

Abstract
Purpose:
Treatment failure from drug resistance is the primary reason for relapse in acute lymphoblastic leukemia. Improving outcomes by targeting mechanisms of drug resistance is a potential solution.
Experimental Design:
We report results investigating the epigenetic modulators decitabine and vorinostat with vincristine, dexamethasone, mitoxantrone and PEG-asparaginase for pediatric patients with relapsed or refractory B-cell ALL (B-ALL). Twenty-three patients, median age 12 years (range, 1 – 21) were treated on this trial.
Results:
The most common Grade 3–4 toxicities included hypokalemia (65%), anemia (78%), febrile neutropenia (57%), hypophosphatemia (43%), leukopenia (61%), hyperbilirubinemia (39%), thrombocytopenia (87%), neutropenia (91%) and hypocalcemia (39%). Three subjects experienced dose limiting toxicities (DLT) which included cholestasis, steatosis, hyperbilirubinemia (n=1); seizure, somnolence, delirium (n=1); and pneumonitis, hypoxia, hyperbilirubinemia (n=1). Infectious complications were common with 17/23 (74%) subjects experiencing Grade ≥3 infections including invasive fungal infections in 35% (8/23). Nine subjects (39%) achieved a complete response (CR + CR without platelet recovery + CR without neutrophil recovery) and 5 had stable disease (22%). Nine (39%) subjects were not evaluable for response, primarily due to treatment-related toxicities. Correlative pharmacodynamics demonstrated potent in vivo modulation of epigenetic marks, and modulation of biologic pathways associated with functional anti-leukemic effects.
Conclusion/Discussion:
Despite encouraging response rates and pharmacodynamics, the combination of decitabine and vorinostat on this intensive chemotherapy backbone was determined not feasible in B-ALL due to the high incidence of significant infectious toxicities. This study is registered at http://www.clinicaltrials.gov as NCT01483690.
Keywords: decitabine, vorinostat, relapse, acute lymphoblastic leukemia, epigenetic
Introduction
Acute lymphoblastic leukemia (ALL) remains the most common pediatric malignancy with 5-year survival rates currently >90%.(1) Despite this accomplishment, approximately 20% of children will relapse.(2) Successful treatment of children with relapsed ALL continues to be a challenge as the majority of patients die of their disease.(3–8) This is particularly true for children with refractory leukemia or in ≥2nd relapse, where remission rates are typically no greater than 45%.(9, 10) Regimens for relapsed disease often fail to eliminate the resistant clone With resistance to chemotherapy the primary reason for treatment failure. Thus, there is an urgent need to develop new strategies to overcome resistance.
Epigenetic alterations are prevalent in ALL and have been identified as foci of treatment resistance and relapse.(11–16) These alterations may involve DNA methylation or histone modification and are potentially reversible, prompting the design of drug therapies targeting epigenetic changes.(17, 18) Collectively, these drugs are referred to as “epigenetic modifying agents” and include histone deacetylase inhibitors (HDACi) and DNA methyltransferase inhibitors (DNMTi). Aberrant epigenetic regulation of a number of genes (e.g. silencing of tumor suppressor genes) have been associated with chemotherapy resistance.(11) Epigenetic changes can be reversible through de-methylation and/or inhibiting histone deacetylation, thereby re-inducing normal gene expression.(17–19) This suggests that incorporating epigenetic modifying agents into ALL therapy to reverse epigenetic silencing may improve clinical outcomes. We therefore developed a pilot study for children, adolescents and young adults (AYA) with refractory or relapsed ALL, combining two classes of epigenetic modifying agents with a multi-agent chemotherapy backbone (T2009–003/ NCT01483690, IND 113393). The primary objective of the trial was to evaluate the feasibility and characterize toxicities of decitabine and vorinostat when used in combination prior to and concurrently with chemotherapy in pediatric and AYA patients with relapsed/refractory ALL. Secondary objectives included exploring the pharmacodynamic effects of decitabine and vorinostat, treatment response, and the minimal residual disease (MRD) response rate.
Methods
The study was conducted in the Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) Consortium, comprised of 34 pediatric centers across North America and Australia. The study was reviewed and approved by the institutional review boards of all participating TACL centers and written informed consent was obtained from all patients, that the studies were conducted, in accordance with the Declaration of Helsinki. Individual and/or parental informed consent was obtained from all eligible subjects as per local and federal requirements. Eligible patients were ages 1 to 25 years with non-Down syndrome ALL (≥25% blasts in the bone marrow), with or without extramedullary disease. Patients must have had a ≥2nd relapse or refractory disease after first or greater relapse and a re-induction attempt or failing to go into remission from original diagnosis after 2 previous induction attempts. Patients who experienced relapse after allogeneic hematopoietic cell transplantation (HCT) were eligible provided they had no evidence of active graft-versus-host-disease and were at least 60 days post-HCT. A Karnofsky (patients >16 years of age) or Lansky (≤16 years of age) score >50% with adequate renal, hepatic, and cardiac function were required at study entry.
Treatment
Decitabine 15mg/m2/dose (intravenously) was administered on days 1–7 and 15–21 and vorinostat 180mg/m2/dose (orally, tablet or suspension) (max dose 400mg daily) was given on days 3–10 and 17–24. The chemotherapy regimen used in this study included block 1 of the UK ALLR3 re-induction regimen consisting of dexamethasone 20mg/m2/dose divided twice daily days 8–12 and 22–26; vincristine 1.5mg/m2/dose (max dose 2mg) days 10, 17, 24 and 31; mitoxantrone 10mg/m2/dose days 8 and 9 and PEG-asparaginase 2500 IU/m2/dose days 10 and 24.(20) Patients who developed an allergy to PEG-asparaginase on study or had a known allergy prior to the start of the study were eligible to substitute with Erwinia asparaginase (25,000 IU/m2 × 6 doses for each dose of PEG-asparaginase). Intrathecal therapy consisted of methotrexate and was dosed by age and central nervous system (CNS) status.
The study was amended after the first 5 patients enrolled due to significant infectious toxicities reported (4 of 5 patients with invasive fungal infections) and two patients experiencing a dose limiting toxicity (DLT). Post-amendment the treatment schema was modified to lower the dose of decitabine from 15mg/m2/dose to 10mg/m2/dose as well as the duration of decitabine from days 1–7 and 15–21 to 1–5 and 15–19 and vorinostat from days 3–10 and 17–24 to 2–7 and 16–21 (Figure 1).
