

Abstract
The Na+-pumping NADH-ubiquinone (UQ) oxidoreductase (Na+-NQR) is present in the respiratory chain of many pathogenic bacteria and is thought to be a promising antibiotic target. Whereas many details of Na+-NQR structure and function are known, the mechanisms of action of potent inhibitors is not well-understood; elucidating the mechanisms would not only advance drug design strategies but might also provide insights on a terminal electron transfer from riboflavin to UQ. To this end, we performed photoaffinity labeling experiments using photoreactive derivatives of two known inhibitors, aurachin and korormicin, on isolated Vibrio cholerae Na+-NQR. The inhibitors labeled the cytoplasmic surface domain of the NqrB subunit including a protruding N-terminal stretch, which may be critical to regulate the UQ reaction in the adjacent NqrA subunit. The labeling was blocked by short-chain UQs such as ubiquinone-2. The photolabile group (2-aryl-5-carboxytetrazole (ACT)) of these inhibitors reacts with nucleophilic amino acids, so we tested mutations of nucleophilic residues in the labeled region of NqrB, such as Asp49 and Asp52 (to Ala), and observed moderate decreases in labeling yields, suggesting that these residues are involved in the interaction with ACT. We conclude that the inhibitors interfere with the UQ reaction in two ways: the first is blocking structural rearrangements at the cytoplasmic interface between NqrA and NqrB, and the second is the direct obstruction of UQ binding at this interfacial area. Unusual competitive behavior between the photoreactive inhibitors and various competitors corroborates our previous proposition that there may be two inhibitor binding sites in Na+-NQR.
Keywords: Na+-pumping NADH-ubiquinone oxidoreductase (Na+-NQR), respiratory enzyme, ubiquinone, aurachin, korormicin, photoaffinity labeling, respiratory chain, chemical biology, enzyme inhibitor
The Na+-pumping NADH-ubiquinone (UQ) oxidoreductase (Na+-NQR) is the first enzyme in the respiratory chain of many marine and pathogenic bacteria, such as Vibrio alginolyticus, Vibrio cholerae, and Hemophilus influenzae (1, 2). This enzyme couples electron transfer from NADH to UQ with Na+-pumping, generating an electrochemical Na+ gradient across the inner bacterial membrane. Na+-NQR is an integral membrane protein complex that consists of six subunits (NqrA–F) encoded by the nqr operon (2). All of the subunits have transmembrane helices except for NqrA, which is peripherally associated to the core assembly of membrane-spanning subunits, from the cytoplasmic side. Studies using a variety of techniques have defined the locations and redox properties of the cofactors in the enzyme (3–12) and led to the consensus that the electron transfer takes place along a pathway consistent of at least five redox cofactors: from NADH to a FAD, a 2Fe-2S center, two covalently bound FMNs, and a riboflavin, before finally reaching UQ. The mechanism responsible for Na+-pumping driven by electron transfer remains largely elusive.
There is an X-ray crystallographic model of V. cholerae Na+-NQR, in an oxidized state, with no bound UQ or inhibitor, which has provided valuable information about the overall structure of the enzyme (13). However, the model is difficult to reconcile with some of the results obtained in earlier biochemical/biophysical studies (5, 11, 14). For example, whereas the sequence of electron transfers through the cofactors of the enzyme has been experimentally determined, the spatial distances between several pairs of redox cofactors in the crystallographic model (e.g. between FMN and riboflavin in NqrB) are too long (29–32 Å) to support physiologically relevant electron transfer (13, 15). Also, the proposed binding position of the UQ head-ring in the NqrA subunit is located ∼20 Å above the cytoplasmic membrane surface, and too far (∼40 Å in a straight line) from its proximal electron donor, riboflavin, which is predicted to lie between NqrB and NqrE. Therefore, as Steuber et al. (13) and others have suggested, the subunits harboring the cofactors and the UQ-binding cavity may need to undergo large conformational rearrangements that reduce the spatial gaps at appropriated times during catalytic turnover. In addition, Steuber et al. (13) proposed that NqrD and NqrE bind a sixth cofactor, a single iron coordinated by four cysteine residues. This extra cofactor would decrease the distance of one of the longest electron transfers (i.e. from the 2Fe-2S center in NqrF to FMN in NqrC), but does not solve the distance-rate problem entirely. Also the presence of this putative iron center, and its redox function, remain to be verified by further experiments because earlier kinetic and spectroscopic studies, including EPR measurements, have not provided evidence for an additional redox center (6, 16, 17).
We previously discovered that aurachin D-42 (Fig. 1), one of numerous aurachins in a chemical library screened in our laboratory, is a very potent inhibitor of V. cholerae Na+-NQR (14). The inhibitory potency of aurachin D-42 (IC50 value ∼2 nm) is comparable with that of korormicin A and is remarkably greater (∼500-fold) than that of HQNO (2-n-heptyl-4-hydroxyquinoline N-oxide), a commercially available aurachin C-type inhibitor, which has been widely used in earlier Na+-NQR research (6–12). In our previous work, we characterized the binding site for UQ as well as aurachin D-42 in V. cholerae Na+-NQR by means of photoaffinity labeling using synthetic photoreactive derivatives (14). That study produced three important findings. First, the UQ head-ring labels a part of the rear wall of the UQ binding cavity in the NqrA subunit (NqrA-Leu32−Met39 and Phe131−Lys138, Fig. 2, A and B), which was predicted by the crystallographic model (13). Casutt et al. (7) also identified NqrA as the binding subunit of the UQ head-ring using a different photoreactive UQ probe. Second, the photoreactive aurachin D-type inhibitors ([125I]PAD-1 and [125I]PAD-2, Fig. 1) label the N-terminal region of NqrB (NqrB-Tyr23−Gly89), which protrudes from the membrane and forms a long stretch that anchors NqrA to the transmembrane subunits (Fig. 2C). Note that only the segment NqrB-Gly38−Gly89 is shown in Fig. 2C because the region Met1−Pro37 was not included in the structural model (13). Although the region labeled by [125I]PAD-1 and [125I]PAD-2 is in NqrB and the region labeled by the UQ probe is in NqrA, Fig. 2, B and C, shows that the two loci are adjacent, or in close proximity, in the assembled structure. Third, the photoaffinity labeling by [125I]PAD-1 and [125I]PAD-2 rather than being competitively suppressed in the presence of an excess of other inhibitors (including their nonradioactive analogs), is enhanced under some experimental conditions (for example, when a molar ratio of 125I-incorporated inhibitor to Na+-NQR is relatively low). Note that we refer to this seemingly paradoxical result as “unusual competitive behavior” throughout this manuscript.
Figure 1.

The structures of aurachin-D42, korormicin A, and their photoreactive derivatives ([125I]PAD-3, [125I]PAD-4, and [125I]PKRD-1) synthesized in this study. [125I]PAD-1 and [125I]PAD-2 are photoreactive aurachin probes used in the previous study (14). The averaged IC50 value of each inhibitor, which was determined with 0.90 nm Na+-NQR, is shown in the parentheses.
Figure 2.
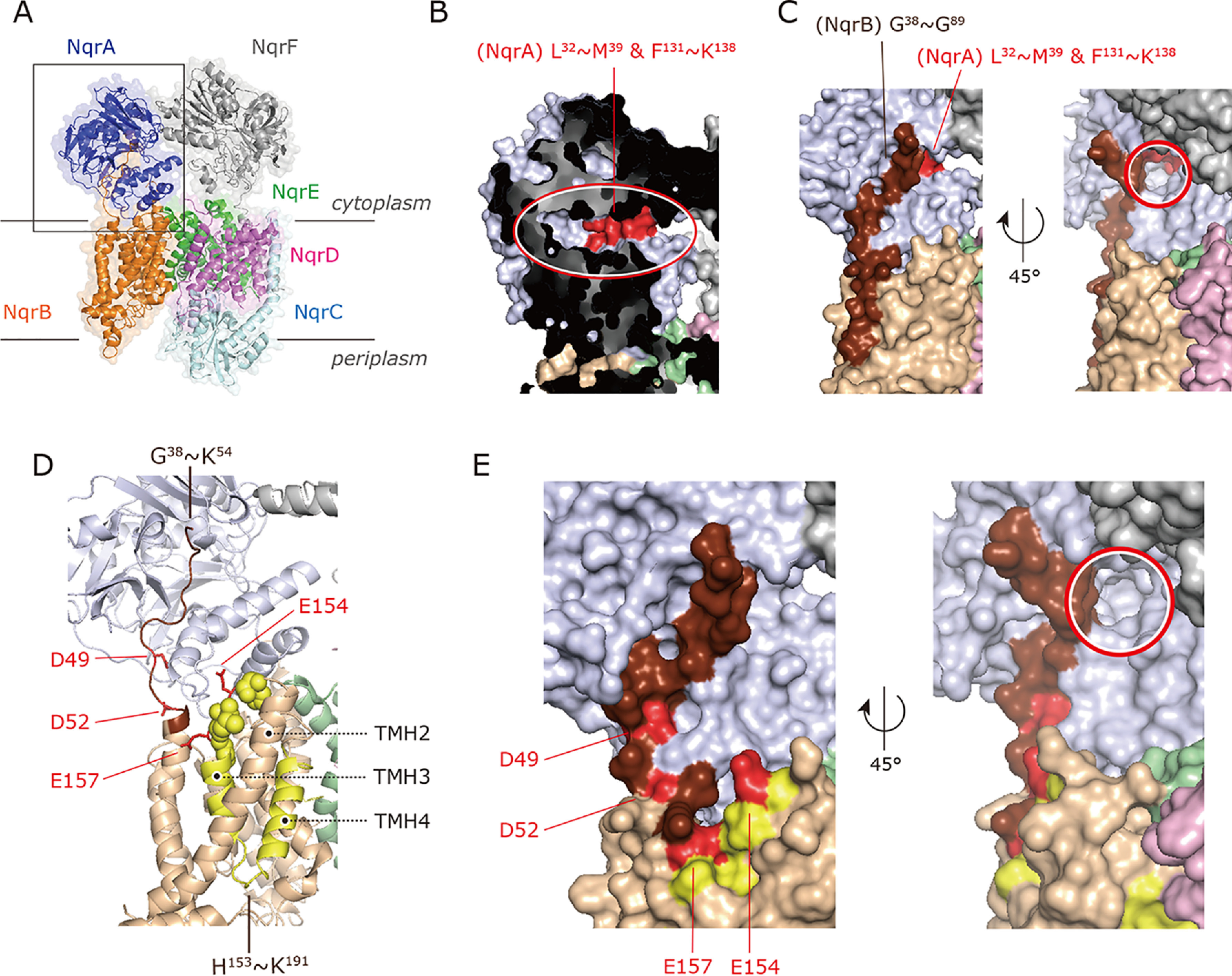
X-ray crystallographic model of V. cholerae Na+-NQR and the binding sites of UQ and inhibitors. A, entire structure of V. cholerae Na+-NQR (PDB entry 4P6V). Note that a part of the N-terminal stretch (Met1–Pro37) of NqrB was not modeled in Ref. 13. B, the area marked by a gray square in panel A was expanded. The binding region (red) of the UQ head-ring in NqrA identified in the previous work (14) is indicated. The putative solvent-accessible cavity is marked by a red oval. C, the binding region of [125I]PAD-1 and [125I]PAD-2 in NqrB (dark brown) identified in the previous work (14). The red circle marks an entrance to the putative binding cavity of the UQ head-ring. D, the binding regions of [125I]PAD-3 (His153−Lys191 in yellow) and [125I]PAD-4/[125I]PKRD-1 (His153−Lys191 and Trp23−Lys54 in dark brown) in NqrB are shown. The loop connecting TMHs 2 − 3 (His153−Gly158) is indicated by yellow spheres. The NqrB-Asp49, -Asp52, -Glu154, and -Glu157 are shown as a red stick model. E, close up view of the binding regions of [125I]PAD-3, [125I]PAD-4, and [125I]PKRD-1 in NqrB. The NqrB-Asp49, -Asp52, -Glu154, and -Glu157 are shown in red. The red circle marks an entrance to the putative binding cavity of the UQ head-ring.
The first and second findings together indicate that the binding positions of the UQ head-ring and aurachin D-type inhibitors are close to each other, but it is not clear whether they actually overlap (Fig. 2, B and C). This notion seems to be consistent with the earlier steady-state kinetic studies, which suggested that neither HQNO nor korormicin A competes directly with UQ1 (9, 11, 18–20). Although these binding regions protrude from the membrane toward the cytoplasm, we cannot rule out the possibility that the crystallographic structure might differ from that of the native enzyme, as described above. The third finding is difficult to reconcile with a simple scenario in which different inhibitors share a common binding pocket. To explain the unusual competitive behavior of aurachin D-type inhibitors, we earlier proposed an equilibrium model for the binding of the 125I-incorporated inhibitors based on the following three suppositions: (i) there are two distinct inhibitor-bound states (i.e. one-inhibitor– and two-inhibitor–bound states), (ii) the inhibitor binding to the second site is almost negligible unless the first site is already occupied by a molecule of the inhibitor, and (iii) the yield of the labeling reaction in the two-inhibitor–bound state is higher than that in the one-inhibitor-bound state (see Ref. 14 for details). Although we cannot exclude other scenarios that could explain the unusual competitive behavior, this model accounts for the consecutive changes in the nature of the competition (from enhancement to suppression) as the concentration of competitor increases. Thus, our previous study provided important insights into the terminal electron transfer step in V. cholerae Na+-NQR (14). Nevertheless, to thoroughly elucidate the mechanisms of action of various potent inhibitors of the enzyme, several issues raised by our previous work still need to be addressed, including the following.
