

Abstract
The discovery of activating epidermal growth factor receptor (EGFR) mutations spurred the use of EGFR tyrosine kinase inhibitors (TKIs), such as erlotinib, as the first-line treatment of lung cancers. We previously reported that differential degradation of TKI-sensitive (e.g. L858R) and resistant (T790M) EGFR mutants upon erlotinib treatment correlates with drug sensitivity. We also reported that SMAD ubiquitination regulatory factor 2 (SMURF2) ligase activity is important in stabilizing EGFR. However, the molecular mechanisms involved remain unclear. Here, using in vitro and in vivo ubiquitination assays, MS, and superresolution microscopy, we show SMURF2–EGFR functional interaction is important for EGFR stability and response to TKI. We demonstrate that L858R/T790M EGFR is preferentially stabilized by SMURF2-UBCH5 (an E3-E2)-mediated polyubiquitination. We identified four lysine residues as the sites of ubiquitination and showed that replacement of one of them with acetylation-mimicking glutamine increases the sensitivity of mutant EGFR to erlotinib-induced degradation. We show that SMURF2 extends membrane retention of EGF-bound EGFR, whereas SMURF2 knockdown increases receptor sorting to lysosomes. In lung cancer cell lines, SMURF2 overexpression increased EGFR levels, improving TKI tolerance, whereas SMURF2 knockdown decreased EGFR steady-state levels and sensitized lung cancer cells. Overall, we propose that SMURF2-mediated polyubiquitination of L858R/T790M EGFR competes with acetylation-mediated receptor internalization that correlates with enhanced receptor stability; therefore, disruption of the E3-E2 complex may be an attractive target to overcome TKI resistance.
Keywords: epidermal growth factor receptor (EGFR), Smad ubiquitination regulatory factor 2 (SMURF2), ubiquitin-conjugating enzyme H5 (UBCH5), protective ubiquitination, tyrosine kinase inhibitor (TKI) resistance, E3 ubiquitin ligase, ubiquitylation (ubiquitination), receptor regulation, tyrosine-protein kinase (tyrosine kinase)
There are two “classical” activating mutations, in-frame deletions in exon 19 and a point mutation in exon 21 (L858R) in EGFR, which drive adenocarcinoma of the lung in the majority of never smokers (1). The presence of these receptor mutations is a marker for sensitivity to treatment with epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs). First-generation TKIs, such as erlotinib and gefitinib, produce responses; however, within a year, tumors develop TKI resistance. In about 50% of these cases, resistance is because of the enrichment of a second point mutation at exon 20, where threonine at the 790 position is mutated to methionine (T790M) (2). To circumvent resistance, a third-generation EGFR inhibitor (AZD9291, osimertinib) has been generated, which is effective for patients carrying the T790M mutation (3). Unfortunately, with time, patients show osimertinib resistance because of multiple mechanisms (4). Despite significant efforts, the molecular mechanisms causing acquired TKI resistance are still poorly understood. In initial studies, differential drug-binding abilities and altered ATP-binding affinities of various EGFR mutants have been proposed to be responsible for differential TKI response (5). In the case of osimertinib resistance, an additional mutation at the tyrosine kinase domain of EGFR (C797S) has been implicated in about one-third of cases (6–8), and MET amplification has also been noted (4). However, these factors together may be only a part of the acquired resistance story.
TKI-resistant cell lines are sensitive to EGFR degradation or knockdown by RNAi, which suggests that the physical presence of EGFR independent of its kinase activity is important in cancer cell survival (9, 10). These findings led us to hypothesize that inducing EGFR degradation overcomes TKI resistance. We previously reported that EGFR degradation determines the response to radiation and chemotherapy and that targeting EGFR for degradation can induce cytotoxicity in cancer cells (11–18). We further elucidated the importance of EGFR degradation in head and neck cancer patients treated with erlotinib (19). Together, these findings led us to hypothesize that EGFR degradation is a key phenomenon in determining cytotoxicity to various chemotherapies, radiation, or TKI targeting EGFR. Differential EGFR degradation may contribute to the dramatic difference in sensitivity to TKIs of patients harboring L858R compared with T790M. Our data demonstrated that TKI-sensitive EGFR delE746-A759 or L858R mutant proteins undergo rapid degradation upon erlotinib treatment, whereas TKI-resistant L858R/T790M protein is highly stable (20).
Ubiquitin ligases and deubiquitinating enzymes, which play an important role in EGFR endocytosis, trafficking, and degradation, regulate EGFR stability (21). The most extensively studied E3 ligase responsible for EGF-mediated EGFR polyubiquitination is c-CBL (22). Additionally, AIP4/ITCH, pVHL, and UBE4B play roles in the polyubiquitination and degradation of EGFR (23, 24). We have reported that the HECT-type E3 ubiquitin ligase, Smad ubiquitination regulatory factor 2 (SMURF2), directly interacts with EGFR, but, unlike several others, SMURF2's ubiquitin ligase activity is critical for stabilizing EGFR (25). Here, we study the mechanistic and functional importance of SMURF2-mediated ubiquitination on different EGFR mutants, identify residues undergoing posttranslational modifications, and determine the relevance of mutant receptor membrane retention and protein stability, which together can predict TKI response. Additionally, we explore the impact of disruption of SMURF2 interaction with its cognate ubiquitin-conjugating enzyme (E2), UBCH5, on EGFR protein stability to overcome TKI resistance.
Results
EGFR (L858R+T790M) is a preferred substrate for SMURF2-UBCH5-mediated ubiquitination
We previously reported that SMURF2 ubiquitin ligase activity is critical in maintaining EGFR protein stability (25). To determine whether the SMURF2 can directly ubiquitinate EGFR and whether such modification can vary depending on the type of EGFR mutations, we performed in vitro ubiquitination assays by incubating either WT or different EGFR (either L858R [L] or L858R/T790M [L+T]) mutants with SMURF2. As SMURF2 partners with either UBCH7 or UBCH5 as the E2, we tested SMURF2 catalytic activity on EGFR in the presence or absence of either one of the E2s. As shown in Fig. 1A, we noted substantial polyubiquitination of L+T mutant EGFR compared with WT and L mutant. Additionally, the polyubiquitinated species formed in the presence of UBCH5 was of higher molecular weight than those formed in the presence of UBCH7; the latter we speculate to be autoubiquitinated SMURF2, as reported earlier (26). As we previously reported for WT EGFR (25), a catalytic-dead (C716A) mutant SMURF2 also failed to polyubiquitinate L+T mutant EGFR, supporting the hypothesis that EGFR is a SMURF2 substrate (Fig. 1B). To decipher whether preferential binding is responsible for SMURF2-mediated mutant EGFR ubiquitination, we performed immunoprecipitation studies. As shown in Fig. 1C, we noted comparable binding of SMURF2 with WT and L and L+T mutant EGFR. To understand the kind of ubiquitin linkage formed on L+T mutant EGFR, we utilized different ubiquitin mutants (K11R, K48R, and K63R) deficient in forming specific linkages. As shown in Fig. 1D, there was a significant reduction of mutant EGFR polyubiquitination only in the presence of K63R recombinant ubiquitin, suggesting SMURF2-mediated EGFR ubiquitination is predominantly K63 linked.
Figure 1.

EGFR (L858R/T790M) is a preferred substrate for SMURF2-UBCH5-mediated ubiquitination. A, purified EGFR proteins, either WT or mutants (L858R [L] and L858R/T790M [L+T]), were subjected to in vitro ubiquitination using recombinant SMURF2 as an E3 in the presence of either UBCH5 or UBCH7 as E2 enzymes. Following completion, reaction mixtures were subjected to immunoprecipitation using the EGFR antibody followed by immunoblotting using the indicated antibodies. B, CHO cells overexpressing (L+T) mutant EGFR alone or in the presence of WT or catalytic-dead (C716A) SMURF2 were immunoprecipitated with EGFR antibody followed by immunoblotting using the indicated antibodies. C, CHO cells, either vector control (−) or overexpressing EGFR (WT, L, and L+T), and SMURF2 were immunoprecipitated with either EGFR or SMURF2 antibodies and immunoblotted for the indicated antibodies. D, SMURF2-UBCH5-mediated in vitro ubiquitination of mutant EGFR (L+T) was performed as described above in the presence of WT or different ubiquitin mutants (K11R, K48R, and K63R) deficient in promoting specific linkages. Higher-molecular-weight ubiquitinated EGFR species were detected following immunoprecipitation and immunoblotting.
Acetylation mimicking K1037Q mutation in L+T mutant EGFR background makes the stable receptor vulnerable to TKI-mediated degradation
To determine the site(s) ubiquitinated in EGFR by SMURF2, we performed MS analysis of immunoprecipitated receptor and identified four lysine residues, K721, K846, K1037, and K1164 (Fig. S1, A–D, and Table S1). Interestingly, K721 and K846 are known to be important for EGFR kinase activity (27–29), and the latter two residues (K1037 and 1164) undergo acetylation necessary for receptor internalization (30), thereby indirectly impacting receptor degradation. Acetylation and ubiquitination counteract each other (31). To better understand the role of the two posttranslational processes in determining EGFR protein stability, we generated site-specific EGFR mutants of K1037, mutated either to arginine (R, can be neither ubiquitinated nor acetylated) or glutamine (Q, mimics acetylated lysine) (32, 33). As shown in Fig. 2A, the incorporation of the K-to-Q mutation in an L+T background converted a stable double mutant to unstable following erlotinib treatment with consequent accumulation of polyubiquitinated species (Fig. 2B). In contrast, conversion of K1037 to R affected the triple mutant EGFR steady-state levels to a lesser extent following erlotinib treatment (Fig. 2A).
