

Abstract
Mycobacterium tuberculosis has evolved numerous type VII secretion (ESX) systems to secrete multiple factors important for both growth and virulence across their cell envelope. ESX-1, ESX-3, and ESX-5 systems have been shown to each secrete a distinct set of substrates, including PE and PPE families of proteins, named for conserved Pro-Glu and Pro-Pro-Glu motifs in their N termini. Proper secretion of the PE–PPE proteins requires the presence of EspG, with each system encoding its own unique copy. There is no cross-talk between any of the ESX systems, and how each EspG recognizes its subset of PE–PPE proteins is currently unknown. The only current structural characterization of PE–PPE–EspG heterotrimers is from the ESX-5 system. Here we present the crystal structure of the PE5mt–PPE4mt–EspG3mm heterotrimer from the ESX-3 system. Our heterotrimer reveals that EspG3mm interacts exclusively with PPE4mt in a similar manner to EspG5, shielding the hydrophobic tip of PPE4mt from solvent. The C-terminal helical domain of EspG3mm is dynamic, alternating between “open” and “closed” forms, and this movement is likely functionally relevant in the unloading of PE–PPE heterodimers at the secretion machinery. In contrast to the previously solved ESX-5 heterotrimers, the PE–PPE heterodimer of our ESX-3 heterotrimer is interacting with its chaperone at a drastically different angle and presents different faces of the PPE protein to the chaperone. We conclude that the PPE–EspG interface from each ESX system has a unique shape complementarity that allows each EspG to discriminate among noncognate PE–PPE pairs.
Keywords: mycobacteria, tuberculosis, X-ray crystallography, protein complex, protein–protein interaction, type VII secretion system, Mycobacterium tuberculosis, protein secretion, chaperone
Tuberculosis is currently the deadliest infectious disease in the world, killing 1.5 million people in 2019 (1). The lack of an effective vaccine against the most prevalent pulmonary form of tuberculosis, as well as the emergence of numerous multidrug-resistant strains of the causative agent, Mycobacterium tuberculosis, highlights the growing need for more effective treatment options. Therefore, a more comprehensive understanding of the M. tuberculosis virulence machinery is needed to aid the development of new therapeutics.
M. tuberculosis, like all mycobacteria, contains a thick hydrophobic cell envelope that aids in protecting the mycobacterium from its environment. To overcome the limited permeability created by this envelope, mycobacteria have evolved specialized secretion systems to export proteins across their cell envelopes, the type VII secretion systems, also known as the ESX systems (2). Five different ESX systems are encoded in the M. tuberculosis genome, and three are known to secrete proteins: ESX-1, ESX-3, and ESX-5 (3). Recently, the structures of the core complex of both ESX-3 (4, 5) and ESX-5 (6) have been solved. The ESX-5 core complex has 6-fold symmetry and sits on the inner membrane (6), whereas the ESX-3 core complex was solved as a dimer that could be modeled onto the 6-fold symmetry of the ESX-5 core complex (4, 5). These systems are not functionally redundant, because their substrates are not rerouted to other ESX systems (7). The ESX systems secrete a variety of different substrates, each containing a general type VII secretion motif of YXXX(D/E) (8). A significant class of substrates being the PE and PPE proteins, named for conserved residues (Pro-Glu for PE and Pro-Pro-Glu for PPE) within their N-terminal domains (9, 10). The N-terminal domains are ∼110 (PE) or ∼180 (PPE) amino acids in length and interact together to form a PE–PPE heterodimer. A cytosolic chaperone, EspG, is required for proper folding and/or stability of the PE–PPE proteins and ultimately their proper secretion (11, 12). Each ESX system secretes a unique subset of PE–PPE heterodimers, and therefore each encodes an EspG that binds to only its corresponding heterodimers (11, 12). The first structural insight into the EspG and PE–PPE interaction was revealed by analysis of the structure of the PE25–PPE41–EspG5 complex, a heterotrimer from ESX-5 (12, 13). EspG5 interacts solely with PPE41 at the tip distal to the PE25 interaction and aids in preventing PE–PPE heterodimer aggregation in part by shielding a conserved hydrophobic tip on the PPE proteins, known as the hh motif (12). The additional structure of the ESX-5–related, PE8–PPE15–EspG5 heterotrimer, revealed similar interactions of the substrate PE–PPE dimer with the EspG5 chaperone (14). Despite high conservation among PPE proteins in the identified Esp5-binding region from PPE41, three residues vary depending on whether the PPE protein is secreted by ESX-1, ESX-3, or ESX-5 (12). Alteration of any or all of these positions in the ESX-5–dependent PPE41 did not disrupt PPE41-EspG5 binding (12). Based on this observation it has been suggested that structural elements outside of the EspG-binding region differentiate the ESX-5–specific PPE proteins from their ESX-1 and ESX-3 homologs to bind EspG5 (12).
This study was initiated to understand the how each EspG from the different ESX systems specifically recognizes its unique subset of cognate PE–PPE heterodimers. Here we present the structure of PE5–PPE4–EspG3 from ESX-3. This structure reveals a novel binding mode of PE–PPE proteins with the EspG chaperone and suggests the molecular mechanism by which the PE–PPE dimers are specifically targeted by cognate chaperones.
Results
EspG3 forms a complex with PE5–PPE4, and binding is conserved across species
To understand the mechanism for the specificity of PE–PPE recognition by cognate chaperones, a high-resolution structure of a heterotrimer produced by the ESX systems, other than ESX-5, was needed. Our efforts have focused on optimizing the ESX-3 PE–PPE–EspG heterotrimer for X-ray structural studies. Constructs of full-length PE5 (Rv0285), the conserved N-terminal PPE domain of PPE4 (Rv0286, residues 1–181), in a complex with the cognate full-length EspG3 (Rv0289) from Mycobacterium tuberculosis (Fig. 1A) never formed high-resolution diffraction quality crystals, despite our best efforts. The difficulty could be due to some heterogeneity in the processing of EspG3mt within the Escherichia coli cell, as seen by the double band in Fig. 1B and Fig. S1a. Numerous variations of PE5–PPE4–EspG3 constructs were screened utilizing multiple mycobacterial species, different fusion approaches, and even mixing PE5–PPE4 dimers with EspG3 chaperones from different species (Table S1). This latter approach was inspired by the work done on the Plasmodium aldolase–thrombospondin–related anonymous protein complex (15) and in the end, produced the best crystals for further diffraction experiments. To ensure that the mixed heterotrimers behaved the same in solution as the WT heterotrimer, a size-exclusion chromatography with multiangle light scattering (MALS) experiment was performed on both the WT PE5mt–PPE4mt–EspG3mt heterotrimer and the mixed PE5mt–PPE4mt–EspG3mm heterotrimer that contained the Mycobacterium marinum EspG3 (MMAR_0548) with 78% sequence identity to EspG3mt (Fig. 1, B and C). Both heterotrimers form a 1:1:1 complex with experimental molecular masses of 56.2 kDa (Fig. 1B) for the full M. tuberculosis heterotrimer (theoretical heterotrimer molecular mass of 58.8 kDa) and 54.6 kDa (Fig. 1C) for the mixed heterotrimer with the M. marinum EspG3 (theoretical heterotrimer molecular mass of 58.1 kDa). Co-purification assays were run with both M. tuberculosis and M. marinum EspG3 with the M. tuberculosis PE4–PPE5, along with EspG3s from Mycolicibacterium smegmatis (MSMEG_0622), Mycolicibacterium hassiacum (MHAS_04631), and Mycobacterium kansasii (MKAN_17015). Because of the His6 tag only being present on PE5mt, EspG3 co-purification required interaction with the PE5mt–PPE4mt heterodimer. Across all species that were tested, EspG3 co-purified with the PE5mt–PPE4mt heterodimer (Fig. S1, a–e). The binding of different EspG3s to the same PE–PPE heterodimer suggests a common protein–protein recognition mechanism within the ESX-3 family.
