), and interleukin 2 (IL2; n = 12 per group), as well as anti-inflammatory interleukin 10 (IL10; n = 12 per group). D, assessment of apoptosis by quantification of TUNEL-positive cells in LV tissue of WT and CatA-TG (n = 3 per group). All values are presented as means ± S.D.
CatA-TG mice demonstrate LV structural and functional remodeling
Body weight did not differ between WT controls and CatA-TG mice (WT, 35.4 ± 4.4 g; CatA-TG, 33.5 ± 5.2 g; n = 15 each; p = 0.304). Heart weight to body weight ratio (n = 15 each) (Fig. 5A), as well as LV weight to body weight ratio, was significantly increased (WT, 3.27 ± 0.19 mg/g; CatA-TG, 4.29 ± 0.56 mg/g; n = 6; p = 0.0019), and LV cardiomyocyte diameter was enlarged (p = 0.024) (Fig. 5C). Gene expression of the hypertrophy marker atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) was elevated in CatA-TG mice compared with that of their WT littermate controls (Fig. 5D). CatA-TG mice also demonstrated increased LV interstitial fibrosis formation (Fig. 6, A and C) and a shift in the ratio of collagen type I (red-yellow fibers) to collagen type III (green fibers), as assessed by polarized light microscopy (Fig. 6, B and D). In the LV of CatA-TG mice, mRNA expression of profibrotic transforming growth factor beta 1 (TGFβ-1) and of the ECM components collagen 1a2 (Col1a2), Col3a, Col5a1, fibronectin (FN), and ECM stabilizing protein lysyl oxidase (Lox), which catalyzes cross-linking of collagen fibrils and elastin (18), were elevated (Fig. 6E). The development of perivascular fibrosis in the heart was unaffected by cardiomyocyte-specific cathepsin A overexpression (WT, 31.3 ± 5.9%; CatA-TG, 34.3 ± 5.4%; p = 0.398; Fig. S6). To assess LV function in this unchallenged phenotype of CatA-TG, we used an isolated working heart preparation to measure functional parameters under standardized hemodynamic conditions. At 6 months of age, isolated CatA-TG hearts demonstrated significantly increased LV end diastolic volume (LVEDV) (WT, 50.8 ± 5.8; CatA-TG, 61.9 ± 6.2 μl; p = 0.018), whereas other functional parameters where unchanged (Table 1). It is worth noting that CatA-TG mice developed a significantly increased left ventricular end systolic pressure at an age of 18 months (Fig. S7).
Figure 5.
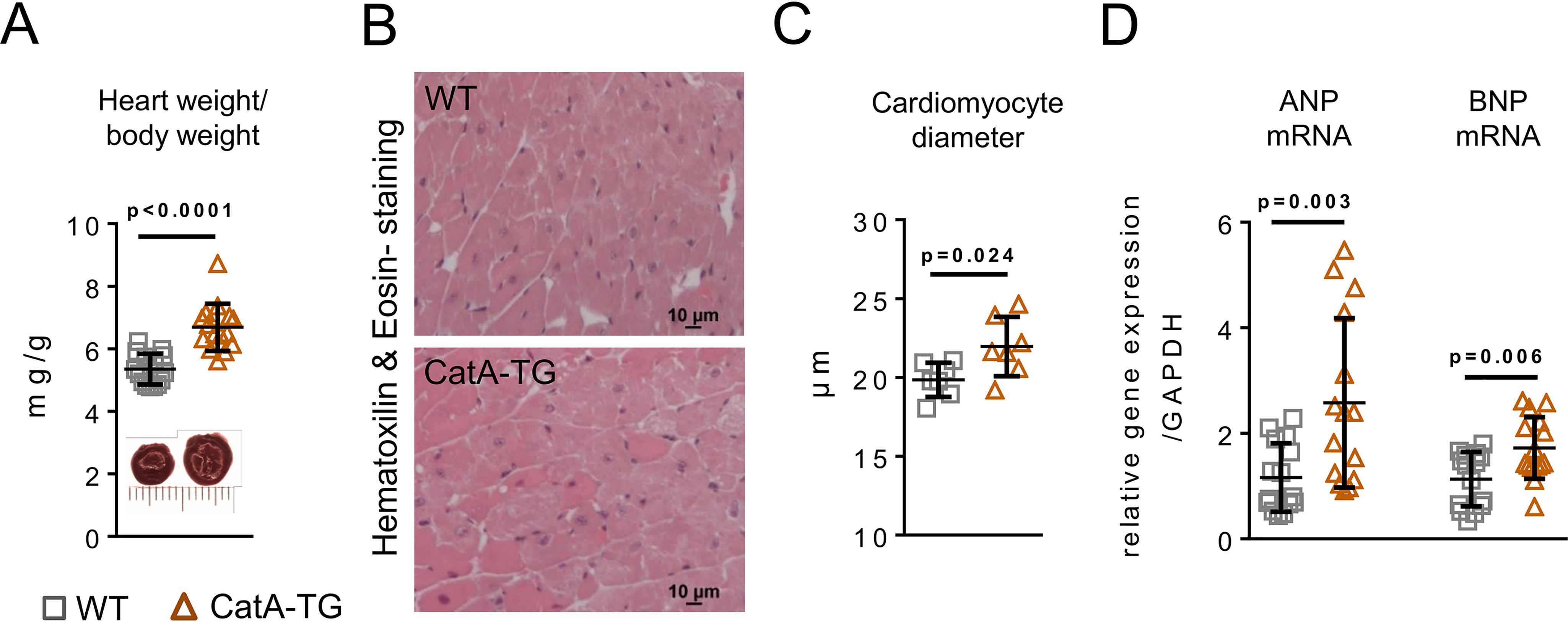
Cardiomyocyte-specific overexpression of CatA and cardiac hypertrophy. A, heart weight to body weight ratio (n = 15 per group). B and C, representative images used for histological analysis (B) and quantification of cardiomyocyte diameter (n = 7 per group) (C). D, mRNA expression of hypertrophy marker genes atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) in WT and CatA-TG mice (n = 15 per group). All values are presented as means ± S.D.
Figure 6.