Figure 2.

Incorporation of a K1037Q mutation in an L+T EGFR background makes stable receptor more vulnerable to TKI-mediated degradation. A, CHO cells overexpressing either WT or different mutants of EGFR (as indicated) were treated with 3 μm erlotinib for 24 h. Cell lysates were then subjected to immunoblot analysis with the indicated antibodies. Erlotinib-induced EGFR downregulation was calculated by quantifying band intensities (arbitrary units) considering the untreated level as 1. B, CHO cells transfected with indicated EGFR mutants were treated with erlotinib as described for panel A followed by immunoprecipitation using EGFR antibody and immunoblotting with the indicated antibodies.
EGFR surface density depends on SMURF2 expression
Ubiquitination and acetylation counteract each other, and acetylation of EGFR has been associated with receptor internalization (30, 31). Therefore, we tested the hypothesis that SMURF2-mediated ubiquitination is important to maintain the surface density of EGFR by enhancing protein stability and thereby reducing internalization. We addressed this question by directly measuring the protein density in the membrane using stochastic optical reconstruction microscopy (STORM) (34, 35). STORM images were quantified using a pair correlation-based analytical method that can correct quantitative artifacts arising from overcounting. This method takes advantage of the observation that the magnitude of self-clustering that arises from overcounting is inversely proportional to the density of the labeled protein, as long as the labeled protein is sampled randomly. We calculated the autocorrelation function for reconstructed images and used the value of the autocorrelation function at different radii to determine the density of protein in the reconstructed image using Equation 1 described in Materials and methods.
We first tested the validity of our approach by quantifying EGFR levels in the membranes of multiple cell lines (CHO with no detectable EGFR and head and neck cancer cell lines UMSCC-11b, UMSCC-1, and UMSCC-29b), which express various amounts of receptor as assessed by conventional immunoblotting (Fig. S2A). As shown in Fig. S2, B and C, there was a high correlation (R2 = 0.9854) observed between the conventional immunoblotting and total internal reflection fluorescence (TIRF) microscopy, demonstrating applicability. To validate the methodology further, we measured the receptor population in a human breast cancer MCF7 cell line with known (1000–5000 per cell) numbers of EGFR (36). As shown in Fig. S2, D and E, we counted n = 900–1771 receptor molecules per cell (assuming a radius of 20–25 μm), consistent with the literature. The mean and standard deviation of cell-to-cell variation of EGFR density in MCF-7 was 0.86 ± 0.40 receptors/µm2.
Having established the reliability of the methodology, we next quantified EGFR surface density in the presence and absence of SMURF2. We conducted this experiment in both the presence and absence of EGF. EGF treatment caused a decrease in surface EGFR population arising from internalization of EGFR (Fig. S2F). The decrease in EGFR surface expression was greater following EGF treatment in SMURF2 siRNA treated cells (∼60% versus ∼40% loss) compared with control siRNA treatment, which was further demonstrated using immunoblotting of the same samples (Fig. S2F, lower). These findings show that there is a role for SMURF2 in maintaining EGFR membrane levels.
EGF treatment promotes EGFR-SMURF2 membrane coclustering
We previously reported a dynamic interaction between EGFR and SMURF2 (25), and here we found that knockdown of SMURF2 alters EGFR density, especially following EGF stimulation. This led us to hypothesize that EGFR-SMURF2 interaction occurs proximal to the plasma membrane. To test this hypothesis, we conducted immunoprecipitation and immunoblot analysis in cell-fractionated isolates from UMSCC-1 cells, using siRNA to alter SMURF2 levels. As expected, we observed EGFR was predominantly present in the plasma membrane, but SMURF2 was primarily cytosolic (Fig. 3A, right) with a small fraction present in the membrane. Interestingly, we found an increased EGFR-SMURF2 interaction in the membrane fraction following EGF treatment (Fig. 3A, left). Next, we conducted two-color STORM experiments to quantify interaction between EGFR and SMURF2 localized proximal to the plasma membrane (Fig. 3B). UMSCC-1 cells were grown normally in serum-containing media, and SMURF2 and EGFR levels were determined in the presence and absence of EGF (10 ng/ml, 30 min). Interestingly, upon stimulation with EGF, we observed an increase (2.9 ± 0.3-fold) in SMURF2 coclustering with EGFR to the plasma membrane (Fig. 3C). SMURF2 density in the membrane went up almost 4-fold in the membrane, in line with the cell fractionation studies.
Figure 3.
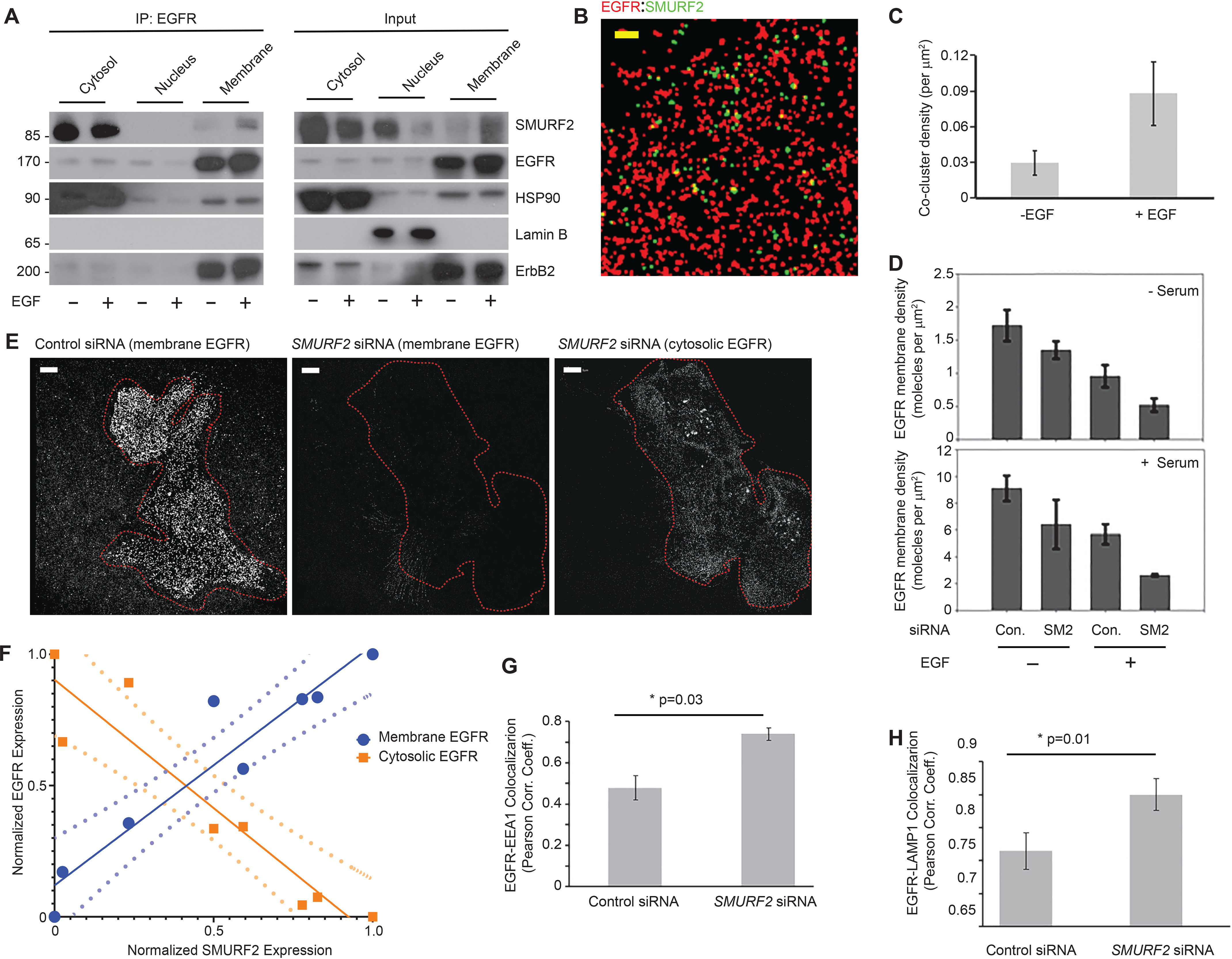
EGFR membrane density depends on SMURF2 expression, and EGF treatment promotes EGFR-SMURF2 coclustering. A, UMSCC-1 cells were either left untreated or treated with EGF (10 ng/ml for 6 h). Cell lysates were then subjected to fractionation into cytosol, nuclear, and membrane fractions. The fractionated samples were then immunoprecipitated using EGFR antibody followed by immunoblotting as indicated. To demonstrate fractionation efficiency, Hsp90, Lamin B, and ErbB2 were used as markers for cytosolic, nuclear, and membrane-bound fractions, respectively. B, representative STORM image showing EGFR and SMURF2 coclustering on UMSCC-1 cell surface (−EGF, +serum) following immunostaining. Scale bar, 5 μm. C, an increase in EGFR-SMURF2 cocluster density was observed following EGF treatment (10 ng/ml for 6 h). D, quantification of EGFR membrane density in UMSCC-1 cells transfected either with control (Con.) or SMURF2 (SM2) siRNA and grown in the presence and absence of serum and treated with EGF (10 ng/ml for 30 min) where indicated. E, membrane-proximal EGFR levels were compared for UMSCC-1 cells transfected with either control or SMURF2 siRNA using immunofluorescence staining 48 h posttransfection and analyzed using superresolution microscopy. Left, reconstructed image showing EGFR density in plasma membrane of a cell transfected with control siRNA. Middle and right, reconstructed images of both plasma membrane and cytosolic EGFR of a cell transfected with SMURF2 siRNA. F, positive and negative correlation of membrane (0.92 [0.12]) and cytosolic (−0.98 [0.14]) EGFR levels, respectively, with the cytosolic expression of SMURF2 in cells. Values in parentheses indicate means (standard deviations). The relative expression of proteins in cells were quantified using epifluorescence images for SMURF2 and EGFR (cytosol) and by imaging the same cell in the TIRF mode for the membrane expression of EGFR. The dotted lines represent the 95% confidence interval when the data were fitted with a linear regression model. G and H, UMSCC-1 cells transfected with either control or SMURF2 siRNA were costained either with EGFR and EEA1 (F) or with EGFR and LAMP1 (G). Following imaging, Pearson correlation coefficients were determined for either EGFR and EEA1 (p = 0.03) or EGFR and LAMP1 (p = 0.01) as a measure of colocalization between EGFR and the marker. Pearson correlation coefficients were calculated by drawing a mask outside the cell, and the two conditions (control versus SMURF2 siRNA) were compared for at least 10 cells for the analysis.
