

Abstract
We identified and evaluated differentially expressed genes (DEGs) by RNA-Sequencing (RNA-Seq) in the intestinal mucosa of two Fayoumi chicken lines, M5.1 and M15.2, that are affected by necrotic enteritis (NE); these chicken lines share the same genetic background but have different major histocompatibility complexes. RNA-Seq generated over 49 and 40 million reads for lines M5.1 and M15.2, respectively. The alignment of these sequences with the Gallus gallus genome database revealed the expression of more than 14,500 genes in two lines, among which 581, 1270, and 1140 DEGs were detected when lines M15.2 and M5.1 were compared with the control and compared between each other. The analysis of all DEGs using the gene ontology database revealed annotations for 111 biological processes, 32 cellular components, and 17 molecular functions, and KEEG pathway mapping indicated that the DEGs were primarily involved in immunity, responses to various stimuli, and signal transduction. In addition, we analyzed 183 innate immune genes that were differentially expressed in NE-induced chicken lines, including 46 CD molecular genes, 89 immune-related genes, and 13 β-defensin genes with 3 lineage-specific duplications. Taken together, the transcriptional profiles showed that line M5.1 was more resistant to NE than line M15.2 and that differential gene expression patterns were associated with host genetic differences in resistance to NE. qRT-PCR and RNA-Seq analyses showed that all the genes examined had similar responses to NE (correlation coefficient R=0.84 to 0.88, p<0.01) in both lines. To the best of our knowledge, this is the first study that describes NE-induced DEGs using RNA-seq in two lines with different levels of susceptibility to NE. These results will lead to increased insights on NE disease resistance mechanisms and the role of host genes in the control of the host immune response.
Keywords: DEGs, Fayoumi chicken, necrotic enteritis, RNA-seq
Introduction
Necrotic enteritis (NE), first reported in chickens by Parish (1961), is an enteric disease caused by Clostridium perfringens, a gram-positive, anaerobic, spore-forming, rod-shaped bacterium. NE is a global problem and costs the international poultry industry over USD 2 billion per year in production losses and control measures (McReynolds et al., 2009). NE is caused by netB toxin-producing C. perfringens type A, and to a lesser extent by type C strains, and it is a major enteric disease of chickens worldwide (Mot et al., 2013). High-throughput RNA sequencing (RNA-Seq) is a powerful tool used to identify differentially expressed transcripts, to classify transcriptomes in different cell types, developmental stages, and conditions, and to detect novel transcripts and genes (Mortazavi et al., 2008; Trapnell et al., 2010, 2012; Wang et al., 2013). Recently, RNA-Seq has been used to analyze genes from different species, including humans (Mortazavi et al., 2008; Wang et al., 2013), mice (Trapnell et al., 2010), Thale Cress (Arabidopsis thaliana) (Lister et al., 2008), cattle (Chitwood et al., 2013), and duck (Huang et al., 2013). In our previous study, transcriptome profiling was used to clarify the molecular mechanisms underlying the pathogenesis of NE and analyze immune gene responses in two inbred chicken lines (6.3 and 7.2) with NE (Truong et al., 2015a, 2015b). However, differentially expressed transcripts in Fayoumi chickens co-infected with Eimeria maxima and C. perfringens have not yet been investigated.
Fayoumi chickens, which originated in Egypt and have been genetically selected into two lines (M5.1 and M15.2), have been reported to be resistant to avian leucosis (Pinard-Van Der Laan et al., 1998). The analysis of genetic diversity in the original M5.1 and M15.2 lines indicated a genetic coefficient of 0.99 with broiler and Leghorn chicken lines (Zhou and Lamont, 1999). These two Fayoumi chicken lines share the same genetic background but have different major histocompatibility complexes (MHC) in the microchromosome (Zhou and Lamont, 1999). Moreover, line M5.1 is more resistant to E. maxima than line M15.2 (Kim et al., 2008, 2009). In particular, the Fayoumi chicken lines show different levels of body weight loss and intestinal lesions upon NE induction, and line M5.1 was more resistant to NE than line M15.2 (Kim et al., 2015). However, the transcriptomes of M5.1 and M15.2 lines with NE have not been evaluated. This study is a RNA-Seq-based genomewide expression analysis of differentially expressed genes (DEGs) in two Fayoumi chicken lines, M5.1 and M15.2, after NE induction to identify host genes associated with resistance to NE. The analysis of the expressed genes in clusters in different functional categories clearly reveals the molecular and cellular events associated with NE.
Materials and Methods
Experimental Animals and Sample Collection
The strain of C. perfringens (Del-1, 1.0×109/mL) and E. maxima (41A, 1.0×104/mL) was prepared according to protocols of the Animal Biosciences and Biotechnology Laboratory of the Agriculture Research Service, United States Department of Agriculture. Fayoumi chicken lines M5.1 and M15.2 were obtained from Iowa State University. The method used to induce NE was described by Kim et al. (2015). Briefly, 20 chickens were randomly selected and infected with E. maxima (1.0×104 oocysts/bird) by oral gavage on days 14 and 18, followed by oral gavage with C. perfringens strain Del-1 (1.0×109 CFU/bird). NE was induced according to protocols of the Beltsville Area Institutional Animal Care and Use Committee, United States Department of Agriculture. Intestinal mucosal samples were collected at 20 days post-hatching (Hong et al., 2012).
RNA Extraction and Quality Analysis
Total RNA was extracted from the intestinal mucosal layer using the TRIzol RNA extraction kit (Invitrogen, Carlsbad, CA, USA), purified using the RNeasy Mini Kit (Qiagen, Germantown, MD, USA), and treated with DNase I (Promega, Madison, WI, USA) according to the manufacturer's instructions. RNA concentration and quality were further determined using the Agilent 2100 bioanalyzer (Agilent Technologies, San Diego CA, USA) and a Tecan F2000 microplate reader (Tecan group Ltd., Männedorf, Switzerland). Samples with RNA integrity >7 and high-quality RNA (28S/18S>1) were used in the experiments.
RNA-Seq, Mapping, and Identification of DEGs
Total RNA of all samples was pooled from each experimental group before library preparation. The mRNA libraries for Illumina sequencing were constructed using total RNA, as previously described by Trapnell et al. (2010). Sequencing was performed at the Theragen Bio Institute (Suwon, Korea) using an Illumina HiSeq 2000 high-throughput sequencer, according to the manufacturer's specifications. RNA-Seq data were analyzed according to a method described previously by Trapnell et al. (2012). Briefly, each read was mapped to the Gallus gallus reference genome (v.4.0), which was obtained from the University of California, Santa Cruz (UCSC) database (UCSC: http://genome.ucsc.edu/), using TopHat v.2.0.3 (http://tophat.cbcb.umd.edu/) and Bowtie v.0.12.8 (http://bowtie-bio.sourceforge.net/index.shtml), both from Illumina iGenomes (http://support.illumina.com/). Transcript abundance and differential gene expression were calculated with the program Cufflinks v.2.0.1 (http://cufflinks.cbcb.umd.edu/), as previously described by Trapnell et al. (2012). To identify DEGs, we used the EdgeR package (Robinson et al., 2010). The data were normalized by the fragments per kilobase of exon model per million mapped reads (Mortazavi et al., 2008), and data on reads per kilobase per million reads were used to quantify the relative gene expression. The threshold for considering a gene as a DEG was a false discovery rate<0.01. FDR<0.01 and |log2-ratio| ≥ 2 were used to validate the DEGs.
Expression Pattern, Gene Ontology, and Pathway Enrichment Analysis of DEGs
The genes identified were subjected to hierarchical clustering using Cluster (MeV v4.9: www.tm4.org) and Java Treeview (http://jtreeview.sourceforge.net/). The hierarchical clustering map for the genes in lines M5.1 and M15.2 was prepared using Euclidean distance, with p-values ≤ 0.01. Samples from the intestinal mucosal layer of the two lines were compared as treatment vs. control, respectively. The gene ontology (GO) functional enrichment analysis was performed using Blast2GO (version 2.7.1) (http://www.blast2go.org/). Kyoto Encyclopedia of Genes and Genomes (KEGG: http://www.kegg.jp/) pathway analysis was performed using DAVID Bioinformatics Resources version 6.7, NIAID/NIH (http://david.abcc.ncifcrf.gov/tools.jsp).