Figure 1:
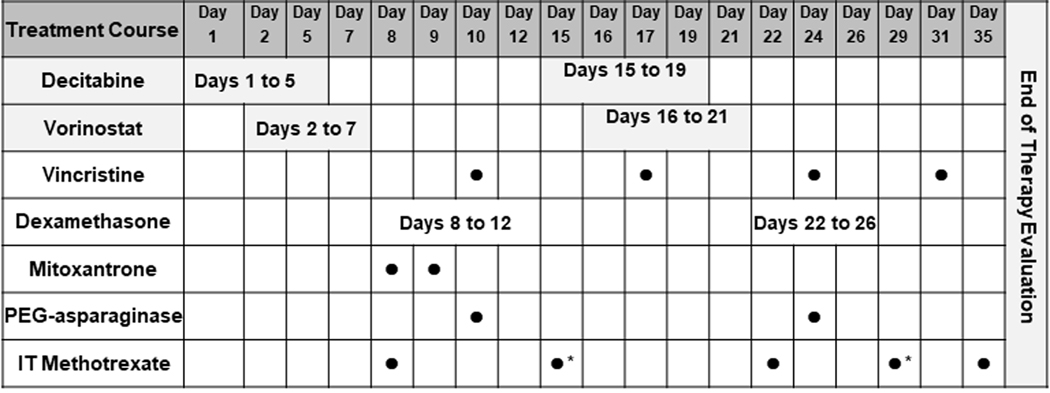
T2009–003 Treatment Schema
IT, intrathecal; *CNS positive patients only
Supportive Care
Anti-bacterial and anti-fungal prophylaxis was recommended during periods of neutropenia (<750/μL). Due to the invasive fungal infections (IFI) identified with this regimen in 4 of the first 5 patients enrolled, the study was amended to require prophylaxis with an anti-fungal agent and excluded patients with a history of a positive fungal culture within 30 days of study enrollment. Patients were strongly recommended to remain hospitalized throughout study therapy until evidence of adequate neutrophil recovery (ANC >500/μL) and were required to start prophylactic anti-fungal therapy on Day 1 of study, using either an echinocandin or amphotericin class of agent.
Toxicity Evaluation
Toxicity was graded according to the Common Terminology Criteria for Adverse Events (CTCAE) version 4.0. DLT was defined as any event that was at least possibly attributed to decitabine and/or vorinostat and was further defined into non-hematologic and hematologic DLT. Non-hematologic DLT was defined as any Grade 3 or 4 non-hematologic toxicity attributed to decitabine and/or vorinostat with the exception of nausea (Grade 3); vomiting (Grade 3); fatigue; fever/infection; elevation of transaminase, alkaline phosphatase or GGT that returned to Grade ≤1 or baseline prior to the end of the course; transient electrolyte abnormalities not associated with clinical sequelae; hypoalbuminemia; and/or anorexia. Hematologic DLT was defined as an absence of peripheral blood count recovery [absolute neutrophil count (ANC) >500/μL and platelet count >20,000/μL] within 6 weeks of starting the first dose of protocol therapy, in patients who achieved remission, as documented by marrow aplasia, not marrow infiltration/persistent disease.
Response Evaluation
A bone marrow evaluation was performed on day 35 of study to evaluate treatment response. A complete response (CR) was defined as attaining an M1 marrow (<5% blasts) with no evidence of circulating blasts or extramedullary disease in addition to recovery of peripheral blood counts (ANC >750/μL and platelet count >75,000/μL). Complete response without platelet recovery (CRp) was defined as attaining an M1 marrow with no evidence of circulating blasts or extramedullary disease in addition to recovery of ANC but insufficient recovery of platelets (<75,000/μL). Complete response with incomplete recovery (CRi) was defined as attaining an M1 marrow with no evidence of circulating blasts or extramedullary disease but insufficient recovery of ANC (<750/μL) with or without sufficient recovery of platelets. Partial response (PR) was defined as no evidence of circulating blasts and achievement of M2 marrow status (5–25% blasts) without new sites of extramedullary disease and with recovery of ANC. Stable disease (SD) was designated for patients who did not meet the criteria for PR, CR, CRp, or CRi. Progressive disease (PD) was defined as an increase of at least 25% in the absolute number of leukemia cells (circulating blasts or marrow), development of new sites of extramedullary disease or other laboratory or clinical evidence of PD. Induction death (ID) was defined as any patient who died after receiving protocol therapy prior to receiving subsequent post-induction therapy. Patients were defined as not evaluable (NE) if they did not satisfy the criterion for PD or ID and either did not have a bone marrow evaluation or had a hypocellular marrow. Centralized MRD testing was performed on bone marrow samples at the end of Induction (Day 35) using multi-parameter flow cytometry performed at the University of Washington Hematopathology Laboratory, Seattle, WA.
Correlative Studies
Sample acquisition and processing
Participation in the correlative studies was optional and required signed informed consent. For subjects who participated, peripheral blood and bone marrow were collected prior to the start of therapy (prior to decitabine and vorinostat) and on study day 8 (after one-week exposure to decitabine and vorinostat therapy). Paired pre- and post-treatment specimens were available for 11 patients (22 paired samples). DNA was isolated from all 11 patients and in 6 of these patients (12 paired samples), there was sufficient material to also isolate RNA. Additionally, in 7 of these patients (14 paired samples), there was sufficient material to perform flow cytometric evaluation for histone deacetylase inhibitor (HDACi) pharmacodynamics. See Supplemental Appendix 1 for methodology of methylation using whole genome bisulfite sequencing, gene expression using RNA-sequencing and flow cytometry evaluating HDAC pharmacodynamics.
Assessing methylation (whole genome bisulfite sequencing and analytics)
Pre- and post-decitabine/vorinostat treatment samples were bisulfite converted and whole genome bisulfite sequencing (WGBS) was performed, aiming for 31X whole-genome coverage on average per sample (~635 million single-end 151bp reads, ~96 billion sequenced bases, per sample) using Illumina HiSeq 4000. We used a custom WGBS analysis pipeline to trim the first 7bp of reads (TrimGalore), align to hg19 genome (Bismark/bowtie2), remove polymerase chain reaction (PCR) duplicates and repeats, and call methylation states at CpG sites. After adapter trimming and alignment, the average coverage per sample was 26.5X (~82.3 billion aligned bases per sample). The methylation level at each CpG site was calculated by counting the number of reads that are methylated at the CpG site (CG dinucleotides that remain CG after bisulfite conversion) and dividing by the total number of reads that cover the CpG site (number of unconverted CG->CGs plus converted CGs->TGs; expressed as percent methylation). Since CpG methylation levels can be locally correlated, we segmented 16 million CpGs into 1.3 million segments using HaarSeg/R. Change in methylation after treatment was calculated by subtracting the methylation level of a CpG or CpG segment after treatment from that of the same CpG or CpG segment before treatment.