To better understand the protective role of SMURF2 in the retention of activated receptor in the cell membrane, we determined changes in EGFR density in the plasma membrane following SMURF2 knockdown both in the presence and absence of EGF. Surface EGFR molecules of UMSCC-1 cells grown in serum-containing medium were 5.3 ± 0.6-fold higher than those of cells grown under serum-free conditions (Fig. 3D). As previously noted, EGF (10 ng/ml for 30 min) treatment caused a decrease of 44.5% ± 3.3% and 37.2% ± 1.2%, respectively, for cells grown in serum-starved and serum-containing media, suggesting that EGF-induced EGFR internalization is independent of the growth conditions. However, the impact of SMURF2 loss was predominant in EGF-treated cells grown in serum-containing media; in the absence of EGF, SMURF2 knockdown caused a decrease of 21.1% ± 2.3% and 29.3% ± 3.4% in serum-starved and serum-containing conditions, respectively. However, upon EGF treatment, we noted 46% ± 3.3% and 53% ± 4.3% decreases in EGFR surface expression depending on serum absence or presence.
As shown in Fig. 3E, loss of SMURF2 resulted in significant loss of surface EGFR. In contrast, there was an increase in the cytosolic EGFR levels when the same cell was imaged using superresolution imaging on a cross-sectional area of the cytosol using a greater penetration depth in the TIRF setup. However, because of clustering of EGFR, we were unable to use STORM microscopy to quantify receptor density in the cytosol. We circumvented this by investigating the relationship between fluorescence intensity of EGFR in the membrane and cytosol to that of SMURF2 in individual cells in nonreconstructed images. Two-color epifluorescence imaging was used to determine the fluorescence intensity of SMURF2 and EGFR in the cytosol, and TIRF imaging was used to measure the membrane fluorescence intensity of EGFR in individual cells. The intensities were then normalized and binned. As shown in Fig. 3F, membrane levels of EGFR are positively correlated with SMURF2 expression, whereas cytosolic EGFR levels are inversely correlated. Together, as the loss of surface EGFR was more prevalent upon SMURF2 knockdown upon EGF treatment, our data indicate that SMURF2 better protects ligand-activated EGFR, which otherwise undergoes receptor internalization.
As EGF treatment promotes endosome-mediated receptor internalization followed by lysosome-mediated degradation, we tested the effects of SMURF2 loss on EGFR cytosolic trafficking. Although TIRF imaging can be used to quantify EGFR membrane expression, clustering of receptors in cytosolic organelles is less reliable. Therefore, we relied on conventional two-color colocalization studies of EGFR with EEA1 (as an endosomal marker) and LAMP1 (as a lysosomal marker). A Pearson correlation coefficient was calculated by drawing a mask of the cell, and the two (control versus SMURF2 siRNA) conditions were compared for at least 10 cells. Upon SMURF2 knockdown, we found an increase in cytosolic EGFR colocalization both with EEA1 (p = 0.03) and LAMP1 (p = 0.01) (Fig. 3, G and H, and Fig. S3, A and B).
SMURF2 levels dictate TKI sensitivity in lung cancer cells
To understand the importance of SMURF2-mediated protection of mutant EGFR in regulating TKI sensitivity, we utilized either SMURF2 overexpression or siRNA-mediated knockdown in multiple lung adenocarcinoma cell lines to test their sensitivity or resistance to TKI, respectively. Erlotinib-sensitive PC9 cells harboring exon 19 deletion mutation in EGFR showed increased erlotinib tolerance (IC50 of 8.42 ± 2.79 nm for parental compared with 66.6 ± 31.4 nm upon SMURF2 overexpression) in clonogenic survival assays (Fig. 4A). In contrast, siRNA-mediated loss of SMURF2 further sensitized PC9 cells (IC50 of 6.0 ± 2.3 nm upon SMURF2 loss compared with 19.6 ± 1.3 nm in control siRNA-treated cells) (Fig. 4B). In erlotinib-resistant NCI-H1975, cells (with L858R/T790M mutations) became slightly more sensitive (IC50 of 4.62 ± 1.0 μm reduced to 2.0 ± 0.4 μm) following siRNA-mediated SMURF2 knockdown (Fig. 4E). A PC9 clone resistant to a third-generation TKI, AZD9291 (PC9-AR), upon SMURF2 overexpression showed increased cell death, with minimal change in AZD9291 toxicity (IC50 of 3.8 ± 1.1 and 4.4 ± 1.02 μm respectively) (Fig. 4G). In contrast, PC9-AR cells became sensitive (IC50 of 1.5 ± 0.08 μm compared with 2.6 ± 0.1 μm) to the drug treatment following siRNA-mediated SMURF2 loss (Fig. 4H). In all these cases, SMURF2 alterations also impacted EGFR steady-state levels (Fig. 4, C, D, F, I, and J) that correlated with drug sensitivity. These data support the critical importance of SMURF2 in maintaining mutant EGFR protein stability and further identify SMURF2 targeting as a novel approach to target mutant EGFR to overcome TKI resistance.
Figure 4.
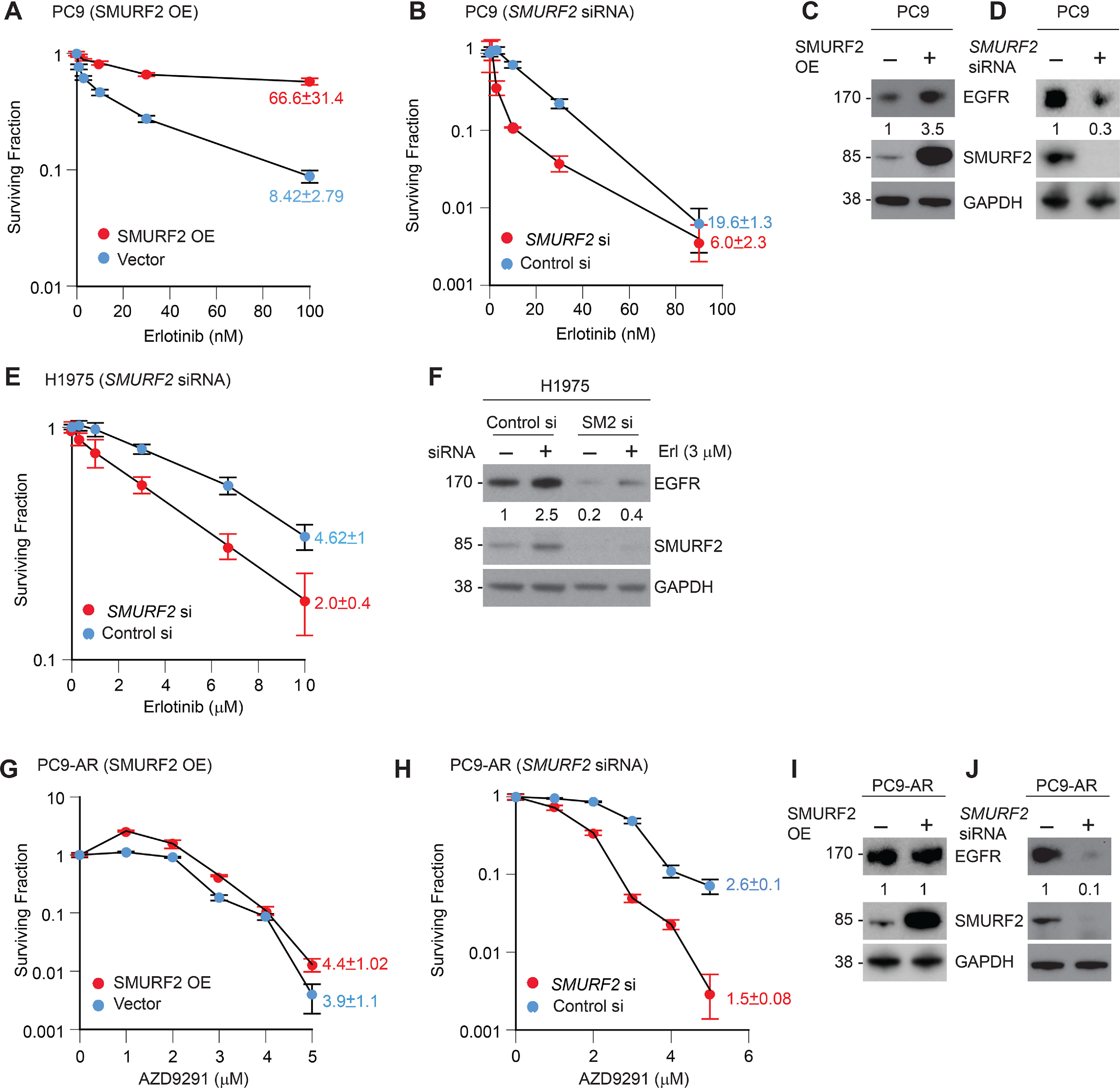
SMURF2 levels dictate TKI sensitivity in lung cancer cells. A and B, SMURF2 was either overexpressed (OE) (A) or subjected to siRNA knockdown (B) in erlotinib-sensitive PC9 lung adenocarcinoma cells (containing EGFR exon 19 deletion), and clonogenic cell survival assays were performed to test erlotinib sensitivity. C and D, immunoblot analysis of PC9 cells overexpressing SMURF2 showing increased EGFR accumulation (C) and loss of EGFR following SMURF2 knockdown (D) after 48 h of transfection. E, clonogenic survival of erlotinib-resistant NCI-H1975 cells following control and SMURF2 siRNA-mediated knockdown. F, corresponding immunoblot analysis showing SMURF2 and corresponding EGFR loss, which correlates with the change in survival. G, SMURF2 was overexpressed in a PC9 clone resistant to a third-generation TKI (AZD9291 resistant, PC9-AR), which resulted in slight resistance to AZD9291 treatment. H, in contrast, PC9-AR cells became sensitive to the drug treatment following siRNA-mediated SMURF2 knockdown. I and J, corresponding immunoblots from the above experiments confirming either SMURF2 overexpression (I) or loss (J) and its impact on EGFR steady-state levels. In panels A, B, E, G, and H, IC50 values were calculated from three independent experiments and presented as means ± S.E. In panels C, D, F, I, and J, fold changes in EGFR levels were calculated based on band intensities (arbitrary units) considering either vector-transfected or control siRNA-treated samples as 1.
