

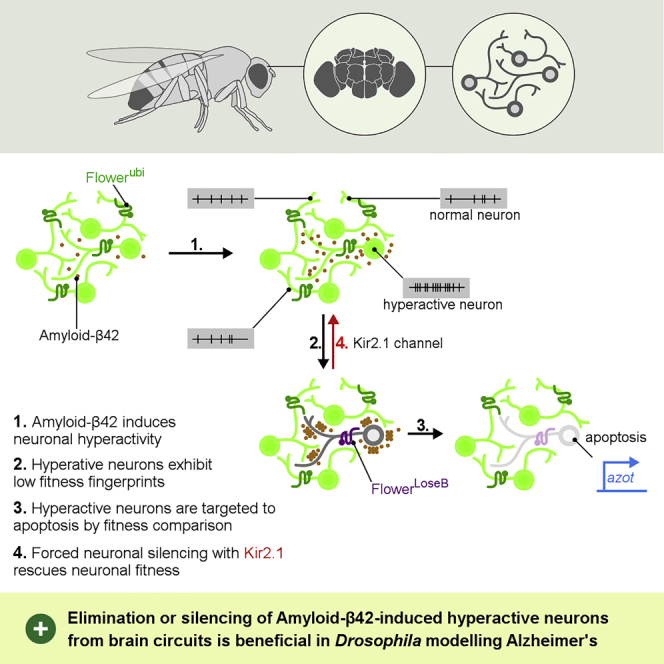
Summary
During adult life, damaged but viable neurons can accumulate in the organism, creating increasingly heterogeneous and dysfunctional neural circuits. One intriguing example is the aberrant increased activity of cerebral networks detected in vulnerable brain regions during preclinical stages of Alzheimer's disease. The pathophysiological contribution of these early functional alterations to the progression of Alzheimer's disease is uncertain. We found that a unique cell selection mechanism based on relative fitness comparison between neurons is able to target and remove aberrantly active neurons generated by heterologous human amyloid-β in Drosophila. Sustained neuronal activity is sufficient to compromise neuronal fitness and upregulate the expression of the low fitness indicators FlowerLoseB and Azot in the fly. Conversely, forced silencing of neurons restores brain fitness and reduces amyloid-β-induced cell death. The manipulation of this cell selection process, which was already proved to be conserved in humans, might be a promising new avenue to treat Alzheimer's.
Subject Areas: Molecular Biology, Neuroscience, Cell Biology
Graphical Abstract
Highlights
-
•
Expression of human Amyloid-β generates hyperactive neurons in the Drosophila brain
-
•
Hyperactive neurons up-regulate the low fitness indicators, FlowerLoseB and Azot
-
•
Fitness comparison between neurons targets hyperactive neurons to apoptosis
-
•
Forced neuronal silencing improves brain fitness in Amyloid-β-flies
Molecular Biology; Neuroscience; Cell Biology
Introduction
Neurons are particularly vulnerable to external and internal insults as they are post-mitotic, long-lived cells with intense vesicle trafficking. During adult life, suboptimal but viable neurons can accumulate in the organism creating increasingly heterogeneous and dysfunctional neural circuits that may originate disease (Frere and Slutsky, 2018; Coelho et al., 2019). One relevant case study is the aberrant activity of individual neurons and complex cerebral networks detected in patients and mouse models at early stages of Alzheimer's disease (AD) (Busche and Konnerth, 2015; Palop and Mucke, 2010; Stargardt et al., 2015; Zott et al., 2018). fMRI studies revealed abnormally increased activity and deactivation defects in brain circuits vulnerable to AD, mainly the hippocampus and the default mode network, which are important for memory encoding and introspective thought, respectively (Bookheimer et al., 2000; Dickerson et al., 2005; Quiroz et al., 2010; Sperling et al., 2009). These functional alterations were detected at preclinical stages, sometimes even decades before the overt onset of symptoms, in groups at risk for AD, including asymptomatic individuals with high amyloid-β burden, pre-symptomatic carriers of familial AD mutations, and subjects with genetic predisposition for late-onset AD (Filippini et al., 2009; Mondadori et al., 2006; Quiroz et al., 2010; Reiman et al., 2012; Sperling et al., 2009). In agreement, two-photon Ca2+ imaging showed a great abundance of hyperactive neurons in the cortex and the hippocampus of mouse models of AD, strikingly clustering near amyloid plaques (Busche et al., 2008; 2012; Grienberger et al., 2012; Rudinskiy et al., 2012;).
In Drosophila, a unique mechanism of active cell selection is able to detect and eliminate suboptimal neurons based on their fitness status (Rhiner et al., 2010; Merino et al., 2013; Moreno et al., 2015). This mechanism generally known as “cell competition” has important physiological consequences for the organism such as to sculpt the visual system during development, to replace old or damaged brain tissue during aging or upon injury, and to protect long-term memory (Merino et al., 2013, 2015, 2016; Moreno et al., 2015; Coelho et al., 2018).
Neurons compare relative fitness among themselves through external clues exhibited at their membranes called “fitness fingerprints.” The different isoforms encoded by the conserved protein Flower compose these “fingerprints” and mediate the elimination of less-fit cells both in Drosophila and in humans (Madan et al., 2019; Petrova et al., 2012; Rhiner et al., 2010). We recently described that overproduction of human amyloid-β, a pathogenic peptide forming extracellular brain plaques in AD (for abbreviation Aβ), impairs neuronal brain fitness and that Aβ-damaged neurons are removed by fitness-mediated apoptosis in Drosophila (Coelho et al., 2018). An important unsolved question in this study, and in AD research in general, is how broad expression of Aβ mounts an insult that predisposes particular neurons to death and damage, whereas other neurons are less vulnerable.
Results
Amyloid-β Generates Hyperactive Neurons in the Drosophila Brain
We hypothesized that Aβ-induced damaged neurons that are eliminated by fitness comparison in Drosophila correspond to dysfunctional neurons with lower fitness status (Coelho et al., 2018). To find and characterize these neurons, we sought out to monitor neural activity with the genetically encoded Ca2+ sensor, GCaMP6s (Chen et al., 2013). We induced the expression of the human 42-aminoacid amyloid-β peptide (Aβ42) under the control of the Mef2-Gal4.247 driver using the UAS-GAL4 system (hereafter referred to as MB247>Aβ42). This driver is specific for mature neurons of the mushroom body (MB), a critical center for learning and memory in flies (Aso et al., 2014). On average, we detected a higher baseline intensity of GCaMP6s fluorescence per neuron and per mushroom body calyx in two-photon images of ex vivo brains from Aβ42-flies compared with control flies (Figures 1A–1C, MB247>+ = 891.4 [n = 141] versus MB247>Aβ42 = 1464 [n = 160]; and Figure S1A, MB247>+ = 1 [n = 15] versus MB247> Aβ42 = 1.38 [n = 16]). When recording videos of Ca2+ imaging in non-stimulated ex vivo brains, we registered that Aβ42-brains show neurons with augmented spontaneous activity (Figures S1B and S1C, n.er neurons with spontaneous activity: MB247>+ = 1/10 neurons versus MB247>Aβ42 = 10/30 neurons; Videos S1 and S2).