Figure 1.

Solution characterization of the PE5–PPE4–EspG3 heterotrimer. A, schematic showing design and molecular masses for constructs used in this study. PE5 from M. tuberculosis (Rv0285) contains an N-terminal His6 tag that is connected to the gene via a TEV protease cleavable linker. PPE4 (Rv0286) was truncated after its N-terminal PPE domain. Full-length copies of both M. tuberculosis (Rv0289) and M. marinum (MMAR_0548) EspG3 were used. B and C, elution profile of PE5mt–PPE4mt–EspG3mt (B) and PE5mt–PPE4mt–EspG3mm (C), with the right y axis showing the MALS-measured molecular mass. The insets show an SDS-PAGE image of the major peak fraction. res., residues.
Overall structure of PE5mt–PPE4mt–EspG3mm
The PE5mt–PPE4mt–EspG3mm heterotrimer was able to form diffraction quality crystals, and two different crystal forms were observed that diffracted to 3.3 Å (I422) and 3.0 Å (P212121) (Table 1). The final refinement and data statistics are shown in Table 1. Overall there is little structural variation between the individual proteins across the copies present in the two crystal forms (Table 2).
Table 1.
Data collection and refinement statistics
| PE5mt–PPE4mt–EspG3mm | PE5mt–PPE4mt–EspG3mm | |
|---|---|---|
| PDB code | 6UUJ | 6VHR |
| Data collection | ||
| Wavelength (Å) | 1.000 | 1.000 |
| Space group | P212121 | I422 |
| Cell dimensions | ||
| a, b, c (Å) | 72.26, 158.63, 209.31 | 219.14, 219.14, 104.44 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 39.51–3.00 (3.08–3.00)a | 35.73–3.30 (3.39–3.30) |
| Rsym | 0.131 (1.56) | 0.087 (2.18) |
| Rpim | 0.070 (0.848) | 0.029 (0.524) |
| CC1/2b | 0.998 (0.590) | 0.999 (0.597) |
| I/σ | 9.45 (1.14) | 15.70 (1.31) |
| Completeness (%) | 99.1 (99.5) | 99.8 (100) |
| Multiplicity | 4.2 (4.4) | 10.3 (9.5) |
| Refinement | ||
| Resolution (Å) | 39.51–3.00 | 35.73–3.30 |
| No. reflections (total/free) | 48463/2462 | 19335/928 |
| Rwork/Rfree | 0.266/0.303 | 0.248/0.266 |
| Number of atoms | ||
| Protein | 14,535 | 3643 |
| Ligand/ion | 0 | 0 |
| Water | 4 | 0 |
| B-factors | ||
| Protein | 101.6 | 173.2 |
| Water | 70.6 | |
| All atoms | 101.6 | 173.2 |
| Wilson B | 87.9 | 147.3 |
| RMSD | ||
| Bond lengths (Å) | 0.002 | 0.002 |
| Bond angles (°) | 0.53 | 0.503 |
| Ramachandran distributionc (%) | ||
| Favored | 96.42 | 91.34 |
| Allowed | 3.58 | 8.04 |
| Outliers | 0 | 0.62 |
| Rotamer outliersc (%) | 0.28 | 0 |
| Clashscored | 7.02 | 6.94 |
| MolProbity scoree | 1.62 | 1.89 |
aThe values in parentheses are for the highest resolution shell.
cCalculated with the MolProbity server (http://molprobity.biochem.duke.edu) (29).
dClashscore is the number of serious steric overlaps (>0.4 Å) per 1000 atoms.
eMolProbity score combines the Clashscore, rotamer, and Ramachandran evaluations into a single score, normalized to be on the same scale as X-ray resolution (29).
Table 2.
Structural variations in copies of PE5mt–PPE4mt–EspG3mm structure in RMSD (Å)
| PE5mt | PPE4mt | EspG3mm | |
|---|---|---|---|
| Aligned to 6UUJ copy 1 | |||
| 6UUJ copy 2 | 0.2 | 0.3 | 0.4 |
| 6UUJ copy 3 | 0.4 | 0.2 | 0.4 |
| 6UUJ copy 4 | 0.3 | 0.2 | 0.4 |
| 6VHR | 0.4 | 0.5 | 0.6 |
| Aligned to 6UUJ copy 2 | |||
| 6UUJ copy 1 | 0.2 | 0.3 | 0.4 |
| 6UUJ copy 3 | 0.3 | 0.3 | 0.3 |
| 6UUJ copy 4 | 0.3 | 0.3 | 0.4 |
| 6VHR | 0.4 | 0.5 | 0.6 |
| Aligned to 6UUJ copy 3 | |||
| 6UUJ copy 1 | 0.4 | 0.2 | 0.4 |
| 6UUJ copy 2 | 0.3 | 0.3 | 0.3 |
| 6UUJ copy 4 | 0.4 | 0.3 | 0.4 |
| 6VHR | 0.5 | 0.5 | 0.6 |
| Aligned to 6UUJ copy 4 | |||
| 6UUJ copy 1 | 0.3 | 0.2 | 0.4 |
| 6UUJ copy 2 | 0.3 | 0.3 | 0.4 |
| 6UUJ copy 3 | 0.3 | 0.3 | 0.4 |
| 6VHR | 0.4 | 0.5 | 0.7 |
For all structural analysis and comparisons, the first copy of the PE5mt–PPE4mt–EspG3mm heterotrimer from the higher resolution P212121 crystal form was used because it diffracted at a higher resolution and has the lowest B-factors from the noncrystallographic copies in the P212121 form. EspG3mm interacts solely with the tip of PPE4mt (Fig. 2), similar to EspG5 in the previously solved ESX-5 heterotrimers (12–14). However, the orientation of PE5mt–PPE4mt relative to EspG3mm is dramatically different from what was observed for either ESX-5 heterotrimer, and the differences between them will be described in later sections. The YXXX(D/E) motif for ESX secretion of PE5mt is accessible for interactions with the rest of the ESX machinery, because it is located distal to the EspG3mm interaction (11). In both crystal forms, this secretion motif is disordered, similar to the motif in PE8mt from the PE8mt–PPE15mt–EspG5mt heterotrimer (14). The individual components of the PE5mt–PPE4mt–EspG3mm heterotrimer align well to the individual components of the previously reported ESX-5 heterotrimers, both PE25mt–PPE41mt–EspG5mt (4KXR and 4W4L) and PE8mt–PPE15mt–EspG5mt (5XFS), with only moderate variations (Table 3).