First, we were unable to examine the inhibition mechanism of korormicin A (Fig. 1), a very potent natural inhibitor of V. cholerae Na+-NQR (19), because no photoreactive derivative was available due to synthetic difficulties related to its complicated structure. Although aurachin-type inhibitors are also able to inhibit other respiratory enzymes such as complex III (21) and the cytochrome bo3- and bd-type oxidases (22, 23), the inhibitory effect of korormicin A is highly specific to Na+-NQR. Because of this high selectivity, korormicin A and its natural analogs are anticipated to be excellent seeds for developing antibiotics against a variety of pathogenic bacteria that have Na+-NQR (24–26). However, the mechanism by which korormicin A inhibits Na+-NQR remains largely unexplored except for steady-state kinetic studies using UQ1 as a quinone substrate (18–20). We have now succeeded in synthesizing the photoreactive korormicin derivative [125I]PKRD-1, which has 2-aryl-5-carboxytetrazole (ACT) as its photolabile group (Fig. 1). This derivative maintains potent inhibitory activity suitable for photoaffinity labeling experiments. Notably, with [125I]PKRD-1 in hand, we were also able to examine another important unsolved question: whether the unusual competitive behavior observed for [125I]PAD-1 and [125I]PAD-2 is specific solely to aurachin-type inhibitors or if it is a more general phenomenon that operates with different types of inhibitors.
Second, the spatial relationship between the location where UQ binds to Na+-NQR and the binding sites of the inhibitors remains to be fully elucidated. Some structural biology studies of respiratory enzymes have successfully modeled the electron density attributable to bound UQ and/or bound inhibitor: for example, in mitochondrial complexes II and III (27–30), heme-copper quinol oxidases (31, 32), and the alternative quinol oxidase (33). Based on such structural modeling, the binding positions of UQ and the inhibitors overlap (at least partially) irrespective of structural similarities between UQ and inhibitor molecules. Our previous study showed that the UQ head-ring binds to NqrA, whereas the aurachin-type inhibitors ([125I]PAD-1 and [125I]PAD-2) bind to the NqrB subunit (14), as described above. Nevertheless, it is still not known whether the binding positions of UQ and the inhibitors are entirely discrete and separate without any overlap on the cytoplasmic surface of Na+-NQR. The reason why we consider this concern is that we cannot exclude the possibility that, whereas the polar head-ring of UQ8 orients toward the predicted binding pocket in NqrA (Fig. 2B) (13), its long isoprenyl chain may anchor it into the membrane part of the enzyme by binding to the adjacent membrane spanning subunit NqrB. If so, there could be some overlapping or steric hindrance between the side chains of UQ and the inhibitor, which could prevent both binding at the same time. Elucidating the spatial relationship between the binding positions of UQ and the inhibitors is the key to understanding how inhibitors interfere with the UQ reaction, and this may, in turn, reveal the mechanism of the terminal electron transfer step in Na+-NQR.
Here, we have investigated the mechanism of action of two different types of potent inhibitors of V. cholerae Na+-NQR (aurachin and korormicin derivatives) by focusing mainly on the unresolved issues described above via photoaffinity labeling experiments in combination with mutated enzymes. Comprehensive interpretation of the results obtained in this study strongly suggests that both aurachins and korormicins block the UQ reaction by binding to the N-terminal stretch of NqrB, which may play a critical role in regulating the reaction of UQ at the adjacent NqrA. We also found that the unusual competitive behavior, which was originally observed for [125I]PAD-1 and [125I]PAD-2, is also observed with korormicin, despite its substantially different chemical framework, suggesting that this may be a general phenomenon. This finding is additional evidence for our previous proposition that there may be two binding sites for inhibitors in Na+-NQR (14). Altogether, the present study reveals that the cytoplasmic interfacial region between NqrA and NqrB is a target spot of the potent inhibitors.
Results
Molecular design of photoreactive aurachin and korormicin derivatives
The two photoreactive aurachin derivatives used in our previous paper (14), [125I]PAD-1 and [125I]PAD-2, both have phenylazido as their photolabile group. The nitrene species that is a photogenerated intermediate of phenylazido is prone to react with any proximal C-H and/or X-H bonds (X = N, O, S). This indiscriminate reactivity is convenient for achieving a high total cross-linking yield, but may obscure the positional specificity of binding, particularly when phenylazido is attached to a flexible part of an inhibitor, as in the case of the side chain of aurachins. To improve the specificity for the current work, we chose ACT, in place of phenylazide, as the photolabile group to incorporate into a new set of inhibitor probes (Fig. 1). ACT, a recently developed photolabile group (34), interacts with a target protein via a unique mechanism: namely, a photogenerated carboxynitrile imine that reacts only with proximal nucleophilic amino acid residues such as Glu, Asp, and Cys (Fig. S1). This more specific reactivity should make the labeling yield sensitive to the kind of structural changes in the microenvironment around the inhibitor bound to Na+-NQR described above. Additionally, this reactivity would enable pinpoint identification of the amino acid cross-linked by ACT when used in combination with mutagenesis of the putative labeled residue(s).
We synthesized two new photoreactive aurachin derivatives ([125I]PAD-3 and [125I]PAD-4, Fig. 1) using aurachin D-42 as a molecular template. The synthetic procedures for [125I]PAD-3 and [125I]PAD-4 are described under the supporting data (Schemes S1 and S2). We introduced ACT into the side chain of aurachin because our earlier work showed that structural modifications of the toxophoric quinolone moiety result in significant decreases in inhibitory potency (PAD-1 versus PAD-2). The length of the spacer connecting the quinolone ring to ACT was varied in anticipation of different cross-linked positions. The parent aurachin D-42 is a very potent inhibitor of the NADH-UQ1 oxidoreductase activity of Na+-NQR with an IC50 value of ∼2 nm (the concentration required to inhibit the activity by 50%). PAD-3 with an IC50 value of 7.0 (± 0.8) nm is almost as potent as aurachin D-42, whereas PAD-4 is a somewhat weaker inhibitor with an IC50 of 380 (± 32) nm. It should be noted, however, that PAD-4 is still a more potent inhibitor than the commercially available HQNO (IC50 = 2,100 nm).
In designing a photoreactive derivative of korormicin A, our preliminary structure-activity studies indicated that the methyl/ethyl branch at the 5S-position, OH group at the 3'R-position, and epoxy group at 9'S/10'R are all critical for the inhibitory activity (Fig. S2). Because of these restrictions, the photolabile ACT was introduced into the terminal end of the side chain ([125I]PKRD-1, Fig. 1). The synthetic procedure is described in the supporting data (Scheme S3). The introduction of ACT resulted in considerable decrease in the inhibitory potency: the IC50 values of natural korormicin A and PKRD-1 are 5.0 (± 0.3) and 83 (± 8) nm, respectively. Here again, as in the case of the aurachin derivatives, the photoreactive korormicin derivative is still a much more potent inhibitor than HQNO.
Photoaffinity labeling of WT Na+-NQR by [125I]PAD-3, [125I]PAD-4, and [125I]PKRD-1
Isolated WT V. cholerae Na+-NQR (900 nm) was photoaffinity labeled by [125I]PAD-3 (5.0 nm) and subjected to direct autoradiography. The radioactivity was exclusively incorporated into a ∼30-kDa band, corresponding to the NqrB subunit (Fig. 3A). We also carried out the reaction in different redox states of Na+-NQR by adding NADH, UQ1, or both. The incorporated radioactivity in the presence of NADH (100 μm) alone was ∼90% lower than in the absence of NADH (Fig. 3A). This result suggests that reduction of the cofactors by NADH induces structural changes in the NqrB subunit that harbors the FMN and riboflavin cofactors. Although the labeling by [125I]PAD-2 also decreased in the presence of NADH (by ∼50%, Fig. 5A in Ref. 14), the extent of decrease in the case of [125I]PAD-3 was significantly greater than that for [125I]PAD-2.
Figure 3.

Photoaffinity labeling of Na+-NQR by [125I]PAD-3, [125I]PAD-4, and [125I]PKRD-1. A, the isolated Na+-NQR (900 nm) was labeled by [125I]PAD-3 (5.0 nm) in the presence of NADH (100 μm) and/or UQ1 (50 μm). B, Na+-NQR (900 nm) was labeled by [125I]PAD-4 (5.0 nm) in the presence of NADH (100 μm) and/or UQ1 (50 μm). C, Na+-NQR (900 nm) was labeled by [125I]PKRD-1 (5.0 nm) in the presence of NADH (100 μm) and/or UQ1 (50 μm). All data are representative of three independent experiments.
Figure 5.
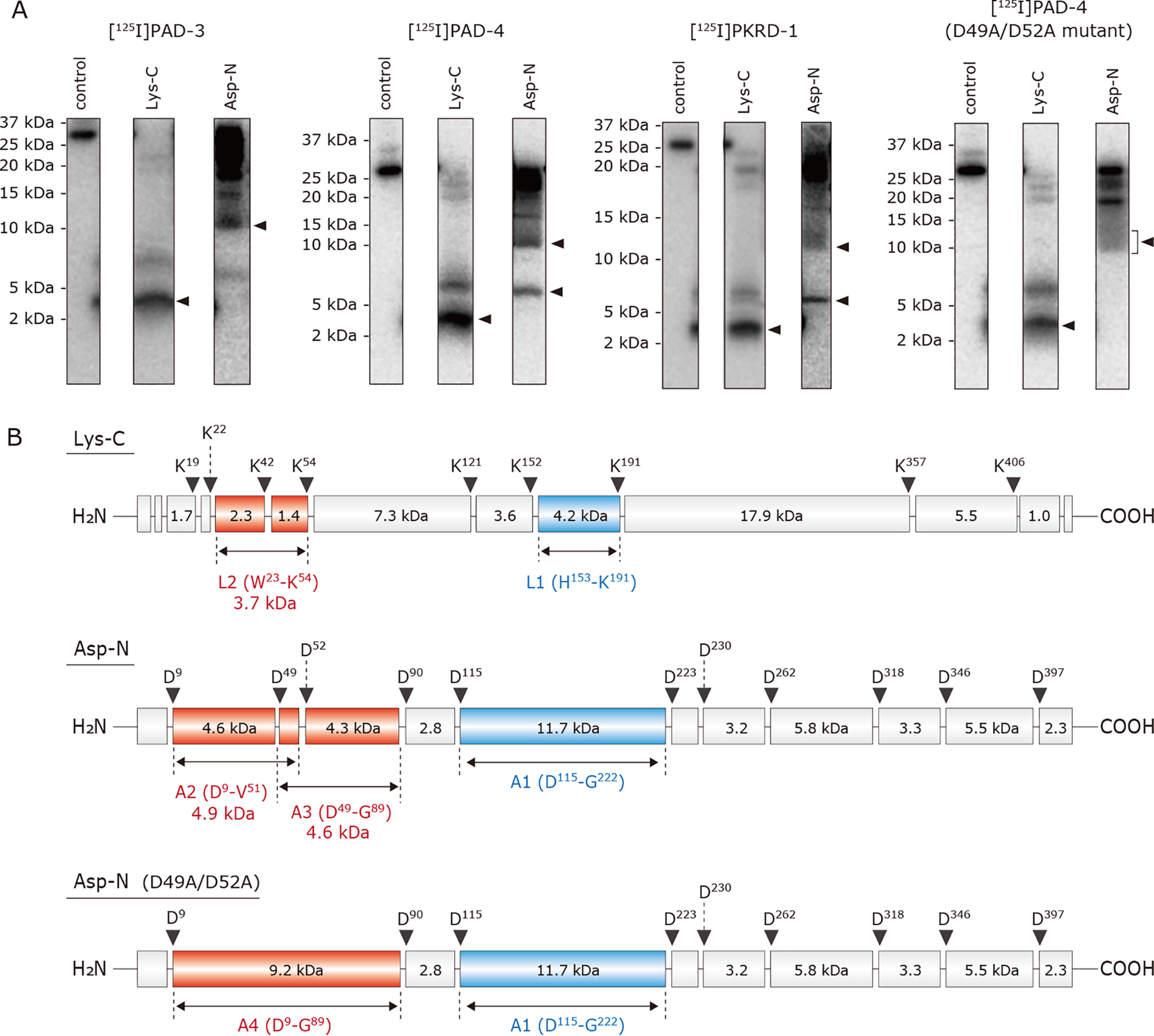
Localization of the region labeled by [125I]PAD-3, [125I]PAD-4, and [125I]PKRD-1 in Na+-NQR. A, the WT Na+-NQR (900 nm), labeled by [125I]PAD-3, [125I]PAD-4, or [125I]PKRD-1 (5.0 nm), was separated on a 16% Schägger-type SDS gel (16% T, 3% C), followed by the partial isolation of the NqrB subunit by electroelution. The D49A/D52A mutant Na+-NQR was also labeled by [125I]PAD-4 under then same experimental conditions. The labeled NqrB subunit was digested with Lys-C or Asp-N, and the digests were resolved by SDS-PAGE using a 16% Schägger-type SDS gel (16% T, 6% C containing 6.0 m urea). The SDS gel was subjected to autoradiography. All data are representative of three independent experiments. B, schematic presentation of the digestion of the NqrB subunit by Lys-C or Asp-N. The predicted cleavage sites are denoted by arrows and marked with their residue numbers in the sequences of V. cholerae NqrB subunit (SwissProt entry Q9KPS2).