Alteration of SMURF2-UBCH5 protein–protein interaction affects mutant EGFR levels
Although the finding that SMURF2 silencing can enhance mutant EGFR degradation to reduce clonogenic survival of erlotinib resistant T790-bearing lung AC cells is encouraging, direct inhibition of SMURF2 activity is not a viable strategy because of the critical importance of SMURF2 during mitosis (37, 38). Therefore, we decided to explore selective antagonism of SMURF2 interaction with its partner E2, UBCH5, to prevent SMURF2-mediated mutant EGFR protection from degradation. To achieve this goal, mutational analyses were carried out to define the critical residues important for SMURF2 and UBCH5 interaction. We found two proline (P) residues in UBCH5, which were critical for allowing binding between the two proteins (39). This is in agreement with the X-ray crystal structure of SMURF2 binding with another E2 partner, UBCH7 (26). To confirm the importance of these residues, we mutated the two proline residues located at positions 61 and 95 of UBCH5 to alanine (A), which surprisingly locked SMURF2–UBCH5 interaction compared with that of the WT E2 (Fig. 5A). However, catalytically, these UBCH5 PA mutants (particularly P95A and P95A/A96S) were found to be less efficient in promoting SMURF2-mediated EGFR (L+T) polyubiquitination (Fig. 5, B and D). A similar observation was previously reported for UBCH5 when only A96 residue was mutated (40). Consequently, overexpression of the proline-mutated UBCH5 caused enhanced EGFR (L+T) protein degradation (Fig. 5C), which was even more pronounced in the case of L+T+K1037Q mutant EGFR (Fig. 5E). These studies suggest a method for developing a novel strategy of targeting TKI-resistant EGFR via altering SMURF2–UBCH5 interaction.
Figure 5.
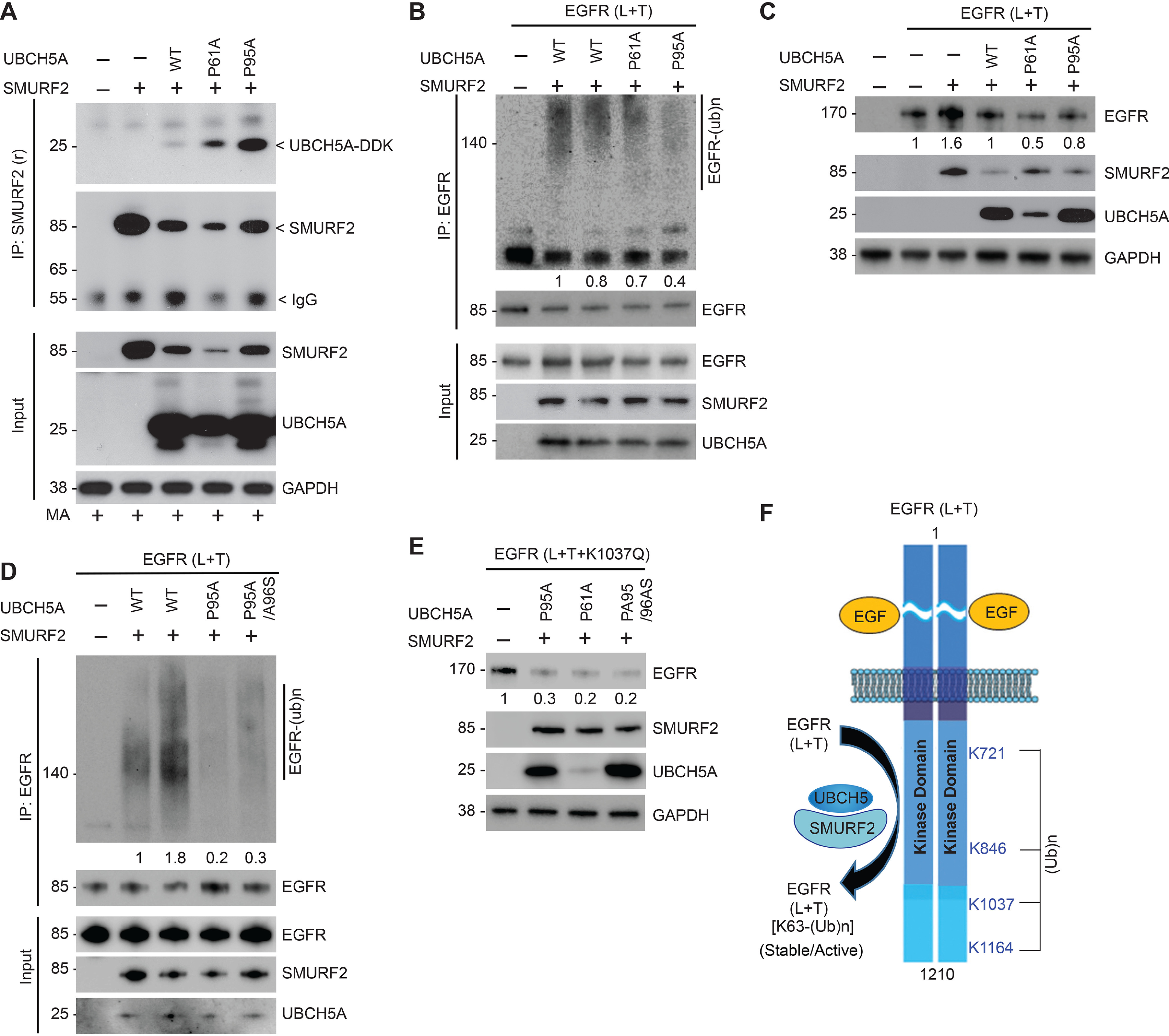
Alteration of SMURF2-UBCH5 protein–protein interaction impacts mutant EGFR levels. A, Flag-tagged SMURF2 and Myc-tagged UBCH5A (WT, P61A, and P95A mutants) were overexpressed in HEK293 cells. Cell lysates were prepared 24 h posttransfection and subjected to immunoprecipitation using SMURF2 antibody followed by immunoblotting using the indicated antibodies. B, to determine catalytic activity of P61A and P95A mutant UBCH5A, E2 proteins were synthesized using a TNT T7 quick-coupled in vitro transcription/translation system, followed by in vitro ubiquitination assay using recombinant EGFR (L+T) and SMURF2 proteins. Reactions were then subjected to immunoprecipitation using EGFR antibody followed by immunoblotting using the indicated antibodies. The total amount and catalytic activity of commercially available UBCH5A (lane 2) were comparable with WT UBCH5A protein synthesized using an in vitro transcription/translation system (lane 3). C, effect of overexpression of UBCH5A (WT and P61A and P95A mutants) along with SMURF2 on L858R/T790M EGFR steady-state levels. D, comparison of in vitro ligase activity of mutant UBCH5A (P95A and P95A/A96S) compared with WT E2 obtained either commercially (lane 2) or synthesized using the TNT system (lane 3) as described above. E, HEK293 cells overexpressing EGFR (L+T+K1037Q) mutant were transfected alone or in combination with the indicated UBCH5A mutants (P95A, P61A, or P95A/A96S), and cell lysates prepared 48 h posttransfection followed by immunoblotting using the indicated antibodies. F, schematic diagram showing lysine (K) residues (K721, K846, K1037, and K1164) as identified residues that underwent SMURF2-mediated ubiquitination as determined using MS analyses. We propose altering the SMURF2-UBCH5 interaction as a future targeting strategy to promote protein destabilization of mutant EGFR to overcome TKI resistance.