Quantitative Real-Time PCR
To confirm the differential expression of genes identified by RNA-Seq, quantitative real-time (qRT)-PCR was used for validation. Primer sequences of 19 cytokine genes and an internal control gene (chicken glyceraldehyde-3-phosphate dehydrogenase: GAPDH) were designed using Lasergene software (DNASTAR Inc. Madison, WI, USA) and synthesized by Genotech Co. Ltd. (Daejeon, South Korea; Table S1). Complementary DNA was synthesized using 3 µg of total RNA treated with DNase I (Thermo Scientific, Waltham, MA, USA), according to the manufacturer's recommendations. RNA was reverse-transcribed using the Maxima First Strand cDNA Synthesis Kit (Thermo Scientific), according to the manufacturer's recommendations. The gene expression of cytokines was quantitated using standard curves generated using log10-diluted cDNA from individual total RNA, as previously described by Hong et al. (2012). Complementary DNA (100 ng) was added to a reaction mixture containing 10 µL of 2X Power SYBR Green Master Mix (Roche, Indianapolis, IN, USA), 0.5 µL of each primer, and RNase-free water to a total volume of 20 µL. qRT-PCR was performed using a LightCycler 96 system (Roche) and a standard cycling program. The relative gene expression was quantitated using the 2−ΔΔCt method after normalization with GAPDH (Livak and Schmittgen, 2001).
Statistical Analysis
Statistical analysis was performed using SPSS software version 21.0 for Windows (IBM, Chicago, IL, USA). Data were expressed as mean±standard deviation. Comparisons were performed using Student's t-test for two-group comparisons, and the level of statistically significant difference was set at p<0.05.
Results and Discussion
Gene Profiling for the NE-afflicted Chicken Lines
We examined global gene expression profiles using the intestinal mucosa transcriptomes of the two NE-induced Fayoumi chicken lines and controls (Table 1). Approximately 439 million paired-end reads were aligned with the merged reference gene set. After gene mapping to the G. gallus reference genome database, 46,030,728, 40,720,918, 49,822,608, and 52,698,770 uniquely mapped reads were acquired from the controls and NE-induced lines M15.2 and M5.1, respectively (Table 1). To assess the quality of RNA-Seq data, several quality-control analyses were performed. The gene coverage method was used to evaluate the RNA-Seq results. Overall, gene coverage and the distribution of distinct reads in each sample showed similar patterns in four RNA-Seq datasets. The analysis of the overall coverage in line M5.1 indicated that 70% and 76% of the genes in the control and NE-induced samples, respectively, covered more than 70% of the chicken genome (Fig. S1). In line M15.2, more than 73% of the sequence reads in the control and treated group covered more than 70% of the chicken genome (Fig. S1). These data indicate the high quality of the RNA-Seq data and were used for the analysis of gene expression in the chicken lines. In our previous study (Truong et al., 2015a), the transcriptomes of two NE-induced inbred chicken lines (MD-resistant line 6.3 and MD-susceptible line 7.2) provided a total of approximately 38.8 and 40.2 million sequence reads, respectively. Of the total reads, more than 70% were successfully mapped on the chicken genome, suggesting that more than 14,500 genes were identified in the intestinal mucosa of lines 6.3 and 7.2. Chitwood et al. (2013) used RNA-Seq to analyze single bovine blastocysts; these authors found that approximately 38 million sequencing reads were generated per embryo and identified the expression of 9,489 known genes. In addition, Lee et al. (2012) reported that, out of approximately 168 million pairs of reads obtained using RNA-Seq from murine hearts, approximately 95 million reads (57%) were uniquely mapped. In this study, we calculated the genes based on the counted reads mapped to the G. gallus reference genome, and 12,183 and 12,797 genes were identified in the intestinal mucosa of NE-induced lines M5.1 and M15.2, respectively; overall, 14,542 genes were transcribed in four intestinal mucosal layers. In general, the overall gene expression patterns of line M15.2 were similar to those of line M5.1. We identified DEGs in these two chicken lines. The expression of 581 and 1,270 genes was significantly altered in lines M15.2 and M5.1, respectively. In line M15.2, 308 out of 581 DEGs (53%) were upregulated and 273 out of 581 DEGs (47%) were downregulated. In line M5.1, 192 out of 1,270 DEGs (15.11 %) were significantly downregulated and 1,078 out of 1,270 DEGs (80.89%) were significantly upregulated (p<0.01, fold-change ≥ 2; Table 1 and Fig. 1). These findings, together with the hierarchical clustering analysis results, showed the occurrence of more appreciable responses in line M5.1 than in line M15.2 and suggested that 1, 140 genes were differentially expressed; in addition, 1065 (93.42%) DEGs were strongly upregulated in line M5. 1 and 75 (6.58%) DEGs were upregulated in line M15.2 (p<0.01, fold-change ≥ 2; Table 1 and Fig. 1). These results clearly indicate that line M5.1 is more resistant than line M15.2.
Table 1. Number of reads and genes detected using RNA sequencing in control and NE induced two chicken lines.
| Group | Total Reads | No. of Expressed genes | No. of uniquely mapped reads |
No. of DEGs1 |
||
|---|---|---|---|---|---|---|
| Genome | Gene | Versus Control | 2 DEG set | |||
| Line M15.2-Control | 103,359,764 | 12,199 | 46,030,728 | 37,084,815 | ||
| Line M15.2-NE | 106,627,968 | 12,183 | 40,720,918 | 34,564,640 | 581 | |
| Line M5.1-Control | 111,112,672 | 12,330 | 49,822,608 | 41,633,677 | ||
| Line M5.1-NE | 119,002,128 | 12,797 | 52,698,770 | 44,821,639 | 1,270 | 1,140 |
DEGs are genes that showed significantly different expression with p<0.01 and fold change ≥ 2.
DEG set are genes that showed significantly different expression in NE induced M5.1 and M15.2 chicken line with p<0.01 and fold change ≥ 2.
Fig. 1.
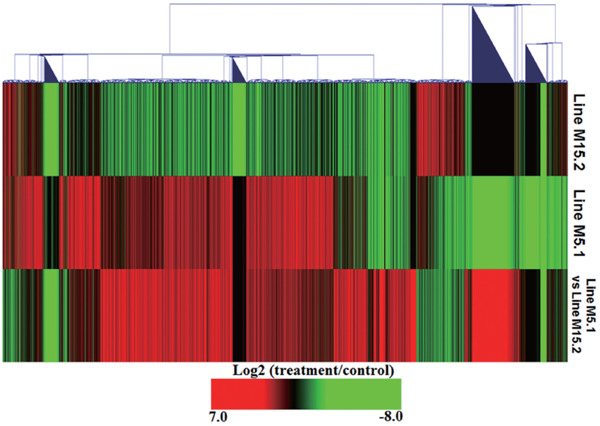
Heatmap of genes that showed significantly different expression in line M5.1 and M15.2 compared to control and line M5.1 as compared line M15.2-NE infected. This heatmap generated from hierarchical cluster analyses of genes (Euclidean Distance correlation). Genes included in this figure showed significantly different gene expression (p<0.01, log2 fold change compared treatment to control). Genes showed in red were upregulated and those showed in green were down regulated in line M5.1 and M15.2 NE induced.
