

Highlights
-
•
Circuit thrombosis complicates CRRT in COVID-19 despite standard heparin-based anticoagulation regimens.
-
•
5 cases of CRRT thrombosis despite heparin-based anticoagulation resolved using a direct thrombin inhibitor, argatroban.
-
•
Changes in fibrinogen levels better reflected response to anticoagulation than did changes in D-dimer levels.
-
•
High fibrinogen levels and decreased anti-thrombin III activity may relate to argatroban superiority in these cases.
Keywords: Thrombotic complications, Fibrinogen, Direct thrombin inhibitor
Progressive respiratory failure and acute kidney injury (AKI) are associated with the highest risk of mortality in SARS-CoV-2-associated Coronavirus disease 2019 (COVID-19) [[1], [2], [3]]. These manifestations are linked to a systemic microvascular thrombosis associated with persistent activation of the complement [4] and coagulation [5,6] cascades. We investigated five individuals with critical COVID-19, including respiratory failure and an AKI complicated by circuit thrombosis during continuous renal replacement therapy (CRRT), who were hospitalized in an intensive care unit (ICU) setting. Thrombosis occurred despite use of standard ICU anti-coagulation regimens [6,7]. This is a frequent complication of COVID-19, noted in 28 of 29 cases in one series of CRRT cases [8]. Appropriate interventions are unknown. Circuit thrombosis was heralded by a marked rise in D-dimer levels in 4 of our 5 cases, and a persistent elevation of fibrinogen in all cases. Although fibrinogen is not an independent risk factor for COVID-19 mortality, its elevation is relevant to CRRT clotting for two reasons: high levels of fibrinogen are associated with heparin resistance [5], and ultrafiltration results in concentration of fibrinogen on dialyzer capillaries [8]. Resolution of the need for repetitive filter replacement and a decline in D-dimer and fibrinogen levels followed substitution of argatroban, a direct thrombin inhibitor here prescribed as an "off-label" medication (see Fig. 1 ).
Fig. 1.
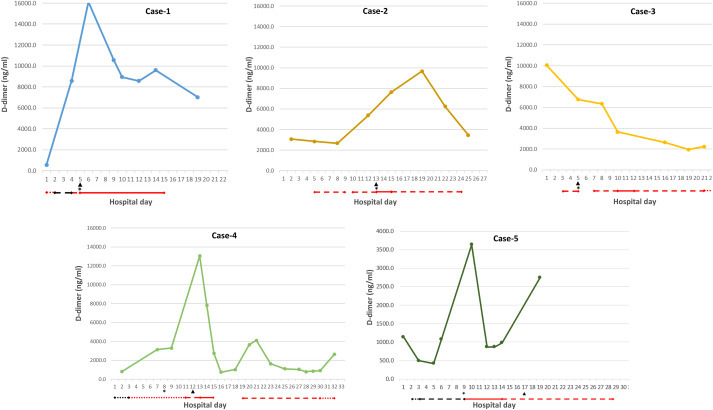
Intervention with a direct thrombin inhibitor in persistent circuit clotting occurring in COVID-19 patients on CRRT. Five patients with SARS-CoV-2 infection and critical disease, involving respiratory failure and acute kidney injury, were followed from the time of ICU admission (hospital day 1). Day of institution of continuous renal replacement therapy (CRRT) is marked by an asterisk. Day of first recognition of a circuit clot, which persisted after dialysis membrane flushes, is marked by an arrow. Anticoagulation regimens are indicated as: argatroban, red solid line; therapeutic intravenous heparin, red dashed line; prophylactic enoxaparin (0.5mg/kg body weight, once daily), black dotted line; intermediate dose enoxaparin (0.5mg/kg body weight twice daily), black dashed line; therapeutic dose enoxaparin (1mg/kg body weight twice daily), black solid line. The gap in anticoagulation for Case 4 on days 15–17 related to concern for a bleed following an acute drop in hemoglobin. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
All five patients had co-morbidities classic for severe/critical COVID-19 as well as elevated lactate dehydrogenase (LDH) and absolute neutrophil counts (ANC) (Table 1 ). Measures of organ and tissue dysfunction consistent with a hypercoaguable state, including liver function abnormalities and skeletal muscle damage, assessed by aspartate transaminase (AST) and creatine kinase (CK), respectively, were also prevalent (Table 1). COVID-19-associated coagulopathies have been shown to correlate with pro-inflammatory cytokines interleukin (IL)-6 and C-reactive protein (CRP) [5]. CRP levels were high in all 5 cases, and IL-6 levels elevated in 3 of the 4 cases tested (Table 1).