Assessing gene expression (RNA-sequencing and analytics)
RNA-sequencing (RNA-seq) was performed on the RNA samples using Illumina HiSeq 4000 (2 × 126bp, 16 Gbp). We used RSEM, STAR and EBSeq software packages for RNA-seq alignment and differential gene expression calls between pre- and post-treatment samples. Genes and transcript annotation were used from GENCODE v19, where only protein-coding genes (N=22,522) were selected as input to RSEM. Three metrics were used from the RSEM and EBSeq gene-based quantitation and differential expression output: Transcripts per million (TPM), log2 posterior fold change, and posterior probability of differential expression (PPDE). The quintiles of expression are based on TPM, the most appropriate metric for comparing relative gene expression within a sample, namely, the expression of all genes within the sample sum to 1 million. The PPDE and log2 fold change produce differential expression values that are not related to differences in TPM, but instead are based on the differential expression model of the EBSeq tool for comparing two samples.(21)
Flow cytometric evaluation of HDAC pharmacodynamics
Flow cytometry was used to assess for known biologic effects of HDAC inhibition (acetylation of tubulin, and induction of gamma H2AX, a marker of DNA damage) using published methods.(22)
Statistical Methods
The primary endpoint of this pilot study was DLT. Any patient who experienced a DLT after receiving at least one dose of decitabine or vorinostat was evaluable. Patients who did not experience DLT had to receive a pre-determined amount of protocol therapy (e.g. >50%) to be evaluable. Patients not evaluable were replaced. Patients were considered evaluable for the secondary endpoint of treatment response if they completed all or part of protocol therapy, had the required end of therapy bone marrow evaluation. Patients who died as a result of toxicity after receiving all or part of protocol therapy prior to their end of therapy evaluation, or who were withdrawn from therapy due to toxicity or physician choice, were considered non-responders in computations of response rate. As serious infection was determined to be a common event in patients with relapsed/refractory ALL, infection did not contribute to DLT. If infection prevented the subject from receiving >50% of protocol therapy, or confounded non-infectious DLT assessment, the subject was replaced.
The study’s accrual goal was 16 evaluable patients. The study design was to first enroll 6 patients according to a 3+3 design. If no more than 1 DLT was observed in the first 6 patients, the study would continue to enroll up to 16 evaluable patients without dose adjustments provided that no more than 3 additional DLTs in total were observed. If ≥2 DLTs were observed within the first 6 patients enrolled, or if ≥4 DLTs were observed at any time during continued enrollment, the study would be suspended for possible dose reduction of decitabine and/or vorinostat. The rate of Grade 3 or 4 toxicity reported during reinduction treatment was computed for all patients as well as by excluding patients with pre-existing Grade 3 or 4 toxicity at study entry. The classical response rate to reinduction therapy for a population of high-risk patients with ≥2nd relapse or refractory ALL for this study was placed at 40%.(9) Nine of 16 responses (CR +CRp + CRi) were required to pursue the regimen further on the basis of response. With this criterion, there was 84% chance of rejecting the treatment if the response rate was <35% and a greater than 86% chance of accepting the treatment if the response rate was >60%. Kaplan-Meier estimate was used for overall survival. The analysis of MRD response among patients with a CR was descriptive where <0.01% MRD was considered negative on study.
Results
Patient Characteristics
The study opened to accrual in December 2011. Twenty-three patients with ≥2nd relapse or refractory B-ALL enrolled with a median age of 12.0 years (range, 1.6 – 21.4; Table 1 and Supplemental Table 1). Six patients (26%) had refractory disease and 11 (48%) had a prior HCT.
Table 1.
Patient Characteristics (n=23)
| Dose Level | |||
|---|---|---|---|
| 15 mg/m2/dose | 10 mg/m2/dose | Total | |
| # of patients | 5 (100%) | 18 (100%) | 23 (100%) |
| Sex | |||
| Male | 3 (60%) | 14 (78%) | 17 (74%) |
| Female | 2 (40%) | 4 (22%) | 6 (26%) |
| Lineage | |||
| B-Cell ALL | 5 (100%) | 18 (100%) | 23 (100%) |
|
Age median (range) |
12.5 (4.9–14.5) |
12.0 (1.6–21.4) |
12.0 (1.6–21.4) |
| Race | |||
| White | 5 (100%) | 10 (56%) | 15 (65%) |
| Black or African American | - | 2 (11%) | 2 (9%) |
| Asian | - | 1 (6%) | 1 (4%) |
| Not Reported | - | 2 (11%) | 2 (9%) |
| Unknown | - | 3 (17%) | 3 (13%) |
| Ethnicity | |||
| Hispanic or Latino | 3 (60%) | 9 (50%) | 12 (52%) |
| Non-Hispanic | 2 (40%) | 9 (50%) | 11 (48%) |
| CNS Status | |||
| CNS1 | 4 (80%) | 14 (78%) | 18 (78%) |
| CNS2 | 1 (20%) | 2 (11%) | 3 (13%) |
| CNS3 | - | 2 (11%) | 2 (9%) |
| Prior HCT | |||
| Yes | 3 (60%) | 8 (44%) | 11 (48%) |
| No | 2 (40%) | 10 (56%) | 12 (52%) |
| Relapse # at enrollment | |||
| 2nd Relapse | 2 (40%) | 13 (72%) | 15 (65%) |
| 3rd Relapse | 1 (20%) | 1 (6%) | 2 (9%) |
| Refractory | 2 (40%) | 4 (22%) | 6 (26%) |
| Final DLT Attribution | |||
| DLT | 2 (40%) | 1 (6%) | 3 (13%) |
| No DLT | 2 (40%) | 12 (67%) | 14 (61%) |
| Not Evaluable | 1 (20%) | 5 (28%) | 6 (26%) |
| Response | |||
| CR | 0 (0%) | 1 (6%) | 1 (4%) |
| CRp | 1 (20%) | 3 (17%) | 4 (17%) |
| CRi | 1 (20%) | 3 (17%) | 4 (17%) |
| ORR (CR + CRp + CRi) | 2 (40%) | 7 (39%) | 9 (39%) |
| SD | 1 (20%) | 4 (22%) | 5 (22%) |
| Not Evaluable | 2 (40%) | 7 (39%) | 9 (39%) |
CNS1= no evidence of leukemia in cerebral spinal fluid (CSF); CNS2= <5 WBC in CSF with blasts present; CNS3= ≥5 WBC in CSF with blasts present. ORR, Overall response rate
Toxicity
Of the 23 patients enrolled on study, 17 were evaluable for DLT. Six patients did not receive a majority of protocol therapy due to infection (n=3), clinical deterioration (n=1), receipt of emergent non-protocol radiation therapy (n=1), and physician decision (n=1), and were not evaluable for DLT. The study was suspended after the first 5 subjects experienced significant infectious toxicities including 2 DLTs. The infections were predominantly IFI (n=4) identified as non-albicans Candida. The 2 DLTs included cholestasis (Grade 3), steatosis (Grade 3) and hyperbilirubinemia (Grade 4) in 1 subject and seizure (Grade 4), delirium (Grade 3) and somnolence (Grade 4) in another; both occurring in the setting of IFI.
The study re-started post-amendment enrolling two subjects with reduced exposure to decitabine and vorinostat. The study was again suspended after another non-albicans Candida IFI occurred in a subject who received anti-fungal prophylaxis with fluconazole instead of an echinocandin/amphotericin agent. The study was amended further to make anti-fungal prophylaxis with an echinocandin or amphotericin agent mandatory in all subjects. The study re-opened and enrolled an additional 16 subjects in which 3 more IFI were reported (See Table 2 for full description of infectious toxicities).
Table 2.