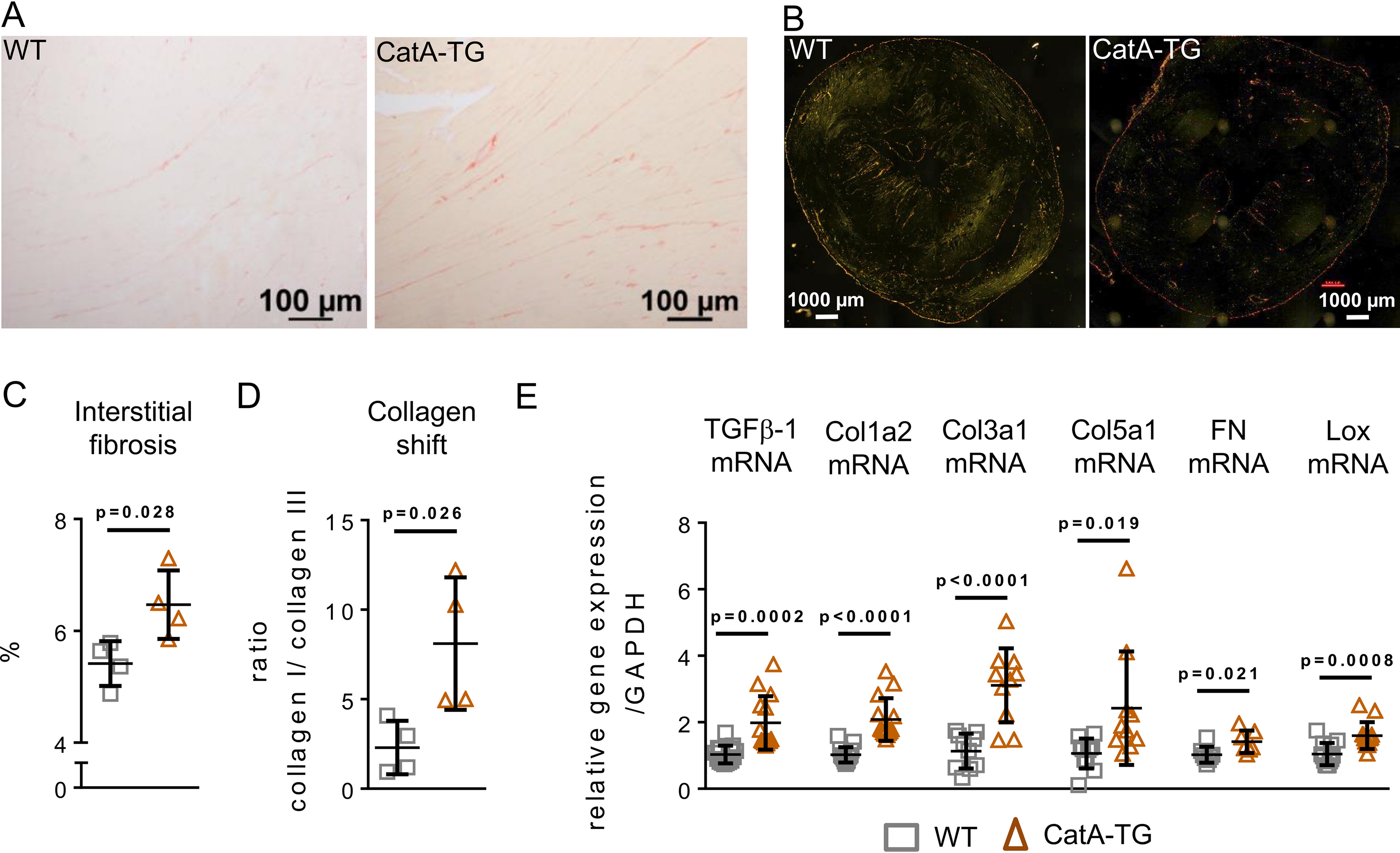
Cardiomyocyte-specific overexpression of CatA causes LV interstitial remodeling. A and B, representative images used for histological analysis (A) and polarized light microscopy (B) of Picro-Sirius Red-stained left ventricular preparations to visualizes collagen type I (red-yellow fibers) and collagen type III (green fibers). C, quantification of LV interstitial fibrosis amount (n = 4 per group). D, quantification of collagen type I to collagen type III ratio. E, transcription levels of profibrotic gene transforming growth factor beta (TGFβ), of collagens Col1a2, Col1a3, and Col5a1, fibronectin (FN), and lysyl oxidase (Lox) in WT and CatA-TG. All values are presented as means ± S.D.
Table 1.
Hemodynamic characterization of CatA transgenic mice in the working heart apparatusa
| Parameter | Value(s) for: |
p value | |
|---|---|---|---|
| WT (n = 5) | CatA-TG (n = 5) | ||
| EF (%) | 51.4 ± 19.2 | 51.4 ± 6.3 | 0.998 |
| LVEDV (µl) | 50.8 ± 5.8 | 61.9 ± 6.2 | 0.018 |
| LVESV (µl) | 25.2 ± 12.0 | 29.8 ± 3.4 | 0.434 |
| SV (µl) | 25.6 ± 7.5 | 32.1 ± 6.3 | 0.172 |
| HR (bpm) | 397 ± 16 | 402 ± 41 | 0.812 |
| dP/dt max (mmHg/s) | 4980.8 ± 1782.0 | 4913.6 ± 565.0 | 0.937 |
| dP/dt min (mmHg/s) | −3446.6 ± 782.3 | −3670.8 ± 493.4 | 0.602 |
| CO (µl/min) | 10.2 ± 3.1 | 13.0 ± 3.2 | 0.206 |
| Tau-weiss (ms) | 14.2 ± 4.2 | 11.9 ± 1.9 | 0.298 |
| Endsystolic pressure (mmHg) | 67.6 ± 6.4 | 70.9 ± 4.7 | 0.381 |
| Enddiastolic pressure (mmHg) | 7.7 ± 3.5 | 7.6 ± 2.5 | 0.952 |
a Cardiac function was characterized in six-month-old CatA transgenic mice (CatA-TG) and their wild-type littermates (WT) using an isolated working heart apparatus. Abbreviations: ejection fraction (EF), left ventricular end-diastolic volume (LVEDV), left ventricular end-systolic volume (LVESV), stroke volume (SV), heart rate (HR), LV contractility (dP/dtmax), relaxation (dP/dtmin), and cardiac output (CO). All values are presented as means ± S.D.
Discussion
The composition and turnover of the ECM is tightly controlled by proteolysis (2–4). Pathophysiological remodeling of the ECM contributes critically to LV dysfunction and progression of heart failure (19, 20). Here, we provide new evidence for the involvement of the serine carboxypeptidase CatA in the degradation of the extracellular antioxidant enzyme EC-SOD and regulation of subsequent ECM remodeling processes. These findings highlight proteolysis as a potential target in cardiovascular diseases and heart failure.
Proteomic analysis of the secretome of adult mouse CFs identified EC-SOD as a novel target and substrate of CatA at slightly acidic pH5.5. As a multifunctional enzyme, CatA exhibits a strong carboxypeptidase activity in an acidic milieu and a de-amidase as well as a weakened enzymatic carboxypeptidase activity at neutral pH (5, 6, 21).
At neutral pH, the ECM is protected against extracellular degradation by secreted or membrane-bound cathepsins. However, in the LV myocardium of CatA-TG mice, the imbalanced expression pattern of pro- and anti-inflammatory marker genes suggests an inflammatory state. Under inflammatory conditions, acidification of the peri- and extracellular space increases the proteolytic activity of cathepsins and further promotes the secretion of lysosomal proteases. Moreover, within the ECM the acidic pH facilitates the processing of secreted inactive procathepsins into catalytically active mature proteases (22). Additionally, binding of cathepsins to heparin or heparan sulfate has been shown to stabilize its enzyme structure and potentiate peptidase activity even at alkaline pH (23). Taken together, these factors may regulate CatA activity to hydrolyze its target proteins after secretion into the interstitial space (6, 7, 21).
EC-SOD represents the main defense mechanism in the ECM against oxidative stress and is the only extracellular enzyme detoxifying superoxide radicals (15). EC-SOD is anchored to the cell surface and the ECM via its C-terminal region, known as the heparin-binding domain. The heparin-binding domain determines the binding affinity for its ligands, including cell surface heparan sulfate proteoglycans, heparin, and type I collagen (15, 24, 25), thereby tightly regulating distribution of EC-SOD in the ECM. Interaction of EC-SOD with ECM components like collagen and syndecan-1 shields its binding partners from oxidative damage, preventing ECM remodeling and attenuating inflammatory responses (15, 25). Subsequently, EC-SOD depletion reduces antioxidant protection, leading to fragmentation of ECM components, oxidant-induced fibrosis formation, and myocyte hypertrophy (15, 25, 26).