Table 1.
Clinical and laboratory dataa.
| Case | Age | Obesity | Co-morbidity | ANC (x103) | LDH (U/L) | Platelets (x109/L) | INR | CRP (mg/dL)/IL-6 (pg/ml) | Fibrinogen (mg/dL) Pre- Post- argatroban | Ferritin (ng/mL) | CK (U/L) | AST/ALT (U/L) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 77 | Overwt. | HTN | 13.3 | 1037 | 183 | 1.2 | >30.4/ND | 815 | 392 | 2793 | 2130 | 461/258 |
| 2 | 55 | Overwt. | Renal transplant, a. fib. | 14.8 | 456 | 114 | 1.2 | 24.4/125 | 585 | 146 | 8699 | 64 | 102/36 |
| 3 | 56 | No | DM, CKD | 25.9 | 2254 | 107 | 1.2 | 22.6/25 | 586 | 242 | 4713 | >7800 | 1027/538 |
| 4 | 65 | Overwt. | HTN, HLD | 22.1 | 772 | 232 | 1.0 | 15.2/<5 | 855 | 397 | 2020 | 87 | 540/181 |
| 5 | 65 | No | HTN, HLD, DM2 | 14.9 | 847 | 152 | 1.2 | 26.2/18 | 672 | ND | 2564 | 1551 | 151/112 |
Laboratory values represent peak values for the period preceding recognition of CCRT circuit thrombosis. Obesity: overweight, body mass index (BMI) > 25 but <30. HTN, hypertension. HLD, hyperlipidemia. DM2, type 2 diabetes. A. fib., atrial fibrillation. CKD, chronic kidney disease. ANC, absolute neutrophil count. LDH, lactate dehydrogenase (nl. 118–230 U/L). Plts, platelet count. INR, International Normalized Ratio. CRP, C-reactive protein (nl ≤ 0.9mg/dL). IL-6, interleukin-6 (nl ≤ 5pg/ml). CR, creatine kinase (nl. 34–145 U/L). AST, aspartate transaminase (nl. ≤34U/L). ALT, alanine transferase (nl. 10–49U/L). Ferritin, nl.10–291ng/ml. Fibrinogen, nl.180–400 mg/dL.
No patients had evidence of disseminated intravascular coagulation (DIC) by International Society on Thrombosis and Hemostasis diagnostic criteria, SOFA (sequential organ system failure assessment) scoring, or elevation of the International Normalized Ratio in the setting of thrombocytopenia (Table 1). Although 4T scores to evaluate the possibility of a heparin-induced thrombocytopenia (HIT) were ≤4 in all cases, indicating a low probability of this phenomenon, ELISAs for anti-platelet factor 4 (PF4) antibodies were performed in Cases 2, 3, and 5 and were negative.
Case 1
Rapid escalation of D-dimers to >16,000ng/ml (normal 0–229ng/ml) occurred despite use of subcutaneous unfractionated heparin (UFH; 5000 units twice daily) followed by intermediate dose enoxaparin, 0.5mg/kg body weight every 12 hours, targeting peak anti-Xa levels of 0.4–0.5 IU/ml. Persistent filter clotting was noted on institution of CRRT, with complete resolution following argatroban initiation ((2 μg/kg/min as a continuous intravenous infusion, targeting an activated partial thromboplastin time (aPTT) ≥2.5 times baseline). This was accompanied by normalization of fibrinogen (Table 1), and a 60% decrease in D-dimers, though persisting >7000ng/ml (Figure).
Case 2
A marked, progressive increase in D-dimers was noted on therapeutic intravenous UFH, targeting anti-Xa levels of 0.4–0.7 IU/ml, correlating with aPTT times of >80 seconds (normal 27.6–36.6 seconds). Persistent filter clotting occurred on institution of CRRT, with complete resolution following argatroban substitution. Fibrinogen levels declined from a peak of 585ng/ml (normal 180–400ng/ml) at the initiation of argatroban to normal levels within 3 days (Table), despite the fact that D-dimers, though declining, remained elevated (Figure). Successful transition back to therapeutic UFH was achieved five days later.