Figure 2.
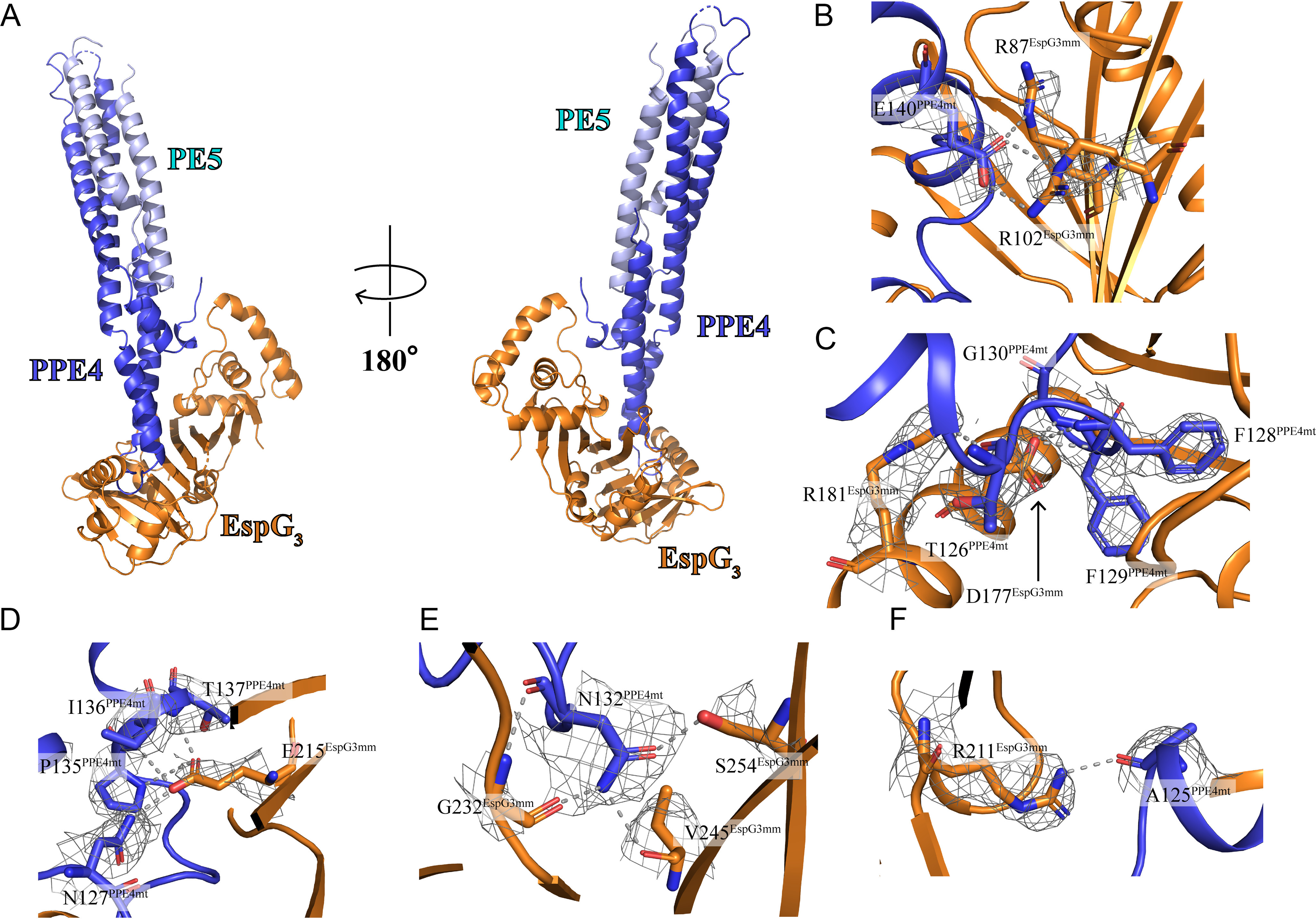
Crystal structure of the PE5mt–PPE4mt–EspG3mm heterotrimer and selected interactions in PPE4mt-EspG3mm interface. A, crystal structure of the PE5mt–PPE4mt–EspG3mm heterotrimer in a cartoon representation with two views related by a rotation of 180°. EspG3 interacts exclusively with the tip PPE4, distal to PE5. B–F, interacting residues are shown with main chain and side chain in stick form with electron density map (2Fo − Fc shown at 1.0 σ) covering side chains and hydrogen bonding in gray dashed lines.
Table 3.
Structural variations between individual components of ESX-3 heterotrimer and the previously published ESX-5 heterotrimers in RMSD (Å)
| PE25mt–PPE41mt–EspG5mt (PDB code 4KXR) | PE25mt–PPE41mt–EspG5mt (PDB code 4W4L) | PE8mt–PPE15mt–EspG5mt (PDB code 5XFS) | |
|---|---|---|---|
| PE5mt | 2.3 | 2.3 | 1.4 |
| PPE4mt | 3.3 | 3.4 | 2.5 |
| EspG3mm | 2.4 | 2.7 | 2.3 |
In a previous study on EspG structures (16), a small-angle X-ray scattering (SAXS) experiment was done on the PE5–PPE4–EspG3 heterotrimer from M. smegmatis. Comparisons between this SAXS analysis and our crystal structure were performed to see whether the solution-based characterization of the heterotrimer matched the X-ray-based characterization. We ran CRYSOL (17) on our crystal structure compared with the experimental scattering data from the M. smegmatis heterotrimer. The overall χ2 is 2.53, which is acceptable given that the heterotrimers are from different species with only 54.0–73.8% sequence identity across the different components (Fig. S2). The main differences are in the extreme high- and low-resolution areas, likely arising from differences in the primary structure between the two samples and from aggregation in the SAXS sample, respectively. Therefore, we are confident that the crystal structure is an appropriate model of the ESX-3 heterotrimer because it exists in solution.