Here, we demonstrated that both CFs and CMs express and secrete CatA, whereas EC-SOD is exclusively expressed and secreted by CFs but not by CMs. In our transgenic mouse model, cardiomyocyte-specific overexpression of CatA induced a significant decrease of LV EC-SOD protein levels. As CMs do not express EC-SOD, our findings suggest a CatA-mediated degradation of EC-SOD protein within the ECM and exclude intracellular digestion of EC-SOD in CMs. Additionally, LV EC-SOD mRNA expression was not affected in CatA-TG mice, which indicates a posttranslational depletion of EC-SOD protein in the ECM.
Oxidative stress highly contributes to the development of cardiac hypertrophy and fibrosis (17). In CatA-TG mice, reduction of EC-SOD was associated with increased oxidative stress, elevated expression of the redox-sensitive profibrotic CTGF (16), and development of a hypertrophic and profibrotic phenotype with increased LV weight and elevated LVEDV, known hallmarks for heart failure (summarized in Fig. 7). Comparable phenotypes have been described previously in EC-SOD knockout mice (25–28). Lack of EC-SOD was associated with exacerbated oxidative stress-induced myocardial apoptosis, LV fibrosis formation, and inflammatory cell infiltration, demonstrating an important protective role of EC-SOD against extracellular oxidative stress (25–28). Vice versa, overexpression of EC-SOD reduced interstitial fibrosis and ventricular dysfunction in a murine model of ischemic cardiomyopathy (29).
Figure 7.

Summary. The antioxidant enzyme EC-SOD is expressed and secreted in abundance into the ECM by CFs. In the ECM, EC-SOD protects against reactive oxygen species by dismutation of .O2− to H2O2 and O2. The carboxypeptidase CatA is expressed by both CFs and CMs and secreted into the extracellular space, where it proteolytically degrades EC-SOD, thereby regulating EC-SOD distribution in the ECM. Cardiomyocyte-specific overexpression of CatA leads to enhanced EC-SOD protein degradation and accelerated depletion from the ECM, subsequently increasing vulnerability to reactive oxygen species, followed by cardiac fibrosis formation with a higher quantity of collagen type I fibers (red fibers) and myocardial hypertrophy.
In the failing heart, major sources of cellular ROS comprise the xanthine oxidase and NAD(P)H oxidases (like Nox2 and Nox4). Dysregulation of these enzymes is involved in cardiovascular disease, hypertension, and heart failure (17). However, in CatA-TG mice, increased superoxide radicals could not be linked to a differential expression of cellular ROS-producing oxidases. Therefore, ECM remodeling in CatA-TG is likely a consequence of CatA-mediated loss of antioxidant protection and subsequent accumulation of extracellularly derived ROS in the ECM.
A recent proteomic profiling analyzed the impact of a pharmacological inhibition of CatA in a mouse model of myocardial infarction, demonstrating a partial rescue of left ventricular proteome alterations associated with myocardial infarction and attenuated elevated levels of cardiac stress response proteins (13). Furthermore, a quantitative proteome comparison of whole heart lysates from CatA transgenic mice and their littermates linked CatA to cardiac oxidative stress response, and CatA overexpression in cultured rat cardiomyoblasts resulted in higher sensitivity to oxidative stress (30).
Potential limitations of this study are as follows. Secretome analysis and transgenic overexpression in mice might not recapitulate physiological levels or cell type-specific expression patterns. Whereas certain proteins may be inaccessible to the protease in a tissue environment, proteomics returned a plethora of other potential substrates of CatA, including decorin. Processed forms of decorin protein core have been shown to regulate the local bioavailability of profibrotic growth factors, like CTGF (31). Analysis of our proteomics data also indicated the possible participation of CatA in the processing of other structural ECM proteins, which may have contributed to the cardiac phenotype observed in CatA-TG mice. A comparable ECM proteolytic activity involving the degradation of laminin, fibronectin, elastin, and fibrillar collagens also has been described for the larger family of cysteine-cathepsins (2, 3). Although our findings in CatA-TG mice are consistent with digestion of EC-SOD by CatA, future biochemical studies are needed to investigate whether CatA degrades ECM proteins directly or via other intermediate molecules being activated or inhibited by CatA.
Conclusions
The present study sheds the first light on a previously unrecognized role of CatA outside the lysosome in the proteolysis of the extracellular antioxidant enzyme EC-SOD. In the heart, CatA-mediated reduction of EC-SOD levels resulted in oxidative stress because of insufficient removal of reactive oxygen species. Thus, EC-SOD degradation represents a plausible link between CatA activation and LV ECM remodeling and implicates CatA as a potential therapeutic target to prevent cardiac ECM remodeling.
Experimental procedures
Study approval
All animal studies were performed in accordance with the German law for the protection of animals. The investigation conforms with Guide for the Care and Use of Laboratory Animals, published by the United States National Institutes of Health (32) and the Declaration of Helsinki and was approved by the local animal ethics committee of the University of the Saarland (no. 21/2014).
Identification of candidate substrates of CatA by a proteomics approach
5-week-old male C57BL/6N mice (n = 4) were anesthetized with 5% isoflurane in 95% O2 and sacrificed by intraperitoneal (i.p.) injection of ketamine hydrochloride (100 mg/kg body weight) and xylazine hydrochloride (10 mg/kg body weight). For isolation and culture of primary CFs, primary mouse CFs were isolated from the hearts of 5-week-old male C57BL/6N mice (n = 4). Hearts were extracted, washed, and diced into small pieces, carefully washed in ice-cold PBS (Sigma-Aldrich) to remove plasma contaminants. The pieces were predigested in collagenase II solution (5 mg/ml) for 10 min. The collagenase II solution was replaced and the tissue pieces were incubated for 45–60 min at 37 °C. The digested tissue pieces were washed in complete medium (DMEM supplemented with 10% FBS, 2 mm l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin) before plating. CFs were cultured on 0.1% gelatin-coated T25 flasks in complete medium at 37 °C in a humidified incubator with 5% CO2 until 80% confluence. Cells were washed 3 times with serum-free medium and then incubated in serum-free medium for 3 days. The conditioned medium was centrifuged at 4000 rpm (3200 × g) for 10 min to remove cell debris. The supernatants were transferred into new tubes and stored at −80 °C until further analysis.
For digestion with CatA, conditioned media were concentrated and the buffer was exchanged to assay buffer (25 mm MES, 5 mm DTT, pH = 5.5) with a 3-kDa molecular weight cutoff spin column (Merck Millipore). Control samples (n = 4) were incubated in buffer with protease inhibitor only (for inhibition of endogenous proteases other than CatA). Treated samples (n = 4) were incubated in buffer with protease inhibitor only (for inhibition of endogenous proteases other than CatA) plus human recombinant CatA (1 μg/ml, Sanofi-Aventis, Deutschland) (10). Both groups were incubated at 37 °C for 24 h with agitation (reaction took place inside the spin column). After the reaction, the samples were centrifuged. The degradation products in the <3-kDa fractions were collected and purified with C18 spin columns (Thermo Fisher Scientific). Peptides were analyzed by LC–MS/MS as previously described (33, 34). Peptides were separated by reverse phase chromatography (Acclaim PepMap100 C18, 75 μm Å by 25 cm) on a nanoflow LC system (Ultimate3000 RSLCnano, Thermo Fisher Scientific) equipped with a trap column (Acclaim PepMap100 C18, 300 μm Å by 5 mm). The chromatographic separation was performed with a mobile phase of HPLC-grade water containing 2% acetonitrile and 0.1% formic aid (eluent A) and a mobile phase of 80% acetonitrile, 20% HPLC-grade water, and 0.1% formic acid (eluent B) with a 70-min gradient (2% to 10% B in 3 min, 10% to 30% B in 34 min, 30% to 40% B in 3 min, 99% B for 10 min, 2% B for 20 min) at a flow rate of 350 nl/min. The column was coupled to an LTQ OrbitrapXL mass spectrometer (Thermo Fisher Scientific) with a nanospray source (Picoview, New Objective, Inc.). The MS acquisition involved 1 full MS scan over a mass-to-charge range encompassing m/z 400–1600 using the Orbitrap analyzer, followed by data-dependent collision-induced dissociation MS/MS scans of the 6 most intense ions detected in the full scan, with dynamic exclusion enabled and rejection of singly charged ions. The data were analyzed using Progenesis LC–MS software (version 4.1, Nonlinear Dynamics). The peak list of MS2 spectra from features with intensity of >2-fold change, p < 0.05 between the CatA treated group and control group, was exported and identified by Mascot (Matrix Science; version 2.3.01) with the following parameters: enzyme, none; database, UniProt/SwissProt mouse database (release 2012_03, 16,520 protein entries); precursor mass tolerance, 10 ppm; fragment mass tolerance, 0.8 Da; fixed modification, none; variable modification, none. Search results were loaded back to Progenesis to match with the significantly changed features. Peptides with a Mascot score of >10 were used for quantification, and the final protein table was exported (Table S1).