Case 3
A steady decline in D-dimers paralleled initiation of therapeutic UFH, with aPTT times >80 seconds (Figure). Despite this salutary trend, fibrinogen levels remained elevated at ≥550mg/dL, and circuit clotting was observed on initiation of CRRT (Figure). Anti-thrombin (AT)III levels, drawn on UFH, were suppressed at 58% (normal 82–136%). With persistent filter clotting over the next four days, UFH was replaced by argatroban on day four. This led to resolution of filter clotting, with successful transition back to prophylactic UFH eight days later. Cessation of circuit clotting was accompanied by normalization of fibrinogen (Table 1), albeit D-dimers remained ~2500ng/ml (Figure).
Case 4
A continued rise in D-dimers occurred despite use of prophylactic subcutaneous UFH. CRRT was initiated and persistent circuit thrombosis, requiring multiple filter flushes and replacement, was noted over the ensuing four days. With a marked peak in D-dimers on day 4 of CRRT use, argatroban was substituted. ATIII levels, drawn in the absence of heparin, were within normal limits (92%). Argatroban led to resolution of filter clotting and, over the next three days, normalization of fibrinogen and a decrease in D-dimers, though persisting at ~1000ng/ml (Figure). Successful transition to therapeutic UFH occurred four days later.
Case 5
Rising D-dimers were noted on intermediate dose enoxaparin. Argatroban was substituted upon detection of a spike in D-dimers on the day CRRT was started, leading to a marked decline in D-dimers (Figure), normal fibrinogen levels (data not shown), and no detectable circuit thrombi or other clotting issues over the subsequent 7 days. Argatroban was then discontinued in favor of therapeutic UFH, with maintenance of aPTT levels >80 seconds. Over the next four days, fibrinogen levels increased to 672mg/dL (Table) in the context of a resurgence of D-dimers (Figure), and circuit clotting and arterial thrombi were noted 4 days later.
The development of persistent circuit thrombosis during CRRT in the context of standard anticoagulation regimens, observed in our five cases, and by others for both CCRT and extracorporeal membrane oxygenation (ECMO), greatly exceed rates typical in the ICU setting [5,8]. This is another example of the fact that resistance to UFH, as well as sub-optimal peak anti-Xa levels following therapeutic low molecular weight heparin (LMWH), appears to be common among COVID-19 patients in the ICU [9]. A direct thrombin inhibitor such as argatroban may prove superior to therapeutic heparins in these situations. Our data also illuminate the need for active surveillance using both D-dimers and fibrinogen in SARS-CoV-2 infection. D-dimer levels declined but remained significantly elevated in all 5 of our cases after initiation of argatroban, while fibrinogen levels normalized. The fact that ultrafiltration leads to concentration of fibrinogen on dialyzer capillaries [8] may explain the greater sensitivity of fibrinogen over D-dimer levels to changes in circuit thrombosis.
What might account for the clinical superiority of a direct thrombin inhibitor in COVID-19? Suppression of ATIII represents one possibility, as argatroban works independently of ATIII [10]. A recent cohort of 10 patients with severe COVID-19, 9 of whom had confirmed thrombosis on hospitalization despite use of prophylactic LMWH, were successfully treated with argatroban [10]. None had circuit thrombosis with CRRT or ECMO while on this therapy. The investigators suggested that heparin resistance was a consequence of low ATIII levels in their cohort, which ranged from 19 to 49 IU/dl, as levels ≥50 IU/dl are considered necessary to achieve an anticoagulant effect with heparin [10]. However, it appears that ATIII was measured while these patients were on heparin. Heparin lowers levels of detectable ATIII antigen and activity detection by ~30%, a phenomenon thought secondary to accelerated clearance, while having no impact on its anticoagulant activity [11]. In fact, a large case series found that ATIII levels rarely fall below 80% in COVID-19, regardless of disease severity [12]. In the two patients from our cohort for whom ATIII levels were measured, both were >50%: Case 4 at 92% (off UFH), and Case 3 at 58% (measured during UFH use).
Changes in ATIII function in vivo in severe COVID-19 remain a possibility, however, as it can be inactivated by metalloproteinases and coagulation proteases, both elevated in the inflammatory milieu of COVID-19,4,5 that cleave its catalytic domain without reducing protein levels [13]. In addition, abnormalities in fibrinolysis may be involved. There is a delicate balance between host coagulation and fibrinolytic pathways. SARS-CoV infection of mice can overwhelm pro-fibrinolytic signaling via the urokinase pathway [14]. A thromboelastography study similarly documented abnormalities of fibrinolysis in severe COVID-19 [15]. Argatroban, unlike heparin, can enhance fibrinolysis by differential inhibition of thrombin-mediated activation of thrombin activatable fibrinolysis inhibitor (TAFI) and a decreased, though persistent, activation of factor XIII [16]. Limitations of this study include the small number of subjects evaluasted, arguing for the need of a controlled clinical trial of direct thrombin inhibitors in the setting of persistent clotting occurring among COVID-19 patients receiving standard prophylactic or therapeutic heparin-based regimens.