Innate Immune Responses to NE-induced Chicken Lines
RNA-Seq analysis provided important information on the differential expression of innate immune genes in the intestinal mucosa of the NE-afflicted chicken lines. In our previous study, we profiled the transcriptome of 149 innate immune-response genes in the NE-afflicted chicken lines 6.3 and 7.2 (Truong et al., 2015a). In this study, we investigated the responses of 183 innate immune genes in the NE-afflicted Fayoumi chicken lines. Herein, the transcriptome analysis of these innate immune genes is presented as fold change in the NE-induced lines compared with the uninfected control (Fig. 2 and Table S2). In line M15. 2, the expression of seven genes (IL-22RA2, IL-20RA, IL-5, IL-19, IL-22, IFIH1, and TLR3) significantly decreased by 2.0- to 3.6-fold whereas the expression of 11 genes (IL-20RB, IL-17REL, TNFRSF13B, TNFAIP2, IL-21, CCLI10, TNFAIP 8L3, TGFBR3, IL-17C, SPP1, and CCR10) significantly increased by 2.3- to 4.2-fold compared NE-afflicted chickens with the control (p<0.01, fold-change ≥ 2; Fig. 2 and Table S2). In addition, the expression of five genes (IL-6, IL-17F, CCLI5, IL-19, and CCR10) significantly decreased by 2.02- to 6.5-fold and the expression of 43 genes (IFNG, IL-10, IL-17B, IL-17C, IL-3, IL-5, IL-9, TLR15, TLR2-1, TLR7, etc.) significantly increased by 2.0- to 5.3-fold in the NE-afflicted line 5.1 (p<0.01, fold-change ≥ 2; Fig. 2 and Table S2). The comparison of lines M5.1 and M15.2, indicated that the expression of five genes (IL17F, SPP2, CCR10, IL19, and CCLI5) was downregulated by 2.31- to 6.34-fold and the expression of 28 genes (IFNG, IL-22, IL-34, IL-5, TLR3, and TLR7, among others) was significantly upregulated by 2.0- to 5.27-fold (p<0.01, fold-change ≥ 2; Fig. 2 and Table S2). Therefore, number of innate immune genes expressed in line M5.1 was considerably higher than that in line M15.2.
Fig. 2.
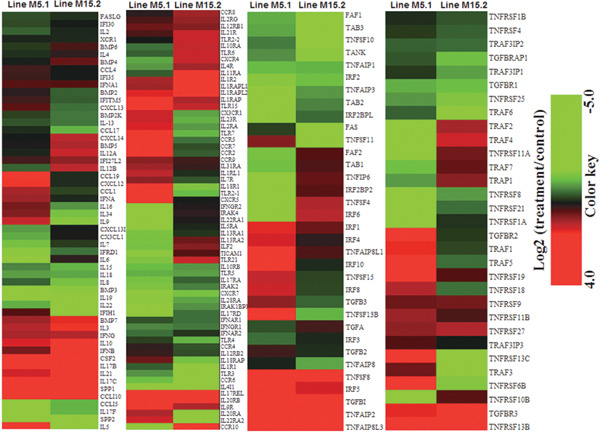
Hierarchical clusters of 183 innate immune genes responsive to E. maxima and C. perfringens coinfection in the intestinal mucosa of two chicken lines were based on Euclidean distance correlation analyses. The genes included here showed significant differences in gene expression (p<0.01, log2 fold change compared treatment to control). The genes shown in red were upregulated and those in green were downregulated.
Th1, Th2, Th17, and pro-inflammatory cytokines have key roles in immune responses to infection (Allam et al., 2011). Of these cytokines, Th1 cytokine genes IFN-γ, IL-21, and IL-2 were upregulated in the two chicken lines: IL-21 was strongly upregulated by 2.78- and 2.56-fold in M5.1 and M15.2 lines, respectively (p<0.01, fold-change ≥ 2; Fig. 2 and Table S2). IL-3 and IL-10 were significantly upregulated by 2.19- and 1.54-fold in line M5.1, respectively, and by 2.62- and 1.73-fold in line M15.2, respectively. In contrast, IL-5 was downregulated by 3.17-fold in line M15.2 and upregulated by 5.3-fold in line M5.1 (p<0.01, fold-change ≥ 2; Fig. 2 and Table S2). These results may indicate that NE induction results in the expression of Th1 and Th2 cytokines in both lines, and the expression levels of Th1 and Th2 cytokine genes were significantly higher in line M5.1 than in line M15.2. We found that four genes that encode cytokine Th17, including IL-17B, IL-17C, IL-17REL, and IL-23R in line M5.1, and IL-17C and IL-17REL in line M15.2, were upregulated by 2.3- to 5.0-fold (p<0.01, fold-change ≥ 2; Fig. 2 and Table S2). Therefore, Th17 and pro-inflammatory cytokines were expressed to a greater extent in line M5.1 than in line M15.2. The analysis of the transcription of pro-inflammatory cytokines or receptors in line M5.1 indicated that the expression of five out of six genes in the IL-1 cytokine family (IL-1R2, IL-1RAP, IL-1RAPL1, IL-1RAPL2, and IL-1RL1), IL-18R1, and of 4 out of 29 genes in the TNF family (TNFAIP2, TNFAIP8L3, TNFRSF13B, and TNFRSF 6B) was upregulated by 2.0-to 3.5-fold (p<0.01, fold-change ≥ 2; Fig. 2 and Table S2). In contrast, in line M15.2, the expression of pro-inflammatory cytokines changed by −1.7- to 1.6-fold in the IL-1 cytokine family and 1.3-fold in IL-18R1; in addition, 3 out of 29 genes of the TNF family (TNFAIP2, TNFAIP8L3, and TNFRSF13B) were upregulated 2.7 to 3.2-fold (Fig. 2 and Table S2). In our previous study, the expression of Th1, Th2, Th17, and pro-inflammatory cytokines was higher in resistant line 6.3 compared with susceptible line 7.2 in response to NE (Truong et al., 2015a). These results suggest that these cytokines may play an important role in different immune responses to NE in lines M5.1 and M15.2.
Similarly, the transcription analysis of genes of the TLR family indicated that the expression of TLR3 was significantly downregulated by 2.0-fold (p<0.01) in line M15.2 whereas the expression of TLR7, TLR15, and TLR2-1 significantly increased by 2.67- to 3.50-fold in line M5.1 (p<0.01; Fig. 2 and Table S2). Chemokines also play an essential role in the immune response to pathogen infections (Kaiser et al., 2005; Khawli et al., 2008). The transcriptome analysis of 23 chemokines, including C-C motif, C-X3-C motif, and C-X-C motif, indicated that the expression of nine chemokines (six genes encoding C-C motif, one gene encoding C-X3-C motif, and two genes encoding C-X-C motif) was markedly upregulated by 2.0- to 5.0-fold and the expression of two chemokines (CCLI5 and CCR10) was significantly downregulated by 2.4- to 3.6-fold in line M5.1 (p<0.01, fold-change ≥ 2; Fig. 2 and Table S2). Unexpectedly, the expression of only two chemokine genes (CCLI10 and CCR10) significantly increased by 3.06- and 4.15-fold in line M15.2, respectively (p<0.01, fold-change ≥ 2; Fig. 2 and Table S2).
