

Abstract
Aims/Introduction
Diabetic hearts are more vulnerable to ischemia‐reperfusion injury (I/RI). The activation of nucleotide‐binding oligomerization domain‐like receptor protein 3 (NLRP3) inflammasome can mediate the inflammatory process, and hence might contribute to myocardial I/RI. Activation of autophagy can eliminate excess reactive oxygen species and alleviate myocardial I/RI in diabetes. The present study aimed to investigate whether the activation of autophagy can alleviate diabetic myocardial I/RI through inhibition of NLRP3 inflammasome activation.
Materials and Methods
A dose of 65 mg/kg streptozotocin was given by tail vein injection to establish a type 1 diabetes model in the rats. The left anterior descending coronary artery was ligated for 30 min followed by reperfusion for 2 h to establish a myocardial I/RI model. H9C2 cardiomyocytes were exposed to high glucose (33 mmol/L) and subjected to hypoxia–reoxygenation (6 h hypoxia followed by 4 h reoxygenation).
Results
The diabetic rats showed significant inhibition of cardiac autophagy (decreased LC3‐II/I and increased p62) that was concomitant with increased activation of NLRP3 inflammasome (increased NLRP3, apoptosis‐related spots protein cleaved caspase‐1, interleukin‐18, interleukin‐1β) and more severe myocardial I/RI (elevated creatine kinase myocardial band, lactate dehydrogenase and larger infarct size). However, administration of rapamycin, an inhibitor of the autophagy, to activate autophagy resulted in the inhibition of NLRP3 inflammasome, and finally alleviated myocardial I/RI. In vitro, high glucose inhibited autophagy, while activating NLRP3 inflammasome in H9C2 cardiomyocytes and aggravating hypoxia–reoxygenation injury, but rapamycin reversed these adverse effects of high glucose.
Conclusion
Activation of autophagy can suppress the formation of NLRP3 inflammasome, which in turn attenuates myocardial ischemia‐reperfusion injury in diabetic rats.
Keywords: Diabetic myocardium, Myocardial ischemia‐reperfusion injury, Nucleotide‐binding oligomerization domain‐like receptor protein 3 inflammasome
Autophagy was inhibited in the myocardium of 8‐week diabetic rats, with activated NLRP3 inflammasome and aggravated myocardial ischemia‐reperfusion injury. Activation of autophagy can reduce the activation of NLRP3 inflammasome and therefore increase the tolerance to myocardial ischemia in diabetes.

Introduction
Reperfusion of the ischemic myocardium is the best way to save the heart, as ischemic heart disease, which raises morbidity and mortality, is the most serious diabetic complication1. However, additional injury has been made by reperfusion itself as ischemic‐reperfusion injury (I/RI)2. Diabetes can aggravate myocardial I/RI. There are persistent inflammatory responses in a diabetic heart. Inflammation plays an important role in the development of diabetic cardiomyopathy3. Previous studies have found that oxidative stress and inflammatory factors in the diabetic myocardium are significantly increased, and myocardial injury is aggravated, while treatment with anti‐oxidant N‐acetylcysteine can reduce the expression of inflammatory factors and reduce myocardial injury4. These results suggest that the inflammation in diabetic myocardium might be a mechanism attributable to the aggravation of myocardial IRI.
Recent studies have found a form of cell death associated with the release of a large number of inflammatory factors, namely pyroptosis, which is characterized by apoptosis and necrosis, and is widely involved in various diseases, such as infectious diseases, diabetes and cardiovascular diseases5, 6. It is mainly regulated by nucleotide‐binding oligomerization domain‐like receptor protein 3 (NLRP3) inflammasome. When cells are exposed to dangerous signals (such as hypoxia or oxidative stress), NCL proteins (nucleotide‐binding oligomerization domain‐like receptor proteins, NLRs, such as NLRP1, NLRP3, IPAF and AIM‐2) and the associated apoptosis‐related spots protein (ASC) and caspase‐1 constitute a protein complex (i.e., inflammasome)4, 7. Activated NLRP3 activates caspase‐1 by cleaving procaspase‐1 into two fragments, p20 and p10. Activated caspase‐1 recruits interleukin (IL)‐1β and IL‐18 precursors, and promotes their maturation and secretion, which in turn activates inflammatory factors that cause cell lysis and chemotaxis of more inflammatory factors8. The NLRP3 inflammasome can be activated by Nigerian toxins, urate crystallization, amyloid‐beta fibrils, and extracellular adenosine triphosphate, K+efflux, lysosomal destabilization, mitochondrial deoxyribonucleic acid and reactive oxygen species (ROS)9, 10. NLRP3 inflammasome plays an important role in many cardiovascular diseases, neurodegenerative diseases and tumorigenesis, including family periodic autoinflammatory response, type II diabetes, Alzheimer’s disease, joint inflammation and atherosclerosis. Recent studies have found that NLRP3 inflammasome activation is involved in diabetic myocardial I/RI4. However, the mechanism initiating or governing the activation of NLRP3 inflammasome in the diabetic myocardium and ,in particular, in the context of diabetic I/RI, is unclear.
Autophagy is a self‐protection mechanism of eukaryotes under normal conditions11. It can remove damaged organelles, misfolded proteins, stress products and so on to achieve cell renewal and energy regeneration, and maintain intracellular homeostasis. Autophagy dysfunction will cause various diseases, such as tumors, neurodegenerative diseases, diabetes, heart disease, inflammation, aging and metabolic syndrome12. Previous studies have shown that autophagy plays a dural role in myocardial I/RI. Activation of autophagy during ischemia can attenuate myocardial I/RI13, 14, whereas, autophagy overactivation during reperfusion can aggravate I/RI15, 16. Autophagy can directly remove inflammasomes. In addition, autophagy can also reduce inflammation by eliminating inflammatory stimuli, such as ROS, mitochondrial deoxyribonucleic acid or damaged organelles, to maintain homeostasis17, 18. Previous observations found that autophagic activity can attenuate cerebral and intestinal I/RI by eliminating mitochondrial deoxyribonucleic acid and mitochondrial ROS, and inhibiting the activation of NLRP3 inflammasome19, 20.