Interface between PPE4mt and EspG3mm
The interface between EspG3mm and PPE4mt contains numerous hydrophobic interactions, multiple hydrogen bonds, and two salt bridges centered around Glu140 of PPE4mt (Fig. 2, B–F). Overall the interface buries 3,121 Å2 of solvent-accessible surface area, as calculated by the PISA server (18), and has the shape correlation Sc value of 0.664 (19). The interface is comprised of 30 total residues from PPE4mt and 49 residues from EspG3mm (Fig. 3). The tip of PPE4mt containing the ends of α4 and α5, and the loop between them is inserted into a groove on EspG3 composed of its central β sheet and C-terminal helical bundle. This bundle shields the hydrophobic tip of PPE4mt, including the hh motif of Phe128-Phe129, from solvent access. The tip of PPE4mt is interacting with EspG3 in such a way that the complex is unlikely to disengage at the ESX secretion machinery without structural rearrangement of the chaperone.
Figure 3.

Interface between PPE4mt and EspG3mm. A, surface representation of PPE4mt and EspG3mm shown in an “open book” view. Interacting residues are colored in blue (PPE4mt) and orange (EspG3mm), with mutated residues highlighted in light orange (PPE4mt) and light blue (EspG3mm). Another view of PPE4mt, related by a 180° rotation, is also shown. B, the same orientations as A but with the surface colored according to surface potential as calculated by APBS (32). The interacting residues are highlighted with a black outline.
Mutations cause disruptions in the PPE4–EspG3 interface
To probe the interface of the crystal structure and test the importance of interacting residues, we made several mutations on both PPE4 and EspG3 sides of the interface and opted to use the cognate PE5mt–PPE4mt–EspG3mt heterotrimer to test our mutations. The PISA output (18) of the interface was analyzed along with sequence alignments of the current known ESX-3 PPE proteins (Fig. S3) and alignments of the EspG3 used in this study (Fig. S4) to select which residues in the interface would be mutated.
PPE4mt is well-conserved along the interface among ESX-3–specific PPE proteins (Fig. S3), and we targeted strictly conserved residues in the interface. We selected Asn127 and Asn132 because they contain buried hydrogen bonds, Phe128 and Phe129 because they are the hh motif and contribute a large amount of solvation energy to the interface according to PISA (18), and Glu140 because it is part of the salt bridges in the interface. We ran co-purification pulldown assays with mutated PPE4mt and EspG3mt (Table 4). As described earlier, EspG3mt is only co-purified with the PE5–PPE4 heterodimer if it forms a complex. The introduction of charges into the buried hydrogen bonds with N127D and N132E was unable to break the PPE4mt–EspG3mt interaction, and neither was the charge reversal of E140R, because all three mutations co-purify with EspG3mt (Fig. S5a). This suggests that disruption of any of these single positions is not sufficient to abolish PPE4mt–EspG3mt interaction. Conversely, the introduction of charged residues into the hh motif with F128R or F129E did disrupt the interface and prevented EspG3mt from being co-purified (Fig. S5a), because it interrupts with the hydrophobic environment deep within the EspG3mt-binding pocket.
Table 4.
Summary of analysis of PE5mt–PPE4mt–EspG3mt interactions in vitro
| Mutations | Maintains Interaction |
|---|---|
| PPE4 mutations | |
| N127D | + |
| F128R | − |
| F129E | − |
| N132E | + |
| E140R | + |
| EspG3 mutations | |
| R87E | + |
| R102E | + |
| R208E | + |
| E212R | − |
| S231Y | − |
The interface of EspG3mt is also well-conserved among the various EspG3s tested in this study (Fig. S4), and again, we targeted strictly conserved residues. We selected Arg208 and Glu212 because they contain buried hydrogen bonds, Arg87 and Arg102 because they form the salt bridge within the interface, and Ser231 because it sits at the top of the groove of EspG3 and could sterically block entrance into the pocket. Neither single mutation of the salt bridge, R87E or R102E, was able to prevent co-purification of EspG3mt (Fig. S5b). Also, the introduction of a charged residue with R208E was unable to prevent the interaction (Fig. S5b). In contrast, E212R was sufficient to prevent co-purification, as well as S231Y (Fig. S5b), because both prevent the hydrophobic tip of PPE4mt from interacting with the binding pocket of EspG3mt either by charge repulsion or steric hindrance. Thus, our mutations on both PPE4mt and EspG3mt highlight the importance of the hydrophobic environment deep within the PPE4mt–EspG3mt interface.
Structure of EspG3 in and out of heterotrimer complex
Our structure is the first of EspG3 solved in complex with a cognate PE–PPE dimer, and thus we wanted to compare it with the previously solved unbound EspG3 structures. In total, there are six available EspG3 structures, four of EspG3ms (PDB codes 4L4W, 4RCL, 5SXL, and 4W4J (13, 16)), one EspG3mt (PDB code 4W4I (13)), and one EspG3mm (PDB code 5DLB (16)). These six structures can be classified into two different forms, an “open” form and a “closed” form. The differentiation between these two forms is the orientation of the C-terminal helical bundle relative to the core β-sheet. The EspG3mm structure (PDB code 5DLB) is representative of the open form, and one of the EspG3ms structures (PDB code 4RCL) is representative of the closed form. Analysis of EspG3mm as it exists in the PE5mt–PPE4mt–EspG3mm heterotrimer was done relative to these two representative structures. The overall alignment of the representative structures to the bound EspG3mm was good with RMSDs of 2.1 and 1.9 Å for the open and closed forms, respectively (Fig. 4A). Inspection of these alignments show the majority of differences to be within the arrangement of the C-terminal helical bundles, with the bound form of EspG3mm being in close to the orientation found in the closed form (Fig. 4, B and C). The bound EspG3mm cannot be any closer to the closed form orientation because the C-terminal helical bundle makes contact with PPE4mt. We hypothesized that this C-terminal helical bundle is dynamic and closes on cognate PPE proteins upon interaction. A comparison between the bound EspG3mm structure and the open EspG3mm was performed with the DynDom server to test this hypothesis (20). DynDom identified a moving domain within the structures that was located in the C-terminal helical bundle (Fig. 4D). DynDom's analysis also performed a whole structure alignment that agreed with the previous Dali alignment in Fig. 4 (A and B). DynDom performed alignments between the fixed domains (residues 11–168 and 189–279) and the moving domains (residues 168–188), which resulted in much better alignments with RMSDs of 1.76 and 0.86 Å, respectively. Therefore, the moving domain, the C-terminal helical bundle, is essentially structurally identical between PPE4mt-bound EspG3mm and the open EspG3mm and its rotation of 30.2° and translation of 0.8 Å is moderately perturbing the fixed domain. Because the moving domain making extensive contact with PPE4mt and PPE4mt would sterically clash with the current orientation of the C-terminal helical bundle, the movement from the closed to the open orientation could be significant in releasing the secreted PE–PPE dimers from the chaperone at the secretion machinery.