Avian β-defensin Genes Are Differentially Expressed to NE-induced Chicken Lines
Defensins are small cysteine-rich cationic peptides with a strong antimicrobial activity against bacteria, fungi, and viruses (Lehrer and Ganz, 2002). Avian β-defensins play a significant role in the avian innate defense system (Sugiarto and Yu, 2004). To date, 14 β-defensin genes have been described in chickens: gallinacin (GAL) 1, 1A, and 2 to 13 (Lynn et al., 2007; van Dijk et al., 2008; Hong et al., 2012), and 14 β-defensin genes with six lineage-specific duplications (LSDs) of AvBD3 (AvDB3A-F) were identified in duck (Huang et al., 2013). In this study, transcriptome analysis indicated that 13 avian defensin genes (AvDB1 to AvDB14, except AvBD7) and three LSDs of these genes (AvDB3ST1, AvDB3ST2, and AvDBST4) were exp ressed in the two chicken lines. Of these, 12 β-defensin genes were markedly upregulated in at least one chicken line compared with the control (p<0.01; Fig. 3). Similar to a previous study that used commercial chickens, AvDB2, AvDB4, AvDB11, and AvDB12 were significantly downregulated (p<0.01) in the two chicken lines compared with the controls (Fig. 3). The comparison of the two lines indicates that the expression of 6 β-defensin genes (AvBD3ST1, AvBD3ST4, AvBD6, AvBD8, AvBD13, and AvBD14) significantly increased by 2.1- to 5.58-fold in M5.1 line (p<0.01, fold-change ≥ 2) whereas the expression of 4 β-defensin genes (AvBD1, AvBD3, AvBD9, and AvBD10) significantly increased by 2.0- to 2.2-fold in line M15.2 (p<0.01, fold-change ≥ 2; Fig. 3). In our previous study, we measured the β-defensin expression in the jejunum and found that AvBD8, AvBD10, and AvBD13 were highly expressed whereas AvBD1, AvBD6, AvBD9, AvBD11, and AvBD12 were moderately expressed in chickens with NE. In addition, β-defensin genes were differentially expressed in commercial chickens in response to NE, which indicates that β-defensins play an important role against NE (Hong et al., 2012). Our results suggest that more β-defensin genes are expressed in line M5.1 than in line M15.2 and that line M5.1 is more resistant to NE than line M15.2.
Fig. 3.

Identification of β-defensins family genes responsive of two chicken lines to NE induced in the intestinal mucosa. The genes included here showed significant differences in gene expression (p<0.01, log2 fold change compared treatment to control). Genes shown in red was upregulated, and those shown in green was down regulated in chicken lines relative to control.
Expression of CDmolecules in the NE-induced Chicken Lines
CD molecules are antigens present in the cell membrane during cell development (Stefanova and Horejsi, 1991; Zola et al., 2007). Most CD antigens are involved in immune functions. We investigated the gene expression patterns of 46 CD molecules in the intestinal mucosa of the two lines using RNA-Seq data. In line M5.1, 12 CD genes (CD4, CD1D, CD226, CD2, CD8B, CD72, CD200, CD28, CD274, CD1B, CD79B, and CD180) were upregulated by 2.1- to 3.8-fold whereas CD36 was downregulated by 2.3-fold (p<0.01, fold-change ≥2; Fig. 4 and Table S3). By contrast, in line M.15.2, the expression of 5 genes (CD180, CD86, CD14, CD 40LG, and CD9) was strongly downregulated by 2.1- to 5.0-fold whereas the expression of 2 genes (CD34 and CD72) significantly increased by 2.5- and 2.8-fold (p<0.01, fold-change ≥2; Fig. 4 and Table S3). The comparison between these chicken lines indicated that 7 CD genes (CD72, CD 109, CD4, CD300LD, CD180, CD79B, and CD9) were up - regulated by 2.4- to 3.4-fold (p<0.01, fold-change≥2; Fig. 4 and Table S3).
Fig. 4.
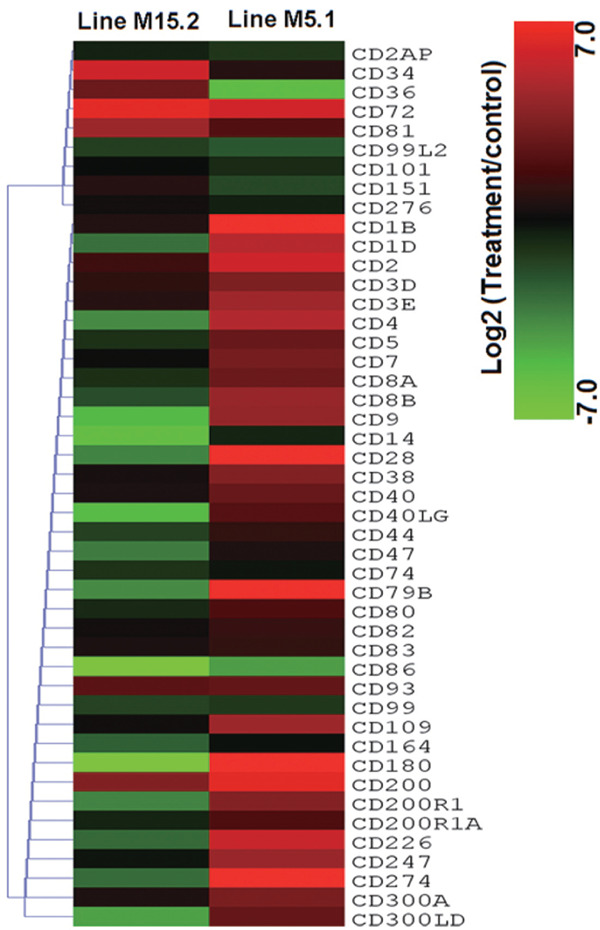
Expression of 46 CD marker genes in NE induced chicken lines. The heatmap was generated from a hierarchical analysis of the 46 CD marker genes that showed significant changes in the NE induced chicken lines. The genes included here showed significant differences in gene expression (p<0.01, log2 fold change compared treatment to control) in at least one experiment. The genes shown in red were upregulated expression and those in green were downregulated in the two NE-induced chicken lines.
Interestingly, CD4, CD8B, CD28, and CD72, which were significantly upregulated in line M5.1, form a co-receptor that assists the T cell receptor in communicating with an antigen-presenting cell and activating T-cells (Ansari-Lari et al., 1996; Suzawa et al., 2007). The expression of CD72 increased in both chicken lines; CD72 protein is associated with the cellular and humoral immune systems of animals and is an important marker for progenitor B-cell leukemia (Schwarting et al., 1992; Ishida et al., 2003). CD72 is expressed in B cells, where it appears to mediate the interaction between B and T cells. Similarly, CD180 belongs to a family of pathogen receptors known as Toll-like receptors (TLRs). CD180/MD-1 works in concert with TLR4 to control the recognition of B cells and signaling of lipopolysaccharide, which is a constituent of the cellular membrane of gram-negative bacteria (Miura et al., 1996). Our results indicated that the CD1 gene family, including CD1B and CD1D, was upregulated in line M5.1 whereas CD1D was downregulated in line M15.2 (Fig. 4 and Table S3). The lipid antigens in the CD1 family and CD2 activate natural killer T (NKT) cells via interaction with the T-cell receptor present on the membrane of NKT cells. When activated, NKT cells rapidly produce Th1 and Th2 cytokines via the synthesis of IFN-γ and IL-4 (Zhou et al., 2006; Bendelac et al., 2007). Furthermore, CD226 is expressed on the surface of NKT cells, monocytes, and a subset of T cells. CD226 is a member of the immunoglobulin superfamily and contains two Ig-like domains of the V-set (Tahara-Hanaoka et al., 2004). Similar to other cytokines and CD molecules, CD226 was upregulated by 2.44-fold in line M5.1 and was downregulated by 1.12-fold in line M15.2 (Fig. 4 and Table S3). These results indicate that CD molecules are more highly expressed in line M5.1 than in M15.2 and may better regulate the response of innate immune genes to NE in line M5.1 compared with M15.2.
Expression of Immune-related Genes in the NE-induced Chicken Lines
On the basis of annotation data from the GO Consortium, 89 immune-related genes were identified in this study. To evaluate the host response to NE and the genetic differences between the two chicken lines, the list of immune-related genes was used to narrow the previously identified DEGs (p<0.01). Using the designated cut-off fold-change of 2.0, four genes in line M15.2 and 15 genes in line M5.1 were found to be differentially expressed after NE induction (Table S4); among them, 11 genes were more highly expressed in line M5.1 than in line M15.2 (p<0.01, fold-change ≥ 2; Table S4). Several of these DEGs play important roles in protein transportation, transmembrane transportation as well as in the interaction between B cells, T-cell immunoglobulin, and BCL2. These genes are probably associated with the degradation and processing of antigens for MHC class I and II molecules (Table S2). Most of the DEGs related to MHC-II antigen-processing pathways, including immunoglobulin, cytokine, and hemoglobin, were significantly upregulated or downregulated (Table S4).