Yet, the possible regulatory relationship between autophagy and NLRP3 inflammasome during diabetic myocardial I/RI is still unknown. The present study aimed to investigate whether the activation of autophagy can alleviate diabetic myocardial I/RI through inhibition of NLRP3 inflammasome activation.
Methods
Establish diabetes mode
Male Sprague–Dawley rats (250 g, aged 6–8 weeks) were used for diabetes induction, as described previously21. Briefly, a dose of 65 mg/kg streptozotocin was given by tail vein injection to establish a type 1 diabetes model in the rats. All rats were housed in the Laboratory Animal Units of University of Hong Kong receiving standard care, and the protocols committed were approved by the Committee on the Use of Live Animals in Teaching and Research, the University of Hong Kong.
In vivo coronary ligation model and infarct size determination
After 8 weeks of diabetic induction, the rats were anesthetized. The chest was opened to expose the heart, then the left anterior descending branch was ligated to make it ischemic for 30 min, and reperfusion for 2 h22. In the sham operated group, the coronary artery was threaded without ligation. Myocardial infarct size was expressed as a percentage of the area at risk. The area at risk was calculated by injecting 5% Evans blue into the right jugular vein to show the non‐ischemic area, followed by excessive pentobarbital injection to euthanize the rat at the end of the experiment. The hearts of the rats were quickly taken out and cut into five pieces with a cross‐section of 1 mm. Sliced samples were incubated in 1% tetrazolium/formazan test at 25°C for 20 min and fixed by 10% formalin overnight.
In vitro models of cardiac H9C2 cells hypoxia–reoxygenation and treatment
The rat cardiac H9C2 cell line was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Then, 10% fetal bovine serum (Gibco, Grand Island, NY, USA), Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA) and 1% penicillin/streptomycin (100 U/mL, Thermo Fisher Scientific) were used during the culture of H9C2. In short, the H9C2 cells were incubated in glucose and serum‐free DMEM medium with 95% N2 and 5% CO2 to mimic ischemia for 6 h4, 22. Then the cells were incubated in complete DMEM medium (DMEM + fetal bovine serum + penicillin/streptomycin) with 5% CO2 , 21% O2 and 74% N2 for 4 h.
Analysis of lactate dehydrogenase leakage and cell viability
We used lactate dehydrogenase (LDH) to evaluate cell damage. According to the manufacturer’s manual, the commercial Lactate Dehydrogenase Kit (Roche, Mannheim, Germany) was used to measure the lactate dehydrogenase in the media. The cell viability of the H9C2 cells was detected by the 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide assay22. In brief, differently treated H9C2 cells were seeded into 96‐well plates and incubated with 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide solution (1 mg/mL; Sigma‐Aldrich, St. Louis, MO, USA) at 37°C for 4 h. We added dimethyl sulfoxide (100 mL/well) to resolve the formazan crystal. The absorbance was measured at 570 nm by Epoch microplate spectrophotometer (BioTek, Winooski, VT, USA).
Western blotting
H9c2 cardiomyocytes were lysed with lysis buffer supplemented with Protease Inhibitor Cocktail and Phosphatase Inhibitor Cocktail, and the total cell lysate was collected. The protein concentration of the cell lysate was determined by Bradford assay. The extracted protein samples were separated by 8–12.5% 12 alkyl sulphate polyacrylamide gel electrophoresis, and transferred to polyvinylidene difluoride membrane, and detected with appropriate antibodies. Primary antibodies against β‐actin antibody (1: 1,000), total LC3 (1:1,000), p62 (1:1,000), NLRP3 (1:1,000) and horseradish peroxidase‐conjugated anti‐rabbit or anti‐mouse secondary antibodies (1: 3,000) were purchased from Cell Signaling Technology. IL‐1, IL‐18 and ASC (1:1,000) were purchased from Abcam (Cambridge, MA, USA), and caspase‐1p20 antibody (1:1,000) was purchased from Santa Cruz (Santa Cruz, CA, USA). Blotted polyvinylidene difluoride membranes were incubated by Clarity ECL Western Blotting Detection Reagent (Bio‐Rad, Hercules, CA, USA) and exposed to X‐ray film (Carestream, Rochester, NY, USA). ImageJ (National Institutes of Health, Bethesda, MA, USA) was used to analyze the optical densities of the immunoreactive bands. β‐Actin expression was used as the loading control.
Terminal deoxynucleotidyl transferase dUTP nick end labeling assay
H9C2 cells were seeded at a density of 2 × 105 cells/mL in eight‐chamber slides (Thermo Fisher Scientific). After treatment, we used terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining to detect the cellular apoptosis using an In‐Situ Cell Death Detection Kit (Roche). With each sample, five images were taken with an Olympus BX41 fluorescence microscope (Olympus, Tokyo, Japan) followed by counting and calculating the TUNEL‐positive cell percentage.
Statistical analysis
All data are expressed as the mean ± standard error of the mean. Comparison between groups was carried out by one‐way anova or two‐way anova followed by Bonferroni’s test, wherever appropriate, using the GraphPad Prism 7.0 software (San Diego, CA, USA). P‐values <0.05 were considered statistically significant differences.
Results
Attenuated cardiac autophagy and activated NLRP3 inflammasome in type 1 diabetic myocardium
As shown in Table 1, 8 weeks after streptozotocin injection, the diabetic rats had notable diabetic symptoms, such as hyperglycemia and weight loss. The bodyweights of the diabetic rats were decreased, but their plasma glucose levels were significantly increased compared with that of non‐diabetic rats (Table 1).
Table 1.