Discussion
This study extends our previous work that defined the critical importance of SMURF2 catalytic activity in promoting TKI resistance via mutant EGFR ubiquitination and stabilization. Here, we have identified SMURF2-mediated preferential polyubiquitination of TKI-resistant (L858R/T790M) mutant EGFR as one of several mechanisms that facilitate increased membrane retention of the mutant receptor, thereby causing TKI resistance. Using biochemical and genetic approaches, we have identified four lysine (K) residues in T790M mutant EGFR that undergo protective ubiquitination. Mutation of one (K1037) of these lysines (a known acetylation site that increases receptor internalization) to acetylation-mimicking glutamine (Q) converts the TKI-resistant stable L858R/T790M mutant EGFR to unstable upon TKI treatment. Consequently, loss of SMURF2 enhanced mutant EGFR degradation to potentiate drug responsiveness, whereas SMURF2 overexpression stabilized the receptor to convert a TKI-sensitive line into a resistant one. These data support the potential of SMURF2 targeting as a novel therapeutic strategy to overcome TKI resistance. We summarized our findings in Fig. 5F.
We previously reported that SMURF2 directly interacts with EGFR; however, unlike several other ubiquitin ligases, SMURF2's ubiquitin ligase activity is critical for maintaining EGFR protein stability (25). Here, we identified that among various mutants, the L858R/T790M mutant EGFR is a better SMURF2 substrate, which, along with its partner, UBCH5 (a ubiquitin-conjugating enzyme, E2), can efficiently polyubiquitinate mutant EGFR at lysine (K) 721, 846, 1037, and 1164 positions. Conversely, siRNA-mediated SMURF2 silencing caused rapid disappearance of membrane-bound EGFR and promoted enhanced internalization and degradation (Fig. 3, Fig. S3). Earlier studies have indicated that K721 and K846 residues of EGFR are important for maintaining EGFR kinase activity (29, 41), whereas K1037 and K1164 are important sites of acetylation required for receptor internalization (30). In another study, Jiang et al. reported that inhibition of K1037 acetylation by an oncogenic protein, sulfiredoxin (Srx), can promote sustained EGFR activation (42). Taken together, these data allow us to hypothesize that SMURF2-UBCH5-mediated ubiquitination at K721 and K846 residues is critical in the constitutive activation of the mutant EGFR, whereas ubiquitination at K1037 and K1164 may be competing with acetylation to balance receptor internalization, membrane retention, and protein stability. Here, we have substantiated our findings by incorporating lysine acetylation mimicking glutamine (Q) substitution only at the K1037 site, which made the erlotinib-resistant L858R/T790M mutant EGFR vulnerable to degradation following drug treatment as well as UBCH5 PA mutant overexpression. EGFR is known to undergo polyubiquitination at multiple lysine (K) residues (43). Some lysine polyubiquitinations promote EGFR degradation, and others are protective. Our study identified polyubiquitination of K1037 as protective. However, this site can also undergo acetylation, which blocks the SMURF2-mediated protective polyubiquitination and enhances receptor internalization and degradation. Consequently, a K1037Q mutant EGFR, which mimics constitutive acetylation and cannot be polyubiquitinated by SMURF2, can still undergo polyubiquitination at other lysine residues by an unknown E3 ligase(s) that enhances receptor degradation. Together, our data indicate that besides K1037, three other Ks (K721, K846, and K1164) are also important in maintaining EGFR protein stability. Upon a decrease of SMURF2 activity (following overexpression of mutant UBCH5A), all four of the protective ubiquitinations of mutant EGFR are compromised. This decrease in protective ubiquitination combined with the more efficient internalization of the polyubiquitinated L+T+K1037Q mutant (Fig. 2B and 5E) leads to faster degradation than that with (L+T) mutant EGFR.
In this study, we further utilized both cell fractionation and superresolution TIRF microscopy to demonstrate the importance of SMURF2 for the activated receptor membrane retention. Our data demonstrated that via SMURF2 targeting, we can reduce EGFR surface expression because activated receptors promptly undergo internalization and degradation via endosomal/lysosomal trafficking.
Although SMURF2 could be an attractive molecular target for anti-EGFR therapy, it is a major mitotic regulator in the spindle assembly checkpoint (37), and forced alterations cause chromosomal instability and aneuploidy as well as promoting enhanced tumor initiation in Smurf2-null mice (44). This suggests that siRNA/shRNA-mediated silencing of SMURF2 expression is not an ideal anticancer strategy. Additionally, in most circumstances, the SMURF2-UBCH7 complex promotes substrate degradation (45, 46), whereas the SMURF2-UBCH5 E3-E2 complex is mostly involved in oncoprotein stability, including EGFR, mutant KRAS, DNA topoisomerase, and others (25, 39, 47). Thus, to increase substrate specificity without compromising SMURF2's normal cellular function, we proposed to disrupt protein–protein interaction between SMURF2-UBCH5. We hypothesized that as SMURF2-UBCH5 complex is specific in polyubiquitinating L858R/T790M mutant EGFR, a strategy altering E3-E2 complex formation should impact mutant EGFR ubiquitination and promote mutant EGFR degradation. Our data revealed that there are two conserved proline (P) residues located at the 61 and 95 positions of UBCH5 that are critical for SMURF2 interaction (Fig. 5A). Mutation of either of the P's to alanine (A) significantly tightened SMURF2–UBCH5 interaction, and, in the process, compromised catalytic activity. Overexpression of these UBCH5 PA mutants compromised SMURF2-mediated L858R/T790M EGFR stabilization (Fig. 5, B–E). E3–E2 interaction is dynamic and often shows moderate to weak interaction (48), but as UBCH5 PA mutants bind tighter with the SMURF2 than with WT E2, our in vitro ubiquitination data showed reduction in SMURF2-mediated EGFR (L+T) ubiquitination. Previously, Levin et al. (40) reported that mutations of F62A and A96D in UBCH5 (which are adjacent to the P residues we mutated) reduced E2 catalytic activity when tested with SspH2, a bacterial E3 from S. typhimurium, supporting our data.
Multiple ubiquitin ligases have been implicated in maintaining EGFR protein stability, including c-CBL and p-VHL (22, 23). Although siRNA-mediated loss of SMURF2, blocking of SMURF2 interaction with its cognate E2 (UBCH5), or mutation of K1037 ubiquitination site in EGFR promoted EGFR degradation upon TKI treatment, in the near future it may be important to identify the ligase(s) involved in L858R/T790M mutant EGFR degradation following SMURF2 targeting. Our previous studies identified SMURF2 as a negative regulator of beta-transducin repeat-containing protein 1 (β-TrCP1), an F-box family ubiquitin ligase (39). Although β-TrCP1 has yet to be implicated in promoting EGFR ubiquitination and degradation, it may be involved in targeting mutant EGFR, particularly in the absence of SMURF2. Our study also identified K721 (the catalytic lysine), K846, and K1064 in EGFR as other sites of ubiquitination; however, the importance of this ubiquitination in EGFR activity and stability remains to be tested in future studies.
We recognize certain limitations of our study. First, we do not know the stoichiometry of ubiquitinated and acetylated species of mutant EGFR molecules in a cell and whether such a posttranslationally modified receptor ratio changes during TKI resistance. Second, a challenge to quantify superresolution imaging stems from the fact that many probes used in these studies either overcount or undercount the number of labeled proteins. Labeled proteins can be undercounted if a large number of genetically encoded fluorophores fail to activate because they are improperly folded or if proteins of interest are inaccessible to labeling antibodies. Overcounting can occur when photoswitchable or photoactivatable probes blink reversibly or when multiple fluorophores decorate labeling antibodies. A pair-correlation-based analytical method was recently developed that can correct, in some instances, quantitative artifacts that arise from overcounting. This method takes advantage of the observation that the magnitude of self-clustering that arises from overcounting is inversely proportional to the density of the labeled protein, as long as the labeled protein is sampled randomly. In this work, we take advantage of overcounting in superresolution images to provide a quantitative measure of receptor density in intact cells, which we further validated utilizing conventional cell fractionation studies followed by immunoprecipitation and immunoblotting, which improved the reliability of data.
In summary, this study improved our biochemical and molecular understanding of TKI resistance and establishes SMURF2-UBCH5-mediated posttranslational modifications of L858R/T790M mutant EGFR as a critical partner in TKI resistance. Furthermore, our project supports the development of a novel strategy capable of overcoming TKI resistance via selectively altering the interaction between SMURF2 and UBCH5 and degrading mutant EGFR.
Materials and methods
Reagents
Anti-EGFR (sc-03) and anti-ubiquitin (P4D1) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). GAPDH, HSP90, ErbB2, Lamin B1, and LAMP1 antibodies were purchased from Cell Signaling (Danvers, MA). Another EGFR antibody (31G7) and Lipofectamine were purchased from Invitrogen (Grand Island, NY). Recombinant active SMURF2 (no. SRP0228) and rabbit polyclonal SMURF2 antibody, human recombinant EGFR (668-1210 amino acids with C-terminal GST tag) purified from baculovirus-infected insect cells expressing either WT (no. 14-531), L858R (no. 14-626), and L858R/T790M (no. 14-721) were purchased from the Millipore Sigma (St Louis, MO). Erlotinib was obtained from Genentech Inc. (San Francisco, CA), and AZD9291 was purchased from Cayman Chemicals (Ann Arbor, MI). A mAb against human SMURF2 has been described previously (45). Anti-UBCH5 (UBE2D1, no. ab66600) and EEA1 (no. ab2900) antibodies were obtained from Abcam (Cambridge, MA). Control (no. D-001810) and SMURF2 (no. D-007194) siRNA were purchased from Thermo Fisher Scientific (Lafayette, CO) as described previously (37).