Infectious Toxicities (Grade ≥3)
| Patient ID | Decitabine Dose Level 1 | Toxicity Name | CTCAE v4.0 Grade | Organism |
|---|---|---|---|---|
| 138 | 15mg/m2 | Sepsis | 4 | Candida Lusitaniae |
| 141 | 15mg/m2 | Infections -other | 3 | Coagulase negative Staphylococus |
| Infections -other | 3 | Enterococcus Faecalis | ||
| Infections -other | 5 | Candida Kruseii | ||
| 146 | 15mg/m2 | Infections -other | 3 | Candida Kruseii |
| 147 | 15mg/m2 | Infections -other | 3 | Enterococcus |
| 149 | 15mg/m2 | Infections -other | 3 | Candida Kruseii |
| Patient ID | Decitabine Dose Level 2 | Toxicity Name | CTCAE v4.0 Grade | Organism |
| 182 | 10mg/m2 | Lip infection | 3 | Herpes Simplex Virus |
| 191 | 10mg/m2 | Infections -other | 3 | Klebsiella pneumonae |
| Enterocolitis | 3 | Clostridium difficile | ||
| sepsis | 4 | Candida guillermondi | ||
| 250 | 10mg/m2 | Enterocolitis | 3 | Adenovirus |
| Soft tissue infection | 3 | Pseudomonas aeruginosa | ||
| sepsis | 4 | Pseudomonas aeruginosa | ||
| 261 | 10mg/m2 | Lung infection | 3 | No organism isolated |
| Soft tissue infection | 4 | No organism isolated | ||
| Sepsis | 5 | Soft tissue infection | ||
| 268 | 10mg/m2 | Lung infection | 3 | No organism isolated |
| 269 | 10mg/m2 | Catheter related | 3 | Clostridium tertium |
| 284 | 10mg/m2 | Lung infection | 3 | Epstein-bar Virus |
| Sepsis | 4 | Rothia mucilaginosa; Streptococcus viridans | ||
| 285 | 10mg/m2 | Infections -other | 4 | Candida parapsilosis |
| Sepsis | 4 | Pseudomonas aeruginosa | ||
| 292 | 10mg/m2 | Catheter related | 3 | Staphylococcus capitis |
| Lung infection | 5 | No organism isolated | ||
| 299 | 10mg/m2 | Infections -other | 4 | Pseudomonas aeruginosa |
| Infections -other | 5 | Necrotizing fasciitis | ||
| 300 | 10mg/m2 | Infections -other | 5 | Bipolaris spicifera |
| 301 | 10mg/m2 | Lung infection | 5 | Fusarium |
Three subjects (13%) experienced a DLT on study, 2 of whom are detailed above, and the third subject developed pneumonitis (Grade 3), hypoxia (Grade 3) and hyperbilirubinemia (Grade 3). Grade ≥3 non-hematologic and hematologic toxicities observed are reported in Supplemental Tables 2 and 3.
Based on the number of serious infectious toxicities observed on this trial and despite the addition of enhanced supportive care measures, the trial was closed to further accrual in August 2015. The study was determined by the study committee and DSMC to not be feasible when using decitabine and vorinostat in combination prior to and concurrently with block 1 of UK ALLR3 chemotherapy.
Response
Fourteen subjects completed protocol therapy and were evaluable for response. A CR was achieved in 1 subject, with 8 subjects achieving either a CRp (n=4) or CRi (n=4). Five subjects were reported as SD. Nine subjects were withdrawn from study treatment, due predominantly to infection-related toxicities, and were not fully evaluable for response. In an intent-to-treat analysis, the overall response rate (CR + CRp + CRi) was 39.1% (9/23), however when considering patients who completed protocol therapy and were evaluable for response, the ORR was 64.3% (9/14). MRD testing was performed on 7/9 subjects who achieved a CR (CR + CRp + CRi). Two patients achieved MRD negativity. The median level of MRD detected in the 7 patients was 0.087% (range, 0.00% to 1.6%).
Overall survival for the 23 subjects at 6 months was 43.5% (95% CI, 23.3 to 61.1) and 27.2% (95% CI, 10.6 to 47.0) at 1-year. Death was reported in 18/23 subjects at a median time of 10 weeks from Day 1 of the study (range, 3–138 weeks). Cause of death was most often attributed to refractory leukemia (n=8) followed by organ failure (n= 5), infection (n=3) and HCT-related toxicity (n=2).
Correlative Studies
Methylation pharmacodynamics (N=11 patients, 22 paired samples)
To most accurately assess the pharmacodynamic effect of decitabine/vorinostat on DNA methylation, we looked specifically at CpG sites that were fully methylated in the pre-treatment sample and were covered to a depth of at least 10X in both the pre- and post-treatment samples. This included a mean of 2.2 million CpGs per case (range 1.4 million to 3.1 million). We found clear evidence of a pharmacodynamic effect, with an average decrease in methylation of 12% (range 6–25%; Supplemental Table 4). As expected, lower pre-treatment methylation levels were associated with lower post-treatment decreases in methylation level (Supplemental Table 5). We examined the differential impact of the pharmacodynamic effect according to CpG topography (i.e., CpG islands, shores, shelves) and gene topography (i.e., gene promoters, gene bodies). Averaged over all 11 cases, for fully methylated CpGs at baseline, the methylation decreases were: genome-wide −13%, CpG islands −9%, CpG shores −11%, CpG shelves −11%, within gene promoters (+- 1kb from TSS) −13%, and within gene bodies (TSS to TES) −12%. Note that CpGs within CpG islands tend to be demethylated, which is the likely explanation for the smaller methylation decreases seen in CpG islands.
Of the 11 patients, 2 were treated prior to the amendment with a dose of decitabine of 15 mg/m2 and 9 were treated after the amendment with a dose of 10 mg/m2. Despite the small sample sizes, there was a significant difference between the two groups in terms of magnitude of decrease in methylation, with the higher dose resulting in a greater decrease (mean 21%, range 16–25% vs. mean 11%, range 6–20%, p=0.03). Of the 11 patients, 10 had an evaluable bone marrow morphologic response, defined as having bone marrow blast percentages by morphology available for both pre-treatment and end-of-course bone marrow evaluation (Supplemental Table 4). There was a trend towards a positive correlation between decrease in methylation and reduction in bone marrow blast percentage with treatment (Pearson correlation coefficient 0.62, p=0.053) (Supplemental Figure 1).
Correlation between promoter methylation and gene expression (N=6 patients, 12 paired samples)
In the 6 cases with paired RNA-seq data, we were able to correlate promoter methylation and gene expression. For each sample, transcript expression levels were grouped into quintiles. For each quintile, the average methylation of the CpGs mapping to the promoter or gene body for all the genes in the quintile was plotted as shown in Figure 2 and Supplemental Figure 2. In each of the pre-treatment samples (Figure 2A and Supplemental Figure 2), there was a striking intra-sample correlation between methylation and gene expression, such that the highly expressed genes (Q1, Q2) had relatively low methylation (<20%) near transcription start sites (TSSs), and relatively high methylation in gene bodies. Unexpressed genes (Q4, Q5) had methylated promoters and gene bodies. In the post-treatment samples (Figure 2B and Supplemental Figure 2), there was a global decrease in methylation across the promoters and gene bodies, and the pattern of strong correlation between gene expression and methylation was maintained.
Figure 2: Methylation correlates strongly with gene expression, both before and after epigenetic treatment.