Isolation of primary adult mouse CMs and CFs for gene expression analysis
Isolation of adult mouse CFs and cardiomyocytes (CMs) was described elsewhere (35, 36). In short, 6-week-old male C57BL/6N mice (n = 10) were anesthetized with 5% isoflurane in 95% O2 and sacrificed by intraperitoneal (i.p.) injection of ketamine hydrochloride (100 mg/kg body weight) and xylazine hydrochloride (10 mg/kg body weight). To isolate primary mouse CFs, hearts were extracted, washed, diced on ice into small pieces, and carefully washed in ice-cold PBS (1× PBS, 137 mmol/L NaCl, 2.7 mmol/L KCl, 10 mmol/L Na2HPO4, 1.8 mmol/L KH2PO4). The pieces were transferred to a sterile glass beaker and digested using a 25-ml collagenase II digestion solution (100 units/ml collagenase II [no. LS004176; Worthington] in Hanks balanced salt solution [HBSS] buffer [0.137 M NaCl, 5.4 mm KCl, 0.25 mm Na2HPO4, 0.1 g glucose, 0.44 mm KH2PO4, 1.3 mm CaCl2, 1.0 mm MgSO4, 4.2 mm NaHCO3] and 2.5% Trypsin [no. L2133, Merck Millipore]) under constant stirring at 37 °C for 10 min. Supernatant was collected in a 50-ml falcon tube containing 2 ml complete medium (M199 + GlutaMax [Gibco, no. 41150-020] supplemented with 10% FBS [Gibco, no. 16170-07], 100 units/ml penicillin, 100 μg/ml streptomycin [Gibco, no. 15140-122]). Digestion was repeated 10 times until tissue was completely dissolved. Cardiomyocytes were collected by sedimentation for 8–10 min, and the pellet was snap-frozen in liquid nitrogen for subsequent RNA isolation. Supernatants were collected as described above, and CFs were pelleted by centrifugation for 5 min at 300 × g at 4 °C. CFs were cultured on 6-well tissue culture dishes (VWR, TPPA92006) in complete medium at 37 °C in a humidified incubator with 5% CO2 until 80% confluence. mRNA was isolated from mouse CMs and mouse CFs using peqGold TriFast (no. 30-2010; PeqLab) extraction reagent by following the manufacturer´s protocol.
Isolation of primary neonatal rat CMs and CFs
Neonatal rat CMs and CFs were isolated from 5-day-old Sprague-Dawley rat hearts (Charles River, Germany) of mixed sex. Hearts were removed, and the ventricles were dissected and digested in ADS buffer containing (in mmol/L) NaCl 116, HEPES 20, Na2HPO4 0.8, glucose 5.6, KCl 5.4, MgSO4 × 7H2O 0.8, pH 7.35, 0.6 mg/ml Pankreatin (Sigma, P-3292), and 0.5 mg/ml collagenase type 2 (Worthington Biochemical; no. LS004176) at 37 °C in a water bath with constant stirring at 80–100 rpm for 5 min. Supernatant (not tissue) was transferred in a 50-ml Falcon tube containing 2 ml neonatal calf serum (NCS; Gibco no. 16010-159) to stop the enzymatic reaction. The procedure was repeated 6 times, all supernatants were collected, and the cells were preplated on 6-well plates (Falcon no. 353846, BD, Franklin Lakes, NJ, USA) in F10 medium (Gibco; with 10% horse serum, 5% FCS, and 1% penicillin/streptomycin) to allow separation of cardiac fibroblasts by adhesion. After 45 min, the still-suspended neonatal cardiomyocytes were removed from the attached cardiac fibroblasts, counted (Neubauer counting chamber), and plated at a density of 1.65 × 106 to 1.75 × 106 cells per 60-mm culture dish (no. 93060, TPP, Switzerland) in complete F10 medium at 37 °C in a humidified incubator with 5% CO2 until 80% confluence. The attached neonatal cardiac fibroblasts were grown in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) fetal calf serum, gentamicin (0.08 mg/ml), and penicillin (100 IU/ml) at 37 °C in a humidified incubator with 5% CO2 until 80% confluence. Afterward, both cell lines were kept in serum-free medium for 48 h. Medium and cells were harvested and stored at −80 °C until further processing.
CatA-TG mouse model
For the generation of transgenic mice that overexpress the human CatA specifically in cardiomyocytes, a vector containing the mouse alpha MHC promoter (5.7 kb) driving the human CatA minigene (cDNA clone ID CLN16325899) was constructed and described elsewhere (11). Kozak translation initiation sequence and SV40pA (249 bp) were introduced to enhance transgene expression. A LoxP-hUBp-em7- neo-loxP cassette (2648 bp) was inserted downstream of the stop codon for selection. VelociMouse® technology (Regeneron) was used to target embryonic stem cells and microinject them into mouse embryos (for details, refer to references 11 and 37). In brief, F1H4 (129S6SvEv/C57BL6F1) embryonic stem cells were electroporated with the linearized vector construct, and positive clones were microinjected into 8-cell-stage mouse C57BL/6N embryos. Pseudopregnant recipient female mice were anesthetized with 2.5% avertin (2,2,2-tribromoethanol; Fluka 90710, Aldrich Chemical) in tert-amyl alcohol administered i.p. at a dose of 0.1 ml/10 g body weight, and embryos were transferred to uteri by microinjection, weaned pups were scored, and high-percentage-chimera males were selected for mating with flp-positive C57B/L6N females to remove the selection cassette, to prove germ-line transmission, and to generate F1 animals for further breeding. Hemizygote animals were identified by genomic tail DNA probed with PCR primers 5'-AATCTCTATGCCCCGTGTGC-3′ (F) and 5′-GGCAGGCGAGTGAAGATGTT-3′ (R). The copy number of the transgene (4 copies) was estimated by a quantitative PCR assay for the inserted transgene that determines the normalized average difference in threshold cycle among the transgenic ES cell clones and the mice derived from them. Mice were kept and bred under specific-pathogen-free conditions (SOPF) in the animal care facility.