General characteristics after streptozotocin injection at termination of study
| Parameters | Ctrl | DM |
|---|---|---|
| Bodyweight (g) | 389.0 ± 29.0 | 196.5 ± 41.7* |
| Water intake (mL/kg/day) | 136.5 ± 5.1 | 781.6 ± 18.3* |
| Food consumption (g/kg/day) | 86.2 ± 4.3 | 193.5 ± 12.8* |
| Glucose (mmol/L) | 5.1 ±1.7 | 29.6 ± 3.4* |
General characteristics after streptozotocin injection at termination of study.
n = 12/group.
P < 0.05 versus non‐diabetic rats (Ctrl). DM, diabetic rats.
Subsequently, we found significantly greater infarct size, higher creatine kinase myocardial band (CK‐MB) and LDH release after myocardial ischemia‐reperfusion in 8‐week‐old diabetic rats, compared with non‐diabetic rats (Figure 1a–c). Interestingly, autophagy‐related protein LC3‐II/I is decreased and p62 is increased in the myocardium of diabetic rats, indicating significantly inhibited autophagy (Figure 1d–f). With the increase of NLRP3 inflammasome constituent proteins, such as NLRP3, ASC and caspase‐1 p20, the release of IL‐1β and IL‐18 were also increased in the myocardium (Figure 1g–l), suggesting that autophagy inhibition and NLRP3 inflammasome activation might be involved in the pathology of the increased susceptibility of diabetic heart to I/RI.
Figure 1.
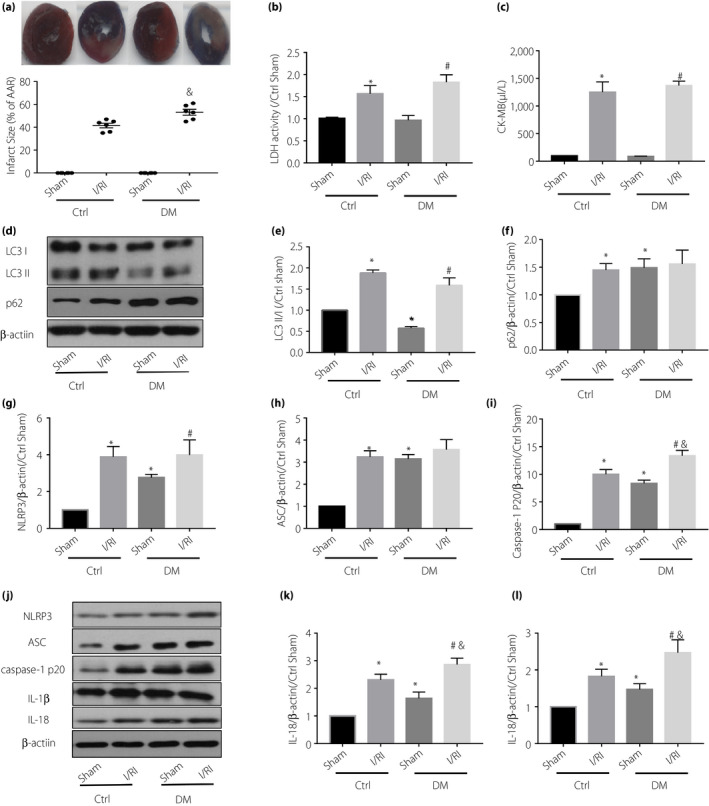
The (a) myocardial infarct size, (b) lactate dehydrogenase (LDH) and (c) creatine kinase myocardial band (CK‐MB) release in diabetic rats (DM) after ischemia‐reperfusion injury (I/RI) were significantly higher than those in non‐diabetic rats (Ctrl). Compared with the non‐diabetic group, the diabetic group had significant inhibition of autophagy, shown as (d,e) decreased LC3‐II/I and (d,f) increased p62, and activation of NLRP3 inflammasome, (g) shown as increased nucleotide‐binding oligomerization domain‐like receptor protein 3 (NLRP3), (h) apoptosis‐related spots protein (ASC), (i) caspase‐1 p20, (k) interleukin (IL)‐1β and (l) IL‐18, in the myocardium. Also, the diabetic group had more severe myocardial I/RI (elevated creatine kinase myocardial band, LDH and larger infarct size). Data are shown as the mean ± standard error of the mean. *P < 0.05 versus Ctrl sham; # P < 0.05 versus DM sham I/RI; and & P < 0.05 versus Ctrl I/RI; n = 6/group.
Activation of cardiac autophagy attenuates myocardial I/RI injury in diabetes through inhibition of NLRP3 inflammasome
To investigate the causal link between cardiac autophagy and NLRP3 inflammasome and their contribution to I/RI in diabetes, the control and diabetic rats were given rapamycin (autophagy activator, 1 mg/kg, i.p. daily for up to 3 days) before being subjected to I/RI.
In both control and diabetic rats, the expression pattern of LC3‐II/I and p62 were increased in the I/RI groups, compared with the sham groups. However, the increase of p62 induced by ischemia and reperfusion in diabetic rats was not significant, suggesting that ischemia‐reperfusion can activate autophagy, but diabetes can inhibit this effect. (Figure 2a–c). As anticipated, treatment with rapamycin significantly enhanced the protein presence of LC3‐II/I and decreased p62, suggesting that rapamycin was effective in activation of cardiac autophagy (Figure 2a–c). Furthermore, treatment with rapamycin reduced the activation of NLRP3 inflammasome (NlRP3, ASC and caspase‐1 p20, as well as increased release of IL‐1β and IL‐18) in the I/RI groups in both control and diabetic rats, suggesting that activated autophagy mediated the inhibition of NLRP3 inflammasome. (Figure 2d–i). In addition, treatment with rapamycin also attenuated myocardial I/RI injury, as evidenced by the reduced infarct size, CK‐MB and LDH level when compared with the control group (Figure 2j,k,l). Given the importance of NLRP3 inflammasome in the I/RI injury in diabetes, these observations suggested that activation of cardiac autophagy might attenuate myocardial I/RI injury in diabetes through inhibition of NLRP3 inflammasome.