The expression of 22,522 genes was averaged between pre- and post- treatment, ranked from highest to lowest, and split into quintiles in order of highest (Q1) to lowest (Q5). (A) The highly expressed genes (Q1, Q2) had low methylation (<20%) at TSSs and expressed genes (Q1-Q3) had increased methylation in gene bodies. Unexpressed genes (Q4, Q5) had methylated promoters and gene bodies. (B) Epigenetic treatment decreased methylation levels across all genomic regions. Shown is patient #300. The remaining 5 patients are shown in Supplemental Figure 2.
To directly assess whether there was an association between changes in promoter CpG methylation and corresponding gene expression, we looked specifically at promoters with pre-treatment methylation levels of at least 40% and CpG density of at least 50 CpG’s near the TSS (+/− 1 kilobase), based on our observation of bimodal distribution patterns for methylation level and CpG density, with the modes separated at 40% and 50, respectively. Combining all 6 cases, we found that there was a correlation between the change in methylation (average decrease 4%, range −1% to 9%) and change in expression (average increase 34%, range −1% to 128%, Pearson correlation coefficient of r=0.13,95% confidence interval 0.10 – 0.16, p<2.2e-16) (Supplementary Table 3, Figure 3). Detailed plots of each individual case are shown in Supplemental Figure 3.
Figure 3: Epigenetic treatment-induced decreases in gene promoter CpG methylation correlate with increases in RNA transcript expression.

We identified gene promoters with at least 50 CpGs within 1 kilobase upstream or downstream of the transcription start site where the CpGs were at least 40% methylated at baseline (pre-treatment). We then calculated the change in methylation after treatment and correlated with the treatment-induced change in transcript expression of the associated gene. For all 6 cases with both methylation and RNA-seq data, the plot demonstrates the highly significant correlation between the change in methylation of promoter CpGs (x-axis) and log2 fold change in gene transcript expression after decitabine and vorinostat treatment (y-axis). Each dot (n=4749) represents one gene/promoter pair and includes composite data from all 6 cases. The color of each dot indicates the baseline (pre-treatment) methylation level of the gene promoter. Promoters with higher levels of baseline methylation (red/orange) show a greater degree of demethylation and increased expression after treatment relative to those with lower levels of baseline methylation (purple/blue). Plots for the contributions from each individual case are shown in Supplemental Figure 3.
Defining biologic pathways affected by epigenetic therapy
We created a rank list of differentially methylated promoters and differentially expressed transcripts. We combined the two rank lists to create an “epigenetically activated gene set” using a composite ranking of each gene’s differential promoter methylation and transcript expression across all 6 paired cases, with the genes at the top of the ranked lists representing the set of genes that were most affected by decitabine and vorinostat treatment. Gene set enrichment analysis (GSEA v3.0 / MSigDB, collection c2) was then performed on this rank list. Normalized enrichment scores reported from GSEA were collected for all gene sets, then combined into a matrix and sorted to rank the gene sets with highest enrichment scores across the cases. Using a false discovery rate (FDR)<25% and p<0.01, 75/4169 gene sets were overrepresented in the epigenetically activated ranked gene list. The top 30 gene sets are shown in (Figure 4 and Supplemental Table 6). Among the top 30 gene sets, there were 3 putative “functional groups” represented by more than one related gene set, including: 1) targets of the polycomb repressive complex 2 (PRC2); 2) targets of the tumor suppressors TP53 and TP63; and 3) extracellular matrix (ECM) associated proteins and secreted factors. Together, these 3 groups accounted for 19 of the top 30 gene sets.
Figure 4: Epigenetically activated pathways include PRC2 and TP53 targets.

Gene set enrichment analysis on 6 cases with paired WGBS and RNA-seq data showed that genes that had highest promoter hypomethylation and gene upregulation after epigenetic treatment were enriched in sets of genes that are: 1) targets of PRC2 components and chromatin remodeling by H3K27ME3 modifications; and 2) targets of TP53 binding. (A) The top 30 gene sets ranked by combined normalized enrichment score (NES, GSEA v3.0) across the 6 cases. The drug-induced activation of PRC2/H3K27ME3 targets suggests that epigenetic treatment of relapsed ALL may induce differentiation, and the activation of TP53 targets suggests that epigenetic treatment can reactivate tumor suppressor genes. (B, C) Enrichment plots for PRC2 and TP53 gene sets, respectively.
Assessing effects of epigenetic therapy on immune target expression
We hypothesized that for critical targets of immunotherapy in ALL, such as CD19, modulation of promoter methylation and chromatin architecture with epigenetic therapy may enhance expression. We therefore examined the differential methylation of the CD19 promoter and the differential expression of CD19 transcripts in the 6 cases. Combining all 6 cases, there was a 10% decrease in methylation of the CD19 promoter from an average of 25% pre-treatment (range 2% to 47%) to 15% (range 1% to 37%) post-treatment, and this was associated with an average increase in CD19 transcript expression of 74% (range −43% to +511%) (Supplemental Table 4). Interestingly, the case with the highest level of CpG methylation in the CD19 promoter at baseline (case #209, 47%) demonstrated a striking decrease in methylation (post-treatment CpG methylation 5%) and a 5.1-fold increase in CD19 transcript expression (Supplemental Figure 4). Indeed, across the 6 samples, there was a clear correlation between baseline CD19 promoter methylation and treatment-induced change in CD19 transcript expression (Pearson correlation coefficient r=0.8, p=0.058) (Supplemental Figure 5).
Histone deacetylase inhibitor pharmacodynamics (N=7 patients, 14 paired samples)
We used flow cytometry to assess for evidence of protein expression changes known to be induced by HDAC inhibition, including tubulin acetylation and induction of gamma H2AX, a marker of DNA damage. Overall, comparing pre-treatment to post-treatment samples. the proportion of leukemic (CD10+, CD19+) cells expressing acetylated tubulin increased by an average of 49.7% (range −11% to +92%, paired t-test p=0.013), and the proportion of leukemic (CD10+, CD19+) cells expressing gamma H2AX increased by an average of 31.3% (range −14% to +82%, paired t-test p=0.03) (Supplemental Table 4).
Discussion
Although there have been clinical studies reporting the combination of a HDACi and a DNMTi in patients with leukemia,(23, 24) this pilot study was the first trial in ALL using two classes of epigenetic modifying agents in decitabine and vorinostat to attempt to synergistically target epigenetic alterations of leukemic blasts prior to and concurrently with intensive chemotherapy. Despite the significant toxicities subjects experienced on this study, the majority of those able to receive their protocol therapy had encouraging response rates. Two subjects achieved MRD negativity (<0.01%), both of whom had relapsed after a prior HCT with one of those subjects being refractory to all re-induction attempts prior to enrolling on this study. However, 9 patients were not evaluable for treatment response, primarily due to significant infection-related toxicities. Thus, it is difficult to judge just how effective this therapeutic approach is, although the overall response rate of 64.3% for the 14 evaluable patients remains encouraging for multiply relapsed/refractory ALL. This trial highlights the challenges of investigating novel agents on an intensive reinduction chemotherapy backbone in ALL where it is difficult to separate the toxicity attributed to new agents with the chemotherapy regimen.