Cell cultures
EGFR-null CHO, MCF-7, and HEK293 cells were purchased from the American Type Culture Collection (ATCC). The human lung adenocarcinoma NCI-H1975 was kindly provided by Dr. J. A. Engelman (Massachusetts General Hospital, Boston). UMSCC-1, -11B, and -29B cell lines were kindly provided by Dr. Thomas Carey (University of Michigan, Ann Abor), and the parental PC9 and AZD9291-resistant PC9-AR cells were kind gifts from Dr. Christine Lovly (Vanderbilt University, Nashville, TN). Cells were cultured in either DMEM or RPMI 1640 supplemented with 10% cosmic calf serum. All the cell lines are routinely tested for pathogens and genotyped at the University of Michigan DNA sequencing core to confirm their authenticity. For plasmid transfection, Lipofectamine and Lipofectamine 2000, and for the siRNA transfection, Lipofectamine RNAimax (Invitrogen, NY, USA), were used according to the manufacturer's instructions.
Protein analyses
Immunoblotting and immunoprecipitation techniques were performed as described previously (25). Briefly, cell lysates were prepared using lysis buffer containing 50 mm HEPES-KOH (pH 7.5), 150 mm NaCl, 1.3 mm CaCl2, 1 mm DTT, 10 mm β-glycerophosphate, 1 mm NaF, 0.1 mm sodium orthovanadate, 10% glycerol, 1% NP-40, and 1× protease inhibitor mixture (Sigma, P8340). For subcellular fractionation studies, cytosolic, nuclear, and membrane fractions were isolated using a compartment protein extraction kit (Millipore, Billerica, MA) by following the manufacturer's instructions. The extracts from these fractions were subjected to immunoprecipitation, and the interaction between EGFR and SMURF2 was assessed by immunoblot analysis.
Clonogenic cell survival assay
Clonogenic survival assays were performed using techniques described previously (49). For this assay, cells were plated at a predetermined plating efficiency, and after 7–9 days the colonies formed were fixed using acetic acid–methanol (1:7) followed by staining using crystal violet (0.5%, w/v) solution. The effects of SMURF2 siRNA, treatment with either erlotinib, or AZD9291 treatments on clonogenic survival of different cell lines were determined by normalizing the survival fraction of control siRNA or vehicle-treated group as 1.
Coupled transcription and translation of UBCH5A
UBCH5A (WT and P61A and P95A mutant) plasmids were subjected to cell-free protein expression using a TnT T7 quick coupled transcription/translation system (no. L1170) from Promega (Madison, WI) according to the manufacturer's protocol and as described previously (50). Following synthesis, expression of full-length protein was confirmed using immunoblotting prior to its use in in vitro ubiquitination assays.
In vitro ubiquitination assay
The in vitro ubiquitination reaction was carried out as described previously (39). Briefly, to reaction buffer (250 mm Tris-HCl, pH 7.5, 50 mm MgCl2, 50 μm DTT, 20 mm ATP), 10 µg of Myc-tagged ubiquitin (no. U-115), 0.35 µg of UBE1 (no. E305), and 0.5 µg of UBCH5 (no. E2-616) (all from Boston Biochemicals, Cambridge, MA) were added. The human recombinant SMURF2 protein and different EGFR (WT, L858R, and L858R/T790M) proteins were then added, and the reaction mixtures were incubated at 37 °C for 2 h. The reactions were then immunoprecipitated using the EGFR antibody, and the pulled immunocomplexes were boiled with 4× gel loading dye. The samples were then resolved and immunoblotted using the indicated antibodies.
MS
The in vitro ubiquitination reaction mix was separated on a polyacrylamide gel, and proteins were visualized with colloidal Coomassie stain. In-gel digestion followed by identification of ubiquitination site mapping was carried out essentially as described previously (39). Briefly, upon trypsin digestion, peptides were resolved on a nanocapillary reverse-phase column and subjected to high-resolution, linear ion-trap MS (LTQ Orbitrap XL, Thermo Fisher). The full MS scan was collected in Orbitrap (resolution, 30,000 at 400 m/z), and data-dependent MS/MS spectra on the nine most intense ions from each full MS scan were acquired. Proteins and peptides were identified by searching the data against the UniProt human protein database (20,353 entries; reviewed only; downloaded on 20 June 2019) using Proteome Discoverer (v 2.4, ThermoScientific) with the following search parameters: MS1 and MS2 tolerances were set to 50 ppm and 0.6 Da, respectively; peptide cleavage specificity was restricted to fully tryptic peptides with up to 2 missed cleavage; carbamidomethylation of cysteines (57.02146 Da) was considered static modification; and oxidation of methionine (15.9949 Da), deamidation of glutamine, and glutamine (0.98401 Da) and diglycine remnant on lysines (114.0292 Da) was considered variable. The Percolator validator module of Proteome Discoverer was used to retain only those peptides that passed ≤1% false discovery rate threshold. Spectral matches to ubiquitinated peptides were manually verified.
Labeling
For the superresolution microscopy, cells were grown in Matek glass-bottom plates. Following all treatments, cells were fixed for 10 min in PBS solution containing 4% paraformaldehyde and 0.1% glutaraldehyde. After fixing, the cells were incubated in a blocking buffer (PBS, 2% fish gelatin, 0.01% sodium azide) for one hour, followed by incubation in the blocking buffer containing the corresponding primary antibodies for one hour. The plates were then washed 3–5 times and then incubated in secondary antibody (Alexa Fluor 532 and 647, 1:1000) for one hour. The plates were then washed again three times with the blocking buffer.
STORM imaging and reconstruction
Samples were imaged in one of two photoconvertible buffers. β-ME buffer (50 mm Tris, 0.1 g/ml glucose, 100 mm NaCl, 40 µg/ml catalase, 500 µg/ml glucose oxidase, 1% β-ME, pH 8.5) was used for one color, whereas 0.1 M cysteamine was used instead of β-ME in the case of two-color STORM. A detailed description of the imaging setup and STORM reconstruction is described elsewhere (51). Briefly, dSTORM was performed on a 100× UAPO TIRF objective (numeric aperture, 1.49) in an Olympus IX81-XDC inverted microscope equipped with a cellTIRF module. A 532-nm laser (Samba 532–150 CW, Cobolt, San Jose, CA) and a 640-nm laser (CUBE 640-74FP; Coherent) were used for excitation of the two fluorophores. Images were captured on an iXon-897 Andor EMCCD camera. For two-color experiments, the emitted light was passed through a DV2 emission splitting system (Photometrics, Tuscon, AZ) to simultaneously image both the near- and far-red channels. To prevent z-drift, an active Z-drift correction (Olympus America, Center Valley, PA) was used. Superresolution images were reconstructed after filtering, localization of individual fluorescent events, correction of stage drift, and postprocessing using our in-house MATLAB program.
Measuring receptor density
To calculate receptor density, the reconstructed STORM image was used. Autocorrelation g(r) and cross-correlation functions were calculated as previously described (52). Autocorrelation g(r) at a given radius, r, gives the probability of finding another protein within r. Thus, in a two-dimensional reconstructed image, the magnitude of the autocorrelation function is determined by two components. First, there is overcounting at short distances because of high antibody labeling and multiple fluorophores per antibody. This increases the probability of finding a second fluorophore. The amplitude of this overcounting effect is inversely dependent on the surface density of labeled proteins when the proteins themselves are not self-clustered according to the relationship
| (Eq. 1) |
where Δr is typically the size of the pixel in a reconstructed image and ρ is the average density of the receptor. This sum is simply the area under the correlation function in two dimensions and in practice is carried out over radii in which g(r) > 1.
Similarly, for a two-color superresolution image, in addition to the autocorrelation function for individual channels, we also calculate the cross-correlation function, which determines the probability of finding a fluorophore of one color within a certain radius of an average fluorophore of the other color. Overcounting does not contribute to cross-correlations, because proteins are labeled with different colors, and observing both colors in the same location arises only when both proteins are present. The cross-correlation amplitude, C(r), gives the probability of coclustering. This is combined with the surface densities obtained from autocorrelation functions (Equation 1) to determine the surface density of coclusters ().
| (Eq. 2) |
Colocalization
The Jacop plugin in Macphotonics ImageJ (ver 1.48) was used to measure Pearson correlation coefficient.
Statistics
Unless noted otherwise, results are presented as mean ± S.E. (standard error of mean estimate) from at least three independent experiments.
Data availability
Data described in the manuscript are in the main text, figures, and the supporting figures. The MS proteomics data have been deposited with the ProteomeXchange Consortium via the PRIDE (53) partner repository with the data set identifier PXD018324.
Supplementary Material
Acknowledgments
We thank Dr. Christine Lovly from the Vanderbilt University for providing AZD9291-resistant PC9 cells (PC9-AR). We also thank Dr. Mary Davis for editorial and Steven Kronenberg for graphic assistance.
This article contains supporting information.