The addition of UQ1 (50 μm) alone slightly reduced the labeling by [125I]PAD-3 (by ∼10−20%, Fig. 3A). The labeling significantly decreased in the presence of NADH plus UQ1 (Fig. 3A), which is equivalent to turnover conditions. This result does not necessarily mean a loss of the binding affinity of [125I]PAD-3 to the enzyme under turnover conditions, because the inhibitory activity of PAD-3 (IC50 = 7.0 nm), which was evaluated in the NADH-UQ1 oxidoreductase assay (i.e. under the turnover conditions), was still very strong. It is instead reasonable to consider that whereas the yield of the ACT labeling reaction markedly decreases due to the predicted structural changes of the NqrB subunit, the binding affinity of the toxophoric quinolone ring moiety to NqrB likely remains almost unchanged.
[125I]PAD-4 and [125I]PKRD-1 (5.0 nm each) also predominantly labeled the NqrB subunit (Fig. 3, B and C, respectively). The effects of NADH and/or UQ1 on the labeling were similar to those observed for [125I]PAD-3 (Fig. 3A) irrespective of the different distances between the quinolone ring and ACT in the case of [125I]PAD-3 and [125I]PAD-4 or different chemical frameworks in the case of [125I]PAD-3 and [125I]PKRD-1. Of note, the cross-linking yields of [125I]PAD-3, [125I]PAD-4, and [125I]PKRD-1, which all have ACT as their photolabile group, were more sensitive to the addition of NADH than that of [125I]PAD-2 that has a phenylazide group. This is consistent with our prediction that the labeling reaction of ACT may be susceptible to structural changes that accompany the reduction of the enzyme because of its higher selective reactivity.
Regarding the effect of UQ1, we previously concluded that the presence of UQ1 (50 μm) alone has no effect on the labeling by [125I]PAD-1 and [125I]PAD-2 (14). However, the effects of UQ1 were examined at only one concentration (50 μm) and its suppressive effects were negligibly small in that study; therefore, we cannot exclude the possibility that the suppressive effects of UQ1 at higher concentrations were overlooked. We therefore re-examined the effects of two short-chain UQs (UQ1 and UQ2) on the labeling by the new inhibitor probes, as described below.
Effects of short-chain UQs on the labeling by [125I]PAD-3, [125I]PAD-4, and [125I]PKRD-1
The photoaffinity labeling of Na+-NQR (900 nm) by each of [125I]PAD-3, [125I]PAD-4, and [125I]PKRD-1 (5.0 nm each) was conducted in the presence of UQ1 or UQ2. Both short-chain UQs suppressed the labeling by the inhibitors in a concentration-dependent manner (Fig. 4, A–C). Interestingly, in every case the suppressive effects of UQ2 were greater than those of UQ1; for example, 100 μm UQ2 almost completely suppressed the labeling by [125I]PKRD-1, whereas ∼70% of the labeling remained in the presence of 100 μm UQ1. Because the binding affinities of UQ1 and UQ2 to the enzyme are similar (averaged Km values of 9.6 and 6.7 μm, respectively, Fig. S3), it is likely that the longer isoprenyl chain of UQ2 may be more effective in obstructing the inhibitor molecules. These results indicate that the binding positions of short-chain UQs and inhibitors are, at least partially, overlapping. The extents of the labeling by the three inhibitors tended to slightly increase in the presence of low concentrations (5.0 and 10 μm) of UQ1 (Figs. 4, A–C). For [125I]PAD-4, the labeling also slightly increased in the presence of low concentrations of UQ2. We have, unfortunately, no definite explanation for this phenomenon at present.
Figure 4.
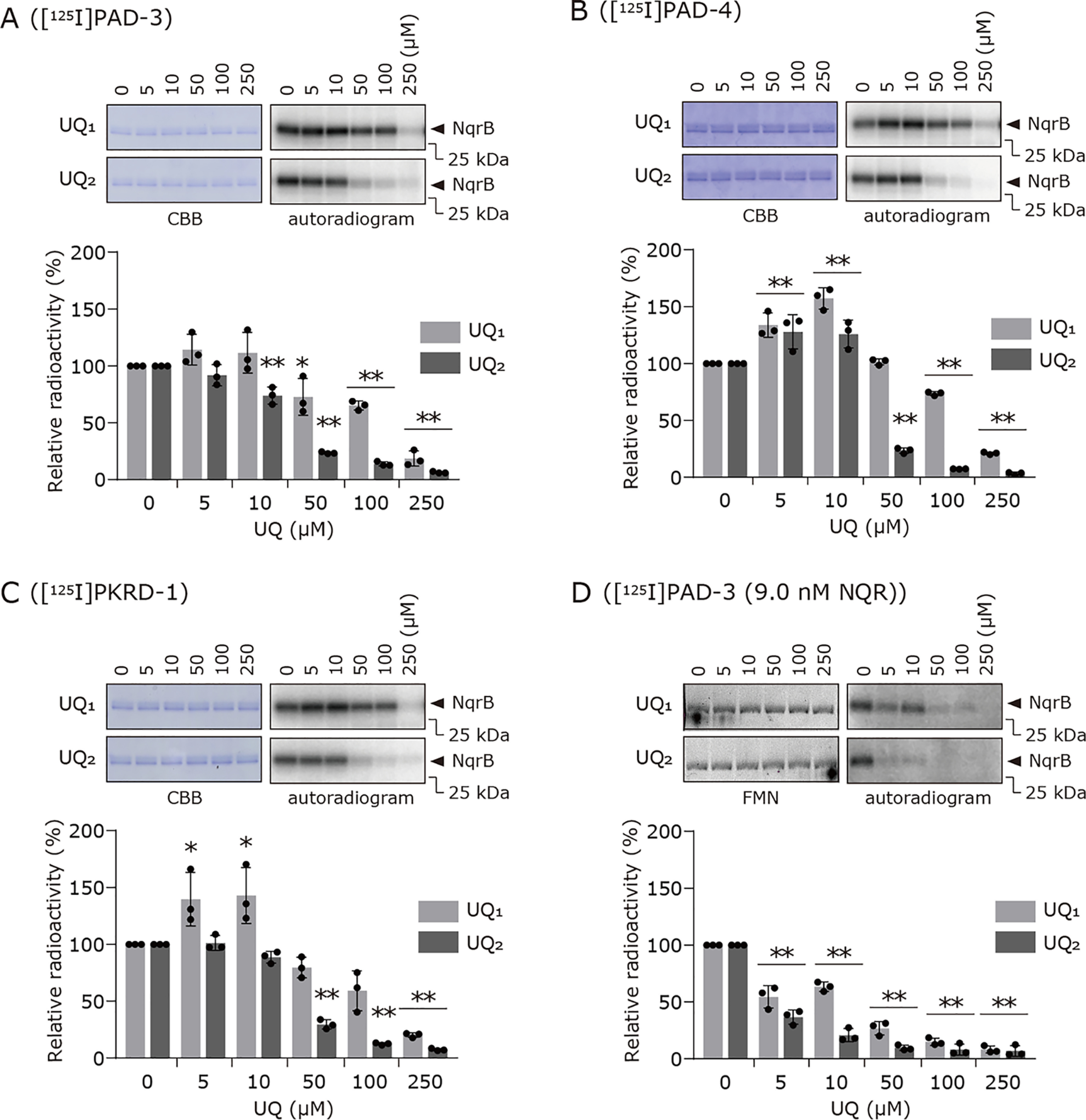
Effects of UQ1 and UQ2 on the labeling of Na+-NQR by [125I]PAD-3, [125I]PAD-4, or [125I]PKRD-1. The photoaffinity labeling of Na+-NQR (900 nm) by [125I]PAD-3 (panel A), [125I]PAD-4 (panel B), or [125I]PKRD-1 (panel C) (5.0 nm each) was conducted in the presence of different concentrations of UQ1 or UQ2. In panel D, the competition tests between [125I]PAD-3 (5.0 nm) and UQs were conducted using one hundredth-fold enzyme (9.0 nm). The FMN-based fluorescence was used as a loading control for SDS-PAGE in place of Coomassie Brilliant Blue (CBB) staining because of a lower concentration of the enzyme used. The extent of labeling in the absence of UQ was used as a control (100%). Values in graphs are means ± S.E. (n = 3). *, p < 0.05; **, p < 0.01 compared with control (one-way analysis of variance followed by Dunnett's test).
It is noteworthy that although the binding affinity of [125I]PAD-3 to the enzyme is significantly higher than that of [125I]PAD-4 (IC50 values: 7.0 and 380 nm, respectively), the labeling by both inhibitor probes was similarly suppressed by UQ1 and UQ2 (Fig. 4, A and B). This finding, along with the above results, suggests that the side chain moiety of the aurachins, not the toxophoric quinolone ring, is responsible for obstruction of the isoprenyl chain of UQs and, consequently, the obstruction is more significant for [125I]PAD-3 that has the longer side chain.
Earlier steady-state kinetic studies using UQ1 as a quinone substrate (presented as double-reciprocal plots of initial velocity versus UQ1 concentration at fixed concentrations of inhibitor) showed that korormicin A and HQNO both act as noncompetitive inhibitors for UQ1 (18–20). We observed a so-called mixed-type inhibition pattern in the double-reciprocal plots, which were described using UQ1 or UQ2 and korormicin A as a quinone substrate and an inhibitor, respectively (Fig. S4). The molar ratios of inhibitor to Na+-NQR in earlier studies and our current kinetic measurements are estimated to be 0.4–2.0, which are much higher than that in the labeling assays in Fig. 4 (the ratio is ∼0.006). Thus, compared with the kinetic studies, our labeling assays were conducted using a much lower concentration of the inhibitor probes relative to the enzyme concentration, although the concentration ranges of UQs were similar in the two lines of experiments (from a few micromolar up to ∼100 μm). Under the experimental conditions of the labeling assay, korormicin A exhibits no apparent inhibition of the catalytic activity of Na+-NQR.
We repeated the competition test between [125I]PAD-3 and UQ1 or UQ2 using one hundredth-fold enzyme (9.0 nm Na+-NQR and 5.0 nm [125I]PAD-3; the molar ratio is ∼0.6). As shown in Fig. 4D, we observed similar results to those obtained using 900 nm Na+-NQR (Fig. 4A); namely, both short-chain UQs suppressed the labeling in a concentration-dependent manner and the suppressive effects of UQ2 were greater than those of UQ1.
Because the binding affinities are remarkably different between our inhibitors and short-chain UQs (probably >102), it may be practically difficult to achieve complete competition (displacement) under the conditions of the kinetic study. In contrast, the direct binding assays using significantly low concentrations of inhibitors may have enabled the competitive behavior to be detectable. Moreover, the native ubiquinone in V. cholerae is UQ8, which is a very hydrophobic molecule. Because the extremely low solubility of UQ8 in water is a significant experimental obstacle in our in vitro competition assay, we did not use UQ8 as a competitor.
Localization of the region labeled by [125I]PAD-3, [125I]PAD-4, and [125I]PKRD-1
The photoaffinity labeling was conducted using an oxidized form of Na+-NQR because the addition of NADH remarkably reduces the labeling yields (Fig. 3). To localize the regions labeled by [125I]PAD-3, [125I]PAD-4, and [125I]PKRD-1 in the NqrB subunit, the labeled NqrB was isolated from the Schägger-type SDS gel and digested by lysylendopeptidase (Lys-C) or endoprotease Asp-N (Asp-N). The Lys-C and Asp-N digests of the NqrB labeled by [125I]PAD-3 gave radioactive bands with apparent molecular masses of ∼4 and ∼12 kDa, respectively (Fig. 5A). On the basis of the theoretical cleavage sites (Fig. 5B), the Lys-C and Asp-N digests may include the peptides His153–Lys191 (4.2 kDa, L1) and Asp115−Gly222 (11.7 kDa, A1), respectively. Therefore, [125I]PAD-3 likely labels in the region His153–Lys191 (L1), shown in yellow in Fig. 2D. The L1 fragment begins with the cytoplasmic loop connecting transmembrane helixes (TMHs) 2−3 and includes TMHs 3 and 4 as well as the loop connecting them. In this region, the labeled residue(s) may lie in the cytoplasmic loop, not in the transmembrane segments, as discussed later.