Figure 1.

Amyloid-β Generates Hyperactive Neurons in the Drosophila Brain
(A and B) Two-photon images of GCaMP6s expressed in the mushroom body calyx of ex vivo brains of MB247>+ flies (A) or MB247>Aβ42 flies (B) at 1 week post eclosion. Scale bar, 15 μm.
(C) Mean intensity fluorescence of GCaMP6s (arbitrary units) measured at baseline in individual neurons of the mushroom body calyx in non-stimulated ex vivo brains of MB247>+ flies or MB247>Aβ42 flies at 1 week post eclosion. ∗∗∗p value <0.001, Mann-Whitney U non-parametric test.
(D) Schematic depicting CaLexA, a bipartite system comprising the chimerical transcription factor lexA::VP16::NFAT and the responsive element lexA operator (lexAOP). Intracellular accumulation of Ca2+ leads to dephosphorylation of the transcription factor NFAT (Nuclear factor of activated T cells) and its shuttling into the nucleus. Once in the nucleus, lexA::VP16::NFAT drives the transcription of GFP downstream of lexAOP.
(E and F) CaLexA activation pattern pseudocolored fire in the mushroom body calyx of MB247>+ (E) or MB247>Aβ42 flies (F) at 10 days post eclosion. Scale bar, 10 μm.
(G) Quantification of the number of saturated pixels for CaLexA fluorescence counted per mushroom body calyx at 10 days post eclosion for the genotypes: MB247>+ or MB247>Aβ42. Pixels displaying a fluorescence intensity value above 75% of the maximum intensity value were assumed to be saturated. ∗∗∗p value <0.001, Mann-Whitney U non-parametric test.
(H and I) CaLexA activation pattern (pseudocolored fire) in the optic lobe of GMR>+ (H) or GMR > Aβ42 (I) flies at 2 weeks old. Scale bar, 5 μm.
(J) Intensity profile of CaLexA fluorescence along the lines represented correspondingly in (H) (GMR>+ flies) and (I) (GMR > Aβ42 flies).
(K) CaLexA-activating Kenyon cells (green) are labeled by the low-fitness marker FlowerLoseB:Myc (red) in MB247>Aβ42 brains (arrows). The mean percentage (± SEM) of co-localization is 12.4 ± 2.12% per mushroom (n = 14). Nuclei labeled by Elav are in blue. Scale bar, 5 μm.
(L) Kenyon cells showing strong activation of CaLexA (green) are labeled by the low-fitness marker Azot:mCherry in MB247>Aβ42 brains (arrow). Nuclei labeled by Elav are in blue. Scale bar, 5 μm.
Data are represented as mean ± SEM. See also Figures S1 and S2, Videos S1 and S2.
Video recorded by two-photon imaging of GCaMP6s using an ex vivo brain preparation from 1-week-old fly. No stimulation was performed.
Video recorded by two-photon imaging of GCaMP6s using an ex vivo brain preparation from 1-week-old fly. No stimulation was performed.
To further characterize dysfunctional neurons in the fly, we employed the CaLexA bipartite system (for calcium-dependent nuclear import of LexA) (Masuyama et al., 2012), which detects increased concentrations of intracellular Ca2+ evoked by sustained neural activity (Figure 1D). We promptly recognized a very intense signal of CaLexA in discrete soma of Kenyon cells in MB247>Aβ42 adult flies at 10 days after eclosion (Figures 1E–1G, mean number of saturated pixels: MB247>+ = 16.75 [n = 16] versus MB247>Aβ42 = 314.8 [n = 12]). We did not observe a specific pattern in the distribution of CaLexA-activating neurons, as different Kenyon cells were randomly labeled in each brain. Taking into account our previous recordings with GCaMP6s showing increased spontaneous neuronal activity in the mushroom body of Aβ42-flies, we think that neurons strongly activating CaLexA correspond to hyperactive neurons. In addition, we confirmed that Aβ42 does not cause expression of the CaLexA sensor in immature (and thus still not active) neurons of the eye disc (Figures S2A and S2B) or pupal retinas (Figures S2C and S2D). This observation supports the idea that CaLexA expression is only induced by increased activity of neurons and not by deregulated Ca2+ fluxes between intracellular compartments, which might be a secondary consequence of Aβ42-caused toxicity.
We also found axons exhibiting high levels of CaLexA in the optic lobe of 15-day-old flies when Aβ42 production was driven by GMR-Gal4, suggesting that Aβ42 can induce hyperactive neurons in different brain regions (Figures 1H–1J). Activation of CaLexA in GMR > Aβ42-flies was accompanied by a 52% increase in the concentration of the excitatory neurotransmitter glutamate in the optic lobe, raising the hypothesis that aberrant neuronal activity promoted by Aβ42 is correlated with an excessive brain excitability (Figures S2E–S2G, mean fluorescence intensity of glutamate: GMR > lacZ = 117.8 [n = 24] versus GMR > Aβ42 = 179.1 [n = 22]).
We then investigated the fitness status of Aβ42-induced hyperactive neurons by checking the expression of the “poor fitness” membrane fingerprint, FlowerLoseB, and the expression of azot, a gene that indispensably targets less-fit cells to death (Merino et al., 2015). Approximately 12.4% of Kenyon cells activating CaLexA displayed cytoplasmic positive signal for the translational reporter FlowerLoseB::Myc (derived from a tagged genomic rescue construct, Yao et al., 2009) (Figure 1K). We also found co-localization between CaLexA and the fluorescent reporter FlowerLoseB:mCherry (produced from a knockin of mCherry into the endogenous flower locus) in the membranes of cell bodies and projections of Kenyon cells (Figure S2H). Moreover, a Azot:mCherry fusion protein was expressed in CaLexA-positive neurons, particularly when CaLexA was highly activated (Figure 1L) or when apoptosis was genetically suppressed by overexpression of UAS-p35 (Figure S2I). These data suggest that Aβ42-induced hyperactive neurons exhibit low fitness markers and might be subjected to negative cell selection via fitness comparison.
Amyloid-β-Induced Hyperactive Neurons Undergo Apoptosis Mediated by Relative Fitness Comparison Using Fitness Fingerprints
To follow the temporal distribution of hyperactive neurons overtime (as done before in Busche et al., 2012 and Hahm et al., 2018), we checked CaLexA expression at three time points: 5, 10, and 15 days post eclosion. We found that the number of hyperactive neurons in the mushroom body increased with age reaching a peak at 10 days old (mean number: 8.35 ± 0.58 neurons per calyx) and decreased in older flies (mean number: 6.08 ± 0.54 neurons per calyx) (Figure 2A). This observation suggests that hyperactive neurons might be undergoing apoptosis overtime. We registered that 8.3% of CaLexA-positive Kenyon cells were marked by the anti-cleaved Drosophila Caspase Protein 1 (DCP1) antibody (Figure 2B). Furthermore, blockage of cell death by overexpression of diap1 resulted in a 47.9% increase in the number of hyperactive neurons in MB247> Aβ42 flies at 10 days old (Figures 2C–2E, MB247>Aβ42/+ = 100% [n = 21] versus MB247>Aβ42/UAS-diap1 = 147.9% [n = 23]). We confirmed that overexpression of UAS-diap1 effectively suppressed apoptosis in this model (Figure 2F, MB247>Aβ42/+ = 100% [n = 25] versus MB247>Aβ42/UAS-diap1 = 57.9% [n = 24]). These findings exclude the possibility that increased Ca2+ levels reported by CaLexA are a consequence of cytoplasmatic Ca2+ overload following execution of the apoptotic pathway and degradation of Ca2+ channels.