The chemotherapy backbone for this study was block 1 of the UKR3 regimen (vincristine, mitoxantrone, dexamethasone and PEG-asparaginase). This re-induction block is part of a relapse regimen (UK ALLR3) reporting survival rates at 3-years of 69% in children with 1st relapse.(20) However, this report by Parker and colleagues did not highlight the toxicity profile of the regimen. As our study was the first trial to combine investigational agents with UK ALLR3, the expected treatment related toxicities were not known. After this study completed, TACL Consortium sites reviewed their collective experience using UK ALLR3 alone to see if similar toxicities were observed.(25) In 59 pediatric patients with ≥1st relapse ALL treated at TACL centers with UK ALLR3 block I, the incidence of Grade ≥3 infection was 92% (54/59) with bacterial infections being most common (68%) followed by fungal (17%) and viral (15%) pathogens. There was no observed difference in the rate of infection between patients in 1st versus ≥2nd relapse and overall the authors associated the UK ALLR3 regimen with a very high risk of life-threatening infections.
That TACL report (28) is consistent with our findings. However, there appears to be a higher incidence of IFI in our study compared to the TACL report. This difference is possibly due to our trial including more heavily pre-treated patients (all having ≥2nd relapse or refractory disease) whereas the TACL report primarily had patients in 1st relapse (72.9%). The only other reported experience combining decitabine with vorinostat followed by re-induction chemotherapy for patients with relapsed/refractory ALL used a different chemotherapy platform with prednisone, vincristine, doxorubicin and PEG-asparaginase. That study did not report similar toxicities.(26) It is possible that the combination of mitoxantrone and dexamethasone may contribute to greater myelosuppression and immunosuppression, therefore placing subjects at higher risk for the infectious toxicities we observed, particularly with regard to IFI.
Our correlative pharmacodynamic studies clearly demonstrate biological activity of decitabine and vorinostat in the subset of patients for whom samples were available. This is the first clinical trial of a demethylating agent and/or HDAC inhibitor in which WGBS and RNA-seq were used to assess epigenetic pharmacodynamics, so the potency of these effects relative to other studies is difficult to assess. Previously published clinical trials have assessed methylation pharmacodynamics using various techniques to measure changes in methylation of LINE-1 sequences, densely methylated repetitive elements that account for about 17% of the human genome. These studies have shown an average reduction of 3–10%.(27–33) In this study, we were able to directly assess the methylation of 16 million CpGs across the genome with average depth of coverage of 25–30X at single base pair resolution. We demonstrated dose-dependent decreases in methylation of 12% on average, which compares favorably with previously published studies in terms of potency. In addition, the CpGs we measured were in non-repetitive, potentially functional regions of the genome rather than in repetitive LINE-1 regions, and so it is reasonable to consider this a more relevant measure of the functional and biologic effect of epigenetic therapy. While the small sample size for the methylation measures and the high rate of subject inevaluability for clinical response limited our ability to assess the impact of methylation on the primary efficacy endpoint of clinical remission rates, we did see an association between a higher magnitude of demethylation and greater reduction in the percentage of leukemic blasts in the bone marrow. This supports our hypothesis that epigenetic therapy may sensitize relapsed ALL cells to cytotoxic chemotherapy.
However, the real power of integrated WGBS and RNA-seq as a pharmacodynamic measure of epigenetic therapy lies in the ability to map nearly every non-repetitive CpG in the genome to its position relative to the regulatory and coding regions of every gene, and then to correlate CpG methylation with gene expression. This is the first clinical trial to harness this power, and while the small sample size was a significant limitation, several intriguing findings emerged. First, when looking simultaneously at all annotated genes in the genome, the expression level of genes correlates strikingly with the level of methylation in two regions, and the direction of this correlation is opposite for the two regions. Near the transcription start site, highly expressed genes are hypomethylated, while within the gene bodies, and especially in the first 1–2 exons, highly expressed genes are hypermethylated. The effect of decitabine and vorinostat does not change this pattern. Rather, it reduces the level of methylation across the entire gene while maintaining the intrasample relationship between methylation and relative expression.
Further, we were able to look across the entire genome and demonstrate a correlation between the magnitude of treatment-induced decrease in promoter CpG methylation with the magnitude of increase in expression of the corresponding gene. While previous studies have looked at specific candidate genes and demonstrated a correlation using targeted assays such as methylation-specific PCR and q-RT-PCR, this is the first study with the scope and resolution to assess this in every gene simultaneously. It is important to note that the magnitude of the effect of demethylation on gene expression when looking across the entire genome is modest (average correlation coefficient of 0.10). This is to be expected, since the control of gene expression is multifactorial - changes in promoter methylation do not exclusively control gene expression. If a promoter is hypomethylated or demethylated, this simply allows the subsequent control of gene expression to occur via transcription factors and other regulatory elements.
Additionally, we report the initial steps to explore these data for biologic insights into the mechanisms of epigenetic therapy. We created a putative “epigenetically activated gene set” using a composite ranking of demethylation and upregulation and used gene set enrichment analysis to discover biologic pathways most impacted by the epigenetic treatment. As a result, a clear pattern emerged from the 6 patients for whom we had paired methylation/expression data, implicating targets of the polycomb repressive complex 2 (PRC2) and the tumor suppressor TP53, and ECM-associated/secreted proteins. In general, targets of PRC2 are repressed in the undifferentiated, stem cell state, and target repression is mediated via epigenetic silencing associated with H3K27 methyltransferase EZH2.(34, 35) Therefore, it is reasonable to hypothesize that epigenetic reversal of this repression may confer anti-leukemic activity by overcoming the block in differentiation that characterizes acute leukemia. TP53 silencing is ubiquitous in cancer, most commonly through mutations in the DNA sequence, and is thought to be a fundamental driver of the phenotypic hallmarks of cancer, such as resistance to apoptosis. Reactivation of TP53 function by various mechanisms has been shown to mediate anti-tumor activity by blocking cell cycle progression and promoting apoptosis.(36) Our demonstration in this study of epigenetic activation of TP53 targets by decitabine and vorinostat suggests that epigenetic therapy may, in part, confer anti-leukemic activity by reversing the oncogenic effects of TP53 silencing. The epigenetic activation of expression of ECM-associated and secreted proteins is interesting and suggests that epigenetic therapy may modulate interactions between leukemic cells and the bone marrow microenvironment.
Given the recent emergence of highly efficacious CD19-targeted immunotherapies, including bispecific T-cell engaging antibodies and chimeric antigen receptor T-cells, and the recognition that degree of expression of immunotherapy targets may be associated with clinical response,(37, 38) we examined the possibility that epigenetic therapy may modulate CD19 expression. While the sample size is too small for firm conclusions, we did see evidence of upregulation of CD19 associated with a decrease in methylation of the CD19 promoter with decitabine and vorinostat treatment, an effect that was more pronounced in samples with higher levels of CD19 promoter methylation prior to treatment. While this is a preliminary finding, it does suggest that epigenetic treatment may be able to sensitize leukemic cells to CD19-targeted immunotherapies, particularly in cases where the CD19 promoter is highly methylated. Of course, a thorough understanding of the effect of epigenetic modulation on both endogenous T-cells and infused CAR T-cells will be critical when considering this use.