Figure 4.

EspG3 exists in multiple structural forms. A, open (PDB code 5DLB, EspG3mm, sand) and closed (PDB code 4RCL, EspG3ms, maroon) conformations of EspG3 were aligned to EspG3mm (PDB code 6UUJ, orange) as it is bound to PPE4mt. Overall the different conformations align well to the bound conformation of EspG3mm with RMSDs of 2.1 Å (open) and 1.9 Å (closed). B and C, closeups highlighting the different orientations of the α5 helices in the open (B) and closed (C) EspG3 structures as compared with EspG3mm bound to PPE4mt. D, movement regions defined in EspG3 as it moves from the open conformation to the bound conformation in two different views related by a 90° rotation. The rotation axis for the moving domain is shown in gray. Each conformation maintains the same coloring as in A, with the hinge between the moving and fixed domains colored blue (open) and light blue (bound).
Comparison of ESX-3 and ESX-5 PE–PPE–EspG heterotrimers
A vastly different binding mode is observed when comparing the ESX-3–specific PE5mt–PPE4mt–EspG3mm heterotrimer to the previously published ESX-5–specific heterotrimers. As mentioned earlier, there is good agreement when comparing individual components of the ESX-3–specific heterotrimer to the available ESX-5–specific heterotrimers (Table 3). The difference between the two sets of heterotrimers became apparent when they were aligned via EspG (Fig. 5, A and B, and Fig. S6) (36). Our results focused on comparisons with the PE25mt–PPE41mt–EspG5mt (PDB code 4KXR) heterotrimer, but the same differences were present with the PE8mt–PPE15mt–EspG5mt (PDB code 5XFS) heterotrimer. The interaction angle of the different PE–PPE heterodimer with EspG is drastically different between the two heterotrimers, with a 30° angle difference (Fig. 5B). Another difference lies within the hh motif loops of PPE25mt (α4-α5 loop) and PPE4mt (α5-α6 loop) (Fig. 5C). In PPE25mt, this loop is seven residues long and undertakes a compact conformation that is not altered during EspG5mt binding (12). In contrast, in PPE4mt, this loop is nine residues long and has an extended conformation. This difference was rapidly apparent when PPE25mt and PPE4mt were aligned (Fig. 5C).
Figure 5.

PE5mt–PPE4mt interacts with EspG3mm chaperone in a unique mode compared with ESX-5 PE–PPE dimers. A, structural alignment of the ESX-3 and ESX-5 heterotrimers via the EspG chaperones (36) reveals a difference in the angle of interaction between the PE–PPE heterodimers with their respective chaperone. B, top view of alignment from A. C, superposition of PPE41mt and PPE4mt highlights difference in hh loop conformations between ESX-3 (PPE4mt) and ESX-5 (PPE41mt). D and E, superposition of PPE alignment from C in context of EspG3mm interaction (D) and EspG5mt interaction (E) shows the incompatibility of each PPE protein with noncognate chaperone binding.
This loop conformation also made each PPE protein incompatible with the other's binding mode. When looking at the PPE alignment in the context of the ESX-3 heterotrimer, the α4-α5 loop of PPE25mt does not align over the central groove of EspG3mm and instead sterically clashes the central β sheet of the chaperone (Fig. 5D). The tip of PPE41mt would have to undergo a drastically new tip confirmation to bind in the opening of EspG3mm. In the context of the ESX-5 heterotrimer, the α5-α6 loop does not align with the central groove of the chaperone, and instead, PPE4mt's hh motif sterically clashes with the C-terminal helical bundle of EspG5mt (Fig. 5E). Also, none of the salt bridges between PPE41mt and EspG5mt are conserved in PPE4mt. Specifically, Asp134-PPE41mt–Lys235-EspG5mt, Asp140-PPE41mt–Arg109-EspG5mt, and Asp144-PPE41mt–Arg27-EspG5mt; that are all replaced with hydrophobic residues in PPE4mt: either Thr137-PPE4mt or Leu138-PPE4mt, Val144-PPE4mt, and Leu147-PPE4mt, respectively.
Discussion
In this work, we present the first structure of the PE5mt–PPE4mt–EspG3mm heterotrimer, which is from the ESX-3 system. Our structure is a mixed heterotrimer, and we presented evidence that EspG3 from numerous mycobacterial species can bind the PE5mt–PPE4mt heterodimer. Conservation of the EspG3s used in this study ranged from 57 to 83% identity, yet an enrichment in conservation is observed within PPE4-interacting residues (Fig. S4). The ability of EspG3 from numerous mycobacterial species to bind PE5mt–PPE4mt suggests that the recognition mechanism is conserved within ESX systems across species. Overall the PE5mt–PPE4mt interaction is similar to the previously reported PE–PPE–EspG heterotrimers (12–14) in that PPE4mt's tip is solely interacting with EspG3mm and the general secretion motif of YXXX(D/E), on PE5mt, is at the distal end of the PE5mt–PPE4mt heterodimer. In all copies of PE5mt, this motif is unstructured because it is in the PE8–PPE15–EspG5 heterotrimer (14), and similarly, Trp63-PPE4mt is pointed away from this secretion motif. This arrangement is distinct from the PE25–PPE41–EspG5 heterotrimers (12, 13) and EspB, an ESX-1 substrate that has a similar structural fold to the PE–PPE heterodimers (21, 22). PE8mt contains an expanded C-terminal domain, and because the secretion motif is located in the linker between the C-terminal domain and the PE domain, the orientation of the secretion motif was unclear (14). PE5mt does not have an expanded C-terminal domain and is just the conserved PE domain, yet its secretion motif is still unstructured in our heterotrimer. Therefore, the exact significance of the structural variations in the ESX secretion motif is still unclear, and further work is needed.