Author contributions—P. R., S. V., T. S. L., M. K. N., and D. R. resources; P. R., K. R., A. A., U. S. A., S. S., V. B., M. K. N., and D. R. data curation; P. R., K. R., A. A., U. S. A., S. S., V. B., S. V., T. S. L., M. K. N., and D. R. formal analysis; P. R., K. R., A. A., U. S. A., S. S., V. B., and D. R. validation; P. R., K. R., A. A., U. S. A., S. S., S. V., M. K. N., and D. R. investigation; P. R., K. R., A. A., U. S. A., S. S., and D. R. visualization; P. R., K. R., A. A., U. S. A., S. S., V. B., S. V., M. K. N., and D. R. methodology; P. R., T. S. L., and D. R. writing-original draft; K. R., S. V., T. S. L., M. K. N., and D. R. conceptualization; K. R., V. B., S. V., and D. R. software; A. A., U. S. A., S. S., V. B., S. V., M. K. N., and D. R. writing-review and editing; S. V., T. S. L., M. K. N., and D. R. supervision; S. V. and D. R. funding acquisition; S. V., M. K. N., and D. R. project administration.
Funding and additional information—This work was supported in part by grants from the National Institutes of Health R01CA160981 (to D. R.), R01CA131290 (to M. K. N.), and R01GM110052 (to S. V.). R. K.'s postdoctoral fellowship is funded by M-Cube and the Fast-Forward Innovation Program at the University of Michigan. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- EGFR
- epidermal growth factor receptor
- TKIs
- tyrosine kinase inhibitors
- STORM
- stochastic optical reconstruction microscopy
- β-ME
- β-mercaptoethanol
- TIRF
- total internal reflection fluorescence.
References
- 1. Sharma S. V., Bell D. W., Settleman J., and Haber D. A. (2007) Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 7, 169–181 10.1038/nrc2088 [DOI] [PubMed] [Google Scholar]
- 2. Costa D. B., Schumer S. T., Tenen D. G., and Kobayashi S. (2008) Differential responses to erlotinib in epidermal growth factor receptor (EGFR)-mutated lung cancers with acquired resistance to gefitinib carrying the L747S or T790M secondary mutations. J. Clin. Oncol. 26, 1182–1184 10.1200/JCO.2007.14.9039 [DOI] [PubMed] [Google Scholar]
- 3. Ramalingam S. S., Vansteenkiste J., Planchard D., Cho B. C., Gray J. E., Ohe Y., Zhou C., Reungwetwattana T., Cheng Y., Chewaskulyong B., Shah R., Cobo M., Lee K. H., Cheema P., Tiseo M., FLAURA Investigators. (2020) Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N. Engl. J. Med. 382, 41–50 10.1056/NEJMoa1913662 [DOI] [PubMed] [Google Scholar]
- 4. Leonetti A., Sharma S., Minari R., Perego P., Giovannetti E., and Tiseo M. (2019) Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 121, 725–737 10.1038/s41416-019-0573-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yun C. H., Mengwasser K. E., Toms A. V., Woo M. S., Greulich H., Wong K. K., Meyerson M., and Eck M. J. (2008) The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. U S A 105, 2070–2075 10.1073/pnas.0709662105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou W., Ercan D., Chen L., Yun C. H., Li D., Capelletti M., Cortot A. B., Chirieac L., Iacob R. E., Padera R., Engen J. R., Wong K. K., Eck M. J., Gray N. S., and Janne P. A. (2009) Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 462, 1070–1074 10.1038/nature08622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Janne P. A., Yang J. C., Kim D. W., Planchard D., Ohe Y., Ramalingam S. S., Ahn M. J., Kim S. W., Su W. C., Horn L., Haggstrom D., Felip E., Kim J. H., Frewer P., Cantarini M., et al. (2015) AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 372, 1689–1699 10.1056/NEJMoa1411817 [DOI] [PubMed] [Google Scholar]
- 8. Thress K. S., Paweletz C. P., Felip E., Cho B. C., Stetson D., Dougherty B., Lai Z., Markovets A., Vivancos A., Kuang Y., Ercan D., Matthews S. E., Cantarini M., Barrett J. C., Janne P. A., et al. (2015) Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 21, 560–562 10.1038/nm.3854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shimamura T., Li D., Ji H., Haringsma H. J., Liniker E., Borgman C. L., Lowell A. M., Minami Y., McNamara K., Perera S. A., Zaghlul S., Thomas R. K., Greulich H., Kobayashi S., Chirieac L. R., et al. (2008) Hsp90 inhibition suppresses mutant EGFR-T790M signaling and overcomes kinase inhibitor resistance. Cancer Res. 68, 5827–5838 10.1158/0008-5472.CAN-07-5428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen G., Kronenberger P., Teugels E., Umelo I. A., and De Greve J. (2012) Targeting the epidermal growth factor receptor in non-small cell lung cancer cells: the effect of combining RNA interference with tyrosine kinase inhibitors or cetuximab. BMC Med. 10, 28 10.1186/1741-7015-10-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chun P. Y., Feng F. Y., Scheurer A. M., Davis M. A., Lawrence T. S., and Nyati M. K. (2006) Synergistic effects of gemcitabine and gefitinib in the treatment of head and neck carcinoma. Cancer Res. 66, 981–988 10.1158/0008-5472.CAN-05-2665 [DOI] [PubMed] [Google Scholar]
- 12. Feng F. Y., Lopez C. A., Normolle D. P., Varambally S., Li X., Chun P. Y., Davis M. A., Lawrence T. S., and Nyati M. K. (2007) Effect of epidermal growth factor receptor inhibitor class in the treatment of head and neck cancer with concurrent radiochemotherapy in vivo. Clin. Cancer Res. 13, 2512–2518 10.1158/1078-0432.CCR-06-2582 [DOI] [PubMed] [Google Scholar]
- 13. Feng F. Y., Varambally S., Tomlins S. A., Chun P. Y., Lopez C. A., Li X., Davis M. A., Chinnaiyan A. M., Lawrence T. S., and Nyati M. K. (2007) Role of epidermal growth factor receptor degradation in gemcitabine-mediated cytotoxicity. Oncogene 26, 3431–3439 10.1038/sj.onc.1210129 [DOI] [PubMed] [Google Scholar]
- 14. Ahsan A., Hiniker S. M., Davis M. A., Lawrence T. S., and Nyati M. K. (2009) Role of cell cycle in epidermal growth factor receptor inhibitor-mediated radiosensitization. Cancer Res. 69, 5108–5114 10.1158/0008-5472.CAN-09-0466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ahsan A., Hiniker S. M., Ramanand S. G., Nyati S., Hegde A., Helman A., Menawat R., Bhojani M. S., Lawrence T. S., and Nyati M. K. (2010) Role of epidermal growth factor receptor degradation in cisplatin-induced cytotoxicity in head and neck cancer. Cancer Res. 70, 2862–2869 10.1158/0008-5472.CAN-09-4294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Argiris A., Duffy A. G., Kummar S., Simone N. L., Arai Y., Kim S. W., Rudy S. F., Kannabiran V. R., Yang X., Jang M., Chen Z., Suksta N., Cooley-Zgela T., Ramanand S. G., Ahsan A., et al. (2011) Early tumor progression associated with enhanced EGFR signaling with bortezomib, cetuximab, and radiotherapy for head and neck cancer. Clin. Cancer Res. 17, 5755–5764 10.1158/1078-0432.CCR-11-0861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ahsan A., Ramanand S. G., Whitehead C., Hiniker S. M., Rehemtulla A., Pratt W. B., Jolly S., Gouveia C., Truong K., Van Waes C., Ray D., Lawrence T. S., and Nyati M. K. (2012) Wild-type EGFR is stabilized by direct interaction with HSP90 in cancer cells and tumors. Neoplasia 14, 670–677 10.1593/neo.12986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ahsan A., Ray D., Ramanand S. G., Hegde A., Whitehead C., Rehemtulla A., Morishima Y., Pratt W. B., Osawa Y., Lawrence T. S., and Nyati M. K. (2013) Destabilization of the epidermal growth factor receptor (EGFR) by a peptide that inhibits EGFR binding to heat shock protein 90 and receptor dimerization. J. Biol. Chem. 288, 26879–26886 10.1074/jbc.M113.492280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsien C. I., Nyati M. K., Ahsan A., Ramanand S. G., Chepeha D. B., Worden F. P., Helman J. I., D'Silva N., Bradford C. R., Wolf G. T., Lawrence T. S., and Eisbruch A. (2013) Effect of erlotinib on epidermal growth factor receptor and downstream signaling in oral cavity squamous cell carcinoma. Head Neck 35, 1323–1330 10.1002/hed.23128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ray P., Tan Y. S., Somnay V., Mehta R., Sitto M., Ahsan A., Nyati S., Naughton J. P., Bridges A., Zhao L., Rehemtulla A., Lawrence T. S., Ray D., and Nyati M. K. (2016) Differential protein stability of EGFR mutants determines responsiveness to tyrosine kinase inhibitors. Oncotarget 7, 68597–68613 10.18632/oncotarget.11860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tomas A., Futter C. E., and Eden E. R. (2014) EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol. 24, 26–34 10.1016/j.tcb.2013.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Levkowitz G., Waterman H., Ettenberg S. A., Katz M., Tsygankov A. Y., Alroy I., Lavi S., Iwai K., Reiss Y., Ciechanover A., Lipkowitz S., and Yarden Y. (1999) Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol. Cell 4, 1029–1040 10.1016/S1097-2765(00)80231-2 [DOI] [PubMed] [Google Scholar]