Figure 2.
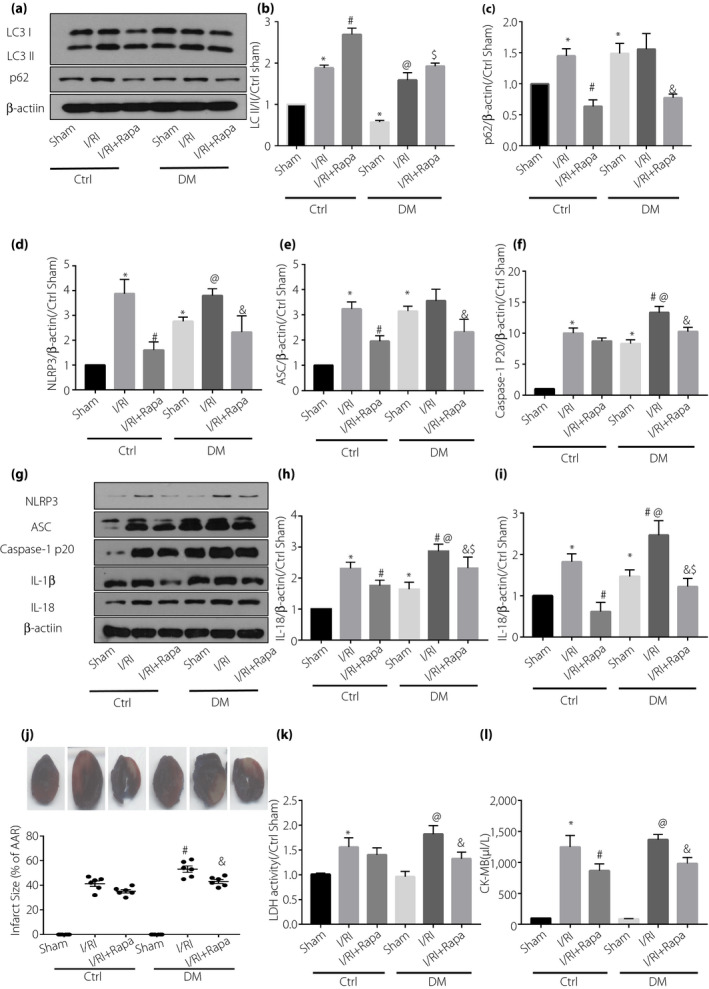
Administration of rapamycin (Rapa) activated autophagy, inhibited domain‐like receptor protein 3 (NLRP3) inflammasome and finally alleviated myocardial after ischemia‐reperfusion injury (I/RI). (a,g) Western blot was used to analyze the expression of (b) LC3‐II/, (c) p62, (d) NLRP3, (e) apoptosis‐related spots protein (ASC), (f) caspase‐1 p20, (h) interleukin (IL)‐1β and (i) IL‐18. (j) Representative images of myocardial infarct size determined by tetrazolium/formazan test and Evans blue staining; post‐ischemic infarct size expressed as the percentage of infarct size (IS) to the area at risk (AAR). (k) Serum lactate dehydrogenase (LDH) level. (l) Serum creatine kinase myocardial band (CK‐MB) level. Data are shown as the mean ± standard error of the mean. *P < 0.05 versus control (Ctrl) sham; # P < 0.05 versus Ctrl sham I/R); @ P < 0.05 versus diabetes (DM) sham; $ P < 0.05 versus Ctrl I/RI + Rapa; & P < 0.05 versus DM I/RI; n = 6/group.
Activation of autophagy can inhibit the activation of NLRP3 inflammasome in high glucose‐treated H9C2 cells
To investigate the effect of high glucose on autophagy and NLRP3 inflammasome in cardiomyocytes, different concentrations of high glucose were used to culture rat cardiomyocyte‐derived cell line H9C2 cells, and then the alterations of autophagy and NLRP3 inflammasome at different time points were determined. We have found that 33 mmol/L high glucose treatment for 24 h significantly inhibited autophagy (reduced LC3‐Ⅱ/Ⅰ, increased p62; Figure 3b,c) and also activated NLRP3 inflammasome (increased the expression of NLRP3, caspase‐1 p20, ASC, IL‐1β and IL‐18; Figure 3d–h).
Figure 3.
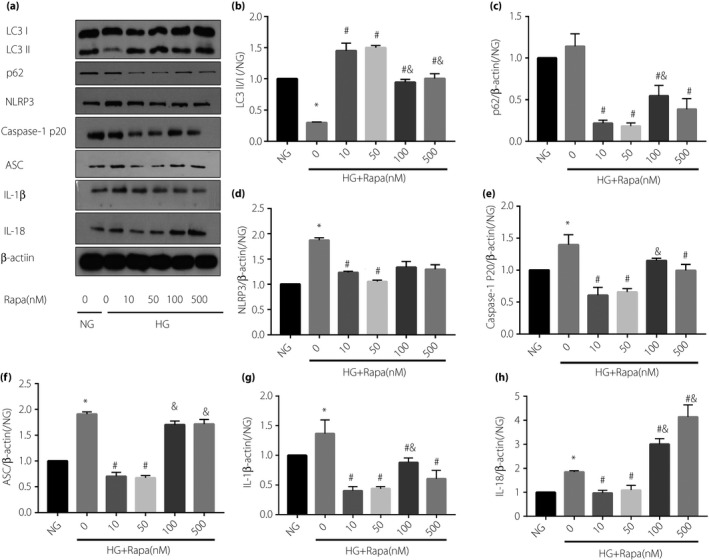
H9C2 cells cultured in high glucose for 24 h inhibited autophagy, as evidenced by (b) reduced LC3‐II/I, (c) increased p62 and (d) significantly enhanced nucleotide‐binding oligomerization domain‐like receptor protein 3 (NLRP3) inflammasome after hypoxia–reoxygenation, (e) caspase‐1 p20 and (f) apoptosis‐related spots protein (ASC), and (g,h) increased the expression of inflammatory factors interleukin (IL)‐1β and IL‐18. Rapamycin (Rapa) can activate autophagy and inhibit the further activation of NLRP3 inflammasome induced by hypoxia–reoxygenation. Data are shown as the mean ± standard error of the mean. *P < 0.05 versus normal glucose (NG, 5.5 mmol/L); # P < 0.05 versus high glucose (HG, 33 mmol/L); & P < 0.05 versus HG + 50 nmol/L Rapa; n = 5/group.