Our structure is the first of EspG3 bound to a cognate PE–PPE heterodimer. In comparisons of the various published EspG3 structures, we identified two different forms that relate to the orientation of the C-terminal helical bundle: an open form and a closed form. EspG3mm, when bound to the PE5mt–PPE4mt heterodimer, is in a conformation slightly different from the closed form because of interactions with the tip of PPE4mt. We also found that the C-terminal helical bundle is a dynamic domain and shifts between the open and closed forms via a hinge movement (Fig. 4D). The functional significance of this domain movement could be 2-fold. First, the plasticity of the C-terminal helical bundle could allow EspG3 to accommodate any variation in the ESX-3–specific PPE tips. Although the tip of ESX-3–specific PPE proteins is mostly conserved (Fig. S3), there is some variations at the end of α5 that could alter the tertiary structure and thus slightly alter the interactions with the EspG3 chaperone and the PPE protein. Second, the movement of the C-terminal helical bundle could be critical to the release of the PE–PPE heterodimers at the ESX-3 secretion machinery. It is unlikely that PPE4mt could be removed from its interactions with EspG3mm without either movement of the C-terminal helical bundle or steric clashes with the C-terminal helical bundle. Movement of this helical bundle and release of PPE4mt would likely require energy input and a candidate to provide that energy is EccA. EccA is an ATPase (2) and interacts with both EspG and PPE proteins in yeast two-hybrid experiments (13, 23). Recent structures of the ESX machinery from both ESX-3 (4) and ESX-5 (6) suggests overall 6-fold symmetry of the core ESX machinery within the inner membrane, and EccA could be acting not only to provide the energy required to uncouple the PE–PPE heterodimers from their EspG chaperone but also to provide a platform for interaction with the core secretion machinery because EccA is likely hexameric when functional.
Previous studies showed that each EspG only recognizes PE–PPE heterodimers from their cognate systems (11, 12). Despite the structures of two different PE–PPE–EspG heterotrimers from ESX-5 (12–14), it was still unclear how EspG5 was differentiating from cognate and noncognate PE–PPE heterodimers. Our structure represents the first PE–PPE–EspG heterotrimer from ESX-3 and allows for direct comparisons between the ESX-3 and ESX-5 heterotrimers. Our structure reveals that PE5mt–PPE4mt interacts with EspG3mm at a different angle of interaction than what was shown for either ESX-5 heterotrimer. This difference in interaction angle presents a different face of PPE4mt to EspG3mm. We hypothesize that this is a conserved feature of the ESX-3 PPE–EspG3 interaction, because both characterized ESX-5 PE–PPE heterodimers (12–14) display the same face to EspG5 despite 33% sequence identity between PPPE41 and PPE15. Therefore, we hypothesize that each ESX system has a unique shape complementarity between its subset of PPE proteins and their cognate EspG chaperone, and these unique shapes are likely not compatiable for interaction with noncognate chaperones. Our structure is also the first of an ESX-3–specific PE–PPE heterodimer. PE5mt–PPE4mt shares the same global conformation as the previously solved PE–PPE heterodimers; however, it differs drastically in PPE4mt in the loop between α5 and α6, which contains the hh motif. This longer, more extended loop interacts deeper in the cleft of EspG3mm and is subsequently much more shielded from solvent. It is possible that the longer, extended loop conformation is a feature of ESX-3 PPE proteins and could play an essential role in EspG3 recognition.
In conclusion, we presented the first structure of a PE–PPE–EspG heterotrimer from the ESX-3 system. This structure allowed us to compare the interactions of EspG3 and a cognate PPE protein to the previously described EspG5–PPE interactions. We hypothesize that shape complementarity is a key feature of distinguishing cognate and noncognate PPE proteins from the EspG chaperones.
Experimental procedures
Bacterial strains and growth conditions
The E. coli Rosetta2 (DE3) strains were grown in Luria–Bertani (LB) medium or on LB agar at 37 °C. When needed, antibiotics were included at the following concentrations: chloramphenicol at 10 μg/ml, streptomycin at 50 μg/ml, and kanamycin at 50 μg/ml.
Expression and purification of PE5–PPE4–EspG3 heterotrimers
Optimized DNA sequences based on the amino acids of full-length PE5 and PPE4 residues 1–180 from M. tuberculosis were obtained from Invitrogen and inserted into a pRSF-NT vector (24) using NcoI and HindIII restriction sites, which contains an N-terminal His6 tag on PE5 that is cleavable by TEV protease. EspG3mm expression plasmid was constructed as described previously (12). Mutations in PPE4mt, EspG3mt, and EspG3mm were introduced with Gibson assembly mutagenesis (SGI-DNA).
Co-expression of all heterotrimers was performed as described previously (12). Briefly, E. coli strains containing the appropriate PE5mt–PPE4mt and EspG3 plasmids were induced with 0.5 mm isopropyl β-d-thiogalactopyranoside when they reached an A600 nm of 0.5–0.8 and then continued to shake at 16 °C for 20 h. The cells were harvested by centrifugation. The cells were then resuspended in lysis buffer (300 mm NaCl, 20 mm Tris, pH 8.0, and 10 mm imidazole) and 1:100 Halt protease inhibitor mixture (Thermo Fisher Scientific, Waltham, MA). The cells were lysed using an EmulsiFlex-C5 homogenizer (Avestin, Ottawa, Canada). The soluble lysate was purified over a nickel–nitrilotriacetic acid column (G-Biosciences, St. Louis, MO, USA). Eluted protein was dialyzed against lysis buffer without imidazole and incubated with 1:20 mg of TEV protease at 4 °C for 20 h before being reapplied to the nickel–nitrilotriacetic acid column. Flow-through and washes were pooled and concentrated for size-exclusion chromatography over a Superdex 200 Increase 10/300 GL column (GE Healthcare Life Sciences, Marlborough, MA) that was equilibrated in buffer A (100 mm NaCl and 20 mm HEPES, pH 7.5).
Crystallization, data collection, and structure solution
Purified protein was concentrated to 4.2 mg/ml. Initial screening was done using the MCSG crystallization suite (Anatrace, Maumee, OH, USA). This initial screening produced the P212121 crystals that were grown in 200 mm NH4 tartrate and 20% PEG 3350. Optimization around three others hits from the initial crystal screening containing NaCl as the precipitant and various buffers ranging from a pH 5.5 to 8.0 produced the I422 crystals, which were grown in 2.0 m NaCl and 100 mm Bis-Tris, pH 6.5. The crystals were transferred to cryoprotectant solution, which contained the crystallization solution supplemented with either 20% (P212121) or 25% (I422) glycerol and then flash-cooled in liquid N2. The data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID Beamline at the Advanced Photon Source, Argonne National Laboratory. The data were processed using XDS and XSCALE (25). Molecular replacement using Phaser (26) was used to solve the structure of both crystal forms. First, the PE25mt–PPE41mt dimer (PDB code 4KXR (12)) and EspG3mm (PDB code 5DLB (16)) were used as search models for the I422 data set. Later, an early model of the I422 structure was used as a search model for the P212121 data set.