Figure 2.

Amyloid-β-Induced Hyperactive Neurons Undergo Apoptosis Mediated by Relative Fitness Comparison Using “Fitness Fingerprints”
(A) Plot showing the number of hyperactive Kenyon cells as labeled by CaLexA at ages 5, 10, and 15 days post eclosion. Mean number of hyperactive cells ± SEM were as follows: 3.09 ± 0.33 (n = 22) at 5 days old, 8.35 ± 0.58 (n = 29) at 10 days old, and 6.08 ± 0.54 (n = 25) at 15 days old. ∗∗∗p value <0.001, ∗∗p value <0.01, one-way ANOVA followed by Holm-Sidak's multiple comparisons test. Note that transgenic flies quantified in (A) carried 3 GFP reporter proteins, LexAop-CD2-GFP (1 × GFP) and LexAop-CD8-GFP-2A-CD8-GFP (2 × GFP), whereas flies used in (C)–(J) carried only one reporter GFP and thus the number of detectable CaLexA-positive cells is lower in the last cases.
(B) Hyperactive neurons identified by CaLexA (green) show immunolabelling of cleaved DCP1 (in red), a marker of apoptosis execution. The mean percentage (± SEM) of co-localization is 8.3 ± 1.94% (n = 39) per mushroom body. Scale bar, 5 μm.
(C and D) Neurons activating CaLexA (green soma, arrow heads) accumulate in the mushroom calyx of MB247> Aβ42 flies when general apoptosis is blocked by overexpression of UAS-diap1 (D) compared with 10-day-old matched controls (C). Scale bar, 10 μm.
(E) Quantification of neurons labeled by CaLexA in the mushroom body of MB247> Aβ42/+ flies or MB247> Aβ42/UAS-diap1 flies at 10 days of age. ∗∗∗p value <0.001, unpaired Student's t test with Welch's correction.
(F) Quantification of neurons stained by anti-DCP1 antibody in the mushroom body of MB247> Aβ42/+ flies or MB247> Aβ42/UAS-diap1 flies at 10 days of age. ∗p value <0.05, Mann-Whitney U non-parametric test.
(G–I) The number of neurons labeled by CaLexA (bright green soma, arrow heads) is increased in the calyx of MB247> Aβ42 brains when fitness mediated-apoptosis is suppressed by silencing of flower with a Long-Hairpin RNAi (H) or with a RNAi transgene from GD library (I), compared with the control genotype expressing β-galactosidase (UAS-lacZ, G) at 10 days post eclosion. Scale bar, 10 μm.
(J) Plot representing the percentage of neurons labeled by CaLexA at 10 days post eclosion in the mushroom body of the following genotypes: MB247> Aβ42/UAS-lacZ; MB247> Aβ42/UAS-flowerLoseA/BLong Hairpin, or MB247> Aβ42/UAS-fwe dsRNA GD line. CaLexA numbers were normalized to the number in MB247> Aβ42/UAS-lacZ genotype in each experimental replicate. ∗∗∗p value <0.001; ∗∗p value <0.01, unpaired Student's t test with Welch's correction. Data are represented as mean ± SEM.
See also Figure S3.
To test the hypothesis that hyperactive neurons are being eliminated from the tissue by fitness-based neuronal selection, we genetically manipulated fitness machinery and used the number of hyperactive neurons marked by CaLexA as a derivative. Downregulation of flower expression is known to prevent fitness comparison and to abrogate neuronal selection in other contexts (Merino et al., 2013; Moreno et al., 2015). To knockdown flower, we used two independent RNAi lines: a Long-Hairpin that specifically silences flowerLoseA/B isoforms and a RNAi from the Vienna Drosophila Resource Center (VDRC) GD library that targets all flower transcripts. Overexpression of the Long-Hairpin caused a drastic 75.5% increase in the number of hyperactive neurons detected by CaLexA in MB247> Aβ42 heterozygous flies at 10 days (Figures 2G, 2H, and 2J MB247>Aβ42/UAS-lacZ = 100% [n = 28] versus MB247>Aβ42/UAS flowerLoseA/B Long Hairpin = 175.5% [n = 34]). Overexpression of the flower dsRNA from VDRC raised by 36.9% the number of CaLexA-positive neurons detected at the same time point (Figures 2G, 2I, and 2J MB247>Aβ42/UAS-lacZ = 100% [n = 46] versus MB247>Aβ42/UAS-fwe dsRNA GD = 136.9% [n = 32]). Altogether, these results indicate that preventing neuronal fitness comparison in the mushroom body partially rescues Aβ42-induced hyperactive neurons from undergoing apoptosis.
The FlowerLoseA isoform, but not the FlowerLoseB isoform, was originally proposed to work as a Ca2+ channel essential for endocytosis of synaptic vesicles at the neuromuscular junction (Yao et al., 2009). However, further evidence supporting the role of Flower as a Ca2+ channel is lacking (Chang et al., 2018; Xue et al., 2012; Madan et al., 2019). Nevertheless, to test if increased Ca2+ levels reported by CaLexA in Aβ42-expressing flies were associated with Ca2+ influx through Flower, we performed two types of experiments using the point mutation FlowerLoseA[E79Q]. In this mutant, a glutamic acid substitution in the predicted Ca2+selective motif blocks putative Ca2+ currents (Yao et al., 2009).
In the first experiment, we induced heat-shocked clones marked by GFP in the wing disc, using an act > y+>Gal4 flip-out cassette. Clones overexpressing the mutant form FlowerLoseA[E79Q] were progressively eliminated from the tissue overtime (Figures S3A–S3C, UAS-lacZ clone number = 11.53 [n = 15], UAS-flowerLoseA[E79Q] clone number = 6.4 [n = 14]). In addition, we detected the expression of Azot:mCherry at clone borders overexpressing FlowerLoseA[E79Q], and DCP1 was more likely expressed at these clone borders than at control borders (Figures S3D and S3E % DCP1 at clone border: UAS-lacZ = 33.46% [n = 15] versus UAS-flowerLoseA[E79Q] = 45.78% [n = 14]). These observations indicate that overexpression of FlowerLoseA[E79Q] is sufficient to target clones for elimination by fitness comparison, independently of a potential Ca2+ influx. In addition, overexpression of FlowerLoseA under the control of the MB247 driver did not induce changes in Ca2+ levels as detected by CaLexA (Figures S3F and S3G), making a solid argument against the idea that Flower working as a putative Ca2+ channel mediates increased Ca2+ fluxes detected by CaLexA.