GO Analysis of DEGs
The analysis of enriched GO terms allowed us to identify significant categories that could be overlooked when evaluating individual genes. The enriched GO terms can help interpret the dominant functions controlled by DEGs (Ashburner et al., 2000). Using GO analysis, approximately 1270 genes with known functions at the logarithm growth phase were identified in Fayoumi line M5.1 and were categorized into 69 terms in the biological processes, in 17 terms in cellular components, and in eight terms in molecular functions. Of the 1270 genes with known functions, 211 genes were assigned to the molecular function category (127 genes were upregulated and 84 genes were downregulated; p<0.01, fold-change ≥2), 141 genes were assigned to the cellular component category (116 genes were upregulated and 25 genes were downregulated; p<0.01, fold-change ≥2), and 918 genes were assigned to the biological process category (628 genes were upregulated and 290 genes were downregulated; p<0.01, fold-change ≥2; Table S5A). Similarly, GO analysis of line M15.2 suggested that 417 genes with known functions at the logarithm growth phase were categorized into 42 terms in biological processes, in 15 terms in cellular components, and in nine terms in molecular functions, and all the genes with 2-fold changes belonged to the aforementioned categories and were differentially expressed (p<0.01). Of the 417 genes with known functions, 53 genes were assigned to the molecular function category (15 genes were upregulated and 38 genes were downregulated; p<0.01, fold-change ≥ 2), 149 genes were assigned to the cellular component category (75 genes were upregulated and 74 genes were downregulated; p<0.01, fold-change ≥ 2), and 215 genes were assigned to the biological process category (120 genes were upregulated and 95 genes were downregulated; p<0.01, fold-change ≥ 2; Table S5B).
Six major categories in GO's biological process in line M5.1 were associated with signal immune response, single-multicellular organism process, regulation of locomotion, monocarboxylic acid metabolism, immune processes, and multi-organism processes. However, GO terms in line M15.2 were included in two major clusters: anion transport and immune response (Fig. S2). In addition, two major clusters for cellular component in line M5.1 were associated with contractile fibers and extracellular region, and three major clusters for cellular component in line M15.2 were associated with the external plasma membrane, high-density lipoprotein particles, and the extracellular region (Fig. S2). The GO term molecular function had four major clusters in line M5.1: iron-ion binding, gram-positive bacteria binding, chemokine activity, and monooxygenase activity, and four major clusters in line M15.2: organic acid transmembrane transporter activity, transporter activity, lysozyme activity, and integrin binding (Fig. S2). Compared with the results of our previous study (Truong et al., 2015a), GO analysis of Fayoumi chicken and of the inbred chicken lines 6.3 and 7.2 indicated the presence of similar DEGs, and the number of genes that GO enriched categories biological process and cellular component differed between the upregulated and downregulated genes. Therefore, the GO molecular functional enrichment analysis suggests different roles and functions for the genes identified in the two Fayoumi chicken lines.
Pathway Analysis of DEGs
The pathway analysis was performed to better understand the biological function of the DEGs in the regulatory system. The information on the molecular networks and enriched pathways of the DEGs allowed us to explore the gene network and the underlying molecular mechanisms between the two chicken lines. Using KEGG pathway mapping, 1851 genes were identified (NCBI Gene ID), in which 1270 and 581 DEGs were identified in lines M5.1 and M15.2 respectively; 1175 genes in line M5.1 and 508 genes in line M15.2 were mapped into several KEGG pathways. The most differentially expressed signaling pathways from each group are listed in Table 2.
Table 2. Distribution of pathways related of two chicken lines with NE induced. KEGG pathway analyses were performed using DAVID Bioinformatics Resources version 6.7, NIAID/NIH (http://david.abcc.ncifcrf.gov/tools.jsp).
| ID | Pathway | Line M5.1 |
Line M15.2 |
||||
|---|---|---|---|---|---|---|---|
| DEGs | % | p-value | DEGs | % | p-value | ||
| gga04080 | Neuroactive ligand-receptor interaction | 60 | 3.9 | 5.2E-13 | 51 | 5.4 | 2E-17 |
| gga04060 | Cytokine-cytokine receptor interaction | 40 | 2.6 | 0.00045 | 19 | 2 | 0.013 |
| gga04020 | Calcium signaling pathway | 33 | 2.2 | 0.000034 | 29 | 3.1 | 5.7E-08 |
| gga04010 | MAPK signaling pathway | 31 | 2 | 0.018 | 11 | 1.2 | 0.039 |
| gga04510 | Focal adhesion | 27 | 1.8 | 0.023 | 14 | 1.5 | 0.024 |
| gga04810 | Regulation of actin cytoskeleton | 27 | 1.8 | 0.049 | 14 | 1.5 | 0.032 |
| gga04514 | Cell adhesion molecules (CAMs) | 22 | 1.4 | 0.0042 | 13 | 1.4 | 0.038 |
| gga04310 | Wnt signaling pathway | 21 | 1.4 | 0.035 | 14 | 1.5 | 0.046 |
| gga04630 | Jak-STAT signaling pathway | 21 | 1.4 | 0.044 | 11 | 1.2 | 0.028 |
| gga04270 | Vascular smooth muscle contraction | 18 | 1.2 | 0.015 | 14 | 1.5 | 0.0047 |
| gga04110 | Cell cycle | 14 | 0.9 | 0.028 | 3 | 0.3 | 0.099 |
| gga04910 | Insulin signaling pathway | 14 | 0.9 | 0.039 | |||
| gga04512 | ECM-receptor interaction | 13 | 0.9 | 0.0156 | 10 | 1.1 | 0.028 |
| gga03320 | PPAR signaling pathway | 12 | 0.8 | 0.033 | 4 | 0.4 | 0.01 |
| gga04916 | Melanogenesis | 12 | 0.8 | 0.023 | 14 | 1.5 | 0.0015 |
| gga00590 | Arachidonic acid metabolism | 11 | 0.7 | 0.02 | 2 | 0.2 | 0.095 |
| gga04370 | VEGF signaling pathway | 11 | 0.7 | 0.011 | 2 | 0.2 | 0.0015 |
| gga04540 | Gapjunction | 11 | 0.7 | 0.024 | 14 | 1.5 | 0.00056 |
| gga04350 | TGF-beta signaling pathway | 10 | 0.7 | 0.034 | 4 | 0.4 | 0.023 |
| gga04012 | ErbB signaling pathway | 9 | 0.6 | 0.048 | 3 | 0.3 | 0.034 |
| gga04914 | Progesterone-mediated oocyte maturation | 9 | 0.6 | 0.047 | |||
| gga04920 | Adipocytokine signaling pathway | 9 | 0.6 | 0.022 | |||
| gga00564 | Glycerophospholipid metabolism | 8 | 0.5 | 0.037 | 5 | 0.5 | 0.047 |
| gga04070 | Phosphatidylinositol signaling system | 8 | 0.5 | 0.046 | |||
| gga00260 | Glycine, serine and threonine metabolism | 6 | 0.4 | 0.012 | 6 | 0.6 | 0.02 |
| gga00512 | O-Glycan biosynthesis | 6 | 0.4 | 0.011 | 3 | 0.3 | 0.046 |
| gga00601 | Glycosphingolipid biosynthesis-Lacto | 6 | 0.4 | 0.019 | 2 | 0.2 | 0.033 |
| gga03030 | DNA replication | 6 | 0.4 | 0.02 | |||
| gga04672 | Intestinal immune network for IgA production | 6 | 0.4 | 0.042 | 5 | 0.5 | 0.024 |
| gga00565 | Ether lipid metabolism | 5 | 0.3 | 0.036 | |||
| gga00592 | alpha-Linolenic acid metabolism | 4 | 0.3 | 0.02 | |||
| gga00603 | Glycosphingolipid biosynthesis | 4 | 0.3 | 0.011 | |||
| gga04320 | Dorso-ventral axis formation | 4 | 0.3 | 0.037 | |||
| gga00604 | Glycosphingolipid biosynthesis | 3 | 0.2 | 0.038 | |||
| gga00910 | Nitrogen metabolism | 4 | 0.4 | 0.011 | |||
| gga04260 | Cardiac muscle contraction | 7 | 0.7 | 0.021 | |||
| gga04340 | Hedgehog signaling pathway | 8 | 0.8 | 0.024 | |||
These data indicate that the elevated expression of genes in the intestinal mucosa of the infected chickens was strongly associated with immunity and defense (cytokine-cytokine receptor interaction), signal transduction (MAPK, JAK-STAT, ErbB, and phosphatidylinositol signaling pathways), extracellular matrix (ECM) (e.g., focal adhesion, cell adhesion, and ECM-receptor interaction), and cell cycle-associated pathways (apoptosis and cell differentiation). Several upregulated genes are involved in immunological responses, such as those associated with the MAPK and JAK-STAT signaling pathways, rather than the downregulated genes in each pathway. These results suggest that new populations of immune cells are recruited in the intestine and replace the original population of epithelial cells. Moreover, these observations are consistent with earlier observations that several innate immunity genes that mediate cellular immunity play a key role in the response to NE (Lee et al., 2011).