Authors’ contributions
All three authors contributed to study design and data analysis. Patient data were collected by M. S. and J.L. J.L. wrote the paper, which has been reviewed by all authors.
Declaration of competing interest
JL has received grants and honoraria from Alexion, Inc. and Omeros, Inc., manufacturers of anti-complement drugs. The remaining authors declare no competing financial interests.
Acknowledgments
This work was supported by National Institutes of Health grant R01 HL148123 (J.A.) and the Angelo Donghia Foundation (J.L.).
References
- 1.Docherty A.B., Harrison E.M., Green C.A. Features of 20 133 UK patients in hospital with covid-19 using the ISARIC WHO Clinical Characterisation Protocol: prospective observational cohort study. BMJ. 2020;369:m1985. doi: 10.1136/bmj.m1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Richardson S., Hirsch J.S., Narasimhan M. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. J. Am. Med. Assoc. 2020;323:2052–2059. doi: 10.1001/jama.2020.6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng Y., Luo R., Wang K. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. 2020;97:829–838. doi: 10.1016/j.kint.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Magro C., Mulvey J.J., Berlin D. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl. Res. 2020;220:1–13. doi: 10.1016/j.trsl.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Connors J., Levy J. COVID-19 and its implications for thrombosis and anticoagulation. Blood. 2020;135:2033–2040. doi: 10.1182/blood.2020006000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spiezia L., Boscolo A., Poletto F. COVID-19-related severe hypercoagulability in patients admitted to intensive care unit for acute respiratory failure. Thromb. Haemostasis. 2020;120:998–1000. doi: 10.1055/s-0040-1710018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alhazzani W., Lim W., Jaeschke R.Z. Heparin thromboprophylaxis in medical-surgical critically ill patients: a systematic review and meta-analysis of randomized trials. Crit. Care Med. 2013;41:2088–2098. doi: 10.1097/CCM.0b013e31828cf104. [DOI] [PubMed] [Google Scholar]
- 8.Helms J., Tacquard C., Severac F. High risk of thrombosis in severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med. 2020;46:1089–1098. doi: 10.1007/s00134-020-06062-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.White D., MacDonald S., Ball T. Heparin resistance in COVID-19 patients in the intensive care unit. J. Thromb. Hemostast. 2020 doi: 10.1007/s11239-020-02145-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arachchillage D.J., Remingtn C., Rosenberg A. Anticoagulation with argatroban in patients with acute antithrombin deficiency in severe COVID-19. Br. J. Haematol. 2020 doi: 10.1111/bjh.16927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khor B., Van Cott E.M. Laboratory tests for antithrombin deficiency. Am. J. Hematol. 2010;85:947–950. doi: 10.1002/ajh.21893. [DOI] [PubMed] [Google Scholar]
- 12.Tang N., Li D., Wang X., Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemostasis. 2020;18:1094–1099. doi: 10.1111/jth.14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rezaie A.R., Giri H. Anticoagulant and signaling functions of antithrombin. J. Thromb. Haemostasis. 2020 doi: 10.1111/JTH.15052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gralinski L.E., Bankhead A., III, Jeng S. Mechanisms of severe acute respiratory syndrome coronavirus-induced acute lung injury. mBio. 2013;4 doi: 10.1128/mBio.00271-13. e00271-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Panigada M., Bottino N., Tagliabue P. Hypercoagulability of COVID-19 patients in intensive care unit. A report of thromboelastography findings and other parameters of hemostasis. J. Thromb. Hemostast. 2020 doi: 10.1111/JTH.14850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nielsen V.G., Kirklin J.K. Argatroban enhances fibrinolysis by differential inhibition of thrombin-mediated activation of thrombin activatable fibrinolysis inhibitor and factor XIII. Blood Coagul. Fibrinolysis. 2008;19:793–800. doi: 10.1097/MBC.0b013e328317f5aa. [DOI] [PubMed] [Google Scholar]