Sustained Activation of Neurons with a Thermosensitive Channel Induces Up-Regulation of the Low Fitness Indicator, FlowerLoseB, and Triggers Apoptosis
Patients with AD are more vulnerable to epileptic seizures, which result from an excessive synchronization of neuronal networks and increased neuronal excitability (Palop, 2009; Palop and Mucke, 2010; Vossel et al., 2013, 2016). To mimic in Drosophila the excessive excitability and hyperactivity observed in patients with AD, we ectopically expressed the excitatory cation channel (TRPA1) (transient receptor potential A1) (Figure 3A). TRPA1 is a thermosensitive channel that promotes depolarization of neurons and action potential firing at elevated temperatures (>25°C) but maintains a closed conformation at lower temperatures (Figure 3A) (Hamada et al., 2008). TRPA1 was driven either under the control of a cholinergic promoter (chat-Gal4) for mosaic expression in the brain or by GMR-Gal4 for expression in the optic lobes (Figure 3B). Sustained activation of neurons with TRPA1 for 4 days at 30°C was sufficient to upregulate FlowerLoseB (Figures 3C–3F, chat > UAS-TRPA1 22°C = 86.4% [n = 30], chat > UAS-lacZ 30°C = 100% [n = 51], chat > UAS-TRPA1 30°C = 191.8% [n = 40]; Figures 3G–3J GMR > UAS-TRPA1 22°C = 37.78% [n = 30], GMR > UAS-lacZ 30°C = 100% [n = 48], GMR > UAS-TRPA1 30°C = 139.8% [n = 50]). Increased brain apoptosis was also detected as a consequence of chronic activation of neurons with TRPA1 (Figures 3K–3M, chat > UAS-lacZ = 100% [n = 13], chat > UAS-TRPA1 = 165.7% [n = 11]; Figures 3N–3P GMR > UAS-lacZ = 100% [n = 14], GMR > UAS-TRPA1 = 237.5% [n = 13]). This result suggests that neuronal hyperactivity is sufficient to decrease neuronal fitness levels in the fly brain, predisposing less-fit neurons to death.
Figure 3.

Sustained Activation of Neurons with a Thermosensitive Channel Induces Up-Regulation of the Low-Fitness Indicator, FlowerLoseB, and Triggers Apoptosis
(A) TRPA1 is a cation channel activated by heat. In the wild, the channel allows the fly to avoid noxious heat and to select for preferred ranges of temperatures. At low temperatures (22°C) TRPA1 is closed but shifts to an open conformation at higher temperatures (30°C), leading to the influx of Na+ and Ca2+ and induction of action potential firing.
(B) Experimental design followed to induce sustained activation of neurons. After eclosion, flies ectopically expressing TRPA1 were either placed at 30°C to trigger action potential firing or at 22°C for TRPA1 inactivation for 4 days. An additional transgene, UAS-lacZ, was expressed at 30°C to serve as a control for expression of Gal4 at high temperatures.
(C–E) The expression of the FlowerLoseB:mCherry reporter (red) was significantly up-regulated by activation of neurons with TRPA1 (E), compared with controls expressing inactive TRPA1 at 22°C (C) or β-galactosidase (lacZ) at 30°C (D). GFP (green) demonstrates the domain of expression for chat-Gal4 in the optic lobe. The nuclei are in blue. Scale bars, 5 μm in the insets.
(F) Quantification of the percentage of FlowerLoseB:mCherry-positive cells in the optic lobes of chat > UAS-lacZ and chat > UAS-trpA1 genotypes (placed at 22°C or 30°C). The mean number of FlowerLoseB:mCherry-positive cells in flies expressing UAS-lacZ was considered as 100%. ∗∗∗p value < 0.001, Kruskal-Wallis test with Dunn's post hoc paired comparisons.
(G–I) The FlowerLoseB:mCherry (red) reporter was significantly up-regulated by activation of neurons with TRPA1 (I), compared with controls expressing inactive TRPA1 at 22°C (G) or β-galactosidase (lacZ) at 30°C (H). GFP (green) demonstrates the domain of expression of GMR-Gal4 in the optic lobe. The nuclei are in blue. Scale bars, 5 μm in the insets.
(J) Quantification of FlowerLoseB:mCherry-positive cells in the optic lobes of the following genotypes: GMR > UAS-lacZ and GMR > UAS-trpA1 (kept at 22°C or 30°C). The number of FlowerLoseB:mCherry-positive cells in flies expressing UAS-lacZ was considered as 100%. ∗∗∗p value < 0.001, ∗p value < 0.05 Kruskal-Wallis test with Dunn's post hoc paired comparisons.
(K and L) Terminal deoxynucleotidyl transferase-mediated dUTP nick-end (TUNEL) labeling of apoptotic cells (white) in the optic lobe of chat > UAS-lacZ (K) or chat > UAS-trpA1 (L) transgenic flies maintained at 30°C after eclosion. DAPI is in blue. Scale bar, 10 μm.
(M) Graph depicts the number of TUNEL-positive cells counted in the optic lobe of chat > UAS-lacZ or chat > UAS-trpA1 flies. The number of TUNEL-positive cells for chat > UAS-lacZ was used as reference. ∗∗p value < 0.01, unpaired Student's t test with Welch's correction.
(N and O) TUNEL labeling of apoptotic cells (white) in the optic lobe of GMR > UAS-lacZ (N) or GMR > UAS-trpA1 (O) flies maintained at 30°C after eclosion. DAPI is in blue. Scale bar, 10 μm.
(P) Graph shows the number of TUNEL-positive cells counted in the optic lobe of GMR > UAS-lacZ or GMR > UAS-trpA1flies. Number of TUNEL-positive cells for GMR > lacZ genotype was assumed as 100%. ∗∗∗p value < 0.001, unpaired Student's t test with Welch's correction. Data are represented as mean ± SEM.
Forced Silencing of Neurons Is Sufficient to Improve Cell Fitness Markers and Reduce Amyloid-β-Induced Cell Death
Based on the previous results, we reasoned that neuronal silencing might have a physiological benefit in the context of Aβ-induced hyperactivity. To test this hypothesis, we expressed a kir2.1 transgene in wild-type and Aβ42-transgenic flies and measured neuronal fitness levels. Kir is an Inward-Rectifier K+ channel that hyperpolarizes neurons and decreases the firing probability of action potentials (Baines et al., 2001). First of all, we confirmed that expression of kir2.1 per se neither disturbed neuronal differentiation during development (Figures S4A and S4B) nor caused increased apoptosis in the adult brain (Figures S4C–S4E, GMR > UAS-lacZ = 100% [n = 8] versus GMR > 10XUAS-kir2.1 = 101% [n = 11]). In particular, the distribution of the neuronal markers Elav (stains nuclei) and Futsch (stains the cytoskeleton) was not affected by kir2.1 in the neuroepithelium of the eye disc in third instar larvae (Figures S4A and S4B).