Working heart
Working heart experiments with isolated hearts of 6-month-old male CatA-TG mice and their male littermates were performed as previously described (38). Mice were anesthetized with 5% isoflurane in 95% O2 and sacrificed by i.p. injection of ketamine hydrochloride (100 mg/kg body weight) and xylazine hydrochloride (10 mg/kg body weight). The aorta was cannulated with an 18-G metal cannula for a Langendorff retrograde perfusion mode (baseline, 80 mmHg perfusion pressure) with Krebs-Henseleit buffer in a working heart apparatus (IH-SR perfusion system type 844/1, Hugo Sachs Elektronik). After establishing coronary perfusion in the Langendorff mode, a “working heart” preparation was continued by cannulating the left atrium through the pulmonary vein with a 16-G steel cannula. This cannula was connected to a preload column, which was water-jacketed and heated to 38 °C, resulting in a myocardial temperature of 37 °C when the heart was operating in the working mode (preload, 10 mmHg; afterload, 60 mmHg). Two platinum pacing electrodes embedded in polyester resin were attached to the right atrium to pace the hearts at about 400 bpm. LV systolic and diastolic function was recorded via a high-fidelity pressure-conductance catheter (Millar 1.4 F SPR-835, Millar) inserted into the LV cavity through a small puncture in the apex made with a 22 1/4 gauge needle. Cardiac inflow and aortic flow were recorded continuously by inline ultrasonic transit time probes, and these measurements were used to calibrate volume measurement of the conductance catheter signal gain. Parallel conductance (conductance signal offset) was determined by the saline dilution method by injecting a 5-ml bolus of hypertonic (5%) saline into the left atrial cannula, causing a transient change in the conductivity of Krebs-Henseleit buffer in the LV cavity.
Detection of superoxide using electron spin resonance spectroscopy
Production of superoxide in the left ventricle was measured using electron spin resonance (ESR) spectroscopy as described previously (39). Briefly, left ventricular tissue was placed in Krebs-HEPES buffer containing 25 μmol/L deferoxamine (Noxygen) and 5 μmol/L diethyldithiocarbamic acid (DETC, Noxygen) and incubated with 1-hydroxy-3-methoxycarbonyl-2,-2,-5,-5-tetramethylpyrrolidine (CMH, 500 μmol/L) as a spin trap for one hour. ESR spectra were recorded using a Bruker e-scan spectrometer (Bruker Biospin) with the following settings: center field, 1.99 g; microwave power, 20mW; modulation amplitude, 2 G; sweep time, 60 s; field sweep, 60 G.
Histology and immunohistochemical staining
Hearts were rapidly removed, trimmed free from noncardiac tissues, and weighed. Thereafter, the heart was fixed in buffered 4% formaldehyde for 24 h and embedded in paraffin for histological evaluation. Tissue sections of 4 μm were fixed at 56 °C overnight, deparaffinized, rehydrated, and stained with hematoxylin and eosin (HE) (hematoxylin, no. 10228.01000, Morphisto GmbH, Frankfurt am Main, Germany and Eosin Y; no. 1.15935.0025, Merck, Darmstadt, Germany) to determine cardiomyocyte diameter. To visualize tissue fibrosis amounts, the sections were stained with Picro-Sirius Red (no. 13422.00500, Morphisto GmbH, Germany). The percentage of LV consisting of interstitial collagen was calculated as the ratio of Picro-Sirius Red positively stained area over total LV tissue area, excluding blood vessels and the epi- and endocardial plane. For LV stained with Picro-Sirius Red, polarization microscopy was performed to visualize collagen type I (yellow-red fibers) and type III (green fibers) based on the birefringence properties of collagen (40). Perivascular fibrosis was evaluated as the ratio of fibrosis surrounding the vessel wall to total vessel area. For the analysis, NIS-Elements (BR 3.2, Nikon instruments) was used.
For immunostaining of CatA, heart tissue was fixed in buffered 4% formaldehyde for 24 h and embedded in paraffin for histological evaluation. Tissue sections of 4 μm were mounted on glass slides, deparaffinized with xylene, and hydrated in descending concentrations of ethanol. Following hydration, sections were incubated for 1 h in 0.05% citraconic anhydride (Sigma-Aldrich) at 98 °C in a water bath and washed afterward in 1× PBS-Tween (PBS containing 0.1% Tween) for 10 min at room temperature. Slides were incubated with anti-cathepsin A antibody (R&D Systems, no. AF1049; dilution, 1:30) in 1× PBS-Tween overnight in a moisture chamber at 4 °C. Sections were washed 3× for 5 min each time with 1× PBS-Tween, and secondary antibody (biotin-labeled anti-goat-IgG; dilution, 1:30) was added for 2 h and incubated in the moisture chamber at 37 °C. Slides were washed 3× for 5 min each time with 1× PBS-Tween, incubated with a tertiary streptavidin-peroxidase antibody (no. SA202; Chemicon/Millipore) in 1× PBS-Tween, and incubated for 20 min in a moisture chamber. After washing as before, slides were incubated shortly with AEC-chromogen (DAKO; no. K3464) at room temperature and rinsed with Aqua Dest, followed by staining with hematoxylin for 5 min and bluing with tap water for 15 min to allow staining. Slides were mounted with Aquatex (Merck) and analyzed.
TUNEL assay
To assess the amount of apoptotic cells, we performed TUNEL (ApopTag®, Chemicon) assay by following the manufacturer's protocol. In short, tissue sections of 4 μm were mounted on glass slides, deparaffinized with xylene, and hydrated in descending concentrations of ethanol. Slices were treated with proteinase K solution (20 μg/ml) for 30 min and washed twice with double-distilled water (ddH2O). Afterward, slices were incubated with 3% hydrogen peroxide solution for 5 min, washed twice with PBS solution, and incubated with working strength TdT enzyme solution for 60 min at 37 °C in a moisture chamber. Slices were washed for 10 min with a stop/wash buffer and incubated with anti-digoxigenin conjugate peroxidase for 30 min. Afterward, slices were stained with AEC-chromogen (3-amino-9-ethylcarbazole; no. K3464 DAKO), washed with ddH2O, and staining with hematoxylin for 5 min and bluing with tap water for 15 min to allow staining. Slides were then mounted with Aquatex (Merck) and analyzed.
For immunostaining of F4/80 and Ly-G6, heart tissue was fixed in buffered 4% formaldehyde for 24 h and embedded in paraffin for histological evaluation. Tissue sections of 4 μm were mounted on glass slides, deparaffinized with xylene, and hydrated in descending concentrations of ethanol. Slides were washed with Aqua Dest for 1 min and incubated in 3% H2O2 (diluted in Aqua Dest) for 5 min and then rinsed again in Aqua Dest and incubated at 95 °C for 20 min in 0.01 mol/L sodium citrate buffer (pH 6.0) containing 0.05% Tween 20. After 10 min of cooling, slides were washed twice with Aqua Dest, followed by 1× PBS for 5 min at room temperature. Afterward, slides were incubated for 30 min in TNB buffer (0.1 mol/L, Tris-HCL [pH 7.5] containing 0.15 mol/L NaCl and 0.5% blocking reagent [Perkin Elmer no. FP1020]) at room temperature. After removal of TNB buffer, primary antibodies were incubated overnight at 4 °C (anti-mouse F4/80, eBioscience, no. 14-4801, diluted 1:50 in TNB buffer, and anti-mouse Ly-6G [Gr-1], eBioscience, no. 14-5931, diluted 1:50 in TNB buffer), for at least 15 h, followed by 1 h of incubation at 37 °C. Slides were washed twice with 1× PBS for 5 min and incubated with secondary antibody conjugated to biotin (Santa Cruz, sc-2041, diluted 1:200 in TNB buffer) for 30 min. Slides were washed twice with 1× PBS for 5 min and incubated with labeled streptavidin-HRP antibody (Perkin Elmer, no. FP1047), diluted 1:100 in TNB buffer, for 30 min, washed as before and incubated for 10 min with biotinyl tyramide diluted (Perkin Elmer, no. FP1019) at 1:50 in amplification diluent (Perkin Elmer, no. FP1050). After washing twice with 1× PBS for 5 min, slides were incubated in SA-fluorophore (Texas red streptavidin, Vector Laboratories, no. SA-5006, diluted 1:50 in HEPES buffer) for 30 min at 37 °C. After washing twice with 1× PBS for 5 min, slides were covered with DAPI-containing mounting medium (Vector Laboratories, Vectorshield, no. H-1500) and analyzed.