Many regulated pathways are interconnected or functionally overlap, including cytokine/cytokine receptor interactions and the JAK-STAT signaling pathway, and immune cells and other cell types are recruited in adhesion molecule interactions, such as those found in MHC class I/II, and TGF-β2 in dendritic cells; the latter interact with CD8, CD4, or CD226 in T cells (Huang et al., 2006). Of note, we also observed an increase in the expression of genes that encode IgA in the intestinal immune network, including genes encoding class II MHC in dendritic cells, genes encoding CD28, CD40L, and ICOS in CD4+ T cells, and genes encoding BAFF in epithelial cells (Table 2). Significant levels of IgA were also found in the gut mucosa in response to infection with E. maxima (Lillehoj and Lillehoj, 2000; Min et al., 2013). IgA is a factor involved in intestinal mucosal immunity and plays a key role in protecting the host against infections by pathogens such as Toxoplasma gondii (Bourguin et al., 1991) and Eimeria tenella (Zhou et al., 2015). We hypothesized that IgA production is essential for chickens to develop protective immunity and plays an important role in NE. In both chicken lines, the gene cluster that included neuroactive ligand-receptor interaction, JAK-STAT signaling pathway, MAPK signaling pathway, regulation of actin cytoskeleton, p53 signaling pathway, cytokine-cytokine receptor interaction, and calcium signaling pathway represented the largest cluster, particularly in line M5.1, which is consistent with the response of these chicken lines to NE.
Two genes of the TGFβ signaling pathway (TGFβ1 and TGFβ3), which also participate in cytokine-cytokine receptor interaction and in other pathways, including MAPK, mTOR, and JAK-STAT signaling pathways, were upregulated in line M5.1 (Table S2). Most immune cells express TGFβ1, which plays an important role in the control of the immune system and shows distinct activity profiles in different cell types and developmental stages (Letterio and Roberts, 1998). TGFβ3 is involved in the differentiation, embryogenesis, and development of cells (Herpin et al., 2005). The stimulation of skeletal muscle and porcine interstitial cells with TGFβ1 increased the mRNA expression of the p38 MAPK family and Mekl/2/Erk1/2 signaling pathways (Das et al., 2013). In this study, the p38 MAPK family (p38β2, p38α, p38γ, and p38δ), JNK1/2/3, and ERK1/2 were differentially expressed in the two chicken lines (data not shown). The expression of TGFβ1 and TGFβ3 was upregulated by 1.12- and 3.39-fold in line M15.2, respectively, and by 2.34- and 1.50-fold in line M5.1, respectively, compared with the controls (p<0.01; Table S2). Therefore, further studies are required to determine how TGFβ1 and TGFβ3 are regulated and controlled in the two chicken lines at the cellular level after NE induction.
Validation of Differential Gene Expression by qRT-PCR
To determine the expression profile obtained using RNA-Seq analysis, 19 randomly selected DEGs with different levels of expression were used for validation by relative qRT-PCR. The expression levels of these 19 genes are shown in Fig. 5. We also conducted a simple linear regression analysis of the relative expression and calculated the RPKM; the coefficient of determination (or R2) was equal to 0.84 for Fayoumi line M5.1 and 0.88 for line M15.2 (Fig. 6). The expression profiles of the selected genes obtained at random using qRT-PCR were consistent with the patterns of expression obtained using RNA-Seq. The results were considered as the technical validation of the DEG analysis.
Fig. 5.
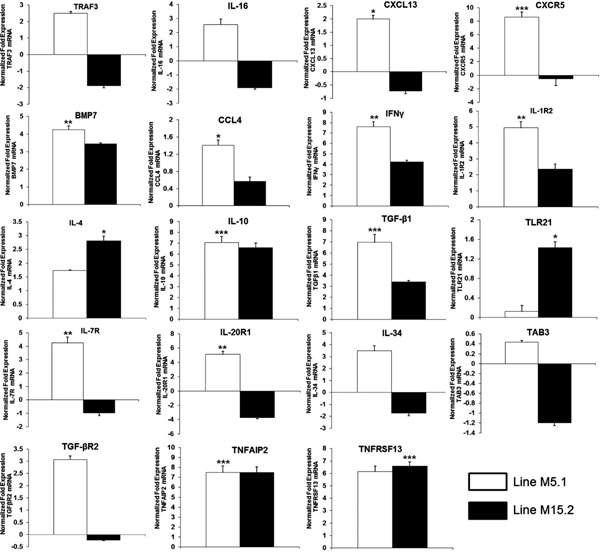
Quantitative expression analysis of up- and down regulated genes in intestinal mucosa derived from NE induced line M5.1 and M15.2 of Fayoumi chicken. Data were expressed as mean values±standard deviation of samples (n=3) and expressed as fold change in mRNA expression: * p<0.05, ** p<0.01, and *** p<0.001.
Fig. 6.
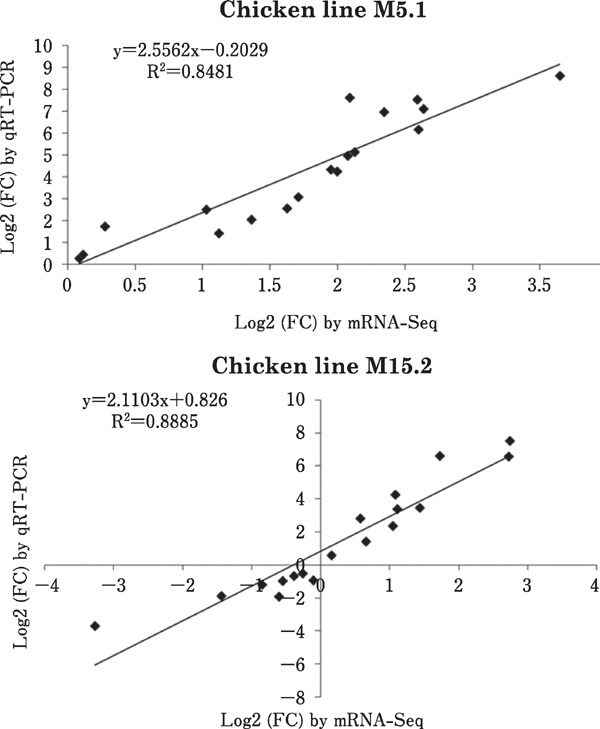
Significant correlations between expressions of qRT-PCR and RNA-Seq in intestinal mucosa of two genetic chicken lines NE induced.