Forced neuronal silencing with kir2.1 downregulated the expression of the FlowerLoseB:mCherry and Azot:mCherry reporters in the brain of GMR > Aβ42 flies compared with controls of the same age (Figures 4A–4E, %FlowerLoseB:mCherry expression: GMR > Aβ42/UAS-lacZ = 183.7% [n = 27] versus GMR > Aβ42/10XUAS-kir2.1 = 95.48% [n = 27]; Figures 4F–4J, % Azot:mCherry expression: GMR > Aβ42/UAS-lacZ = 153.8% [n = 32] versus GMR > Aβ42/UAS-kir2.1 = 75.4% [n = 27]). Moreover, silencing of neurons had no impact on FlowerLoseB:mCherry or Azot:mCherry expression measured during normal adulthood, confirming that the rescue of neuronal fitness promoted by kir2.1 is specific to Aβ42-induced hyperactivity (Figures 4A,4B and 4E, FlowerLoseB:mCherry expression: GMR > UAS-lacZ = 100% [n = 29] versus GMR > 10XUAS-kir2.1 = 103.5% [n = 31]; Figures 4F,4G and 4J, Azot:mCherry expression: GMR > UAS-lacZ = 100% [n = 12] versus GMR > UAS-kir2.1 = 98.5% [n = 8]).
Figure 4.

Forced Silencing of Neurons Is Sufficient to Improve Cell Fitness Markers and Reduce Amyloid-β-Induced Cell Death
(A–D) Expression of the FlowerLoseB:mCherry reporter (red) in the optic lobe of heterozygous 2-week-old-flies of the following genotypes: GMR > UAS-lacZ (A), GMR > 10XUAS-kir2.1 (B), GMR > Aβ42 (C), and GMR > Aβ42/10XUAS-kir2.1 (D). DAPI shows nuclei (blue). Scale bar, 10 μm.
(E) Quantification of FlowerLoseB:mCherry-positive cells (red) for the indicated genotypes. GMR > UAS-lacZ was used as reference and 100% corresponds to the mean number of FlowerLoseB:mCherry-positive cells counted in this genotype. ∗∗p value < 0.01, Ns: not significant, Kruskal-Wallis test with Dunn's post hoc paired multiple comparisons.
(F–I) Expression of the Azot:mCherry reporter (red) in the optic lobes of 2-week-old flies of the following genotypes: GMR > UAS-lacZ (F), GMR > UAS-kir2.1(G), GMR > Aβ42 (H), and GMR > Aβ42/UAS-kir2.1 (I). DAPI shows nuclei (blue). Scale bar, 10 μm.
(J) Quantification of Azot:mCherry-positive cells (red) for the genotypes listed before in (F–I). The mean number of Azot:mCherry-positive cells counted in GMR > UAS-lacZ flies was assumed to be 100%. ∗∗∗p value < 0.001, Ns: not significant, Kruskal-Wallis test with Dunn's post hoc paired multiple comparisons.
(K and L) TUNEL labeling of apoptotic cells (white) in the optic lobe of GMR > Aβ42/UAS-lacZ (K) or GMR > Aβ42/UAS-kir2.1 flies (L). DAPI is in blue. Scale bar, 20 or 10 μm (inset).
(M) The number of apoptotic cells labeled by TUNEL in the optic lobe of GMR > Aβ42/UAS-lacZ or GMR > Aβ42/10XUAS-kir2.1 flies was quantified and potted as showed. ∗∗∗p value < 0.001, Mann-Whitney U non-parametric test.
(N) Schematic summarizing our main findings. Aβ42 generates hyperactive neurons in the Drosophila brain. These dysfunctional hyperactive neurons correspond to less-fit cells that are detected by the Flower-Azot pathway. Their elimination is beneficial against Aβ-induced degenerative phenotypes. Data are represented as mean ± SEM.
See also Figure S4.
Remarkably, cell death was also reduced in GMR > Aβ42 flies upon neuronal silencing with kir2.1. This was tested by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) of apoptotic cells in the adult brain (Figures 4K–4M, GMR > Aβ42/UAS-lacZ = 100% [n = 25] versus GMR > Aβ42/10XUAS-kir2.1 = 60.5% [n = 22]) and by analyzing the degenerative phenotype induced by Aβ42 in the eye (Figures S4F–S4H, number of necrotic patches: GMR > Aβ42/UAS-lacZ = 11 [n = 26], GMR > Aβ42/10XUAS-kir2.1 = 4.8 [n = 32]).
To prevent any possible developmental defect, we restricted kir2.1 expression to adulthood by using a thermosensitive repressor (tubGAL80ts), which blocks Gal4 activity at lower temperature, 18°C, but allows its expression at higher temperatures (above 29°C) (McGuire et al., 2004). Again, using conditional expression of kir2.1 in the adult brain, the number of apoptotic cells present in GMR > Aβ42 brains was significantly reduced (Figures S4I–S4K, GMR > Aβ42/UAS-tubGal80ts = 100% [n = 47] versus GMR > Aβ42/UAS-tubGal80ts,UAS-kir2.1 = 67.23% [n = 44]).
Our previous findings demonstrated that Aβ42 reduces the fitness status of neurons and that less-fit neurons displaying poor fitness fingerprints are eliminated by apoptosis in Drosophila (Figure 4N) (Coelho et al., 2018). Data presented here extend this model and suggest that these unfit neurons that are targeted by fitness comparison correspond to aberrantly active neurons (Figure 4N). Forced silencing of these neurons rescues brain fitness fingerprints and suppresses Aβ42-induced cell death. Altogether, removal of dysfunctional hyperactive neurons is beneficial in Drosophila modelling AD, prolonging lifespan, rescuing motor coordination, and protecting against memory loss.
Discussion
Building upon previous work, we found that a unique cell selection mechanism based on relative fitness comparison is able to detect and remove “excessively active neurons” generated by Aβ42 in Drosophila. These findings were surprising since events of non-cell autonomous selection (broadly known as cell competition) were originally described in the fly to remove slow proliferating and/or deficient cells from epithelial tissues (Igaki et al., 2009; Menéndez et al., 2010; Moreno et al., 2002; Vincent et al., 2011; Rodrigues et al., 2012; Moreno and Basler, 2004). In epithelia, genetic factors that downregulated fitness status and trigger cell selection through fitness fingerprints were already identified, including ribosomal gene mutations, low dMyc levels, scribble defects, and reduced Dpp signaling (Rhiner et al., 2010; Merino et al., 2015). Equivalent genetic modulators that initiate neuronal selection via fitness comparison are yet unknown in the neural tissue.
At the moment, it is not understood how network alterations detected in preclinical stages of AD contribute to the progression of the disease. Nevertheless, growing evidence has established a correlation between abnormally increased network activity and poor cognitive performance in risk groups for AD (Bakker et al., 2012; Kunz et al., 2015; Miller et al., 2008; Putcha et al., 2011). Work done by our group and others suggests that limiting neuronal hyperactivity by pharmacological or genetic means might have staggering beneficial effects against AD progression. Treatment with the anti-epileptic drug levetiracetam was sufficient to improve cognitive performance upon reversing network and synaptic abnormalities in the hippocampus in patients at risk to develop AD and in a mouse model of AD (Bakker et al., 2012, 2015; Sanchez et al., 2012). In a Drosophila AD model, restoring the wild-type levels of a conserved A-type K+ channel (which was targeted to degradation by Aβ) decreased neuronal excitability and was sufficient to attenuate both locomotor and learning defects and to delay neurodegeneration (Ping et al., 2015). However, the same study failed to rescue Aβ-induced neurodegenerative phenotypes by generally decreasing neuronal excitability with a EKO channel, a result that might be explained by EKO being a relatively weak tool to silence neurons when compared with the potent Inward-Rectifier Kir2.1 channel, for example (Ping et al., 2015; White, 2009). Our findings show that counteracting Aβ-induced hyperactivity by expression of the Kir2.1 channel halts neurodegeneration and enhances brain fitness markers; in alternative, promoting the removal of hyperactive neurons by fitness machinery can even rescue motor and memory decline (Coelho et al., 2018).