Density modification and original model
The starting model for both forms was then iteratively rebuilt and refined using Coot and phenix.refine (27, 28). The final structure for both crystal forms was refined in phenix.refine, with the P212121 form using noncrystallographic symmetry restraints. All data collection and refinement statistics are listed in Table 1. The final model was assessed using Coot and the MolProbity server (29) for quality.
Size-exclusion chromatography with MALS
Proteins were expressed and purified as described and then passed over an AKTA pure with an inline Superdex 200 Increase 10/300 GL column (GE Healthcare Life Sciences), miniDAWN TREOS, and Optilab T-rEX (Wyatt Technologies, Santa Barbara, CA, USA). The system was equilibrated and run in buffer A. The samples were loaded at a volume of 500 μl at a concentration of 2–4 mg/ml, and the system was run at 0.5 ml/min. Analysis of light scattering data was done using Astra (Wyatt Technologies). Molecular mass determination was done by analyzing peaks at one-half their maximum. The graphics were prepared using Prism (GraphPad Software, La Jolla, CA, USA).
Sequence analysis
Sequence analysis was performed using the EMBL-EBL analysis tools, specifically the Clustal Omega program (30). Rendering of sequence analysis was done with the ESPript server (31).
Structural analysis
All structural figures were generated using PyMOL (http://www.pymol.org) and Chimera/ChimeraX (37). Electrostatic surface potentials were calculated using the APBS Electrostatics plugin in PyMol (32). Structural alignments were performed with the Dali server (33).
SAXS data comparison and ab initio model reconstruction
PE5ms–PPE4ms–EspG3ms heterotrimer SAXS data (SASDDX2) (16) was compared with a single copy of the mixed PE5mt–PPE4mt–EspG3mm heterotrimer structure (PDB code 6UUJ) using CRYSOL (17). Ab initio reconstruction of the envelope was completed using GASBOR (34). Monomeric symmetry was used as a constraint for GASBOR. Twenty ab initio models were generated and averaged using the DAMAVER software package (35). DAMSEL rejected only one model.
Data availability
The coordinates and structure factors were deposited in the Protein Data Bank with accession codes 6UUJ (P212121) and 6VHR (I422). All other data generated during this study are included in the article and the supporting information.
Supplementary Material
This article contains supporting information.
Author contributions—Z. A. W., C. T. C., and K. V. K. formal analysis; Z. A. W. and K. V. K. validation; Z. A. W., C. T. C., W. A. C., N. K., and K. V. K. investigation; Z. A. W. and K. V. K. visualization; Z. A. W. writing-original draft; C. T. C., N. K., and K. V. K. writing-review and editing; N. K. and K. V. K. conceptualization; K. V. K. supervision; K. V. K. funding acquisition.
Funding and additional information—This work was supported by an Institutional Development Award from the NIGMS, National Institutes of Health, by National Institutes of Health Grants P20GM103486 and P30GM110787, and by the NIAID, National Institutes of Health Grant R01AI119022 (to K. V. K.). W.A.C. was supported by National Science Foundation Research Experiences for Undergraduates Grant 1358627. Use of SER-CAT is supported by its member institutions and Equipment Grants S10_RR25528 and S10_RR028976 from the National Institutes of Health. Use of the Advanced Photon Source was supported by the U.S. Dept. of Energy, Office of Science, Office of Basic Energy Sciences under Contract W-31-109-Eng-38. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- ESX system
- type VII secretion system
- PDB
- Protein Data Bank
- RMSD
- root-mean-square deviation
- SAXS
- small-angle X-ray scattering
- MALS
- multiangle light scattering.
References
- 1. World Health Organization (2019) Tuberculosis Fact Sheet 2019, World Health Organization, Geneva, Switzerland [Google Scholar]
- 2. Bitter W., Houben E. N., Bottai D., Brodin P., Brown E. J., Cox J. S., Derbyshire K., Fortune S. M., Gao L. Y., Liu J., Gey van Pittius N. C., Pym A. S., Rubin E. J., Sherman D. R., Cole S. T., et al. (2009) Systematic genetic nomenclature for type VII secretion systems. PLoS Pathog. 5, e1000507 10.1371/journal.ppat.1000507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gröschel M. I., Sayes F., Simeone R., Majlessi L., and Brosch R. (2016) ESX secretion systems: mycobacterial evolution to counter host immunity. Nat. Rev. Microbiol. 14, 677–691 10.1038/nrmicro.2016.131 [DOI] [PubMed] [Google Scholar]
- 4. Famelis N., Rivera-Calzada A., Degliesposti G., Wingender M., Mietrach N., Skehel J. M., Fernandez-Leiro R., Böttcher B., Schlosser A., Llorca O., and Geibel S. (2019) Architecture of the mycobacterial type VII secretion system. Nature 576, 321–325 10.1038/s41586-019-1633-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Poweleit N., Czudnochowski N., Nakagawa R., Trinidad D. D., Murphy K. C., Sassetti C. M., and Rosenberg O. S. (2019) The structure of the endogenous ESX-3 secretion system. Elife 8, e52983 10.7554/eLife.52983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beckham K. S., Ciccarelli L., Bunduc C. M., Mertens H. D., Ummels R., Lugmayr W., Mayr J., Rettel M., Savitski M. M., Svergun D. I., Bitter W., Wilmanns M., Marlovits T. C., Parret A. H., and Houben E. N. (2017) Structure of the mycobacterial ESX-5 type VII secretion system membrane complex by single-particle analysis. Nat. Microbiol. 2, 17047 10.1038/nmicrobiol.2017.47 [DOI] [PubMed] [Google Scholar]
- 7. Abdallah A. M., Gey van Pittius N. C., Champion P. A., Cox J., Luirink J., Vandenbroucke-Grauls C. M., Appelmelk B. J., and Bitter W. (2007) Type VII secretion–mycobacteria show the way. Nat. Rev. Microbiol. 5, 883–891 10.1038/nrmicro1773 [DOI] [PubMed] [Google Scholar]
- 8. Daleke M. H., Ummels R., Bawono P., Heringa J., Vandenbroucke-Grauls C. M., Luirink J., and Bitter W. (2012) General secretion signal for the mycobacterial type VII secretion pathway. Proc. Natl. Acad. Sci. U.S.A. 109, 11342–11347 10.1073/pnas.1119453109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ates L. S. (2020) New insights into the mycobacterial PE and PPE proteins provide a framework for future research. Mol. Microbiol. 113, 4–21 10.1111/mmi.14409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Delogu G., Brennan M. J., and Manganelli R. (2017) PE and PPE genes: a tale of conservation and diversity. Adv. Exp. Med. Biol. 1019, 191–207 10.1007/978-3-319-64371-7_10 [DOI] [PubMed] [Google Scholar]