The Asp-N digestion of the NqrB labeled by [125I]PAD-4 or [125I]PKRD-1 gave radioactive bands of ∼12 and ∼6 kDa (Fig. 5A). The ∼6-kDa band may contain the peptide Asp9–Val51 (4.9 kDa, A2) or Asp49–Gly89 (4.6 kDa, A3) or both (Fig. 5B), although the two peptides are indistinguishable on the SDS gel. This assignment is supported by the fact that the ∼6-kDa band disappeared when the NqrB-D49A/D52A mutant enzyme labeled by [125I]PAD-4 was digested by Asp-N, although the expected new band (9.2 kDa, A4) was not distinctly detected (Fig. 5A, far right). Note that we will describe the reason why we prepared this mutant enzyme in the next section. Thus, because [125I]PAD-4 may label the N terminus of NqrB, it is reasonable to consider that the observed ∼4-kDa band of the Lys-C digestion contains the peptide Trp23–Lys54 (3.7 kDa, L2) in addition to His153–Lys191 (4.2 kDa, L1) that is identical to the region labeled by [125I]PAD-3 (above). Taken together, [125I]PAD-4 and [125I]PKRD-1 label the two regions, the N terminus (possible fragments: Trp23–Lys54 (L2), Trp23–Val51, or Asp49–Lys54) and His153–Lys191 (L1). We cannot unequivocally assign the labeled region to one of the three peptides in the N terminus (Trp23–Lys54 (L2), Trp23–Val51, or Asp49–Lys54) because the possible Asp-N digests (Asp9–Val51 (A2) and Asp49–Gly89 (A3)) are indistinguishable. In any case, the three peptides (Trp23–Lys54 (L2), Trp23–Val51, and Asp49–Lys54) are all located in the protruding N-terminal stretch of NqrB that anchors NqrA to NqrB. We have tentatively assigned the longest peptide Trp23–Lys54 (L2) as the labeled region in the stretch and highlighted this sequence in Fig. 2D (dark brown). Note that in Fig. 2D, the fragment L2 is indicated as Gly38–Lys54 (rather than the complete Trp23–Lys54) because a portion of L2 (Tyr23–Pro37) is not present in the structural model. These results, taken together, indicate that [125I]PAD-3 labels L1 (His153–Lys191), whereas [125I]PAD-4 and [125I]PKRD-1 label both L1 and L2 (Trp23–Lys54). These two regions are adjacent to each other at the cytoplasmic area of NqrB.
Photoaffinity labeling of Na+-NQR mutants by [125I]PAD-3, [125I]PAD-4, and [125I]PKRD-1
The present results, together with those of the previous study (14) indicate that the potent inhibitors (aurachin D-42 and korormicin) bind to an area on the cytoplasmic surface of NqrB, which is close to, or in contact with the binding pocket for the UQ head-ring in NqrA (Fig. 2, C–E). To search for potential reaction partner(s) of ACT in this region, we tentatively focused on four nucleophilic carboxylic residues: NqrB-Asp49, Asp52, Glu154, and Glu157 (Fig. 2, D and E). Asp49 and Asp52 are located in the protruding N-terminal stretch and are highly conserved across many bacterial species (Fig. S5). There are four other carboxylic residues in the N-terminal stretch (Asp9, Glu15, Glu21, and Glu28), which we did not consider because they are in the region Met1−Pro37 that was not structurally modeled (13). Glu154 and Glu157, which are located in the loop connecting TMHs 2−3, are close to the root of the N-terminal stretch. We constructed five mutants (D49A, D52A, D49A/D52A, E154A, and E157A) and carried out photoaffinity labeling experiments on each of them.
Before conducting the photoaffinity labeling, we measured the NADH-UQ1 oxidoreductase activities of the isolated mutant enzymes (Table 1). A UQ1 concentration of 50 μm was chosen because this results in an apparently maximum activity. Although all mutations affected the enzyme activity, Asp49 and Asp52, which reside in the N-terminal stretch, appear to be important for the catalytic activity. Because earlier mutagenesis studies (35–38) had the goal of identifying the pathway for Na+ translocation, they focused on the transmembrane segments of the subunits NqrB, NqrD, and NqrE. The present results are the first to use mutagenesis to show the importance of residues in the N-terminal stretch of NqrB for the catalytic activity of the enzyme.
Table 1.
Relative catalytic activity of the mutated enzymes
| Enzyme | Relative enzyme activity (%)a |
|---|---|
| WT | 100 |
| NqrB-D49A | 44 (± 5) |
| NqrB-D52A | 76 (± 8) |
| NqrB-D49A/D52A | 41 (± 3) |
| NqrB-E154A | 157 (± 18) |
| NqrB-E157A | 44 (± 7) |
a The catalytic enzyme activity was determined in the NADH-UQ1 oxidoreductase assay. The averaged activity of the WT enzyme was 5.1 (± 0.3) mmol of UQ1/min/µmol of Na+-NQR. The concentrations of Na+-NQR, NADH, and UQ1 were set to 0.20 µg/ml (0.90 nm), 100 μm, and 50 μm, respectively. Values are mean ± S.E. (n = 3–4).
Next, each of the isolated mutants (900 nm) was labeled by [125I]PAD-3 or [125I]PAD-4 (5.0 nm each); the extents of labeling are summarized in Fig. 6. No mutant completely lost the reactivity with [125I]PAD-3 and [125I]PAD-4, indicating that none of the four residues is the sole reaction partner of the ACT group in any of the inhibitors. Decreases in the labeling of the D52A and D49A/D52A mutants were relatively significant among the five mutants. In the case of [125I]PKRD-1, decreases in the labeling of the D49A and D52A mutants were relatively significant (Fig. 6). The D49A/D52A mutant does not appear to decrease labeling significantly from the single mutants. This is probably because the flexible side chains of the inhibitors would enable ACT to react with multiple nucleophilic amino acid residues (in addition to Asp49 and Asp52), which reside on the cytoplasmic surface of the NqrB subunit including the protruding N-terminal stretch.
Figure 6.
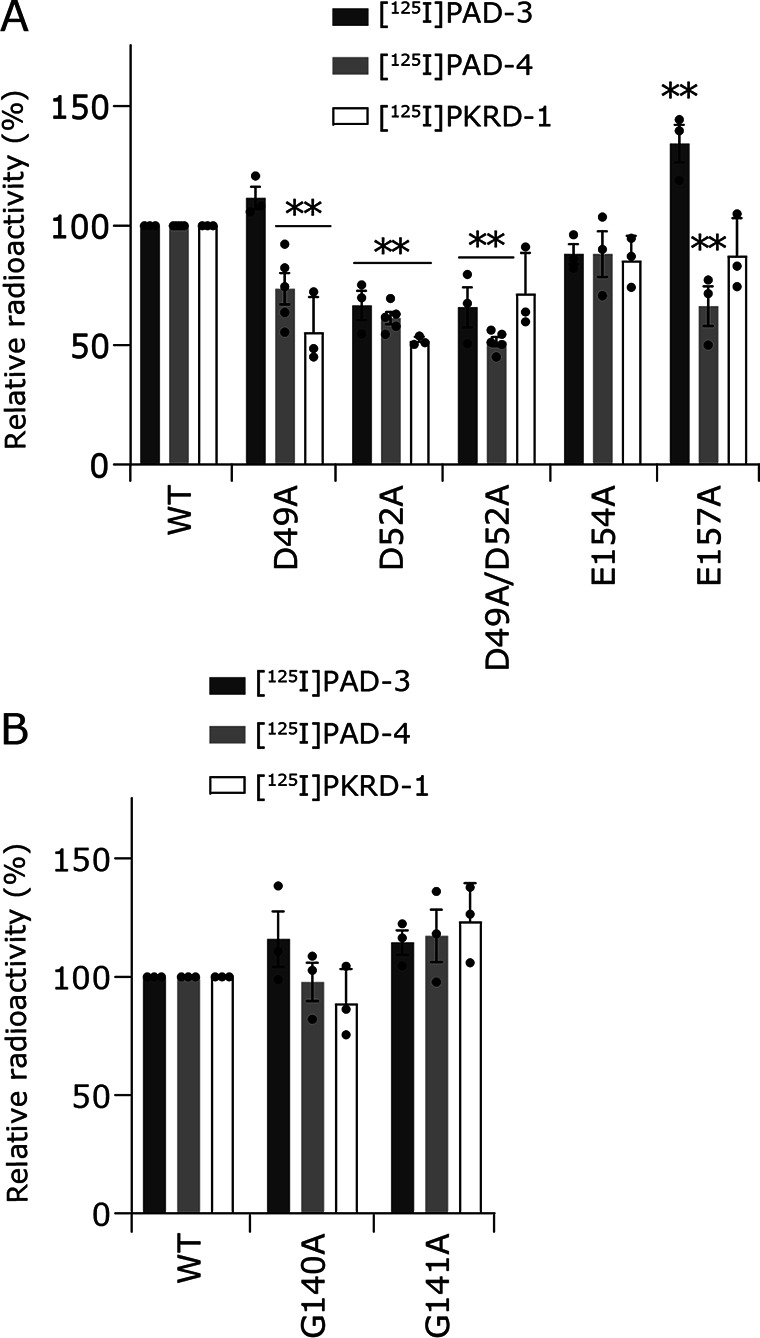
Photoaffinity labeling of Na+-NQR mutants by [125I]PAD-3, [125I]PAD-4, or [125I]PKRD-1. The five mutants (NqrB-D49A, -D52A, -D49A/D52A, -E154A, and -E157A, 900 nm each) (panel A) and the two mutants (NqrB-G140A and -G141A, 900 nm each) (panel B) were labeled by each inhibitor probe (5.0 nm). The experimental conditions are the same as those in Fig. 3. The extent of labeling of the WT enzyme was used as a control (100%). Values in graphs are mean ± S.E. (n = 3 − 5). **, p < 0.01 compared with control (one-way analysis of variance followed by Dunnett's test).
As is sometimes the case with this type of experiments using mutated enzymes, it is difficult to discriminate whether the decrease in the labeling yield is due to deletion of the reaction partner or due to reduction in the binding affinity of inhibitor itself because of some structural change in its binding environment brought about by the mutation. Accordingly, whereas we cannot simply assign Asp49 and/or Asp52 as the reaction partners of the ACT group, together with the results of the photoaffinity labeling (Fig. 5), it is highly likely that the inhibitor molecules interact directly with the protruding N-terminal stretch in NqrB.
Effects of korormicin on the labeling of cysteines in NqrB by eosin-5-maleimide
There are only two cysteines in the NqrB subunit (Cys148 and Cys368) of a total of 21 cysteines in V. cholerae Na+-NQR. NqrB-Cys148 is located on the cytoplasmic surface, adjacent to the inhibitor binding region identified above (i.e. the loop connecting TMHs 2−3) (Fig. S6). On the other hand, NqrB-Cys368, which resides in TMH 9 is surrounded by several other TMHs and is far from the inhibitor-binding region (Fig. S6). To confirm that korormicin binds to the cytoplasmic surface of NqrB, we examined the effects of korormicin on the labeling of both cysteines by the fluorescent SH-reagent eosin-5-maleimide (EMA).
In the absence of inhibitors, EMA (5.0 μm) nonselectively labeled the cysteine residues of Na+-NQR (900 nm) (Fig. 7A). Note that the reactivity of each of the cysteines may vary depending on its local environment particularly because EMA is a bulky SH-reagent. Korormicin and aurachin D-42 (5.0 μm each) significantly suppressed the labeling of NqrB (by ∼60%, Fig. 7A), whereas the labeling of other subunits was barely influenced. However, solely from this result, we cannot know whether the inhibitors suppressed labeling of Cys148, Cys368, or both.
Figure 7.