Conclusion
In the present study, genome-wide gene expression profiles of the host response to NE induction in the intestinal mucosa of two Fayoumi chicken lines were evaluated using RNA-Seq. We drew four noteworthy conclusions from our results. First, using RNA-Seq, we generated the first draft of approximately 52 and 40 million reads for M5.1 and M15.2 lines, respectively, one of which is a natural host for NE. Second, we used RNA-Seq transcriptome analysis to characterize the DEG profiles and identify genes responsive to NE. Overall, genes of line M5.1 were expressed to a greater extent than those of line M15.2 after NE induction. Third, we detected 89 immune-related and 46 CD molecular genes differentially expressed in the two lines. In addition, approximately 183 innate immune genes were differentially expressed in the two lines. Fourth, we found 13 β-defensin genes with 3 LSDs of AvBD3, and these genes might be involved in the host immune response to NE in these chicken lines. Our results suggest that line M5.1 is more resistant to NE than line 15.2; furthermore, they helped elucidate the mechanisms underlying disease resistance to NE and will foster the development of effective control strategies against these enteric pathogens.
Acknowledgments
This study was partially supported by the Next-Generation BioGreen 21 Program (No. PJ00808401), Rural Development Administration, and Chung-Ang University Excellent Student Scholarship in 2015, Republic of Korea. We are thank to Dr. Susan J. Lamont (Iowa State University) for Fayoumi chicken.
Conflict of Interest
The authors have declared that no competing interests exist.
Sequence Data Availability
All raw Illumina sequence data can be obtained freely by contacting the Department of Animal Science and Technology, Chung-Ang University. The AMG_Gallus database has been uploaded in the National Agricultural Biotechnology Information Center (NABIC, http://nabic.rda.go.kr/) [ID: NN-1820-000001, NN-1821-000001, NN-1823-000001, and NN-1824-000001].
Supplemental Information
Fig. S1. Evaluation of sequencing quality. Gene coverage statistics.
Fig. S2. The “TreeMap” view of difference expressed in terms of biological process, cellular component, and molecular function for lines M5.1 and M15.2. Each rectangle is a single cluster representative. The representatives are joined into “superclusters” of loosely related terms and visualized with different colors. Size of the rectangles may be adjusted to reflect p-value.
Table S1. Oligonucleotide primers used for quantitative RT-PCR.
Table S2. Description of genes responsive to NE induced in the intestinal mucosa of 2 chicken lines. These data show significant changes in gene expression for 183 innate immune genes in the chicken lines. The genes included here show significant differences in gene expression (p<0.01, log2-fold change when treatment was compared with the control).
Table S3. Description of CD molecular genes responsive to NE induced in the intestinal mucosa of 2 chicken lines. These data show significant changes in gene expression for 46 CD molecular genes in the chicken lines. The genes included here show significant differences in gene expression (p<0.01, log2-fold change when treatment was compared with the control).
Table S4. Differentially expressed genes related to the antigen processing and presentation pathways in the intestinal mucosa of 2 chicken lines (p<0.01, log2-fold change when treatment was compared with the control).
Table S5A. Gene ontology (GO) for DEGs in NE-induced line M5.1 (CC: Cellular Component; BP: Biological Process, and MF: Molecular Function; p<0.01; log2-fold change when treatment was compared with the control ≥ 2 or ≤ -2). The p values were calculated using the right-tailed Fisher's exact test. GO functional enrichment analysis was performed using Blast2GO (version 2.3.5) (http://www.blast2go.org/).
Table S5B. Gene ontology (GO) for DEGs in NE-induced line M15.2 (CC: Cellular Component; BP: Biological Process, and MF: Molecular Function; p<0.01; log2-fold change when treatment was compared with the control ≥ 2 or ≤ -2). The p values were calculated using the right-tailed Fisher's exact test. GO functional enrichment analysis was performed using Blast2GO (version 2.3.5) (http://www.blast2go.org/).
References
- Allam JP, Duan Y, Winter J, Stojanovski G, Fronhoffs F, Wenghoefer M, Bieber T, Peng WM, Novak N. Tolerogenic T cells, Th1/Th17 cytokines and TLR2/TLR4 expressing dendritic cells predominate the microenvironment within distinct oral mucosal sites. European Journal of Allergy and Clinical Immunology, 66: 532-539. 2011. [DOI] [PubMed] [Google Scholar]
- Ansari-Lari MA, Muzny DM, Lu J, Lu F, Lilley CE, Spanos S, Malley T, Gibbs RA. A gene-rich cluster between the CD4 and triosephosphate isomerase genes at human chromosome 12p13. Genome Research, 6: 314-326. 1996. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstei D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium, Nature Genetics, 25: 25-29. 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annual Review of Immunology, 25: 297-336. 2007. [DOI] [PubMed] [Google Scholar]
- Bourguin I, Chardes T, Mevelec MN, Woodman JP, Bout D. Amplification of the secretory IgA response to Toxoplasma gondii using cholera toxin. FEMS Microbiology Letters, 65: 265-271. 1991. [DOI] [PubMed] [Google Scholar]
- Chitwood JL, Rincon G, Kaiser GG, Medran JF, Ross PJ. RNA-seq analysis of single bovine blastocysts. BMC Genomics, 14: 350 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das D, Holmes A, Murphy GA, Mishra K, Rosenkranz AC, Horowitz JD, Kennedy JA. TGF-beta1-Induced MAPK activation promotes collagen synthesis, nodule formation, redox stress and cellular senescence in porcine aortic valve interstitial cells. Journal of Heart Valve Disease, 22: 621-630. 2013. [PubMed] [Google Scholar]
- Herpin A, Lelong C, Becker T, Rosa FM, Favrel P, Cunningham C. Structural and functional evidences for a type 1 TGF-beta sensu stricto receptor in the lophotrochozoan Crassostrea gigas suggest conserved molecular mechanisms controlling mesodermal patterning across bilateria. Mechanisms of Development, 122: 695-705. 2005. [DOI] [PubMed] [Google Scholar]
- Hong YH, Song W, Lee SH, Lillehoj HS. Differential gene expression profiles of beta-defensins in the crop, intestine, and spleen using a necrotic enteritis model in 2 commercial broiler chicken lines. Poultry Science, 91: 1081-1088. 2012. [DOI] [PubMed] [Google Scholar]
- Huang R, Wallqvist A, Covell DG. Comprehensive analysis of pathway or functionally related gene expression in the National Cancer Institute's anticancer screen. Genomics, 87: 315-328. 2006. [DOI] [PubMed] [Google Scholar]
- Huang Y, Li Y, Burt DW, Chen H, Zhang Y, Qian W, Kim H, Gan S, Zhao Y, Li J, Yi K, Feng H, Zhu P, Li B, Liu Q, Fairley S, Magor KE, Du Z, Hu X, Goodman L, Tafer H, Vignal A, Lee T, Kim KW, Sheng Z, An Y, Searle S, Herrero J, Groenen MA, Crooijmans RP, Faraut T, Cai Q, Webster RG, Aldridge JR, Warren WC, Bartschat S, Kehr S, Marz M, Stadler PF, Smith J, Kraus RH, Zhao Y, Ren L, Fei J, Morisson M, Kaiser P, Griffin DK, Rao M, Pitel F, Wang J, Li N. The duck genome and transcriptome provide insight into an avian influenza virus reservoir species. Nature Genetics, 45: 776-783. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida I, Kumanogoh A, Suzuki K, Akahani S, Noda K, Kikutani H. Involvement of CD100, a lymphocyte semaphorin, in the activation of the human immune system via CD72: implications for the regulation of immune and inflammatory responses. International Immunology, 15: 1027-1034. 2003. [DOI] [PubMed] [Google Scholar]