The report of aberrantly active neurons and circuits both in humans and animal models at early AD stages since 15 years ago has been surprising. Full-blown AD is typically characterized by neuronal silencing and death as well as synaptic dysfunction in vulnerable brains regions. The recent literature now seems to conciliate these two apparently contradictory observations by supporting a view wherein late neuronal silencing/damage might be a maladaptive consequence of early neuronal hyperactivity. A Ca2+ imaging study performed in a murine model of AD revealed that the fraction of hyperactive neurons found in the hippocampus decreased with age, whereas inversely the fraction of silent neurons increased in the same brain region, suggesting a progressive compensatory inhibition of the circuit (Busche et al., 2012). In another relevant study using a Drosophila transgenic line, Aβ42-mediated early neuronal hyperactivity promoted pre-synaptic remodeling and later inhibition of the α7 nicotinic acetylcholine receptor signaling (Hahm et al., 2018). Our discoveries expand this general view by revealing that neuronal death might be another detrimental consequence of Aβ42-induced hyperactivity, which was overlooked by other studies so far. We found that hyperactive neurons displaying high Ca2+ influx are targeted to cell death. Ca2+ overload has long been associated with necrotic and apoptotic neuronal death as a consequence of toxic stimulation of NMDA receptors or leaky mutant glutamate receptors (Dong et al., 2009; Esposito et al., 2013; Selimi et al., 2000; Zuo et al., 1997).
The fitness fingerprints mediating cell selection were recently proved to be conserved in humans, promoting competitive growth in cancer (Madan et al., 2019). In the future, it will be important to address the role of the human fitness machinery in the central nervous system during disease and aging since its manipulation might originate promising new therapeutic approaches.
Limitations of the Study
This study was conducted on a single organism, Drosophila melanogaster, that has critical limitations to model AD. Drosophila shows a very simple brain anatomy and homolog structures and circuits of the human brain are not present. Moreover, complex behaviors that are impaired in AD, such as cognitive functions encoded in the hippocampus, cannot be found in the fly. The main conclusions of this study that identifies Kir2.1 as an effective repressor of Aβ42-induced neuronal death and hyperactivity need to be subjected to genetic and pharmacologic validation in mammalian systems and humans. Although the Flower protein is conserved in humans, its role as a mediator of neuronal selection in the human brain is still completely unknown. This work extensively used CaLexA expression as readout for neuronal hyperactivity. The activation of CaLexA depends on the nuclear translocation of a Ca2+-responsive transcription factor and the transcriptional activation of reporter GFP molecules; therefore, this Ca2+ indicator has a slow response dynamics and poor temporal resolution. CaLexA signal is weaker and sparser than the signal of other transcriptional reporters of neuronal activity available in Drosophila, such as TRIC (Gao et al., 2015). As a consequence, the number of hyperactive neurons detected by CaLexA in the MB247>Aβ42 model used in this work might be underrepresented. Neuronal activity was also tested with GCaMP6. Ca2+ imaging with GCaMP6 relies on rapid conformational changes and increased brightness of the modified circular GFP molecule induced by Ca2+ binding (Chen et al., 2013). GCaMP6 is characterized by high sensitivity and fast kinetics, detecting subtle changes in intracellular free Ca2+, allowing the visualization of fast neuronal activity at a very short time scale (Chen et al., 2013). Owing to all these contrasting features between the two Ca2+ sensors employed, the number of hyperactive neurons detected by each of them might seem divergent.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Eduardo Moreno (eduardo.moreno@research.fchampalimaud.org)
Materials Availability
This study did not generate any new reagent. The authors will provide upon request Drosophila strains generated previously by the laboratory or unavailable from public Drosophila centers. Antibodies and chemicals are available from commercial suppliers or public resources and their catalog numbers are given in the Transparent Methods section.
Data and Code Availability
This study did not generate new datasets or code. The original images presented here as maximum projections and image quantifications obtained are available upon request.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Vasconcelos lab at Champalimaud Research for sharing equipment; Nélia Varela for assistance at the two-photon microscope; Gil Costa for help with diagrams; the Champalimaud Fly Platform for help with stock ordering and maintenance; the fly community at Champalimaud Research for sharing reagents and scientific discussions, DSHB for antibodies; the Bloomington Stock Center and the VDRC for Drosophila stocks. This study was funded by Fundação D. Anna de Sommer Champalimaud e Dr. Carlos Montez Champalimaud and the European Research Council (Consolidator Grant to E.M.: “Active Mechanisms of Cell Selection: From Cell Competition to Cell Fitness”). Fly platform was funded by CONGENTO LISBOA-01-0145-FEDER-022170, co-financed by FCT (Portugal) and Lisboa2020, under the PORTUGAL2020 agreement (European Regional Development Fund).
Author Contributions
D.S.C. and E.M. designed the experiments. D.S.C. conducted the experiments and analyzed data.
Declaration of Interests
Authors declare no competing interests.
Published: September 25, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101468.