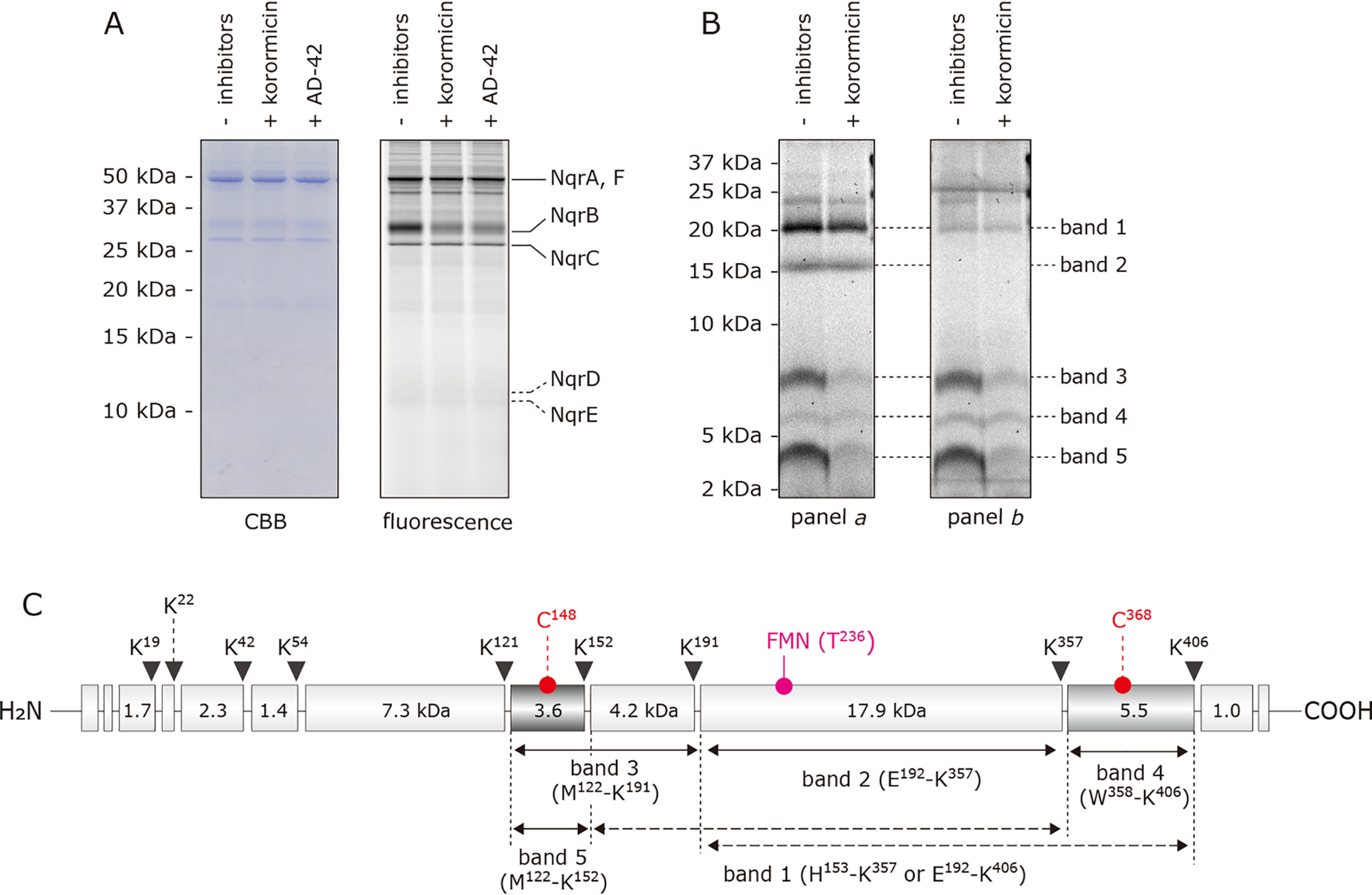
Effect of korormicin on the labeling of cysteines in NqrB by EMA. A, Na+-NQR (900 nm) was labeled by EMA (5.0 μm) in the presence or absence of korormicin or aurachin-D42 (5.0 μm each). The labeled enzyme was separated on a 16% Schägger-type SDS gel (16% T, 3% C). The SDS gel was subjected to fluorescent gel imaging for EMA with a FLA-5100 imaging system (FUJIFILM) using 532 nm laser and LPG filter (emission wavelengths shorter than 575 nm is cut-off). B, the labeled enzyme was separated on a 16% Schägger-type SDS gel (16% T, 3% C), followed by the partial isolation of NqrB by electroelution. The labeled NqrB subunit was digested with Lys-C, and the digests were resolved by SDS-PAGE using a 16% Schägger-type SDS gel (16% T, 6% C containing 6.0 m urea). The migration pattern of the digests was visualized by fluorescence of FMN (panel a) and EMA (panel b). In panels a and b, fluorescence was detected using 473 nm laser and LPB filter (emission wavelengths shorter than 510 nm are cut-off) and 532 nm laser and LPG filter (emission wavelengths shorter than 575 nm are cut-off), respectively. All data are representative of three independent experiments. C, schematic presentation of the digestion of the NqrB subunit by Lys-C. The positions of Cys148, Cys368, and FMN phosphoryl threonine (Thr236) are indicated. The predicted cleavage sites are denoted by arrows and marked with their residue numbers in the sequences of V. cholerae NqrB subunit (SwissProt entry Q9KPS2).
To discriminate the effect of the inhibitors on labeling of the two cysteines, the labeled NqrB subunit was subjected to proteolytic digestion. The bands corresponding to the labeled NqrB were isolated from the Schägger-type SDS gel, digested with Lys-C and the fragments were separated on a second gel (Fig. 7B). To assign the bands on the gel, corresponding to the proteolytic fragments, we were able to use the fact that NqrB contains a natural fluorophore, the FMN cofactor covalently attached to Thr236 (39, 40). To discriminate between the fluorescence from FMN and EMA, the SDS gel was read using a FLA-5100 imaging system (FUJIFILM, Tokyo) under two different scanning conditions: A: 473 nm laser and LPB filter (emission wavelengths shorter than 510 nm are cut-off) and B: 532 nm laser and LPG filter (emission wavelengths shorter than 575 nm are cut-off). Because the maximum emission wavelength of FMN (λex 448 nm and λem 514 nm) is shorter than that of EMA (λex 524 nm and λem 545 nm) (41), the fluorescence from the EMA is expected to be dominant under condition B (Fig. 7B, panel b). On the other hand, fluorescence both from FMN and EMA can be detected in condition A (Fig. 7B, panel a).
The Lys-C digestion gave five main fluorescent bands with approximate sizes of 20 kDa (band 1), 16 kDa (band 2), 8 kDa (band 3), 6 kDa (band 4), and 4 kDa (band 5) (Fig. 7B, panels a and b). The intensity of bands 1 and 2 drops significantly between scanning conditions A and B, indicating that the fluorescence is predominantly due to FMN. In contrast, the intensity of bands 3, 4, and 5 is approximately the same in both conditions, suggesting that the fluorescence is predominantly due to EMA. On the basis of theoretical cleavage sites (Fig. 7C), band 1 is likely to be either His153–Lys357 (22.1 kDa) or Glu192–Lys406 (23.4 kDa), whereas band 2 can be assigned as Glu192–Lys357 (17.9 kDa). Bands 3, 4, and 5, which are labeled by EMA but do not contain FMN, can be assigned as Met122–Lys191 (7.8 kDa, containing Cys148), Trp358–Lys406 (5.5 kDa, containing Cys368), and Met122–Lys152 (3.6 kDa, containing Cys148), respectively. The fluorescence intensity of band 4 is significantly weaker than that of bands 3 and 5. This is probably because Cys368 is buried among multiple TMHs (Fig. S6) and, hence, its reactivity to EMA is masked. When korormicin was introduced into the labeling reaction, the fluorescence intensities of bands 3 and 5 decreased markedly, whereas the intensity of band 4 was almost unchanged. Altogether, these results indicate that korormicin predominantly suppresses the labeling of NqrB-Cys148, corroborating that this inhibitor binds near the cytoplasmic surface of the NqrB subunit.
Competition between [125I]PAD-3, [125I]PAD-4, or [125I]PKRD-1 and other inhibitors
We previously reported that the extents of the photoaffinity labeling of V. cholerae Na+-NQR by [125I]PAD-1 or [125I]PAD-2 are enhanced in the presence of other inhibitors under some experimental conditions, rather than being competitively suppressed (14). Notably, because this unusual competitive behavior was observed even in the presence of an excess of PAD-1 or PAD-2 (i.e. the nonradioactive forms of the same compounds), we cannot explain this phenomenon by a simple scenario in which [125I]PAD-1 or [125I]PAD-2 and various competitors share a common binding pocket in the enzyme. Therefore, we proposed an equilibrium state model assuming two distinct inhibitor-bound states (i.e. one-inhibitor- and two-inhibitor-bound states), as briefly described in the Introduction. To examine whether this unusual competitive behavior could be a more general phenomenon, rather than being dependent on the specific chemical frameworks of different inhibitor probes and photolabile groups, we carried out competition tests using [125I]PAD-3, [125I]PAD-4, or [125I]PKRD-1.
First, the labeling of Na+-NQR (900 nm) by [125I]PAD-3 (5.0 nm) was conducted in the presence of a wide concentration range (2–5,000 nm) of other inhibitors (PAD-3, aurachin D-42, or korormicin A). In this, and the following competition tests, we included a nonradioactive form of each inhibitor probe, which can also cross-link to the enzyme, as one of the competitors. When PAD-3 was used as the competitor, ∼40% of the radioactivity in the control lane remained even in the presence of a 100-fold excess (500 nm) of the nonradioactive species, whereas ∼20% remained even with a 1000-fold excess (5000 nm) (Fig. 8A). In the case of aurachin D-42, the extent of labeling was almost unchanged in the presence of aurachin D-42 up to 5000 nm (Fig. 8A). In the presence of a 1000-fold excess of korormicin A, the extent of labeling decreased to ∼30% of the control lane (Fig. 8A).
Figure 8.
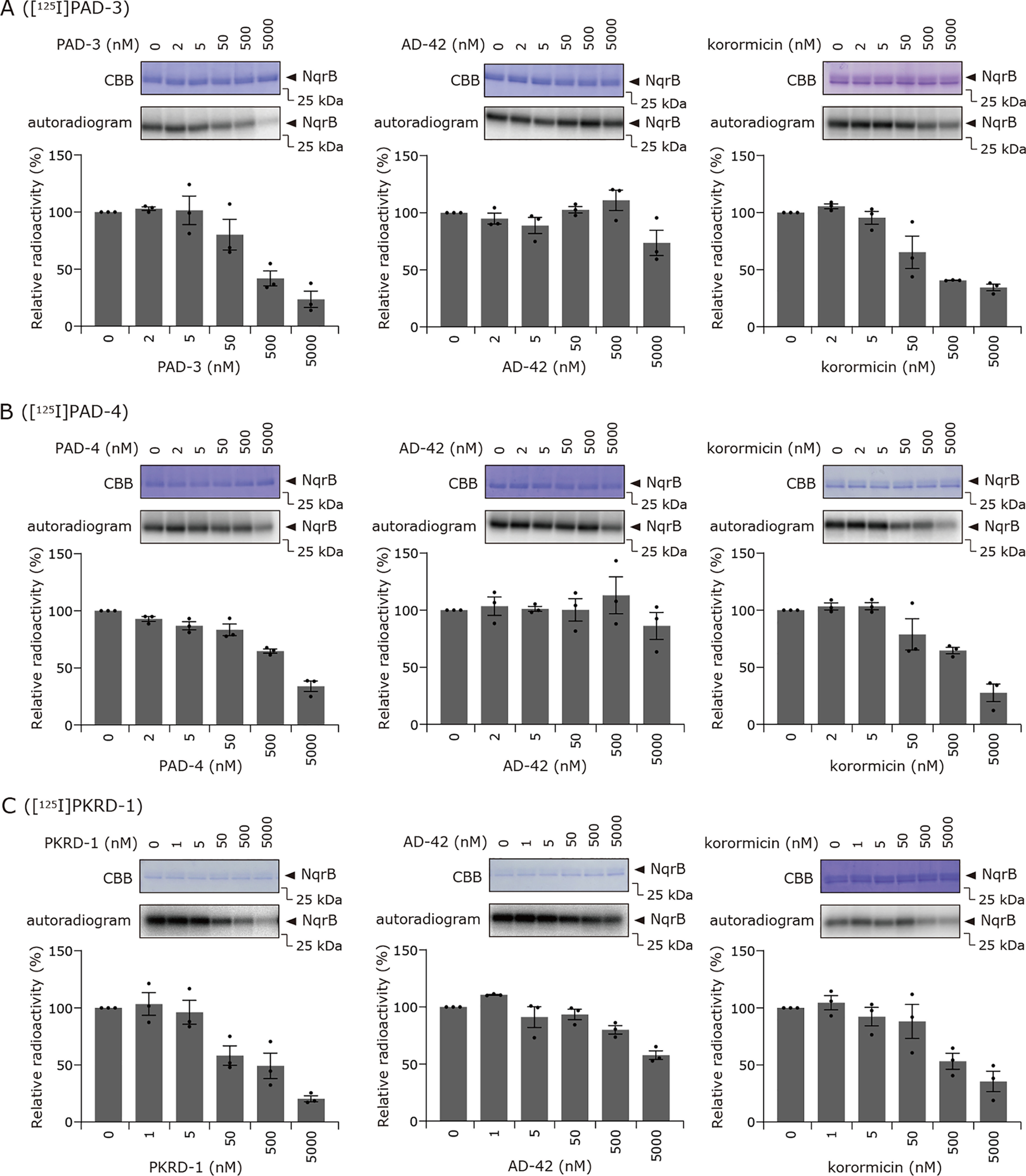
Competition tests between [125I]PAD-3, [125I]PAD-4, or [125I]PKRD-1 and other inhibitors. A, the labeling of Na+-NQR (900 nm) by [125I]PAD-3 (5.0 nm) was conducted in the presence of PAD-3, aurachin D-42, or korormicin A. B, the labeling by [125I]PAD-4 (5.0 nm) was conducted in the presence of PAD-4, aurachin D-42, or korormicin A. C, the labeling by [125I]PKRD-1 (5.0 nm) was performed in the presence of PKRD-1, aurachin D-42, or korormicin A. Values in graphs are mean ± S.E. (n = 3).