Finally, selective HDAC inhibitor-induced pharmacodynamic effects (acetylation of tubulin protein and induction of gamma-H2AX) were demonstrable in 6 of the 7 patients with paired samples available for flow cytometric assessment. Thus, integrated WGBS/RNA-seq/flow cytometry represents a powerful tool for pharmacodynamic evaluation of epigenetic therapy. Incorporating similar assays into larger clinical trials, particularly trials with lower rates of serious adverse events and higher rates of sample submission, could provide more conclusive mechanistic insights.
In summary, the toxicity of decitabine and vorinostat when given with UK ALLR3 reinduction was not acceptable in pediatric patients with relapse/refractory B-ALL but did demonstrate potent pharmacodynamic modulation of biological pathways associated with anti-leukemic effects. If future leukemia trials are to include epigenetic reprogramming agents, they may need to be used either not concurrently with chemotherapy and/or in combination with chemotherapy regimens with less intense myelosuppression than the UK ALLR3 regimen. A study such as the Children’s Oncology Group infant ALL trial AALL15P1 has successfully incorporated the DNMTi 5-azacytidine into INTEFANT therapy as an example (NCT02828358). Combining epigenetic therapy with CD19-targeted immunotherapy may represent another strategy for decreasing the intensity of myelosuppressive chemotherapy required to induce clinical responses in this population. Future studies are needed to determine if epigenetic modifying therapies can be successfully combined with multi-agent chemotherapy and other therapies for children with multiply relapsed leukemia.
Supplementary Material
Translational Relevance.
This pilot study was the first trial in ALL using two classes of epigenetic modifying agents in decitabine and vorinostat to attempt to synergistically target epigenetic alterations of leukemic blasts prior to and concurrently with intensive chemotherapy. Although the toxicity of decitabine and vorinostat when given with UK ALLR3 re-induction was not acceptable in pediatric patients with relapse/refractory B-ALL, they demonstrated potent pharmacodynamic modulation of biological pathways associated with anti-leukemic effects. Thus, future studies are needed to determine if epigenetic modifying therapies can be successfully combined with multi-agent chemotherapy for children with multiply relapsed leukemia, within the context of a different chemotherapy backbone.
Acknowledgments:
This trial was supported by grants from the NCI R21CA161688–01, research supported by Stand Up to Cancer-American Association for Cancer Research Team Translational Cancer Research Great, Grant number SU2C-AACR-DT0109, Rally Foundation for Childhood Cancer Research, Alex’s Lemonade Stand Foundation, Children’s Cancer Research Fund and the Kids Cancer Alliance. This work was also supported in part by NCI award P30CA014089.
Footnotes
Conflict-of-interest disclosure: M.J.B. has received honoraria from Shire Pharmaceuticals, Jazz Pharmaceuticals and Amgen. L.G. has received consulting fees from Amgen, Bristol-Myers Squibb, Celgene, Novartis and Genentech/Roche. T.W.L. has consulted for Novartis, Loxo Oncology, Bayer, and Eli Lilly. J.E.O has received honoraria from Shire Pharmaceuticals. S.G.D. has consulted for Loxo Oncology and has received travel expenses from Loxo Oncology and Roche. R.A.G. has received honoraria from Novartis. A.S.W. has received research funding from MedImmune, Kite Pharma, Institut de Recherches Internationales Servier (Servier), and Spectrum Pharmaceuticals and has served on an advisory committee for Servier.
References
- 1.Hunger SP, Loh ML, Whitlock JA, Winick NJ, Carroll WL, Devidas M, et al. Children’s Oncology Group’s 2013 blueprint for research: acute lymphoblastic leukemia. Pediatric blood & cancer. 2013;60(6):957–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Einsiedel HG, von Stackelberg A, Hartmann R, Fengler R, Schrappe M, Janka-Schaub G, et al. Long-term outcome in children with relapsed ALL by risk-stratified salvage therapy: results of trial acute lymphoblastic leukemia-relapse study of the Berlin-Frankfurt-Munster Group 87. J Clin Oncol. 2005;23(31):7942–50. [DOI] [PubMed] [Google Scholar]
- 3.Tallen G, Ratei R, Mann G, Kaspers G, Niggli F, Karachunsky A, et al. Long-term outcome in children with relapsed acute lymphoblastic leukemia after time-point and site-of-relapse stratification and intensified short-course multidrug chemotherapy: results of trial ALL-REZ BFM 90. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28(14):2339–47. [DOI] [PubMed] [Google Scholar]
- 4.Feig SA, Harris RE, Sather HN. Bone marrow transplantation versus chemotherapy for maintenance of second remission of childhood acute lymphoblastic leukemia: a study of the Children’s Cancer Group (CCG-1884). Medical and pediatric oncology. 1997;29(6):534–40. [DOI] [PubMed] [Google Scholar]
- 5.Harrison G, Richards S, Lawson S, Darbyshire P, Pinkerton R, Stevens R, et al. Comparison of allogeneic transplant versus chemotherapy for relapsed childhood acute lymphoblastic leukaemia in the MRC UKALL R1 trial. MRC Childhood Leukaemia Working Party. Ann Oncol. 2000;11(8):999–1006. [DOI] [PubMed] [Google Scholar]
- 6.Lawson SE, Harrison G, Richards S, Oakhill A, Stevens R, Eden OB, et al. The UK experience in treating relapsed childhood acute lymphoblastic leukaemia: a report on the medical research council UKALLR1 study. British journal of haematology. 2000;108(3):531–43. [DOI] [PubMed] [Google Scholar]
- 7.Chessells JM, Veys P, Kempski H, Henley P, Leiper A, Webb D, et al. Long-term follow-up of relapsed childhood acute lymphoblastic leukaemia. Br J Haematol. 2003;123(3):396–405. [DOI] [PubMed] [Google Scholar]
- 8.Barrett AJ, Horowitz MM, Pollock BH, Zhang MJ, Bortin MM, Buchanan GR, et al. Bone marrow transplants from HLA-identical siblings as compared with chemotherapy for children with acute lymphoblastic leukemia in a second remission. The New England journal of medicine. 1994;331(19):1253–8. [DOI] [PubMed] [Google Scholar]
- 9.Ko RH, Ji L, Barnette P, Bostrom B, Hutchinson R, Raetz E, et al. Outcome of Patients Treated for Relapsed or Refractory Acute Lymphoblastic Leukemia (ALL)—A Therapeutic Advances in Childhood Leukemia (TACL) Consortium Study. Journal of Clinical Oncology. 2009;In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun W, Malvar J, Sposto R, Verma A, Wilkes JJ, Dennis R, et al. Outcome of children with multiply relapsed B-cell acute lymphoblastic leukemia: a therapeutic advances in childhood leukemia & lymphoma study. Leukemia. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Manero G, Bueso-Ramos C, Daniel J, Williamson J, Kantarjian HM, Issa JP. DNA methylation patterns at relapse in adult acute lymphocytic leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2002;8(6):1897–903. [PubMed] [Google Scholar]