To clarify whether activation of autophagy can inhibit high glucose‐induced activation of NLRP3 inflammasome at the cellular level, the high glucose cultured H9C2 cardiomyocytes were pretreated with autophagy activator, rapamycin, and analyzed by western blotting with antibodies specific for autophagy and NLRP3 inflammasome. As expected, administration of rapamycin (from 10 nmol/L to 500 nmol/L) reversed high glucose‐induced inhibition of autophagy in H9C2 cardiomyocytes (reduced LC3‐II/I, increased p62; Figure 3b,c). Furthermore, rapamycin treatment decreased the expression of NLRP3, ASC and caspase‐1 p20, and also inhibited the expression of IL‐1β and IL‐18; Figure 3d–h). The inhibitory effect of rapamycin on NLRP3 inflammasome was the most robust at the concentration of 50 nmol/L (Figure 3d–h). Therefore, in the following experiments we selected 50 nmol/L rapamycin to treat cardiomyocytes. Taken together, these results suggest that 24 h of incubation with high glucose (33 nmol/L) can inhibit autophagy and activate NLRP3 inflammasome in H9C2 cardiomyocytes, and that the activation of autophagy could inhibit high glucose‐induced activation of NLRP3 inflammasome.
Activation of autophagy can attenuate hypoxia–reoxygenation injury in H9C2 cells treated with high glucose by inhibiting NLRP3 inflammasome
Studies have shown that NLRP3 inflammasome activation is involved in myocardial I/RI, and that NLRP3 inflammasome activation could aggravate the injury in diabetic myocardium23. To clarify whether or not autophagy activation and NLRP3 inflammasome inhibition could attenuate hypoxia–reoxygenation (HR) injury in H9C2 cardiomyocytes treated with high glucose, we cultured H9C2 cells with 33 mmol/L high glucose for 24 h, followed by oxygen and glucose deprivation for 6 h, and subsequent reoxygenation for 4 h.
In normal glucose (NG, 5.5 mmol/L) groups, HR upregulated the expression of LC3‐II/I and p62, and also increased NLRP3, ASC, caspase‐1 p20 IL‐1β and IL8 (Figure 4a–h). These showed that HR activated autophagy and NLRP3 inflammasome. Similarly, autophagy and NLRP3 inflammasome‐related proteins were increased in high glucose groups (HG) after HR, except for p62 (slightly decreased; Figure 4a–h), suggesting that high glucose‐suppressed autophagy could be restored by HR injury.
Figure 4.
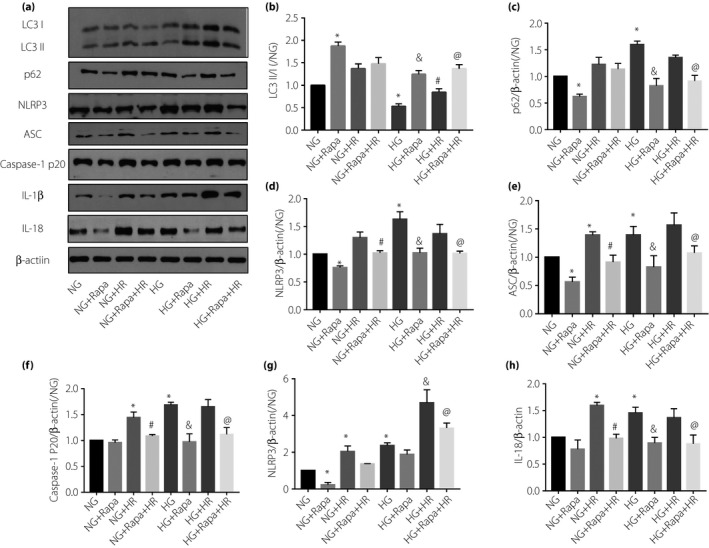
Activated autophagy can attenuate high glucose and aggravate hypoxia–reoxygenation (HR) injury in H9C2 cardiomyocytes by inhibiting nucleotide‐binding oligomerization domain‐like receptor protein 3 (NLRP3) inflammasome. (a) Western blot was used to analyze the expression of (b) LC3‐II/I, (c) p62, (d) NLRP3, (e) ASC, (f) caspase‐1 p20, (g) interleukin (IL)‐1β and (h) IL‐18. Data are shown as the mean ± standard error of the mean. *P < 0.05 versus normal glucose (NG, 5.5 mmol/L); & P < 0.05 versus high glucose (HG, 33 mmol/L); # P < 0.05 versus NG + HR; @ P < 0.05 versus HG + HR, P < 0.05; n = 5/group.
Furthermore, pre‐administration of rapamycin, an autophagy activator, significantly decreased NLRP3, ASC, caspase‐1 p20, IL‐1 and IL‐18 levels in both the NG + HR group and HG + HR group (Figure 4a–h). These results suggested that activation of autophagy can attenuate HR‐induced NLRP3 inflammasome activation. In addition, we also evaluated HR injury in H9C2 by detecting LDH, 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide and TUNEL. We found that HG did not affect cell viability at the basal level, but exacerbated HR injury. As shown in Figure 5, there were no significant differences in LDH, cell viability and TUNEL results between the HG group and the NG group. However, LDH and TUNEL‐positive cells in the NG group and the HG group increased after HR, and cell viability decreased. Although there was no significant change in LDH and cell viability between NG + HR and HG + HR, there were more TUNEL‐positive cells in HG + HR than NG + HR. After administration of rapamycin, LDH and TUNEL‐positive cells in NG + HR and HG + HR were decreased, cell viability increased, but the difference between the two groups disappeared. Rapamycin attenuates the HR injury in HG groups (by increasing cell viability, reducing LDH release and reducing cell death). These results show that autophagy activation can inhibit the activation of NLRP3 inflammasome, and thus alleviating high glucose‐aggravated myocardial I/RI.