- 11. Daleke M. H., van der Woude A. D., Parret A. H., Ummels R., de Groot A. M., Watson D., Piersma S. R., Jiménez C. R., Luirink J., Bitter W., and Houben E. N. (2012) Specific chaperones for the type VII protein secretion pathway. J. Biol. Chem. 287, 31939–31947 10.1074/jbc.M112.397596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Korotkova N., Freire D., Phan T. H., Ummels R., Creekmore C. C., Evans T. J., Wilmanns M., Bitter W., Parret A. H., Houben E. N., and Korotkov K. V. (2014) Structure of the Mycobacterium tuberculosis type VII secretion system chaperone EspG5 in complex with PE25–PPE41 dimer. Mol. Microbiol. 94, 367–382 10.1111/mmi.12770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ekiert D. C., and Cox J. S. (2014) Structure of a PE–PPE–EspG complex from Mycobacterium tuberculosis reveals molecular specificity of ESX protein secretion. Proc. Natl. Acad. Sci. U.S.A. 111, 14758–14763 10.1073/pnas.1409345111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen X., Cheng H. F., Zhou J., Chan C. Y., Lau K. F., Tsui S. K., and Au S. W. (2017) Structural basis of the PE–PPE protein interaction in Mycobacterium tuberculosis. J. Biol. Chem. 292, 16880–16890 10.1074/jbc.M117.802645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bosch J., Buscaglia C. A., Krumm B., Ingason B. P., Lucas R., Roach C., Cardozo T., Nussenzweig V., and Hol W. G. (2007) Aldolase provides an unusual binding site for thrombospondin-related anonymous protein in the invasion machinery of the malaria parasite. Proc. Natl. Acad. Sci. U.S.A. 104, 7015–7020 10.1073/pnas.0605301104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tuukkanen A. T., Freire D., Chan S., Arbing M. A., Reed R. W., Evans T. J., Zenkeviciutė G., Kim J., Kahng S., Sawaya M. R., Chaton C. T., Wilmanns M., Eisenberg D., Parret A. H. A., and Korotkov K. V. (2019) Structural variability of EspG chaperones from mycobacterial ESX-1, ESX-3, and ESX-5 type VII secretion systems. J. Mol. Biol. 431, 289–307 10.1016/j.jmb.2018.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Svergun D., Barberato C., and Koch M. H. J. (1995) CRYSOL: a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 10.1107/S0021889895007047 [DOI] [Google Scholar]
- 18. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 19. Lawrence M. C., and Colman P. M. (1993) Shape complementarity at protein/protein interfaces. J. Mol. Biol. 234, 946–950 10.1006/jmbi.1993.1648 [DOI] [PubMed] [Google Scholar]
- 20. Hayward S., and Berendsen H. J. (1998) Systematic analysis of domain motions in proteins from conformational change: new results on citrate synthase and T4 lysozyme. Proteins 30, 144–154 [DOI] [PubMed] [Google Scholar]
- 21. Korotkova N., Piton J., Wagner J. M., Boy-Röttger S., Japaridze A., Evans T. J., Cole S. T., Pojer F., and Korotkov K. V. (2015) Structure of EspB, a secreted substrate of the ESX-1 secretion system of Mycobacterium tuberculosis. J. Struct. Biol. 191, 236–244 10.1016/j.jsb.2015.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Solomonson M., Setiaputra D., Makepeace K. A. T., Lameignere E., Petrotchenko E. V., Conrady D. G., Bergeron J. R., Vuckovic M., DiMaio F., Borchers C. H., Yip C. K., and Strynadka N. C. J. (2015) Structure of EspB from the ESX-1 type VII secretion system and insights into its export mechanism. Structure 23, 571–583 10.1016/j.str.2015.01.002 [DOI] [PubMed] [Google Scholar]
- 23. Teutschbein J., Schumann G., Möllmann U., Grabley S., Cole S. T., and Munder T. (2009) A protein linkage map of the ESAT-6 secretion system 1 (ESX-1) of Mycobacterium tuberculosis. Microbiol. Res. 164, 253–259 10.1016/j.micres.2006.11.016 [DOI] [PubMed] [Google Scholar]
- 24. Korotkov K. V., Delarosa J. R., and Hol W. G. J. (2013) A dodecameric ring-like structure of the N0 domain of the type II secretin from enterotoxigenic Escherichia coli. J. Struct. Biol. 183, 354–362 10.1016/j.jsb.2013.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 28. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Williams C. J., Headd J. J., Moriarty N. W., Prisant M. G., Videau L. L., Deis L. N., Verma V., Keedy D. A., Hintze B. J., Chen V. B., Jain S., Lewis S. M., Arendall W. B. 3rd, Snoeyink J., Adams P. D., et al. (2018) MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 10.1002/pro.3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Madeira F., Park Y. M., Lee J., Buso N., Gur T., Madhusoodanan N., Basutkar P., Tivey A. R. N., Potter S. C., Finn R. D., and Lopez R. (2019) The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641 10.1093/nar/gkz268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Robert X., and Gouet P. (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 10.1093/nar/gku316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jurrus E., Engel D., Star K., Monson K., Brandi J., Felberg L. E., Brookes D. H., Wilson L., Chen J., Liles K., Chun M., Li P., Gohara D. W., Dolinsky T., Konecny R., et al. (2018) Improvements to the APBS biomolecular solvation software suite. Protein Sci. 27, 112–128 10.1002/pro.3280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Holm L. (2019) Benchmarking fold detection by DaliLite v. Bioinformatics 35, 5326–5327 10.1093/bioinformatics/btz536 [DOI] [PubMed] [Google Scholar]
- 34. Svergun D. I., Petoukhov M. V., and Koch M. H. (2001) Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 80, 2946–2953 10.1016/S0006-3495(01)76260-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Volkov V. V., and Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 10.1107/S0021889803000268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Holm L. (2020) DALI and the persistence of protein shape. Protein Sci. 29, 128–140 10.1002/pro.3749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Goddard T. D., Huang C. C., Meng E. C., Pettersen E. F., Couch G. S., Morris J. H., and Ferrin T. E. (2018) UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 10.1002/pro.3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Karplus P. A., and Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 10.1126/science.1218231 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The coordinates and structure factors were deposited in the Protein Data Bank with accession codes 6UUJ (P212121) and 6VHR (I422). All other data generated during this study are included in the article and the supporting information.