PCR
Gene expression analysis was performed by real-time PCR. Total RNA was extracted from left ventricular tissue of mouse using peqGold TriFast (no. 30-2010; Peqlab) extraction reagent per the manufacturer´s protocol. Genomic DNA impurities were removed by DNase treatment (Peqlab), and cDNA was synthesized by reverse transcription using the HighCap cDNA RT kit (no. 4368814; Applied Biosystems) according to the manufacturer´s protocol. TaqMan PCR was conducted in a StepOne plus thermocycler (Applied Biosystems) using TaqMan GenEx Mastermix (no. 4369016, Applied Biosystems). Signals were normalized to corresponding glyceraldehyde-3-phosphate dehydrogenase (GAPDH) controls. No-template controls were used to monitor for contaminating amplifications. The ΔCT was used for statistical analysis and 2−ΔΔCT for data presentation. Mouse probes used to amplify the transcripts were as the following (purchased by Applied Biosystems Life Technologies): GAPDH (Mm99999915_g1), cathepsin A (Mm00447197_m1), TGFb1 (Mm03024053_m1), CTGF (Mm01192931_g1), Col5a1 (Mm00489342_m1), Col1a2 (Mm01165187_m1), Col3a1 (Mm01254476_m1), fibronectin 1 (Mm01256734_m1), Lox (Mm00495386_m1), ANP (Mm01255747_g1), BNP (Mm01255770_g1), EC-SOD (Sod3: Mm01213380_s1), TNFα (Mm00443258_m1), interleukin 6 (Mm00446190_m1), interleukin 2 (Mm00434256_m1), interleukin 10 (Mm00439614_m1), interleukin 1 beta (Mm00434228_m1), Bcl-2 (Mm00477631_m1), Bak-1 (Mm00432045_m1), Bax (Mm00432051_m1), alpha MHC (Mhy6: Mm00440359_m1), Neu1 (Mm00456846_m1), and lamp1 (Mm00495262_m1). For analysis of human cathepsin A, Hs00264902_m1 (CTSA) was used.
Western blot analysis
Isolated cardiomyocytes and cardiac fibroblasts, as well as left ventricular tissue, were homogenized in lysis buffer (Tris-HCl 100 mmol/L, 4% SDS, 20% glycine) containing complete protease inhibitors (no. 11873580001; Roche) and 1 mm PMSF and centrifuged at 16,000 × g for 10 min. 50 μg of protein was separated on 10% SDS-PAGE and electrophoretically transferred to nitrocellulose membranes (0.2-μm pore size, Schleicher and Schuell). Membranes were blocked in TBS containing 5% nonfat dry milk for at least 120 min at room temperature and exposed to the following primary antibodies overnight: anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Millipore, MAB374), anti-superoxide dismutase-1 (SOD1; Santa Cruz Biotechnology, sc-11407), anti-superoxide dismutase 2 (SOD2; Santa Cruz Biotechnology, sc-30080), anti-superoxide dismutase 3 (EC-SOD, SOD3, Santa Cruz Biotechnology, sc-32222), anti-rat SOD3 (rat EC-SOD, R&D Systems, AF4817), anti-cathepsin A (R&D Systems, AF1049). For rats, we used anti-CTSA (Sigma, HPA031068), anti-collagen 1a2 (Santa Cruz Biotechnologies, sc-393573), anti-catalase (Cell Signaling Technology, no. 14097), anti-Nox2/gp91phox (ERP6991) (abcam; ab129068), anti-NADPH oxidase 4 (abcam ab154244), anti-xanthine oxidase (Santa Cruz Biotechnologies, sc-398548), anti-caspase 3 (Cell Signaling Technology, no. 9662), anti-caspase 1 (Cell Signaling, no. 24232), anti-Bcl-2 (Santa Cruz Biotechnology, sc-492), and anti-Bax (Santa Cruz Biotechnology, Sc-7480). Respective secondary antibodies (purchased from Sigma; anti-mouse, no. A5278; anti-rabbit, no. A6154; anti-goat, A5420) were incubated for 60 min at room temperature. Proteins were visualized by enhanced chemiluminescence according to the manufacturer´s guidelines (no. RPN2106, Amersham Biosciences) and analyzed using the Fusion SL gel documentation system (Peqlab). Data are presented as intensity optical density (IOD).
Cathepsin A activity assay
CatA activity was measured using the fluorogenic peptide substrate V (R&D Systems; no. ES005) according to the manufacturer´s protocol. Degradation of the fluorogenic peptide substrate Mca-R-P-P-G-F-S-A-F-K(Dnp)-OH by CatA was monitoring in a spectrofluorometer (Tecan®; Germany) at 330-nm excitation and 390-nm emission.
Statistical analysis
All data are expressed as means ± S.D. Statistical analysis was carried out using Prism software (Graph Pad version 7) or Progenesis® LC-MC software (Nonlinear Dynamics) for proteomics. An unpaired Student´s t test (two-tailed) was used for statistical analyses comparing 2 groups. p values of <0.05 and fold change of >2 (for proteomics) were considered statistically significant.
Data availability
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (41) partner repository with the data set identifiers PXD019895. All other data are contained within the article (and supplemental information).
Supplementary Material
Acknowledgments
We thank Jeannette Zimolong, Nina Rebmann, Julia Weber, Sarah Triem, and Claudia Noll for excellent technical support. We thank Sanofi for providing CatA-TG mice.
This article contains supporting information.
Author contributions—M. H., M. M., C. G., S. R., T. H., C. M., M. B., T. Sadowski, and D. L. conceptualization; M. H., M. M., C. M., M. B., T. Sadowski, and D. L. supervision; M. H., M. M., J. B. B., X. Y., T. Speer, J.-C. R., C. M., and M. B. validation; M. H., L. L., A. G. N., J. B. B., X. Y., T. Speer, S. R. S., K. E., C. F.-T., J.-C. R., and B. L. investigation; M. H. and D. L. writing-original draft; M. M. software; M. M. methodology; S. R. S. visualization; C. G. writing-review and editing; S. R., T. H., T. Sadowski, and D. L. resources; T. Sadowski and D. L. funding acquisition; M. H. performed experiments.
Funding and additional information—This work was supported by the German Society of Cardiology (DGK0914), German Heart Foundation (F0315), Else-Kröner-Fresenius Foundation (2014A306), and German Research Foundation (SFB TRR219-M02/M04/S02/C09/C02). Lisa Lang received a scholarship by the Stiftung Begabtenförderung berufliche Bildung (SBB) GmbH. Professor Manuel Mayr is a British Heart Foundation (BHF) Chair Holder (CH/16/3/32406) with BHF program grant support (RG/16/14/32397) and member of a network on “Defining the Roles of Smooth Muscle Cells and Other Extracellular Matrix Producing Cells in Late Stage Atherosclerotic Plaque Pathogenesis,” funded by the Foundation Leducq.