Figure 5.
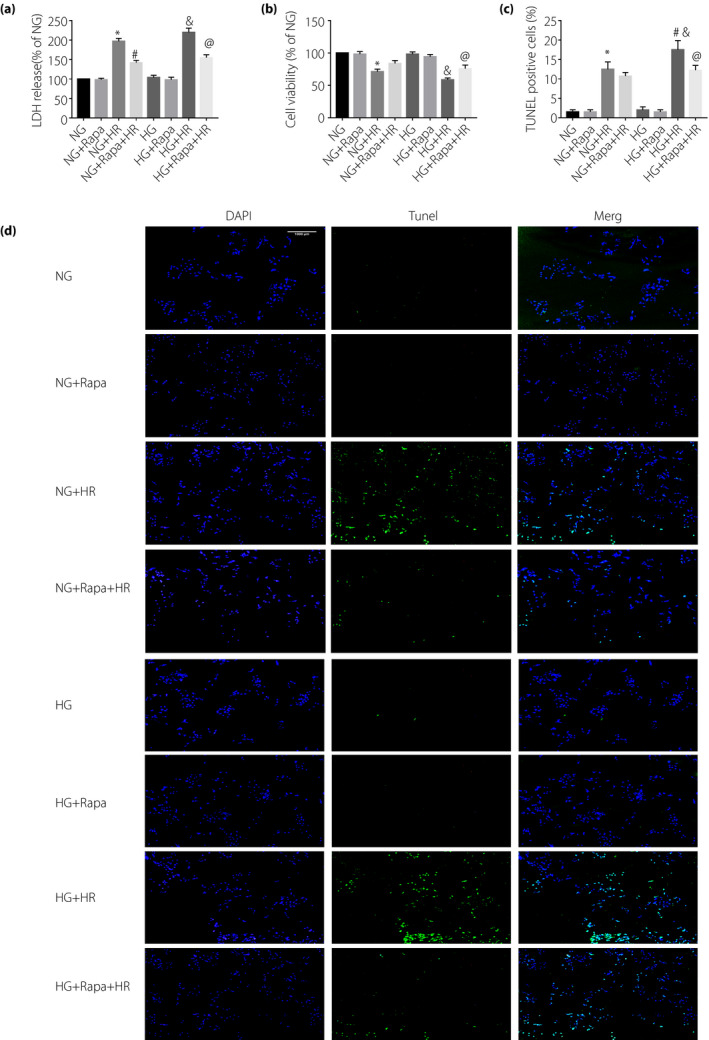
High glucose (HG, 33 mmol/L) can (a) increase lactate dehydrogenase (LDH) release, (b) decrease cell viability and (c,d) increase cell death, as evidenced by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)‐positive apoptotic cells, in H9C2 cells induced by hypoxia–reoxygenation (HR). Rapamycin (Rapa) can alleviate the damage of H9C2 cells induced by hypoxia and reoxygenation. Data are shown as the mean ± standard error of the mean. *P < 0.05 versus normal glucose (NG,5.5 mmol/L); & P < 0.05 versus HG; # P < 0.05 versus NG + HR; @ P < 0.05 versus HG + HR; n = 5/group.
Discussion
The present study showed that autophagy was significantly inhibited in the myocardium of 8‐week‐old diabetic rats, accompanied by NLRP3 inflammasome activation, including increased expression of NLRP3, ASC and caspase‐1 p20, and increased release of inflammatory factors IL‐1β and IL‐18. Myocardial I/RI was aggravated in diabetic rats (increased infarct size, increased release of LDH and CK‐MB). Administration of autophagy activator, rapamycin, could activate autophagy, inhibit the activation of NLRP3 inflammasome and finally reduce myocardial I/RI in diabetic rats. In vitro experiments also showed that high glucose would inhibit autophagy, activate NLRP3 inflammasome in H9C2 cardiomyocytes and aggravate HR injury. Rapamycin could reverse this effect. These results suggest that autophagy inhibition and NLRP3 inflammasome activation in diabetic myocardium might be responsible for increased sensitivity to myocardial ischemia in diabetes. Activation of autophagy and the subsequent inhibition of NLRP3 inflammasome could be an option for the treatment of diabetic myocardial I/RI.
Previous studies have confirmed the involvement of NLRP3 inflammasome activation in myocardial I/IR. For example, selective inhibition of NLRP3 inflammasome can reduce myocardial infarct size and maintain cardiac function in pigs24. Inhibition of NLRP3 inflammasome early in reperfusion (1 h) can alleviate myocardial ischemia‐reperfusion‐induced inflammation and infarct size in mice25. Compared with the wild type, NLRP3‐deficient mice showed significant improvement in cardiac function and reduced hypoxia injury during isolated ischemia‐reperfusion23. Interestingly, the present study found that NLRP3 inflammasome was activated with increased release of inflammatory factors IL‐1β and IL‐18 in the myocardium of diabetic rats. Myocardial I/RI in diabetic rats was greatly aggravated. In vitro experiments also showed that high glucose could activate NLRP3 inflammasome in H9C2 cardiomyocytes, meanwhile aggravating HR injury. These results suggested that NLRP3 inflammasome activation and the subsequent inflammatory response in diabetic myocardium might explain its increased sensitivity to myocardial I/RI. Furthermore, multiple lines of evidence support NLRP3 inflammasome activation to increase sensitivity to myocardial I/RI in diabetes, as shows by the findings that: (i) NLRP3 gene silencing therapy can reduce inflammation and pyroptosis, and improve cardiac function in diabetic myocardium26; and (ii) it has been reported that administration of anti‐oxidant NAC can reduce ROS production, inhibit NLRP3‐induced pyroptosis and finally alleviate myocardial I/RI in diabetes4. Consequently, these studies suggest that inhibition of NLRP3 inflammasome might be one option for the treatment of diabetic myocardial I/RI.