Supplemental Information
References
- Aso Y., Hattori D., Yu Y., Johnston R.M., Iyer N.A., Ngo T.-T.B., Dionne H., Abbott L.F., Axel R., Tanimoto H. The neuronal architecture of the mushroom body provides a logic for associative learning. Elife. 2014;3:e04577. doi: 10.7554/eLife.04577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines R.A., Uhler J.P., Thompson A., Sweeney S.T., Bate M. Altered electrical properties in Drosophila neurons developing without synaptic transmission. J. Neurosci. 2001;21:1523–1531. doi: 10.1523/JNEUROSCI.21-05-01523.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker A., Krauss G.L., Albert M.S., Speck C.L., Jones L.R., Stark C.E., Yassa M.A., Bassett S.S., Shelton A.L., Gallagher M. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 2012;74:467–474. doi: 10.1016/j.neuron.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker A., Albert M.S., Krauss G., Speck C.L., Gallagher M. Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. Neuroimage Clin. 2015;7:688–698. doi: 10.1016/j.nicl.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bookheimer S.Y., Strojwas M.H., Cohen M.S., Saunders A.M., Pericak-Vance M.A., Mazziotta J.C., Small G.W. Patterns of brain activation in people at risk for Alzheimer’s disease. N. Engl. J. Med. 2000;343:450–456. doi: 10.1056/NEJM200008173430701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche M.A., Eichhoff G., Adelsberger H., Abramowski D., Wiederhold K.-H., Haass C., Staufenbiel M., Konnerth A., Garaschuk O. Clusters of Hyperactive Neurons Near Amyloid Plaques in a Mouse Model of Alzheimer’s Disease. Science. 2008;321:1686–1689. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- Busche M.A., Konnerth A. Neuronal hyperactivity - a key defect in Alzheimer’s disease?: neuronal hyperactivity. BioEssays. 2015;37:624–632. doi: 10.1002/bies.201500004. [DOI] [PubMed] [Google Scholar]
- Busche M.A., Chen X., Henning H.A., Reichwald J., Staufenbiel M., Sakmann B., Konnerth A. Critical role of soluble amyloid- for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A. 2012;109:8740–8745. doi: 10.1073/pnas.1206171109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H.-F., Mannebach S., Beck A., Ravichandran K., Krause E., Frohnweiler K., Fecher-Trost C., Schirra C., Pattu V., Flockerzi V. Cytotoxic granule endocytosis depends on the Flower protein. J. Cell Biol. 2018;217:667–683. doi: 10.1083/jcb.201706053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T.-W., Wardill T.J., Sun Y., Pulver S.R., Renninger S.L., Baohan A., Schreiter E.R., Kerr R.A., Orger M.B., Jayaraman V. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho D.S., Moreno E. Emerging links between cell competition and Alzheimer’s disease. J. Cell Sci. 2019;132:jcs231258. doi: 10.1242/jcs.231258. [DOI] [PubMed] [Google Scholar]
- Coelho D.S., Schwartz S., Merino M.M., Hauert B., Topfel B., Tieche C., Rhiner C., Moreno E. Culling less fit neurons protects against amyloid-β-induced brain damage and cognitive and motor decline. Cell Rep. 2018;25:3661–3673.e3. doi: 10.1016/j.celrep.2018.11.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson B.C., Salat D.H., Greve D.N., Chua E.F., Rand-Giovannetti E., Rentz D.M., Bertram L., Mullin K., Tanzi R.E., Blacker D. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology. 2005;65:404–411. doi: 10.1212/01.wnl.0000171450.97464.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X., Wang Y., Qin Z. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009;30:379–387. doi: 10.1038/aps.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito Z., Belli L., Toniolo S., Sancesario G., Bianconi C., Martorana A. Amyloid β, glutamate, excitotoxicity in Alzheimer’s disease: are we on the right track? CNS Neurosci. Ther. 2013;19:549–555. doi: 10.1111/cns.12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippini N., MacIntosh B.J., Hough M.G., Goodwin G.M., Frisoni G.B., Smith S.M., Matthews P.M., Beckmann C.F., Mackay C.E. Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc. Natl. Acad. Sci. U S A. 2009;106:7209–7214. doi: 10.1073/pnas.0811879106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frere S., Slutsky I. Alzheimer’s disease: from firing instability to homeostasis network collapse. Neuron. 2018;97:32–58. doi: 10.1016/j.neuron.2017.11.028. [DOI] [PubMed] [Google Scholar]
- Gao X.J., Riabinina O., Li J., Potter C.J., Clandinin T.R., Luo L. A transcriptional reporter of intracellular Ca2+ in Drosophila. Nat. Neurosci. 2015;18:917–925. doi: 10.1038/nn.4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grienberger C., Rochefort N.L., Adelsberger H., Henning H.A., Hill D.N., Reichwald J., Staufenbiel M., Konnerth A. Staged decline of neuronal function in vivo in an animal model of Alzheimer’s disease. Nat. Commun. 2012;3 doi: 10.1038/ncomms1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahm E.-T., Nagaraja R.Y., Waro G., Tsunoda S. Cholinergic homeostatic synaptic plasticity drives the progression of Aβ-induced changes in neural activity. Cell Rep. 2018;24:342–354. doi: 10.1016/j.celrep.2018.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada F.N., Rosenzweig M., Kang K., Pulver S.R., Ghezzi A., Jegla T.J., Garrity P.A. An internal thermal sensor controlling temperature preference in Drosophila. Nature. 2008;454:217–220. doi: 10.1038/nature07001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaki T., Pastor-Pareja J.C., Aonuma H., Miura M., Xu T. Intrinsic tumor suppression and epithelial maintenance by endocytic activation of Eiger/TNF signaling in Drosophila. Dev. Cell. 2009;16:458–465. doi: 10.1016/j.devcel.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz L., Schröder T.N., Lee H., Montag C., Lachmann B., Sariyska R., Reuter M., Stirnberg R., Stöcker T., Messing-Floeter P.C. Reduced grid-cell-like representations in adults at genetic risk for Alzheimer’s disease. Science. 2015;350:430–433. doi: 10.1126/science.aac8128. [DOI] [PubMed] [Google Scholar]
- Madan E., Pelham C.J., Nagane M., Parker T.M., Canas-Marques R., Fazio K., Shaik K., Yuan Y., Henriques V., Galzerano A. Flower isoforms promote competitive growth in cancer. Nature. 2019;572:260–264. doi: 10.1038/s41586-019-1429-3. [DOI] [PubMed] [Google Scholar]
- Masuyama K., Zhang Y., Rao Y., Wang J.W. Mapping neural circuits with activity-dependent nuclear import of a transcription factor. J. Neurogenet. 2012;26:89–102. doi: 10.3109/01677063.2011.642910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire S.E., Mao Z., Davis R.L. Spatiotemporal gene expression targeting with the TARGET and gene-switch systems in Drosophila. Sci. STKE. 2004;2004:pl6. doi: 10.1126/stke.2202004pl6. [DOI] [PubMed] [Google Scholar]
- Menéndez J., Pérez-Garijo A., Calleja M., Morata G. A tumor-suppressing mechanism in Drosophila involving cell competition and the Hippo pathway. Proc. Natl. Acad. Sci. U S A. 2010;107:14651–14656. doi: 10.1073/pnas.1009376107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino M.M., Rhiner C., Portela M., Moreno E. “Fitness fingerprints” mediate physiological culling of unwanted neurons in Drosophila. Curr. Biol. 2013;23:1300–1309. doi: 10.1016/j.cub.2013.05.053. [DOI] [PubMed] [Google Scholar]
- Merino M.M., Rhiner C., Lopez-Gay J.M., Buechel D., Hauert B., Moreno E. Elimination of unfit cells maintains tissue health and prolongs lifespan. Cell. 2015;160:461–476. doi: 10.1016/j.cell.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino M.M., Levayer R., Moreno E. Survival of the fittest: essential roles of cell competition in development, aging, and cancer. Trends Cell Biol. 2016;26:776–788. doi: 10.1016/j.tcb.2016.05.009. [DOI] [PubMed] [Google Scholar]