- 12.Garcia-Manero G, Daniel J, Smith TL, Kornblau SM, Lee MS, Kantarjian HM, et al. DNA methylation of multiple promoter-associated CpG islands in adult acute lymphocytic leukemia. Clin Cancer Res. 2002;8(7):2217–24. [PubMed] [Google Scholar]
- 13.Shen L, Kondo Y, Issa JP, Garcia-Manero G. Lack of p21(CIP1) DNA methylation in acute lymphocytic leukemia. Blood. 2002;100(9):3432–3; author reply 3–4. [DOI] [PubMed] [Google Scholar]
- 14.Schafer E, Irizarry R, Negi S, McIntyre E, Small D, Figueroa ME, et al. Promoter hypermethylation in MLL-r infant acute lymphoblastic leukemia: biology and therapeutic targeting. Blood. 2010;115(23):4798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agirre X, Vilas-Zornoza A, Jimenez-Velasco A, Martin-Subero JI, Cordeu L, Garate L, et al. Epigenetic silencing of the tumor suppressor microRNA Hsa-miR-124a regulates CDK6 expression and confers a poor prognosis in acute lymphoblastic leukemia. Cancer Res. 2009;69(10):4443–53. [DOI] [PubMed] [Google Scholar]
- 16.Roman-Gomez J, Agirre X, Jimenez-Velasco A, Arqueros V, Vilas-Zornoza A, Rodriguez-Otero P, et al. Epigenetic regulation of microRNAs in acute lymphoblastic leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27(8):1316–22. [DOI] [PubMed] [Google Scholar]
- 17.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qin T, Youssef EM, Jelinek J, Chen R, Yang AS, Garcia-Manero G, et al. Effect of cytarabine and decitabine in combination in human leukemic cell lines. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13(14):4225–32. [DOI] [PubMed] [Google Scholar]
- 19.Yang H, Hoshino K, Sanchez-Gonzalez B, Kantarjian H, Garcia-Manero G. Antileukemia activity of the combination of 5-aza-2’-deoxycytidine with valproic acid. Leukemia research. 2005;29(7):739–48. [DOI] [PubMed] [Google Scholar]
- 20.Parker C, Waters R, Leighton C, Hancock J, Sutton R, Moorman AV, et al. Effect of mitoxantrone on outcome of children with first relapse of acute lymphoblastic leukaemia (ALL R3): an open-label randomised trial. Lancet. 2010;376(9757):2009–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leng N, Dawson JA, Thomson JA, Ruotti V, Rissman AI, Smits BM, et al. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics. 2013;29(8):1035–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chung EJ, Lee S, Sausville EA, Ryan Q, Karp JE, Gojo I, et al. Histone deacetylase inhibitor pharmacodynamic analysis by multiparameter flow cytometry. Ann Clin Lab Sci. 2005;35(4):397–406. [PubMed] [Google Scholar]
- 23.Burke MJ, Lamba JK, Pounds S, Cao X, Ghodke-Puranik Y, Lindgren BR, et al. A therapeutic trial of decitabine and vorinostat in combination with chemotherapy for relapsed/refractory acute lymphoblastic leukemia. American journal of hematology. 2014;89(9):889–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kirschbaum M, Gojo I, Goldberg SL, Bredeson C, Kujawski LA, Yang A, et al. A phase 1 clinical trial of vorinostat in combination with decitabine in patients with acute myeloid leukaemia or myelodysplastic syndrome. British journal of haematology. 2014;167(2):185–93. [DOI] [PubMed] [Google Scholar]
- 25.Sun W, Smith AM, Etan O, Sposto R, Wilkes JJ, Gardner RA, et al. The UK ALLR3 Chemotherapy Regimen for Relapsed/Refractory Acute Lymphoblastic Leukemia of Childhood: A Multi-Institutional Retrospective Study of Treatment Related Adverse Events. Blood. 2014;124(21):3647-. [Google Scholar]
- 26.Burke MJ, Lamba J, Weigel B, Bachanova V, Verneris MR, Miller JS. A Phase II Trial of Decitabine and Vorinostat in Combination with Chemotherapy for Relapsed/Refractory Acute Lymphoblastic Leukemia. ASH Annual Meeting Abstracts. 2012;120(21):4307-. [Google Scholar]
- 27.Braiteh F, Soriano AO, Garcia-Manero G, Hong D, Johnson MM, Silva Lde P, et al. Phase I study of epigenetic modulation with 5-azacytidine and valproic acid in patients with advanced cancers. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14(19):6296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sonpavde G, Aparicio AM, Zhan F, North B, Delaune R, Garbo LE, et al. Azacitidine favorably modulates PSA kinetics correlating with plasma DNA LINE-1 hypomethylation in men with chemonaive castration-resistant prostate cancer. Urol Oncol. 2011;29(6):682–9. [DOI] [PubMed] [Google Scholar]
- 29.Fang F, Balch C, Schilder J, Breen T, Zhang S, Shen C, et al. A phase 1 and pharmacodynamic study of decitabine in combination with carboplatin in patients with recurrent, platinum-resistant, epithelial ovarian cancer. Cancer. 2010;116(17):4043–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu C, Goggin TK, Su XY, Taverna P, Oganesian A, Lowder JN, et al. Simultaneous Modeling of Biomarker and Toxicity Response Predicted Optimal Regimen of Guadecitabine (SGI-110) in Myeloid Malignancies. CPT Pharmacometrics Syst Pharmacol. 2017;6(10):712–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gong Y, Zhang Z. Alternative pathway approach for automating analysis and validation of cell perturbation networks and design of perturbation experiments. Ann N Y Acad Sci. 2007;1115:267–85. [DOI] [PubMed] [Google Scholar]
- 32.Glasspool RM, Brown R, Gore ME, Rustin GJ, McNeish IA, Wilson RH, et al. A randomised, phase II trial of the DNA-hypomethylating agent 5-aza-2’-deoxycytidine (decitabine) in combination with carboplatin vs carboplatin alone in patients with recurrent, partially platinum-sensitive ovarian cancer. British journal of cancer. 2014;110(8):1923–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang AS, Doshi KD, Choi SW, Mason JB, Mannari RK, Gharybian V, et al. DNA methylation changes after 5-aza-2’-deoxycytidine therapy in patients with leukemia. Cancer research. 2006;66(10):5495–503. [DOI] [PubMed] [Google Scholar]
- 34.Laugesen A, Hojfeldt JW, Helin K. Role of the Polycomb Repressive Complex 2 (PRC2) in Transcriptional Regulation and Cancer. Cold Spring Harb Perspect Med. 2016;6(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nature reviews Cancer. 2006;6(11):846–56. [DOI] [PubMed] [Google Scholar]
- 36.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137(3):413–31. [DOI] [PubMed] [Google Scholar]
- 37.Salter AI, Pont MJ, Riddell SR. Chimeric antigen receptor-modified T cells: CD19 and the road beyond. Blood. 2018;131(24):2621–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aldoss I, Song J, Stiller T, Nguyen T, Palmer J, O’Donnell M, et al. Correlates of resistance and relapse during blinatumomab therapy for relapsed/refractory acute lymphoblastic leukemia. American journal of hematology. 2017;92(9):858–65. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.