It should be noted that NLRP3 inflammasomes are mainly activated by ROS and K+ efflux10, 27. However, autophagy can help eliminate damaged organelles, misfolded proteins, stress products, such as ROS, and so on. Autophagy itself, under normal circumstances, is a kind of protective mechanism for the body17, 28. In pathological conditions, such as diabetes, I/RI or cardiac hypertrophy, autophagy is impaired (inhibited or excessive autophagy) 29, 30. Studies have reported that cardiac autophagy is inhibited in type 1 diabetes31. The present results showed that autophagy was inhibited in 8‐week‐old diabetic rats, which agreed with the former study. However, other studies reported that different types of diabetes would have different impairment of autophagy. Autophagy is inhibited in type 2 diabetes mellitus, whereas it is activated in type 1 diabetes mellitus32. The reasons for these inconsistencies are as follows: (i) different methods of autophagy detection lead to different interpretations of the results33; and (ii) the impairment of autophagy varies at different stages of diabetes. In the preliminary experiment, we also found that autophagy was inhibited with 24 h high glucose treatment, but overactivated if treated with high glucose for 72 h. This might be the reason why autophagy has different roles in diabetes. Of course, further research is still required on how autophagy changes in the pathological course of diabetic cardiomyopathy. Apart from that, autophagy also plays different roles in I/RI. It has been reported that autophagy activation during ischemia could reduce the injury, and that inhibition of excessive autophagy during reperfusion would also be protective for the myocardium34, 35. In the present study, autophagy was activated primarily before ischemia.
In vitro activation of autophagy can eliminate ROS produced by oxidative stress and inhibit NLRP3 inflammasome activation36, 37. It has been reported that PINK1‐Parkin‐mediated mitochondrial autophagy played a protective role in CI‐AKI by reducing NLRP3 inflammasome activation38. Autophagy, by inhibiting NLRP3 inflammasome activation, could alleviate intestinal I/RI and the inflammatory response20. The present study showed that autophagy in the myocardium of 8‐week‐old diabetic rats was significantly inhibited, accompanied by activation of NLRP3 inflammasome. The administration of autophagy activator, rapamycin, could inhibit such effect and help increase the tolerance to myocardial ischemia. Similar results were also obtained in in vitro experiments. These results suggest that the activation of autophagy could attenuate diabetic myocardial I/RI by inhibiting the activation of NLRP3 inflammasome. To the best of our knowledge, the present study is the first to report that re‐activation of autophagy can present the activation of NLRP3 inflammasome in the diabetic myocardium and subsequently attenuate myocardial I/RI in diabetes.
In conclusion, the present study showed that autophagy was inhibited in the myocardium of 8‐week‐old diabetic rats, with activated NLRP3 inflammasome and aggravated myocardial I/RI. Activation of autophagy can reduce the activation of NLRP3 inflammasome and therefore increase the tolerance to myocardial ischemia in diabetes. Autophagy activation and the following NLRP3 inflammasome inhibition might be a potential therapeutic strategy for the protection of diabetic myocardium.
DISCLOSURE
The authors declare no conflict of interest.
Acknowledgments
We gratefully acknowledge Shenzhen IVY‐Valued Biotechnology Co. Ltd. for language editing service. The study was supported by Guangdong Natural Science Foundation (2018A030313535, 2018A0303130297, Guangdong, China), NSFC grant (81670770) and in part by HMRF (05161826) of Hong Kong.
J Diabetes Investig 2020; 11: 1126–1136
REFERENCES
- 1. Rawshani A, Rawshani A, Franzen S, et al Risk Factors, Mortality, and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med 2018; 379: 633–644. [DOI] [PubMed] [Google Scholar]
- 2. Davidson SM, Ferdinandy P, Andreadou I, et al Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J Am Coll Cardiol 2019; 73: 89–99. [DOI] [PubMed] [Google Scholar]
- 3. Luo B, Huang F, Liu Y, et al NLRP3 Inflammasome as a Molecular Marker in Diabetic Cardiomyopathy. Front Physiol 2017; 8: 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Qiu Z, Lei S, Zhao B, et al NLRP3 Inflammasome Activation‐Mediated Pyroptosis Aggravates Myocardial Ischemia/Reperfusion Injury in Diabetic Rats. Oxid Med Cell Longev 2017; 2017: 9743280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol 2018; 15: 203–214. [DOI] [PubMed] [Google Scholar]
- 6. Yu SY, Dong B, Tang L, et al LncRNA MALAT1 sponges miR‐133 to promote NLRP3 inflammasome expression in ischemia‐reperfusion injured heart. Int J Cardiol 2018; 254: 50. [DOI] [PubMed] [Google Scholar]
- 7. Hughes MM, O'Neill L. Metabolic regulation of NLRP3. Immunol Rev 2018; 281: 88–98. [DOI] [PubMed] [Google Scholar]
- 8. Samir P, Kesavardhana S, Patmore DM, et al DDX3X acts as a live‐or‐die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 2019; 573: 590–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. An N, Gao Y, Si Z, et al Regulatory Mechanisms of the NLRP3 Inflammasome, a Novel Immune‐Inflammatory Marker in Cardiovascular Diseases. Front Immunol 2019; 10: 1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gross CJ, Mishra R, Schneider KS, et al K(+) Efflux‐Independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria. Immunity 2016; 45: 761–773. [DOI] [PubMed] [Google Scholar]