- Miller S.L., Celone K., DePeau K., Diamond E., Dickerson B.C., Rentz D., Pihlajamäki M., Sperling R.A. Age-related memory impairment associated with loss of parietal deactivation but preserved hippocampal activation. Proc. Natl. Acad. Sci. U S A. 2008;105:2181–2186. doi: 10.1073/pnas.0706818105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondadori C.R.A., Buchmann A., Mustovic H., Schmidt C.F., Boesiger P., Nitsch R.M., Hock C., Streffer J., Henke K. Enhanced brain activity may precede the diagnosis of Alzheimer’s disease by 30 years. Brain. 2006;129:2908–2922. doi: 10.1093/brain/awl266. [DOI] [PubMed] [Google Scholar]
- Moreno E., Basler K., Morata G. Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature. 2002;416:755–759. doi: 10.1038/416755a. [DOI] [PubMed] [Google Scholar]
- Moreno E., Basler K. dMyc transforms cells into super-competitors. Cell. 2004;117:117–129. doi: 10.1016/s0092-8674(04)00262-4. [DOI] [PubMed] [Google Scholar]
- Moreno E., Fernandez-Marrero Y., Meyer P., Rhiner C. Brain regeneration in Drosophila involves comparison of neuronal fitness. Curr. Biol. 2015;25:955–963. doi: 10.1016/j.cub.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop J.J. Epilepsy and cognitive impairments in Alzheimer disease. Arch. Neurol. 2009;66:435. doi: 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop J.J., Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat. Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova E., López-Gay J.M., Rhiner C., Moreno E. Flower-deficient mice have reduced susceptibility to skin papilloma formation. Dis. Model Mech. 2012;5:553–561. doi: 10.1242/dmm.008623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping Y., Hahm E.-T., Waro G., Song Q., Vo-Ba D.-A., Licursi A., Bao H., Ganoe L., Finch K., Tsunoda S. Linking aβ42-induced hyperexcitability to neurodegeneration, learning and motor deficits, and a shorter lifespan in an Alzheimer’s model. PLoS Genet. 2015;11:e1005025. doi: 10.1371/journal.pgen.1005025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha D., Brickhouse M., O’Keefe K., Sullivan C., Rentz D., Marshall G., Dickerson B., Sperling R. Hippocampal hyperactivation associated with cortical thinning in Alzheimer’s disease signature regions in non-demented elderly adults. J. Neurosci. 2011;31:17680–17688. doi: 10.1523/JNEUROSCI.4740-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroz Y.T., Budson A.E., Celone K., Ruiz A., Newmark R., Castrillón G., Lopera F., Stern C.E. Hippocampal hyperactivation in presymptomatic familial Alzheimer’s disease. Ann. Neurol. 2010;68:865–875. doi: 10.1002/ana.22105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman E.M., Quiroz Y.T., Fleisher A.S., Chen K., Velez-Pardo C., Jimenez-Del-Rio M., Fagan A.M., Shah A.R., Alvarez S., Arbelaez A. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol. 2012;11:1048–1056. doi: 10.1016/S1474-4422(12)70228-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues A.B., Zoranovic T., Ayala-Camargo A., Grewal S., Reyes-Robles T., Krasny M., Wu D.C., Johnston L.A., Bach E.A. Activated STAT regulates growth and induces competitive interactions independently of Myc, Yorkie, Wingless and ribosome biogenesis. Development. 2012;139:4051–4061. doi: 10.1242/dev.076760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhiner C., López-Gay J.M., Soldini D., Casas-Tinto S., Martín F.A., Lombardía L., Moreno E. Flower forms an extracellular code that reveals the fitness of a cell to its neighbors in Drosophila. Dev. Cell. 2010;18:985–998. doi: 10.1016/j.devcel.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Rudinskiy N., Hawkes J.M., Betensky R.A., Eguchi M., Yamaguchi S., Spires-Jones T.L., Hyman B.T. Orchestrated experience-driven Arc responses are disrupted in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2012;15:1422–1429. doi: 10.1038/nn.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez P.E., Zhu L., Verret L., Vossel K.A., Orr A.G., Cirrito J.R., Devidze N., Ho K., Yu G.-Q., Palop J.J. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. U S A. 2012;109:E2895–E2903. doi: 10.1073/pnas.1121081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selimi F., Doughty M., Delhaye-Bouchaud N., Mariani J. Target-Related and intrinsic neuronal death in lurcher mutant mice are both mediated by caspase-3 activation. J. Neurosci. 2000;20:992–1000. doi: 10.1523/JNEUROSCI.20-03-00992.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R.A., LaViolette P.S., O’Keefe K., O’Brien J., Rentz D.M., Pihlajamaki M., Marshall G., Hyman B.T., Selkoe D.J., Hedden T. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63:178–188. doi: 10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stargardt A., Swaab D.F., Bossers K. Storm before the quiet: neuronal hyperactivity and Aβ in the presymptomatic stages of Alzheimer’s disease. Neurobiol. Aging. 2015;36:1–11. doi: 10.1016/j.neurobiolaging.2014.08.014. [DOI] [PubMed] [Google Scholar]
- Vincent J.-P., Kolahgar G., Gagliardi M., Piddini E. Steep differences in wingless signaling trigger Myc-independent competitive cell interactions. Dev. Cell. 2011;21:366–374. doi: 10.1016/j.devcel.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel K.A., Beagle A.J., Rabinovici G.D., Shu H., Lee S.E., Naasan G., Hegde M., Cornes S.B., Henry M.L., Nelson A.B. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 2013;70:1158–1166. doi: 10.1001/jamaneurol.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel K.A., Ranasinghe K.G., Beagle A.J., Mizuiri D., Honma S.M., Dowling A.F., Darwish S.M., Van Berlo V., Barnes D.E., Mantle M. Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Ann. Neurol. 2016;80:858–870. doi: 10.1002/ana.24794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White B. Neurotrapping: cellular screens to identify the neural substrates of behavior in Drosophila. Front. Mol. Neurosci. 2009;2:20. doi: 10.3389/neuro.02.020.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L., Zhang Z., McNeil B.D., Luo F., Wu X.-S., Sheng J., Shin W., Wu L.-G. Voltage-dependent calcium channels at the plasma membrane, but not vesicular channels, couple exocytosis to endocytosis. Cell Rep. 2012;1:632–638. doi: 10.1016/j.celrep.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao C.-K., Lin Y.Q., Ly C.V., Ohyama T., Haueter C.M., Moiseenkova-Bell V.Y., Wensel T.G., Bellen H.J. A synaptic vesicle-associated Ca2+ channel promotes endocytosis and couples exocytosis to endocytosis. Cell. 2009;138:947–960. doi: 10.1016/j.cell.2009.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zott B., Busche M.A., Sperling R.A., Konnerth A. What happens with the circuit in Alzheimer’s disease in mice and humans? Annu. Rev. Neurosci. 2018;41:277–297. doi: 10.1146/annurev-neuro-080317-061725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo J., De Jager P.L., Takahashi K.A., Jiang W., Linden D.J., Heintz N. Neurodegeneration in Lurcher mice caused by mutation in δ2 glutamate receptor gene. Nature. 1997;388:769–773. doi: 10.1038/42009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video recorded by two-photon imaging of GCaMP6s using an ex vivo brain preparation from 1-week-old fly. No stimulation was performed.
Video recorded by two-photon imaging of GCaMP6s using an ex vivo brain preparation from 1-week-old fly. No stimulation was performed.
Data Availability Statement
This study did not generate new datasets or code. The original images presented here as maximum projections and image quantifications obtained are available upon request.