Next, we conducted competition tests between [125I]PAD-4 and other inhibitors (PAD-4, aurachin D-42, or korormicin A) under the same experimental conditions. As shown in Fig. 8B, similar results to those for [125I]PAD-3 were observed. Clearly, the competition behavior between [125I]PAD-3 or [125I]PAD-4 and the various competitors is inconsistent with the pattern anticipated when different inhibitors share a common binding site. We conclude, therefore, that the unusual competitive behavior is also the case for [125I]PAD-3 and [125I]PAD-4 irrespective of the different reactivities of the photolabile groups (ACT versus phenylazido). When competition tests were carried out with [125I]PAD-1 and [125I]PAD-2 under the same experimental conditions, as the concentration of the competitor was increased, the labeling yield initially increased before decreasing at even higher concentrations of competitor (14). This initial increase in labeling yield was never observed for [125I]PAD-3 or [125I]PAD-4. This is not inconsistent with our equilibrium state model (14), in which the competition profiles are determined by multiple parameters that reflect physicochemical properties of the inhibitor probes, such as binding affinity and cross-linking yield.
Finally, we conducted competition tests between the korormicin A derivative [125I]PKRD-1 (5.0 nm) and other inhibitors (PKRD-1, korormicin A, and aurachin D-42). Substantial labeling (∼50–60%) was retained even in the presence of a 100-fold excess of PKRD-1 or korormicin A (500 nm) (Fig. 8C), whereas almost complete suppression of labeling was achieved with 1,000-fold excess (5.0 μm) of either competitor. When aurachin D-42 was used as a competitor, ∼60% of the radioactivity remained in the presence of a 1,000-fold excess (5.0 μm). Altogether, these results indicate that the unusual competitive behavior is not limited to aurachin derivatives but is a more general phenomenon that is independent of the different chemical frameworks of the inhibitor probes (aurachins versus korormicins).
Discussion
Na+-NQR is anticipated to be a promising target of antibiotics because it is an exclusively prokaryotic enzyme, not found in eukaryotes, and it plays an important role in both bioenergetics and homeostasis in many pathogenic bacteria (24–26). However, in contrast to extensive studies on numerous inhibitors of mitochondrial respiratory enzymes (both from basic and applied scientific points of view), studies on Na+-NQR inhibitors have been highly limited. Here we investigated the mechanisms of action of several potent inhibitors of V. cholerae Na+-NQR mainly by addressing some unsettled issues in our previous work (14). New information obtained in the present study can be summarized into the following three points: (i) korormicin A, like aurachin-D type inhibitors, binds to the cytoplasmic surface of the NqrB subunit including the protruding N-terminal stretch, (ii) the binding locations of UQ and the inhibitors partially overlap in the interfacial region between NqrA and NqrB close to the cytoplasmic surface, and (iii) the unusual competitive behavior, which was first observed for [125I]PAD-1 and [125I]PAD-2 (14), is observed with all inhibitors tested irrespective of their different chemical frameworks, corroborating our previous proposal that there are two binding sites for inhibitors in the enzyme (14). We will discuss each of these findings below.
First, we found that the korormicin derivative [125I]PKRD-1 labels two regions in NqrB: one is Trp23–Lys54 (L2, Fig. 2, D and E, dark brown) in the protruding N-terminal stretch and the other is His153−Lys191 (L1) that includes the cytoplasmic loop connecting TMHs 2−3 and TMHs 3 and 4 (Figs. 2, D and E, yellow). [125I]PAD-4 labeled the same two regions, whereas [125I]PAD-3 predominantly labeled one of the two regions, His153−Lys191 (L1). Considering that [125I]PAD-1 and [125I]PAD-2 also labeled the N-terminal stretch (Fig. 2C) and that all of the aurachin derivatives ([125I]PAD-1−[125I]PAD-4) have a quinolone ring as a common toxophore, it is reasonable to assume that the binding positions of their quinolone rings overlap somewhere on the N-terminal stretch irrespective of their different side chain structures. Given this, we reason that the residue(s) labeled by [125I]PAD-3, [125I]PAD-4, and [125I]PKRD-1 in the region His153−Lys191 (L1) lies in the cytoplasmic loop connecting TMHs 2−3 (His153−Gly158 (yellow spheres), Fig. 2D), not at transmembrane segments because this loop is the closest area in the region to the protruding stretch. Collectively, we conclude that different types of inhibitors all bind to the cytoplasmic surface regions of NqrB in a similar manner such that the toxophoric moiety (i.e. the lactone and quinolone rings of korormicin and aurachins, respectively) orients to the N-terminal stretch, but their side chain moieties are flexibly positioned in the cytoplasmic surface area. The significant suppressive effect of korormicin on the labeling of NqrB-Cys148 by EMA (Fig. 7) is consistent with this notion.
Although the crystallographic positions of the two labeled regions in NqrB (Trp23–Lys54 (L2) and the loop (His153−Gly158)) appear to be somewhat distant from one another (Fig. 2D), we cannot exclude the possibility that the conformations of NqrB (including the protruding N-terminal stretch) in the crystallographic structure are not exactly the same as those in the native enzyme. In support of this, there is a consensus that for electron transfer to occur at a physiologically relevant rate from riboflavinNqrB/E to UQ in the predicted cavity in NqrA, a large spatial gap (∼40 Å) between the two redox centers must be reduced (13). To do so, the cytoplasmic interfacial area between NqrA and NqrB would need to undergo substantial structural rearrangements. Taken together, we propose that the potent inhibitors block the structural rearrangements required for facilitating reduction of UQ in the adjacent NqrA. In addition to this effect, direct interference in UQ binding by inhibitors should not be ignored, as discussed later.
Hayashi et al. (20) reported that a spontaneous mutation at Gly140 (G140V) in the NqrB subunit of V. alginolyticus Na+-NQR confers remarkable resistance against korormicin A compared with the WT enzyme. We previously showed that the corresponding mutant in V. cholerae NqrB-G141A gives moderate (∼160-fold) resistance to korormicin A, whereas a substitution at the adjacent residue, G140A, confers very significant (>10,000-fold) resistance (14). Based on the crystallographic structure (13), these residues reside in TMH 2 under the loop described above (His153−Gly158) (Fig. S7), and are some distance from the binding region identified in this study. Earlier FT-IR and biochemical studies suggested that a change in the size of the residue at positions 140 or 141 in the V. cholerae NqrB sequence has an indirect effect on the UQ reaction in some way, and that the residues are not actually part of the binding site for UQ (8, 10). To know whether korormicin A retains its binding affinity to the V. cholerae NqrB-G140A and -G141A mutants, we carried out photoaffinity labeling experiments on these mutated enzymes using [125I]PKRD-1. In this case, the labeling efficiencies were essentially unchanged compared with the WT enzyme (Fig. 6B); that is, there was no loss of binding affinity to the enzyme. We previously observed similar results using [125I]PAD-2; namely, although NqrB-G140A conferred significant resistance to PAD-2, the labeling of this mutated enzyme by [125I]PAD-2 did not decrease compared with the WT enzyme (14). Taking these findings together, it is likely that the resistance against korormicin A is not due to diminished binding affinity toward the mutated enzyme. It remains to be elucidated why the ability of korormicin A to inhibit electron transfer from riboflavinNqrB/E to UQ significantly decreased in the G140A and G141A mutants.
Next, we consider the binding locations of UQ and the inhibitors and how they are spatially related. This is critical for understanding how inhibitors interfere with the UQ reaction. The crystallographic study suggested that the NqrA subunit includes a pocket that is large enough to accommodate the UQ head-ring, although the pocket is ∼20 Å above the membrane surface (13). We previously showed that the head-ring of a short-chain UQ photoaffinity probe (PUQ-3) indeed binds to this pocket (14). The protruding N-terminal stretch of NqrB may be flexible because the tip (Met1–Pro37) of the stretch is apparently disordered (13). This stretch seems to anchor a domain of NqrA, which includes the pocket, to NqrB. Given these structural characteristics, we cannot rule out the possibility that whereas the polar head-ring of UQ8 orients to the NqrA subunit, its long isoprenyl chain could anchor into the membrane-embedded part of the enzyme by binding to the adjacent transmembrane subunit NqrB. This role of the isoprenyl chain, along with the predicted flexibility of the protruding stretch of NqrB, could reduce the distance between riboflavinNqrB/E and the UQ head-ring bound in NqrA, allowing electron transfer to take place. If this is the case, there may be some overlapping (or obstruction) between UQ8 and inhibitor molecules at the cytoplasmic interface between NqrA and NqrB. The results of the competition between short-chain UQs and the inhibitor probes corroborate this notion (Fig. 4). Altogether, whereas the UQ head-ring and the toxophoric ring of inhibitors do not directly overlap, their side chain moieties may interfere with each other. We conclude that the inhibitors interfere with the UQ reaction in two ways: one is blocking structural rearrangements at the cytoplasmic interface between NqrA and NqrB and the other is the direct obstruction of the binding of UQ at this interfacial area.
We next discuss an alternative proposal about the catalytic binding position for the UQ head-ring in Na+-NQR. Based on alanine scanning mutagenesis of amino acid residues in membrane subunits NqrB and NqrD of V. cholerae Na+-NQR, Raba et al. (42, 43) proposed that the catalytic site for the UQ head-ring is located at the interface of transmembrane segments of NqrB and NqrD (Fig. S8), where the key residues forming the reaction pocket for the UQ head-ring and also for the quinolone ring of HQNO are Phe185 and Phe211 in TMHs 4 and 5 of NqrB, respectively, and Pro185, Leu190, and Phe193 in TMH 6 of NqrD. In contrast, photoaffinity labeling studies using different photolabile UQs showed that the UQ head-ring binds to the NqrA subunit (7, 14). Because the studies by Raba et al. (42, 43) are largely based on Michaelis-Menten-type kinetic analyses of the activities of site-directed mutant enzymes, it is difficult to determine whether the proposed critical residues are directly involved in the interaction with the UQ ring or whether the mutations indirectly affect the UQ reaction by inducing long-range structural changes. It should be remembered that during catalytic turnover the subunits harboring the cofactors and the UQ-binding pocket need to undergo substantial conformational rearrangements that reduce the long distances for electron transfer (13); therefore the likelihood of the mutations having such indirect effects is high. More importantly, the UQ-binding pocket proposed by Raba et al. (42, 43) is not only located between FMNNqrB (at the periplasmic surface) and riboflavinNqrB/E (at the cytoplasmic surface) but is also spatially closer to the former (∼21 Å) than the latter (∼36 Å) (Fig. S8). This positioning of the UQ head-ring seems difficult to reconcile with the current consensus on the electron transfer pathway in the enzyme (based on stopped-flow kinetics and cofactor-deletion mutants), in which electrons move from FMNNqrB to riboflavinNqrB/E, and from riboflavinNqrB/E to UQ (2, 4, 11, 13). As revealed in this study, it is reasonable to consider that the binding positions of UQ and inhibitors are located at the cytoplasmic interface of the NqrA and NqrB subunits, where these sites overlap partially.
Finally, we discuss the unusual competitive behavior between the photoreactive inhibitor probes and competitors. The present study revealed that this phenomenon is general irrespective of different chemical frameworks of the inhibitor probes and different reactivities of their photolabile groups. Nevertheless, the competition profiles varied somewhat among different pairs of inhibitor/competitor tested. In particular, increases in labeling in the presence of some concentrations of competitor, as observed for [125I]PAD-1 and [125I]PAD-2 (14), were not observed for [125I]PAD-3, [125I]PAD-4, and [125I]PKRD-1. These results are not inconsistent with our equilibrium model (14), in which the competition profiles are determined by multiple parameters that vary depending on physicochemical properties of the paired inhibitor/competitor, such as the relative binding affinities of the inhibitors and cross-linking yields in the two inhibitor-bound states (i.e. one- and two-inhibitor-bound states). To obtain a mechanistic clue to the unusual competitive behavior, we conducted competition tests between [125I]PAD-3 and PAD-3 in the presence of UQ1 (50 μm), which partially suppresses the labeling by [125I]PAD-3 (Fig. 4). We designed this experiment anticipating that if UQ1 masks one of the two inhibitor-binding sites, typical competition would be restored. However, the competition profile remained unchanged compared with that observed in the absence of UQ1, except for overall slight decreases in the labeling yields (Fig. S9). Thus, the presence of UQ has no influence on the unusual competitive behavior. Further studies using varying approaches are needed to elucidate the mechanistic details of this phenomenon.