Conflict of interest—Sven Ruf, Thomas Hübschle, and Thorsten Sadowski are employees of Sanofi-Aventis Deutschland GmbH. The other authors have not conflicts of interest.
- ECM
- extracellular matrix
- CMs
- cardiomyocytes
- LV
- left ventricular
- EC-SOD
- extracellular antioxidant enzyme superoxide dismutase
- CTGF
- connective tissue growth factor
- ROS
- reactive oxygen species
- ANP
- atrial natriuretic peptide
- BNP
- brain natriuretic peptide
- ESR
- electron spin resonance
- LVEDV
- LV end diastolic volume.
References
- 1. Souders C. A., Bowers S. L., and Baudino T. A. (2009) Cardiac fibroblast: the renaissance cell. Circ. Res. 105, 1164–1176 10.1161/CIRCRESAHA.109.209809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Müller A. L., and Dhalla N. S. (2012) Role of various proteases in cardiac remodeling and progression of heart failure. Heart Fail. Rev. 17, 395–409 10.1007/s10741-011-9269-8 [DOI] [PubMed] [Google Scholar]
- 3. Cheng X. W., Shi G. P., Kuzuya M., Sasaki T., Okumura K., and Murohara T. (2012) Role for cysteine protease cathepsins in heart disease: focus on biology and mechanisms with clinical implication. Circulation 125, 1551–1562 10.1161/CIRCULATIONAHA.111.066712 [DOI] [PubMed] [Google Scholar]
- 4. Everts V., van der Zee E., Creemers L., and Beertsen W. (1996) Phagocytosis and intracellular digestion of collagen, its role in turnover and remodeling. Histochem. J. 28, 229–245 10.1007/BF02409011 [DOI] [PubMed] [Google Scholar]
- 5. Timur Z. K., Demir A., and Seyrantepe V. (2016) Lysosomal cathepsin A plays a significant role in the processing of endogenous bioactive peptides. Front. Mol. Biosci. 3, 68 10.3389/fmolb.2016.00068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jackman H. L., Massad M. G., Sekosan M., Tan F., Brovkovych V., Marcic B. M., and Erdos E. G. (2002) Angiotensin 1-9 and 1-7 release in human heart: role of cathepsin A. Hypertension 39, 976–981 10.1161/01.hyp.0000017283.67962.02 [DOI] [PubMed] [Google Scholar]
- 7. Hiraiwa M. (1999) Cathepsin A/protective protein: an unusual lysosomal multifunctional protein. Cell Mol. Life Sci. 56, 894–907 10.1007/s000180050482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou X. Y., Galjart N. J., Willemsen R., Gillemans N., Galjaard H., and d'Azzo A. (1991) A mutation in a mild form of galactosialidosis impairs dimerization of the protective protein and renders it unstable. EMBO J. 10, 4041–4048 10.1002/j.1460-2075.1991.tb04980.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seyrantepe V., Hinek A., Peng J., Fedjaev M., Ernest S., Kadota Y., Canuel M., Itoh K., Morales C. R., Lavoie J., Tremblay J., and Pshezhetsky A. V. (2008) Enzymatic activity of lysosomal carboxypeptidase (cathepsin) A is required for proper elastic fiber formation and inactivation of endothelin-1. Circulation 117, 1973–1981 10.1161/CIRCULATIONAHA.107.733212 [DOI] [PubMed] [Google Scholar]
- 10. Ruf S., Buning C., Schreuder H., Horstick G., Linz W., Olpp T., Pernerstorfer J., Hiss K., Kroll K., Kannt A., Kohlmann M., Linz D., Hübschle T., Rütten H., Wirth K., et al. (2012) Novel ß-amino acid derivatives as inhibitors of cathepsin A. J. Med. Chem. 55, 7636–7649 10.1021/jm300663n [DOI] [PubMed] [Google Scholar]
- 11. Linz D., Hohl M., Dhein S., Ruf S., Reil J. C., Kabiri M., Wohlfart P., Verheule S., Böhm M., Sadowski T., and Schotten U. (2016) Cathepsin A mediates susceptibility to atrial tachyarrhythmia and impairment of atrial emptying function in zucker diabetic fatty rats. Cardiovasc. Res. 110, 371–380 10.1093/cvr/cvw071 [DOI] [PubMed] [Google Scholar]
- 12. Hohl M., Erb K., Lang L., Ruf S., Hübschle T., Dhein S., Linz W., Elliott A. D., Sanders P., Zamyatkin O., Böhm M., Schotten U., Sadowski T., and Linz D. (2019) Cathepsin A mediates ventricular remote remodeling and atrial cardiomyopathy in rats with ventricular ischemia/reperfusion. JACC Basic Transl. Sci. 4, 332–344 10.1016/j.jacbts.2019.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Petrera A., Gassenhuber J., Ruf S., Gunasekaran D., Esser J., Shahinian J. H., Hübschle T., Rütten H., Sadowski T., and Schilling O. (2016) Cathepsin A inhibition attenuates myocardial infarction-induced heart failure on the functional and proteomic levels. J. Transl. Med. 14, 153 10.1186/s12967-016-0907-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Abonnenc M., Nabeebaccus A. A., Mayr U., Barallobre-Barreiro J., Dong X., Cuello F., Sur S., Drozdov I., Langley S. R., Lu R., Stathopoulou K., Didangelos A., Yin X., Zimmermann W. H., Shah A. M., et al. (2013) Extracellular matrix secretion by cardiac fibroblasts: role of microRNA-29b and microRNA-30c. Circ. Res. 113, 1138–1147 10.1161/CIRCRESAHA.113.302400 [DOI] [PubMed] [Google Scholar]
- 15. Petersen S. V., Oury T. D., Ostergaard L., Valnickova Z., Wegrzyn J., Thøgersen I. B., Jacobsen C., Bowler R. P., Fattman C. L., Crapo J. D., and Enghild J. J. (2004) Extracellular superoxide dismutase (EC-SOD) binds to type I collagen and protects against oxidative fragmentation. J. Biol. Chem. 279, 13705–13710 10.1074/jbc.M310217200 [DOI] [PubMed] [Google Scholar]
- 16. Park S. K., Kim J., Seomun Y., Choi J., Kim D. H., Han I. O., Lee E. H., Chung S. K., and Joo C. K. (2001) Hydrogen peroxide is a novel inducer of connective tissue growth factor. Biochem. Biophys. Res. Commun. 284, 966–971 10.1006/bbrc.2001.5058 [DOI] [PubMed] [Google Scholar]
- 17. Sirker A., Zhang M., and Shah A. M. (2011) NADPH oxidases in cardiovascular disease: insights from in vivo models and clinical studies. Basic Res. Cardiol. 106, 735–747 10.1007/s00395-011-0190-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sivakumar P., Gupta S., Sarkar S., and Sen S. (2008) Upregulation of lysyl oxidase and MMPs during cardiac remodeling in human dilated cardiomyopathy. Mol. Cell. Biochem. 307, 159–167 10.1007/s11010-007-9595-2 [DOI] [PubMed] [Google Scholar]
- 19. Frangogiannis N. G. (2017) The extracellular matrix in myocardial injury, repair, and remodeling. J. Clin. Invest. 127, 1600–1612 10.1172/JCI87491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fan D., Takawale A., Lee J., and Kassiri Z. (2012) Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair 5, 15 10.1186/1755-1536-5-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jackman H. L., Tan F. L., Tamei H., Beurling-Harbury C., Li X. Y., Skidgel R. A., and Erdös E. G. (1990) A peptidase in human platelets that deamidates tachykinins. Probable identity with the lysosomal “protective protein.” J. Biol. Chem. 265, 11265–11272 [PubMed] [Google Scholar]