- Kaiser P, Poh TY, Rothwell L, Avery S, Balu S, Pathania US, Hughes S, Goodchild M, Morrell S, Watson M, Bumstead N, Kaufman J, Young JR. A genomic analysis of chicken cytokines and chemokines. Journal of Interferon & Cytokine Research, 25: 467-484. 2005. [DOI] [PubMed] [Google Scholar]
- Khawli LA, Hu P, Epstein AL. Cytokine, chemokine, and costimulatory fusion proteins for the immunotherapy of solid tumors. Handbook of Experimental Pharmacology, 181: 291-328. 2008. [DOI] [PubMed] [Google Scholar]
- Kim DK, Lillehoj HS, Hong YH, Park DW, Lamont SJ, Han JY, Lillehoj EP. Immune-related gene expression in two B-complex disparate genetically inbred Fayoumi chicken lines following Eimeria maxima infection. Poultry Science, 87: 433-443. 2008. [DOI] [PubMed] [Google Scholar]
- Kim DK, Kim CH, Lamont SJ, Keeler CL, Lillehoj HS. Gene expression profiles of two B-complex disparate, genetically inbred Fayoumi chicken lines that differ in susceptibility to Eimeria maxima. Poultry Science, 88: 1565-1579. 2009. [DOI] [PubMed] [Google Scholar]
- Kim DK, Lillehoj HS, Jang SI, Lee SH, Hong YH, Lamont SJ. Genetically Disparate Fayoumi chicken lines show different response to avian necrotic enteritis. Journal of Poultry Science, 52: 245-252. 2015. [Google Scholar]
- Lee JH, Gao C, Peng G, Greer C, Ren S, Wang Y, Xiao X. Analysis of transcriptome complexity through RNA sequencing in normal and failing murine hearts. Circulation Research, 109: 1332-1341. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Lillehoj HS, Jeong W, Jeoung HY, An DJ. Avian necrotic enteritis: experimental models, host immunity, pathogenesis, risk factors, and vaccine development. Poultry Science, 90: 1381-1390. 2011. [DOI] [PubMed] [Google Scholar]
- Lehrer RI, Ganz T. Defensins of vertebrate animals. Current Opinion in Immunology, 14: 96-102. 2002. [DOI] [PubMed] [Google Scholar]
- Letterio JJ, Roberts AB. Regulation of immune responses by TGF-β. Annual Review of Immunology, 16: 137-161. 1998. [DOI] [PubMed] [Google Scholar]
- Lillehoj HS, Lillehoj EP. Avian coccidiosis: A review of acquired intestinal immunity and vaccination strategies. Avian Diseases, 44: 408-425. 2000. [PubMed] [Google Scholar]
- Lister R, O'Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell, 133: 523-536. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT Method. Methods, 25: 402-408. 2001. [DOI] [PubMed] [Google Scholar]
- Lynn DJ, Higgs R, Lloyd AT, O'Farrelly C, Herve-Grepinet V, Nys Y, Brinkman FS, Yu PL, Soulier A, Kaiser P, Zhang G, Lehrer RI. Avian β-defensin nomenclature: a community proposed update. Immunology Letters, 110: 86-89. 2007. [DOI] [PubMed] [Google Scholar]
- McReynolds J, Waneck C, Byrd J, Genovese K, Duke S, Nisbet D. Efficacy of multistrain direct-fed microbial and phytogenetic products in reducing necrotic enteritis in commercial broilers. Poultry Science, 88: 2075-2080. 2009. [DOI] [PubMed] [Google Scholar]
- Min W, Kim WH, Lillehoj EP, Lillehoj HS. Recent progress in host immunity to avian coccidiosis: IL-17 family cytokines as sentinels of the intestinal mucosa. Developmental and Comparative Immunology, 41: 418-428. 2013. [DOI] [PubMed] [Google Scholar]
- Miura Y, Miyake K, Yamashita Y, Shimazu R, Copeland NG, Gilbert DJ, Jenkins NA, Inazawa J, Abe T, Kimoto M. Molecular cloning of a human RP105 homologue and chromosomal localization of the mouse and human RP105 genes (Ly64 and LY64). Genomics, 38: 299-304. 1996. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods, 5: 621-628. 2008. [DOI] [PubMed] [Google Scholar]
- Mot D, Timbermont L, Delezie E, Haesebrouck F, Ducatelle R, Van Immerseel F. Day-of-hatch vaccination is not protective against necrotic enteritis in broiler chickens. Avian Pathology, 42: 179-184. 2013. [DOI] [PubMed] [Google Scholar]
- Parish WE. Necrotic enteritis in the fowl (Gallus gallus domesticus) I Histopathology of the disease and isolation of a strain of Clostridium welchii. Journal of Comparative Pathology, 71: 377-393. 1961. [PubMed] [Google Scholar]
- Pinard-Van Der Laan MH, Monvoisin JL, Pery P, Hamet N, Thomas M. Comparison of outbred lines of chickens for resistance to experimental infection with coccidiosis (Eimeria tenella). Poultry Science, 77: 185-191. 1998. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26: 139-140. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarting R, Castello R, Moldenhauer G, Pezzutto A, von Hoegen I, Ludwig WD, Parnes JR, Dorken B. Human Lyb-2 homolog CD72 is a marker for progenitor B-cell leukemias. American Journal of Hematology, 41: 151-158. 1992. [DOI] [PubMed] [Google Scholar]
- Stefanova I, Horejsi V. Association of the CD59 and CD55 cell surface glycoproteins with other membrane molecules. Journal of Immunology, 147: 1587-1592. 1991. [PubMed] [Google Scholar]
- Sugiarto H, Yu PL. Avian antimicrobial peptides: the defense role of β-defensins. Biochemical and Biophysical Research Communications, 323: 721-727. 2004. [DOI] [PubMed] [Google Scholar]
- Suzawa K, Kobayashi M, Sakai Y, Hoshino H, Watanabe M, Harada O, Ohtani H, Fukuda M, Nakayama J. Preferential induction of peripheral lymph node addressin on high endothelial venule-like vessels in the active phase of ulcerative colitis. The American Journal of Gastroenterology, 102: 1499-1509. 2007. [DOI] [PubMed] [Google Scholar]
- Tahara-Hanaoka S, Shibuya K, Onoda Y, Zhang H, Yamazaki S, Miyamoto A, Honda S, Lanier LL, Shibuya A. Functional characterization of DNAM-1 (CD226) interaction with its ligands PVR (CD155) and nectin-2 (PRR-2/CD112). International immunology, 16: 533-538. 2004. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology, 28: 511-515. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols, 7: 562-578. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong AD, Hong YH, Lillehoj HS. High-throughput sequencing reveals differing immune responses in the intestinal mucosa of two inbred lines afflicted with necrotic enteritis. Veterinary Immunology and Immunopathology, 166: 116-124. 2015. a. [DOI] [PubMed] [Google Scholar]
- Truong AD, Hong YH, Lillehoj HS. RNA-seq Profiles of Immune Related Genes in the Spleen of Necrotic Enteritis-afflicted Chicken Lines. Asian-Australasian Journal of Animal Sciences, 28: 1496-1511. 2015. b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk A, Veldhuizen EJ, Haagsman HP. Avian defensins. Veterinary Immunology and Immunopathology, 124: 1-18. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Qin Z, Feng Z, Wang X, Zhang X. Identifying differentially spliced genes from two groups of RNA-seq samples. Gene, 518: 164-170. 2013. [DOI] [PubMed] [Google Scholar]
- Zhou BH, Liu LL, Liu J, Yuan FW, Tian EJ, Wang HW. Effect of diclazuril on the bursa of Fabricius morphology and SIgA expression in chickens infected with Eimeria tenella. Korean Journal of Parasitology, 53: 675-682 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D. The immunological function of iGb3. Current Protein & Peptide Science, 7: 325-333. 2006. [DOI] [PubMed] [Google Scholar]
- Zhou H, Lamont SJ. Genetic characterization of biodiversity in highly inbred chicken lines by microsatellite markers. Animal Genetics, 30: 256-264. 1999. [DOI] [PubMed] [Google Scholar]
- Zola H, Swart B, Banham A, Barry S, Beare A, Bensussan A, Boumsell L, D Buckley C, Buhring HJ, Clark G, Engel P, Fo D, Jin BQ, Macardle PJ, Malavasi F, Mason D, Stockinger H, Yang X. CD molecules 2006-human cell differentiation molecules. Journal of Immunological Methods, 319: 1-5. 2007. [DOI] [PubMed] [Google Scholar]