Experimental procedures
Materials
UQ1 and UQ2 were kind gifts from Eisai (Tokyo, Japan). Protein standards (Precision Plus Protein Standards Dual Xtra) for SDS-PAGE were purchased from Bio-Rad. EMA was purchased from Life Technologies, Inc. [125I]NaI was purchased from PerkinElmer Life Sciences. n-Dodecyl-β-d-maltoside (DDM) and lauryldimethylamine N-oxide (LDAO) were purchased from Dojindo (Kumamoto, Japan) and Sigma-Aldrich, respectively. Other reagents were all of analytical grade.
Syntheses of PAD-3, [125I]PAD-3, PAD-4, [125I]PAD-4, PKRD-1, and [125I]PKRD-1
The synthetic procedures for PAD-3, PAD-4, and PKRD-1 together with their 125I-incorporated derivatives ([125I]PAD-3, [125I]PAD-4, and [125I]PKRD-1) are described in the supporting data (Schemes S1–S3). All compounds were characterized by 1H and 13C NMR spectroscopy and MS.
Plasmid construction and purification of Na+-NQR
Site-directed mutants were obtained using the QuikChange Lightning and QuikChange II XL mutagenesis kits (Agilent) as reported before (44). The sequences of the forward primers are listed in Table 2.
Table 2.
Primers used in this study
| Forward primer | Sequence 5′ to 3′ |
|---|---|
| NqrB-D49A | GCTTGGCGAAGAGGCCGTTGGCATGCGTC |
| NqrB-D52A | GGCGAAGAGTACGTTGTCATGCGTCCTACTATG |
| NqrB-D49A/D52A | GAAGCTCGCACGTTCGTGCTAGCGTTGCTCTAAAACGTATCAT |
| NqrB-E154A | GTATGGTGCGCAAGCATGCTGTCAACGAAGGTTTCTTC |
| NqrB-E157A | CGCAAGCATGAAGTCAACGCTGGTTTCTTCGTTACCTCTAT |
Recombinant WT and mutant Na+-NQR strains were grown in LB (Miller) medium as reported before (44) in 30-liter fermenters (New Brunswick BF-5000, Microbiology Core Facilty, CBIS, RPI) at 37 °C with constant agitation (300 rpm). The expression of the nqr operon was induced by adding arabinose. Cells were harvested, washed, and broken in the buffer (50 mm Tris-HCl (pH 8.0), 300 mm NaCl, 10 mm MgCl2) in the presence of DNase and protease inhibitors. Membranes were obtained by ultracentrifugation (100,000 × g) and washed with a medium containing 5 mm imidazole, 300 mm NaCl, and 0.05% glycerol.
WT and mutant Na+-NQRs were purified by nickel-nitrilotriacetic acid affinity chromatography and anion exchange DEAE chromatography as reported before (14, 45). For the preparation of LDAO-washed Na+-NQR (10, 14), the enzyme stock solution (50 µl) was diluted in a 20-fold volume with a washing buffer (50 mm Tris-HCl, 1.0 mm EDTA, 5% glycerol, and 0.05% LDAO, pH 8.0), concentrated with a centrifugal filter Amicon Ultra 100 K (Merck-Millipore, Billerica, MA), diluted 20-fold again, and re-concentrated.
Measurement of the electron transfer activity of Na+-NQR
The NADH-ubiquinone oxidoreductase activity of purified Na+-NQR was determined by following reduction of UQ1 or UQ2 at 282 nm (ε = 14.5 mm−1 cm−1) with a Shimadzu UV-3000 instrument at 30 ˚C (14). The reaction medium (2.5 ml) contained 5% glycerol, 0.05% DDM, 1.0 mm EDTA, 100 mm NaCl, and 50 mm Tris-HCl (pH 8.0). The final enzyme concentration was set to 0.90 nm. The reaction was started by the addition of 100 μm NADH after the incubation of the enzyme with ubiquinone for 1 min. The kinetic parameters of ubiquinone reduction were determined by fitting the experimental data to the Michaelis-Menten equation using Prism (version 8, GraphPad, La Jolla, CA). When the effects of inhibitor were examined, the enzyme was incubated with inhibitor for 1 min before the addition of ubiquinone.
Photoaffinity labeling of Na+-NQR by [125I]PAD-3, [125I]PAD-4, and [125I]PKRD-1
LDAO-washed Na+-NQR was suspended in the buffer (100 mm NaCl, 5% glycerol, 0.05% LDAO, and 50 mm Tris-HCl, pH 8.0). The enzyme solution (900 nm, 20 µl) was incubated in a 1.5-ml Eppendorf tube with [125I]PAD-3, [125I]PAD-4, or [125I]PKRD-1 (5.0 nm each) for 5 min at room temperature. Then, the sample was irradiated with a 100 W Xe lamp (LAX-103, Asahi Spectra, Tokyo, Japan) through an UVA mirror module (300−400 nm) for 5 min on ice, positioned 10 cm from the light source (14). When we conducted competition tests between [125I]PAD-3, [125I]PAD-4, or [125I]PKRD-1 and various competitors, the competitor was added to the enzyme and incubated for 5 min at room temperature prior to the treatment with the 125I-incorporated inhibitor.
The labeling reaction was quenched by addition of an appropriate volume of 4× Laemmli's sample buffer, followed by the separation of the subunits on a 15% Laemmli-type SDS gel containing 6.0 M urea (46). The gel was stained with Coomassie Brilliant Blue, dried, exposed to an imaging plate (BAS-MS2040, FUJIFILM, Tokyo, Japan) for 12–48 h, and visualized with the FLA-5100 Bio-Imaging analyzer (FUJIFILM). The radioactivity of each band was quantified with Multi-Gauge (FUJIFILM). As Na+-NQR containing covalently bound FMN in NqrB and NqrC was able to be visualized using a 473-nm light source and an LPB emission filter (510 nm), the FMN-based fluorescence was used as a loading control for SDS-PAGE throughout this study (39, 40).
Labeling of cysteines in Na+-NQR by EMA
LDAO-washed Na+-NQR was suspended in the buffer (100 mm NaCl, 5% glycerol, 0.05% LDAO, and 50 mm Tris-HCl, pH 8.0). The enzyme solution (900 nm, 20 µl) was incubated with EMA (5.0 μm) for 5 min on ice. The reaction was terminated by the addition of DTT (40 mm, 1 µl), followed by the separation of the proteins on a 16% Schägger-type SDS gel (16% T, 3% C) (47). The proteins modified by EMA were visualized using the model FLA-5100 Bio-imaging analyzer (FUJIFILM) with a 473-nm light source and LPB filter or 532-nm light source and LPG filter.
Proteomic analysis
For proteomic analyses of the NqrB subunit labeled by [125I]PAD-3, [125I]PAD-4, or [125I]PKRD-1, NqrB was recovered from the SDS gel by direct diffusion. The slice of unfixed SDS gel was vigorously stirred for 12–16 h in a buffer containing 0.1 m NH4HCO3, 0.1% (w/v) SDS, and 1% 2-mercaptoethanol. The purified subunit was digested with lysylendopeptidase (Lys-C, Wako Pure Chemicals, Osaka, Japan) or endoprotease Asp-N (Roche Applied Science) in 50 mm Tris-HCl buffer containing 0.1% SDS or 50 mm NaPi buffer containing 0.01% SDS, respectively. These digests were separated on a Schägger-type SDS gel (16.5% T, 6% C containing 6.0 m urea) (47).
Data availability
All data described in the manuscript are contained within the manuscript and supporting data.
Supplementary Material
Acknowledgments
We acknowledge the Radioisotope Research Center, Kyoto University, for the technical support in the radioisotope experiments. We also acknowledge the Microbiology Core Facility in Rensselaer Polytechnic Institute.
This article contains supporting information.
Author contributions—Specific contributions to this work are as follows: T. M., Y. S., M. M., B. B., and H. M. designed the research; T. M., Y. S., H. T., N. B., T. I., and T. T. performed the research; T. M., Y. S., J. E. M., M. M., B. B. and H. M. analyzed data; H. M. directed the project and wrote the paper with T. M., J. E. M., M. M., and B. B.
Funding and additional information—This work was supported by JSPS KAKENHI Grants JP18H02147 and JP19K22278 (to H. M.), JP18K05458 (to M. M.), 20K15457 (to T. M.), National Science Foundation Grant 1616674 (to B. B.), and the National Institutes of Health, NIAID Grant R01AI132580 (to B. B.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- UQ
- ubiquinone
- Asp-N
- endoprotease Asp-N
- DDM
- n-dodecyl-β-D-maltoside
- EMA
- eosin-5-maleimide
- Lys-C
- lysylendopeptidase
- HQNO
- 2-n-heptyl-4-hydroxyquinoline N-oxide
- Na+-NQR
- Na+-pumping NADH-ubiquinone oxidoreductase
- TMH
- transmembrane helix
- UQn
- ubiquinone-n
- ACT
- 2-aryl-5-carboxytetrazole
- LDAO
- lauryldimethylamine N-oxide.
References
- 1. Hayashi M., Nakayama Y., and Unemoto T. (2001) Recent progress in the Na+-translocating NADH-quinone reductase from the marine Vibrio alginolyticus. Biochim. Biophys. Acta 1505, 37–44 10.1016/S0005-2728(00)00275-9 [DOI] [PubMed] [Google Scholar]
- 2. Juárez O., and Barquera B. (2012) Insights into the mechanism of electron transfer and sodium translocation of the Na+-pumping NADH-quinone oxidoreductase. Biochim. Biophys. Acta 1817, 1823–1832 10.1016/j.bbabio.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barquera B., Zhou W., Morgan J. E., and Gennis R. B. (2002) Riboflavin is a component of the Na+-pumping NADH–quinone oxidoreductase from Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A. 99, 10322–10324 10.1073/pnas.162361299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Juárez O., Morgan J. E., and Barquera B. (2009) The electron transfer pathway of the Na+-pumping NADH:quinone oxidoreductase from Vibrio cholerae. J. Biol. Chem. 284, 8963–8972 10.1074/jbc.M809395200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Juárez O., Morgan J. E., Nilges M. J., and Barquera B. (2010) Energy transducing redox steps of the Na+-pumping NADH:quinone oxidoreductase from Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A. 107, 12505–12510 10.1073/pnas.1002866107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bogachev A. V., Bloch D. A., Bertsova Y. V., and Verkhovsky M. I. (2009) Redox properties of the prosthetic groups of Na+-translocating NADH:quinone oxidoreductase: 2. study of the enzyme by optical spectroscopy. Biochemistry 48, 6299–6304 10.1021/bi900525v [DOI] [PubMed] [Google Scholar]
- 7. Casutt M. S., Nedielkov R., Wendelspiess S., Vossler S., Gerken U., Murai M., Miyoshi H., Möller H., and Steuber J. (2011) Localization of UQ-8 in the Na+-pumping NADH:quinone oxidoreductase from Vibrio cholerae. J. Biol. Chem. 286, 40075–40082 10.1074/jbc.M111.224980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Juárez O., Neehaul Y., Turk E., Chahboun N., DeMicco J. M., Hellwig P., and Barquera B. (2012) The role of glycine residues 140 and 141 of subunit B in the functional UQ binding site of the Na+-pumping NADH-quinone oxidoreductase from Vibrio cholerae. J. Biol. Chem. 287, 25678–25685 10.1074/jbc.M112.366088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nedielkov R., Steffen W., Steuber J., and Möller H. M. (2013) NMR reveals double occupancy of quinone-type ligands in the catalytic quinone binding site of the Na+-translocating NADH:quinone oxidoreductase from Vibrio cholerae. J. Biol. Chem. 288, 30597–30606 10.1074/jbc.M112.435750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Strickland M., Juárez O., Neehaul Y., Cook D. A., Barquera B., and Hellwig P. (2014) The conformational changes induced by UQ binding in the Na+-pumping NADH:UQ oxidoreductase (Na+-NQR) are kinetically controlled by conserved glycines 140 and 141 of the NqrB subunit. J. Biol. Chem. 289, 23723–23733 10.1074/jbc.M114.574640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tuz K., Mezic K. G., Xu T., Barquera B., and Juárez O. (2015) The kinetic reaction mechanism of the Vibrio cholerae sodium-dependent NADH dehydrogenase. J. Biol. Chem. 290, 20009–20021 10.1074/jbc.M115.658773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Belevich N. P., Bertsova Y. V., Verkhovskaya M. L., Baykov A. A., and Bogachev A. V. (2016) Identification of the coupling step in Na+-translocating NADH:quinone oxidoreductase from real-time kinetics of electron transfer. Biochim. Biophys. Acta 1857, 141–149 10.1016/j.bbabio.2015.12.001 [DOI] [PubMed] [Google Scholar]