- 22. Brömme D., and Wilson S. (2011) Role of cysteine cathepsins in extracellular proteolysis. in Extracellular Matrix Degradation. Biology of Extracellular Matrix, (Parks W., Mecham R., eds) vol 2 Springer, Berlin, Germany. [Google Scholar]
- 23. P. C., Nantes I. L., Chagas J. R., Rizzi C. C., Faljoni-Alario A., Carmona E., Juliano L., Nader H. B., and Tersariol I. L. (2001) Cathepsin B activity regulation. Heparin-like glycosaminogylcans protect human cathepsin B from alkaline pH-induced inactivation. J. Biol. Chem. 276, 944–951 10.1074/jbc.M003820200 [DOI] [PubMed] [Google Scholar]
- 24. Olsen D. A., Petersen S. V., Oury T. D., Valnickova Z., Thøgersen I. B., Kristensen T., Bowler R. P., Crapo J. D., and Enghild J. J. (2004) The intracellular proteolytic processing of extracellular superoxide dismutase (EC-SOD) is a two-step event. J. Biol. Chem. 279, 22152–22157 10.1074/jbc.M401180200 [DOI] [PubMed] [Google Scholar]
- 25. Kliment C. R., and Oury T. D. (2011) Extracellular superoxide dismutase protects cardiovascular syndecan-1 from oxidative shedding. Free Radic. Biol. Med. 50, 1075–1080 10.1016/j.freeradbiomed.2011.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lu Z., Xu X., Hu X., Zhu G., Zhang P., van Deel E. D., French J. P., Fassett J. T., Oury T. D., Bache R. J., and Chen Y. (2008) Extracellular superoxide dismutase deficiency exacerbates pressure overload-induced left ventricular hypertrophy and dysfunction. Hypertension 51, 19–25 10.1161/HYPERTENSIONAHA.107.098186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van Deel E. D., Lu Z., Xu X., Zhu G., Hu X., Oury T. D., Bache R. J., Duncker D. J., and Chen Y. (2008) Extracellular superoxide dismutase protects the heart against oxidative stress and hypertrophy after myocardial infarction. Free Radic. Biol. Med. 44, 1305–1313 10.1016/j.freeradbiomed.2007.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kliment C. R., Suliman H. B., Tobolewski J. M., Reynolds C. M., Day B. J., Zhu X., McTiernan C. F., McGaffin K. R., Piantadosi C. A., and Oury T. D. (2009) Extracellular superoxide dismutase regulates cardiac function and fibrosis. J. Mol. Cell. Cardiol. 47, 730–742 10.1016/j.yjmcc.2009.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dewald O., Frangogiannis N. G., Zoerlein M., Duerr G. D., Klemm C., Knuefermann P., Taffet G., Michael L. H., Crapo J. D., Welz A., and Entman M. L. (2003) Development of murine ischemic cardiomyopathy is associated with a transient inflammatory reaction and depends on reactive oxygen species. Proc. Natl. Acad. Sci. U S A 100, 2700–2705 10.1073/pnas.0438035100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Petrera A., Kern U., Linz D., Gomez-Auli A., Hohl M., Gassenhuber J., Sadowski T., and Schilling O. (2016) Proteomic profiling of cardiomyocyte-specific cathepsin A overexpression links cathepsin A to the oxidative stress response. J. Proteome Res. 15, 3188–3195 10.1021/acs.jproteome.6b00413 [DOI] [PubMed] [Google Scholar]
- 31. Barallobre-Barreiro J., Gupta S. K., Zoccarato A., Kitazume-Taneike R., Fava M., Yin X., Werner T., Hirt M. N., Zampetaki A., Viviano A., Chong M., Bern M., Kourliouros A., Domenech N., Willeit P., et al. (2016) Glycoproteomics reveals decorin peptides with anti-myostatin activity in human atrial fibrillation. Circulation 134, 817–832 10.1161/CIRCULATIONAHA.115.016423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC [Google Scholar]
- 33. Yin X., Cuello F., Mayr U., Hao Z., Hornshaw M., Ehler E., Avkiran M., and Mayr M. (2010) Proteomics analysis of the cardiac myofilament subproteome reveals dynamic alterations in phosphatase subunit distribution. Mol. Cell. Proteomics 9, 497–509 10.1074/mcp.M900275-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barallobre-Barreiro J., Didangelos A., Schoendube F. A., Drozdov I., Yin X., Fernández-Caggiano M., Willeit P., Puntmann V. O., Aldama-López G., Shah A. M., Doménech N., and Mayr M. (2012) Proteomics analysis of cardiac extracellular matrix remodeling in a porcine model of ischemia/reperfusion injury. Circulation 125, 789–802 10.1161/CIRCULATIONAHA.111.056952 [DOI] [PubMed] [Google Scholar]
- 35. Zafiriou M. P., Noack C., Unsöld B., Didie M., Pavlova E., Fischer H. J., Reichardt H. M., Bergmann M. W., El-Armouche A., Zimmermann W. H., and Zelarayan L. C. (2014) Erythropoietin responsive cardiomyogenic cells contribute to heart repair post myocardial infarction. Stem Cells 32, 2480–2491 10.1002/stem.1741 [DOI] [PubMed] [Google Scholar]
- 36. Zafeiriou M. P., Noack C., and Zelarayan L. C. (2016) Isolation and primary culture of adult mouse cardiac fibroblasts. BioProtoc 6, 10.21769/BioProtoc.1860 [DOI] [Google Scholar]
- 37. DeChiara T. M., Poueymirou W. T., Auerbach W., Frendewey D., Yancopoulos G. D., and Valenzuela D. M. (2010) Producing fully ES cell-derived mice from eight-cell stage embryo injections. Methods Enzymol. 476, 285–293 10.1016/S0076-6879(10)76016-X [DOI] [PubMed] [Google Scholar]
- 38. Reil J. C., Hohl M., Reil G. H., Granzier H. L., Kratz M. T., Kazakov A., Fries P., Müller A., Lenski M., Custodis F., Gräber S., Fröhlig G., Steendijk P., Neuberger H. R., and Böhm M. (2013) Heart rate reduction by If-inhibition improves vascular stiffness and left ventricular systolic and diastolic function in a mouse model of heart failure with preserved ejection fraction. Eur. Heart J. 34, 2839–2849 10.1093/eurheartj/ehs218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nickel A. G., von Hardenberg A., Hohl M., Löffler J. R., Kohlhaas M., Becker J., Reil J. C., Kazakov A., Bonnekoh J., Stadelmaier M., Puhl S. L., Wagner M., Bogeski I., Cortassa S., Kappl R., Pasieka B., et al. (2015) Mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab. 22, 472–484 10.1016/j.cmet.2015.07.008 [DOI] [PubMed] [Google Scholar]
- 40. Nagaraju C. K., Dries E., Popovic N., Singh A. A., Haemers P., Roderick H. L., Claus P., Sipido K. R., and Driesen R. B. (2017) Global fibroblast activation throughout the left ventricle but localized fibrosis after myocardial infarction. Sci. Rep. 7, 10801 10.1038/s41598-017-09790-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D. J., Inuganti A., Griss J., Mayer G., Eisenacher M., Pérez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yilmaz S., et al. (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 10.1093/nar/gky1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (41) partner repository with the data set identifiers PXD019895. All other data are contained within the article (and supplemental information).