- 23. Zhou L., and Yang H. (2011) The von Hippel-Lindau tumor suppressor protein promotes c-Cbl-independent poly-ubiquitylation and degradation of the activated EGFR. PLoS One 6, e23936 10.1371/journal.pone.0023936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sirisaengtaksin N., Gireud M., Yan Q., Kubota Y., Meza D., Waymire J. C., Zage P. E., and Bean A. J. (2014) UBE4B protein couples ubiquitination and sorting machineries to enable epidermal growth factor receptor (EGFR) degradation. J. Biol. Chem. 289, 3026–3039 10.1074/jbc.M113.495671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ray D., Ahsan A., Helman A., Chen G., Hegde A., Gurjar S. R., Zhao L., Kiyokawa H., Beer D. G., Lawrence T. S., and Nyati M. K. (2011) Regulation of EGFR protein stability by the HECT-type ubiquitin ligase SMURF2. Neoplasia 13, 570–578 10.1593/neo.11632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ogunjimi A. A., Briant D. J., Pece-Barbara N., Le Roy C., Di Guglielmo G. M., Kavsak P., Rasmussen R. K., Seet B. T., Sicheri F., and Wrana J. L. (2005) Regulation of Smurf2 ubiquitin ligase activity by anchoring the E2 to the HECT domain. Mol. Cell 19, 297–308 10.1016/j.molcel.2005.06.028 [DOI] [PubMed] [Google Scholar]
- 27. Russo M. W., Lukas T. J., Cohen S., and Staros J. V. (1985) Identification of residues in the nucleotide binding site of the epidermal growth factor receptor/kinase. J. Biol. Chem. 260, 5205–5208 10.1074/jbc.271.33.19656 [DOI] [PubMed] [Google Scholar]
- 28. Gullick W. J., Downward J., Foulkes J. G., and Waterfield M. D. (1986) Antibodies to the ATP-binding site of the human epidermal growth factor (EGF) receptor as specific inhibitors of EGF-stimulated protein-tyrosine kinase activity. Eur. J. Biochem. 158, 245–253 10.1111/j.1432-1033.1986.tb09744.x [DOI] [PubMed] [Google Scholar]
- 29. Purba E. R., Saita E. I., and Maruyama I. N. (2017) Activation of the EGF receptor by ligand binding and oncogenic mutations: the rotation model. Cells 6, 13 10.3390/cells6020013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Goh L. K., Huang F., Kim W., Gygi S., and Sorkin A. (2010) Multiple mechanisms collectively regulate clathrin-mediated endocytosis of the epidermal growth factor receptor. J. Cell Biol. 189, 871–883 10.1083/jcb.201001008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Caron C., Boyault C., and Khochbin S. (2005) Regulatory cross-talk between lysine acetylation and ubiquitination: role in the control of protein stability. Bioessays 27, 408–415 10.1002/bies.20210 [DOI] [PubMed] [Google Scholar]
- 32. Wang X., and Hayes J. J. (2008) Acetylation mimics within individual core histone tail domains indicate distinct roles in regulating the stability of higher-order chromatin structure. Mol. Cell. Biol. 28, 227–236 10.1128/MCB.01245-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li M., Luo J., Brooks C. L., and Gu W. (2002) Acetylation of p53 inhibits its ubiquitination by Mdm2. J. Biol. Chem. 277, 50607–50611 10.1074/jbc.C200578200 [DOI] [PubMed] [Google Scholar]
- 34. Rust M. J., Bates M., and Zhuang X. (2006) Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793–795 10.1038/nmeth929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Heilemann M., van de Linde S., Schuttpelz M., Kasper R., Seefeldt B., Mukherjee A., Tinnefeld P., and Sauer M. (2008) Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew Chem. Int. Ed. Engl. 47, 6172–6176 10.1002/anie.200802376 [DOI] [PubMed] [Google Scholar]
- 36. Song L., Falzone N., and Vallis K. A. (2016) EGF-coated gold nanoparticles provide an efficient nano-scale delivery system for the molecular radiotherapy of EGFR-positive cancer. Int. J. Radiat. Biol. 92, 716–723 10.3109/09553002.2016.1145360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Osmundson E. C., Ray D., Moore F. E., Gao Q., Thomsen G. H., and Kiyokawa H. (2008) The HECT E3 ligase Smurf2 is required for Mad2-dependent spindle assembly checkpoint. J. Cell Biol. 183, 267–277 10.1083/jcb.200801049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Moore F. E., Osmundson E. C., Koblinski J., Pugacheva E., Golemis E. A., Ray D., and Kiyokawa H. (2010) The WW-HECT protein Smurf2 interacts with the docking protein NEDD9/HEF1 for Aurora A activation. Cell Division 5, 22 10.1186/1747-1028-5-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shukla S., Allam U. S., Ahsan A., Chen G., Krishnamurthy P. M., Marsh K., Rumschlag M., Shankar S., Whitehead C., Schipper M., Basrur V., Southworth D. R., Chinnaiyan A. M., Rehemtulla A., Beer D. G., et al. (2014) KRAS protein stability is regulated through SMURF2: UBCH5 complex-mediated beta-TrCP1 degradation. Neoplasia 16, 115–IN115 10.1593/neo.14184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Levin I., Eakin C., Blanc M. P., Klevit R. E., Miller S. I., and Brzovic P. S. (2010) Identification of an unconventional E3 binding surface on the UbcH5 ∼ Ub conjugate recognized by a pathogenic bacterial E3 ligase. Proc. Natl. Acad. Sci. U S A 107, 2848–2853 10.1073/pnas.0914821107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shan Y., Eastwood M. P., Zhang X., Kim E. T., Arkhipov A., Dror R. O., Jumper J., Kuriyan J., and Shaw D. E. (2012) Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell 149, 860–870 10.1016/j.cell.2012.02.063 [DOI] [PubMed] [Google Scholar]
- 42. Jiang H., Wu L., Chen J., Mishra M., Chawsheen H. A., Zhu H., and Wei Q. (2015) Sulfiredoxin promotes colorectal cancer cell invasion and metastasis through a novel mechanism of enhancing EGFR signaling. Mol. Cancer Res. 13, 1554–1566 10.1158/1541-7786.MCR-15-0240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huang F., Kirkpatrick D., Jiang X., Gygi S., and Sorkin A. (2006) Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Mol. Cell 21, 737–748 10.1016/j.molcel.2006.02.018 [DOI] [PubMed] [Google Scholar]
- 44. Blank M., Tang Y., Yamashita M., Burkett S. S., Cheng S. Y., and Zhang Y. E. (2012) A tumor suppressor function of Smurf2 associated with controlling chromatin landscape and genome stability through RNF20. Nat. Med. 18, 227–234 10.1038/nm.2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kavsak P., Rasmussen R. K., Causing C. G., Bonni S., Zhu H., Thomsen G. H., and Wrana J. L. (2000) Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol. Cell 6, 1365–1375 10.1016/S1097-2765(00)00134-9 [DOI] [PubMed] [Google Scholar]
- 46. Pan Y., Li R., Meng J. L., Mao H. T., Zhang Y., and Zhang J. (2014) Smurf2 negatively modulates RIG-I-dependent antiviral response by targeting VISA/MAVS for ubiquitination and degradation. J. Immunol. 192, 4758–4764 10.4049/jimmunol.1302632 [DOI] [PubMed] [Google Scholar]
- 47. Emanuelli A., Borroni A. P., Apel-Sarid L., Shah P. A., Ayyathan D. M., Koganti P., Levy-Cohen G., and Blank M. (2017) Smurf2-mediated stabilization of DNA topoisomerase IIalpha controls genomic integrity. Cancer Res. 77, 4217–4227 10.1158/0008-5472.CAN-16-2828 [DOI] [PubMed] [Google Scholar]
- 48. Stewart M. D., Ritterhoff T., Klevit R. E., and Brzovic P. S. (2016) E2 enzymes: more than just middle men. Cell Res. 26, 423–440 10.1038/cr.2016.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nyati M. K., Maheshwari D., Hanasoge S., Sreekumar A., Rynkiewicz S. D., Chinnaiyan A. M., Leopold W. R., Ethier S. P., and Lawrence T. S. (2004) Radiosensitization by pan ErbB inhibitor CI-1033 in vitro and in vivo. Clin. Cancer Res. 10, 691–700 10.1158/1078-0432.CCR-1041-03 [DOI] [PubMed] [Google Scholar]
- 50. Ray D., Ray P., Ferrer-Torres D., Wang Z., Nancarrow D., Yoon H. W., San Martinho M., Hinton T., Owens S., Thomas D., Jiang H., Lawrence T. S., Lin J., Lagisetty K., Chang A. C., et al. (2020) Isoforms of RNF128 regulate the stability of mutant P53 in Barrett's esophageal cells. Gastroenterology 158, 583–597 10.1053/j.gastro.2019.10.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stone M. B., Shelby S. A., Nunez M. F., Wisser K., and Veatch S. L. (2017) Protein sorting by lipid phase-like domains supports emergent signaling function in B lymphocyte plasma membranes. Elife 6, e19891 10.7554/eLife.19891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Veatch S. L., Machta B. B., Shelby S. A., Chiang E. N., Holowka D. A., and Baird B. A. (2012) Correlation functions quantify super-resolution images and estimate apparent clustering due to over-counting. PLoS One 7, e31457 10.1371/journal.pone.0031457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D. J., Inuganti A., Griss J., Mayer G., Eisenacher M., Perez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yilmaz S., et al. (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47, D442–D450 10.1093/nar/gky1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data described in the manuscript are in the main text, figures, and the supporting figures. The MS proteomics data have been deposited with the ProteomeXchange Consortium via the PRIDE (53) partner repository with the data set identifier PXD018324.