- 11. Zhao YG, Zhang H. Core autophagy genes and human diseases. Curr Opin Cell Biol 2019; 61: 117–125. [DOI] [PubMed] [Google Scholar]
- 12. Mizushima N. A brief history of autophagy from cell biology to physiology and disease. Nat Cell Biol 2018; 20: 521–527. [DOI] [PubMed] [Google Scholar]
- 13. Wang B, Yang Q, Sun YY, et al Resveratrol‐enhanced autophagic flux ameliorates myocardial oxidative stress injury in diabetic mice. J Cell Mol Med 2014; 18: 1599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Foglio E, Puddighinu G, Germani A, et al HMGB1 Inhibits Apoptosis Following MI and Induces Autophagy via mTORC1 Inhibition. J Cell Physiol 2017; 232: 1135–1143. [DOI] [PubMed] [Google Scholar]
- 15. Liu CY, Zhang YH, Li RB, et al LncRNA CAIF inhibits autophagy and attenuates myocardial infarction by blocking p53‐mediated myocardin transcription. Nat Commun 2018; 9: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xiao C, Wang K, Xu Y, et al Transplanted Mesenchymal Stem Cells Reduce Autophagic Flux in Infarcted Hearts via the Exosomal Transfer of miR‐125b. Circ Res 2018; 123: 564–578. [DOI] [PubMed] [Google Scholar]
- 17. Chen S, Wang Y, Zhang H, et al The Antioxidant MitoQ Protects Against CSE‐Induced Endothelial Barrier Injury and Inflammation by Inhibiting ROS and Autophagy in Human Umbilical Vein Endothelial Cells. Int J Biol Sci 2019; 15: 1440–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He B, Wang X, Zhu J, et al Autophagy protects murine macrophages from beta‐cypermethrin‐induced mitochondrial dysfunction and cytotoxicity via the reduction of oxidation stress. Environ Pollut 2019; 250: 416–425. [DOI] [PubMed] [Google Scholar]
- 19. Wang Y, Meng C, Zhang J, et al Inhibition of GSK‐3beta alleviates cerebral ischemia/reperfusion injury in rats by suppressing NLRP3 inflammasome activation through autophagy. Int Immunopharmacol 2019; 68: 234–241. [DOI] [PubMed] [Google Scholar]
- 20. Wang Z, Li Z, Feng D, et al Autophagy Induction Ameliorates Inflammatory Responses in Intestinal Ischemia‐Reperfusion Through Inhibiting NLRP3 Inflammasome Activation. Shock 2019; 52: 387–395. [DOI] [PubMed] [Google Scholar]
- 21. Wang S, Wang C, Yan F, et al N‐Acetylcysteine Attenuates Diabetic Myocardial Ischemia Reperfusion Injury through Inhibiting Excessive Autophagy. Mediators Inflamm 2017; 2017: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ge L, Cai Y, Ying F, et al miR‐181c‐5p Exacerbates Hypoxia/Reoxygenation‐Induced Cardiomyocyte Apoptosis via Targeting PTPN4. Oxid Med Cell Longev 2019; 2019: 1957920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sandanger O, Ranheim T, Vinge LE, et al The NLRP3 inflammasome is up‐regulated in cardiac fibroblasts and mediates myocardial ischaemia‐reperfusion injury. Cardiovasc Res 2013; 99: 164–74. [DOI] [PubMed] [Google Scholar]
- 24. van Hout GP, Bosch L, Ellenbroek GH, et al The selective NLRP3‐inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur Heart J 2017; 38: 828–836. [DOI] [PubMed] [Google Scholar]
- 25. Toldo S, Marchetti C, Mauro AG, et al Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia‐reperfusion in the mouse. Int J Cardiol 2016; 209: 215–20. [DOI] [PubMed] [Google Scholar]
- 26. Luo B, Li B, Wang W, et al NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS ONE 2014; 9: e104771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou R, Yazdi AS, Menu P, et al A role for mitochondria in NLRP3 inflammasome activation. Nature 2011; 469: 221–5. [DOI] [PubMed] [Google Scholar]
- 28. Wang S, Xia P, Rehm M, et al Autophagy and cell reprogramming. Cell Mol Life Sci 2015; 72: 1699–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang Y, Whaley‐Connell AT, Sowers JR, et al Autophagy as an emerging target in cardiorenal metabolic disease: From pathophysiology to management. Pharmacol Ther 2018; 191: 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nishida K, Otsu K. Autophagy during cardiac remodeling. J Mol Cell Cardiol 2016; 95: 11–8. [DOI] [PubMed] [Google Scholar]
- 31. Xu X, Kobayashi S, Chen K, et al Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem 2013; 288: 18077–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kanamori H, Takemura G, Goto K, et al Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy 2015; 11: 1146–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang Y, Liang B, Lau WB, et al Restoring diabetes‐induced autophagic flux arrest in ischemic/reperfused heart by ADIPOR (adiponectin receptor) activation involves both AMPK‐dependent and AMPK‐independent signaling. Autophagy 2017; 13: 1855–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu D, Zhang K, Hu P. The Role of Autophagy in Acute Myocardial Infarction. Front Pharmacol 2019; 10: 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ma S, Wang Y, Chen Y, et al The role of the autophagy in myocardial ischemia/reperfusion injury. Biochim Biophys Acta 2015; 1852: 271–6. [DOI] [PubMed] [Google Scholar]
- 36. Cosin‐Roger J, Simmen S, Melhem H, et al Hypoxia ameliorates intestinal inflammation through NLRP3/mTOR downregulation and autophagy activation. Nat Commun 2017; 8: 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Spalinger MR, Lang S, Gottier C, et al PTPN22 regulates NLRP3‐mediated IL1B secretion in an autophagy‐dependent manner. Autophagy 2017; 13: 1590–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lin Q, Li S, Jiang N, et al PINK1‐parkin pathway of mitophagy protects against contrast‐induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol 2019; 26: 101254. [DOI] [PMC free article] [PubMed] [Google Scholar]