- 13. Steuber J., Vohl G., Casutt M. S., Vorburger T., Diederichs K., and Fritz G. (2014) Structure of the V. cholerae Na+-pumping NADH:quinone oxidoreductase. Nature 516, 62–67 10.1038/nature14003 [DOI] [PubMed] [Google Scholar]
- 14. Ito T., Murai M., Ninokura S., Kitazumi Y., Mezic K. G., Cress B. F., Koffas M. A. G., Morgan J. E., Barquera B., and Miyoshi H. (2017) Identification of the binding sites for UQ and inhibitors in the Na+-pumping NADH-UQ oxidoreductase of Vibrio cholerae by photoaffinity labeling. J. Biol. Chem. 292, 7727–7742 10.1074/jbc.M117.781393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Page C. C., Moser C. C., Chen X., and Dutton P. L. (1999) Natural engineering principles of electron tunneling in biological oxidation-reduction. Nature 402, 47–52 10.1038/46972 [DOI] [PubMed] [Google Scholar]
- 16. Barquera B., Morgan J. E., Lukoyanov D., Scholes C. P., Gennis R. B., Mark J., and Nilges M. J. (2003) X- and W-band EPR and Q-band ENDOR studies of the flavin radical in the Na+-translocating NADH:quinone oxidoreductase from Vibrio cholerae. J. Am. Chem. Soc. 125, 265–275 10.1021/ja0207201 [DOI] [PubMed] [Google Scholar]
- 17. Bogachev A. V., Kulik L. V., Bloch D. A., Bertsova Y. V., Fadeeva M. S., and Verkhovsky M. I. (2009) Redox properties of the prosthetic groups of Na+-translocating NADH:quinone oxidoreductase. 1. Electron paramagnetic resonance study of the enzyme. Biochemistry 48, 6291–6298 10.1021/bi900524m [DOI] [PubMed] [Google Scholar]
- 18. Nakayama Y., Hayashi M., Yoshikawa K., Mochida K., and Unemoto T. (1999) Inhibitor studies of a new antibiotic korormicin, 2-n-heptyl-4-hydroxyquinoline N-oxide and Ag+ toward the Na+-translocating NADH-quinone reductase from the marine Vibrio alginolyticus. Biol. Pharm. Bull. 22, 1064–1067 10.1248/bpb.22.1064 [DOI] [PubMed] [Google Scholar]
- 19. Yoshikawa K., Nakayama Y., Hayashi M., Unemoto T., and Mochida K. (1999) Korormicin, an antibiotic specific for Gram-negative marine bacteria, strongly inhibits the respiratory chain-linked Na+-translocating NADH:quinone reductase from the marine Vibrio alginolyticus. J. Antibiot. 52, 182–185 10.7164/antibiotics.52.182 [DOI] [PubMed] [Google Scholar]
- 20. Hayashi M., Shibata N., Nakayama Y., Yoshikawa K., and Unemoto T. (2002) Korormicin insensitivity in Vibrio alginolyticus is correlated with a single point mutation of Gly-140 in the NqrB subunit of the Na+-translocating NADH-quinone reductase. Arch. Biochem. Biophys 401, 173–177 10.1016/S0003-9861(02)00007-3 [DOI] [PubMed] [Google Scholar]
- 21. Von Jagow G., and Link T. A. (1986) Use of specific inhibitors on the mitochondrial bc1 complex. Methods Enzymol. 126, 253–271 10.1016/S0076-6879(86)26026-7 [DOI] [PubMed] [Google Scholar]
- 22. Ek M. S., and Brzezinski P. (1997) Oxidation of ubiquinol by cytochrome bo3 from Escherichia coli: kinetics of electron and proton transfer. Biochemistry 36, 5425–5431 10.1021/bi962478e [DOI] [PubMed] [Google Scholar]
- 23. Kita K., Konishi K., and Anraku Y. (1984) Terminal oxidase of Escherichia coli aerobic respiratory chain: II purification and properties of cytochrome b558-d complex from cells grown with limited oxygen and evidence of branched electron-carrying systems. J. Biol. Chem. 259, 3375–3381 [PubMed] [Google Scholar]
- 24. Dibrov P., Dibrov E., Maddaford T. G., Kenneth M., Nelson J., Resch C., and Pierce G. N. (2017) Development of a novel rationally designed antibiotic to inhibit a nontraditional bacterial target. Can. J. Physiol. Pharmacol. 95, 595–603 10.1139/cjpp-2016-0505 [DOI] [PubMed] [Google Scholar]
- 25. Dibrov P., Dibrov E., and Pierce G. N. (2017) Na+-NQR (Na+-translocating NADH:uniquinone oxidoreductase) as a novel target for antibiotics. FEMS Microbiol. Rev. 41, 653–671 10.1093/femsre/fux032 [DOI] [PubMed] [Google Scholar]
- 26. Maynard A., Butler N., Ito T., da Silva A., Murai M., Chen T., Koffas M., Miyoshi H., and Barquera B. (2019) The antibiotic korormicin A kills bacteria by producing reactive oxygen species. J. Bacteriol. 201, e00718–18 10.1128/JB.00718-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yankovskaya V., Horsefield R., Törnroth S., Luna-Chavez C., Miyoshi H., Léger C., Byrne B., Cecchini G., and Iwata S. (2003) Architecture of succinate dehydrogenase and reactive oxygen species generation. Science 299, 700–704 10.1126/science.1079605 [DOI] [PubMed] [Google Scholar]
- 28. Horsefield R., Yankovskaya V., Sexton G., Whittingham W., Shiomi K., Ōmura S., Byrne B., Cecchini G., and Iwata S. (2006) Structural and computational analysis of the quinone-binding site of complex II (succinate-UQ oxidoreductase). J. Biol. Chem. 281, 7309–7316 10.1074/jbc.M508173200 [DOI] [PubMed] [Google Scholar]
- 29. Huang L. S., Cobessi D., Tung E. Y., and Berry E. A. (2005) Binding of the respiratory chain inhibitor antimycin to the mitochondrial bc1 complex: a new crystal structure reveals an altered intramolecular hydrogen-binding pattern. J. Mol. Biol. 351, 573–597 10.1016/j.jmb.2005.05.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Esser L., Quinn B., Li Y. F., Zhang M., Elberry M., Yu L., Yu C. A., and Xia D. (2004) Crystallographic studies of quinol oxidation site inhibitors: a modified classification of inhibitors for the cytochrome bc1 complex. J. Mol. Biol. 341, 281–302 10.1016/j.jmb.2004.05.065 [DOI] [PubMed] [Google Scholar]
- 31. Safarian S., Hahn A., Mills D. J., Radloff M., Eisinger M. L., Nikolaev A., Meier-Credo J., Melin F., Miyoshi H., Gennis R. B., Sakamoto J., Langer J. D., Hellwig P., Kühlbrandt W., and Michel H. (2019) Active site rearrangement and structural divergence in prokaryotic respiratory oxidases. Science 366, 100–104 10.1126/science.aay0967 [DOI] [PubMed] [Google Scholar]
- 32. Xu J., Ding Z., Liu B., Yi S. M., Li J., Zhang Z., Liu Y., Li J., Liu L., Zhou A., Gennis R. B., and Zhu J. (2020) Structure of the cytochrome aa3-600 heme-copper menaquinol oxidase bound to inhibitor HQNO shows TM0 is part of the quinol binding site. Proc. Natl. Acad. Sci. U.S.A. 117, 872–876 10.1073/pnas.1915013117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shiba T., Kido Y., Sakamoto K., Inaoka D. K., Tsuge C., Tatsumi R., Takahashi G., Balogun E. O., Nara T., Aoki T., Honma T., Tanaka A., Inoue M., Matsuoka S., Saimoto H., et al. (2013) Structure of the trypanosome cyanide-insensitive alternative oxidase. Proc. Natl. Acad. Sci. U.S.A. 110, 4580–4585 10.1073/pnas.1218386110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Herner A., Marjanovic J., Lewandowski T. M., Marin V., Patterson M., Miesbauer L., Ready D., Williams J., Vasudevan A., and Lin Q. (2016) 2-Aryl-5-carboxytetrazole as a new photoaffinity label for drug target identification. J. Am. Chem. Soc. 138, 14609–14615 10.1021/jacs.6b06645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Juárez O., Shea M. E., Makhatadze G. I., and Barquera B. (2011) The role and specificity of the catalytic and regulatory cation binding sites of the Na+-pumping NADH:quinone oxidoreductase from Vibrio cholerae. J. Biol. Chem. 286, 26383–26390 10.1074/jbc.M111.257873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shea M. E., Juárez O., Cho J., and Barquera B. (2013) Aspartic acid 397 in subunit B of the Na+-pumping NADH:quinone oxidoreductase from Vibrio cholerae forms part of a sodium-binding site, is involved in cation selectivity, and affects cation-binding site cooperativity. J. Biol. Chem. 288, 31241–31249 10.1074/jbc.M113.510776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Neehaul Y., Juárez O., Barquera B., and Hellwig P. (2013) Infrared spectroscopic evidence of a redox-dependent conformational change involving ion binding residue NqrB-D397 in the Na+-pumping NADH:quinone oxidoreductase from Vibrio cholerae. Biochemistry 52, 3085–3093 10.1021/bi4000386 [DOI] [PubMed] [Google Scholar]
- 38. Mezic K. G., Juárez O., Neehaul Y., Cho J., Cook D., Hellwig P., and Barquera B. (2019) Glutamate 95 in NqrE is an essential residue for the translocation of cations in Na+-NQR. Biochemistry 58, 2167–2175 10.1021/acs.biochem.8b01294 [DOI] [PubMed] [Google Scholar]
- 39. Hayashi M., Nakayama Y., Yasui M., Maeda M., Furuishi K., and Unemoto T. (2001) FMN is covalently attached to a threonine residue in the NqrB and NqrC subunits of Na+-translocating NADH-quinone reductase from Vibrio alginolyticus. FEBS Lett. 488, 5–8 10.1016/S0014-5793(00)02404-2 [DOI] [PubMed] [Google Scholar]
- 40. Casutt M. S., Schlosser A., Buckel W., and Steuber J. (2012) The single qrB and NqrC subunits in the Na+-translocating NADH:quinone oxidoreductase (Na+-NQR) from Vibrio cholerae each carry one covalently attached FMN. Biochim. Biophys. Acta 1817, 1817–1822 10.1016/j.bbabio.2012.02.012 [DOI] [PubMed] [Google Scholar]
- 41. Nakayama Y., Yasui M., Sugahara K., Hayashi M., and Unemoto T. (2000) Covalently bound flavin in the NqrB and NqrC subunits of Na+-translocating NADH-quinone reductase from Vibrio alginolyticus. FEBS Lett. 474, 165–168 10.1016/S0014-5793(00)01595-7 [DOI] [PubMed] [Google Scholar]
- 42. Tuz K., Li C., Fang X., Raba D. A., Liang P., Minh D. D. L., and Juárez O. (2017) Identification of the catalytic ubiquinone-binding site of Vibrio cholerae sodium-dependent NADH dehydrogenase: a novel ubiquinone-binding motif. J. Biol. Chem. 292, 3039–3048 10.1074/jbc.M116.770982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Raba D. A., Yuan M., Fang X., Menzer W. M., Xie B., Liang P., Tuz K., Minh D. D. L., and Juárez O. (2019) Role of subunit D in ubiquinone-binding site of Vibrio cholerae NQR: pocket flexibility and inhibitor resistance. ACS Omega 4, 19324–19331 10.1021/acsomega.9b02707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Barquera B., Nilges M. J., Morgan J. E., Ramirez-Silva L., Zhou W., and Gennis R. B. (2004) Mutagenesis study of the 2Fe-2S center and the FAD binding site of the Na+-translocating NADH:ubiquinone oxidoreductase from Vibrio cholerae. Biochemistry 43, 12322–12330 10.1021/bi048689y [DOI] [PubMed] [Google Scholar]
- 45. Juárez O., Athearn K., Gillespie P., and Barquera B. (2009) Acid residues in the transmembrane helices of the Na+-pumping NADH:quinone oxidoreductase from Vibrio cholerae involved in sodium translocation. Biochemistry 48, 9516–9524 10.1021/bi900845y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 47. Schägger H. (2006) Tricine-SDS-PAGE. Nat. Protoc. 1, 16–22 10.1038/nprot.2006.4 [DOI] [PubMed] [Google Scholar]
- 48. Enomoto M., Kitagawa W., Yasutake Y., and Shimizu H. (2014) Total synthesis of aurachins C, D, and L, and a structurally simplified analog of aurachin C. Biosci. Biotech. Biochem. 78, 1324–1327 10.1080/09168451.2014.918494 [DOI] [PubMed] [Google Scholar]
- 49. Kobayashi Y., Yoshida S., and Nakayama Y. (2001) Total synthesis of korormicin. Eur. J. Org. Chem. 2001, 1873–1881 [DOI] [Google Scholar]
- 50. Uehara H., Oishi T., Yoshikawa K., Mochida K., and Hirama M. (1999) The absolute configuration and total synthesis of korormicin. Tetrahedron Lett. 40, 8641–8645 10.1016/S0040-4039(99)01812-2 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data described in the manuscript are contained within the manuscript and supporting data.