

Abstract
Purpose of Review
The goal of this review is to evaluate recent advances in understanding the pivotal roles of Cullin-3 (CUL3) in blood pressure regulation with a focus on its actions in the kidney and blood vessels.
Recent Findings
Cul3-based ubiquitin ligase regulates renal electrolyte transport, vascular tone, and redox homeostasis by facilitating the normal turnover of 1) with-no-lysine kinases in the distal nephron, 2) RhoA and phosphodiesterase 5 in the vascular smooth muscle, and 3) nuclear factor-E2-related factor 2 in antioxidant responses. CUL3 mutations identified in familial hyperkalemic hypertension (FHHt) yield a mutant protein lacking exon 9 (CUL3Δ9) which displays dual gain and loss of function. CUL3Δ9 acts in a dominant manner to impair CUL3-mediated substrate ubiquitylation and degradation. The consequent accumulation of substrates and over-activation of downstream signaling causes FHHt through increased sodium reabsorption, enhanced vasoconstriction, and decreased vasodilation.
Summary
CUL3 ubiquitin ligase maintains normal cardiovascular and renal physiology through post-translational modification of key substrates which regulate blood pressure. Interference with CUL3 disturbs these key downstream pathways. Further understanding the spatial and temporal specificity of how CUL3 functions in these pathways is necessary to identify novel therapeutic targets for hypertension.
Keywords: Cullin3, ubiquitylation, with-no-lysine kinases, RhoA, phosphodiesterase 5, blood pressure
1. Introduction
Cullin-3 (CUL3) is a scaffold subunit of the CUL3-RING (really interesting new gene)-E3 ubiquitin Ligase (CRL3) complex. CUL3 bridges the interaction between the RING protein RBX1 and substrate adaptors which deliver targets for ubiquitylation and proteasomal degradation. This post-translational modification is an important mechanism that regulates protein turnover and is essential for normal cell biology and organ function. Ubiquitylation and proteasomal degradation is an important component in cell cycle [1], circadian rhythm [2], immune cell development [3], vascular tone [4–7], renal sodium transport [8–11], and redox homeostasis[12], among many others. Mounting genetic and physiological evidence support a critical role of CUL3 in controlling arterial blood pressure (BP).
It is no longer controversial that CUL3 mutations cause familial hyperkalemic hypertension (FHHt, a rare genetic disease also known as Pseudohypoaldosteronism or Gordon Syndrome) through both renal and vascular mechanisms [13••]. However, CUL3 is ubiquitously expressed, and the physiological significance of CUL3 in other tissues and organs and the consequences of these dominant mutations are largely unknown. The purpose of this review is to highlight the mechanisms by which CUL3 and its adaptor proteins regulate the ubiquitylation and turnover of specific targets which play a role in kidney and vascular to regulate BP.
1.1. Molecular Structure and Organization of the CRL3 Complex
In the CRL3 complex, CUL3 serves as a molecular scaffold or bridge which supports both substrate binding on one terminus and E3 ubiquitin ligase activity on the other (Figure 1). The C-terminus of CUL3 interacts with an E3 ubiquitin ligase that attaches ubiquitin moieties to the lysine residues on target proteins through isopeptidic bonds [14]. The N-terminus of CUL3 interacts with the BTB (Broad Complex, Tramtrack, Bric-a-Brac) domain of substrate adaptors, the best known of which are the BTB-BACK-Kelch domain proteins, including the KLHL (Kelch-like) family and KEAP1 (Kelch-like ECH-associated protein 1). These adaptor proteins recognize their substrates through a β-propeller motif in the Kelch domain to deliver a plethora of substrates to the CRL3 catalytic core [15, 16]. The exact structure of CRL3 assembly is not fully understood, but crystallography has revealed that CUL3-based ubiquitin ligase complexes exist as homodimers [14]. Interestingly, the dimerization interface has been reported to be formed between the BTB domains of the substrate adaptors, not CUL3 which constitutes the periphery of the dimer complex [16].
Figure 1. The Structure and Function of CUL3 Complexes.
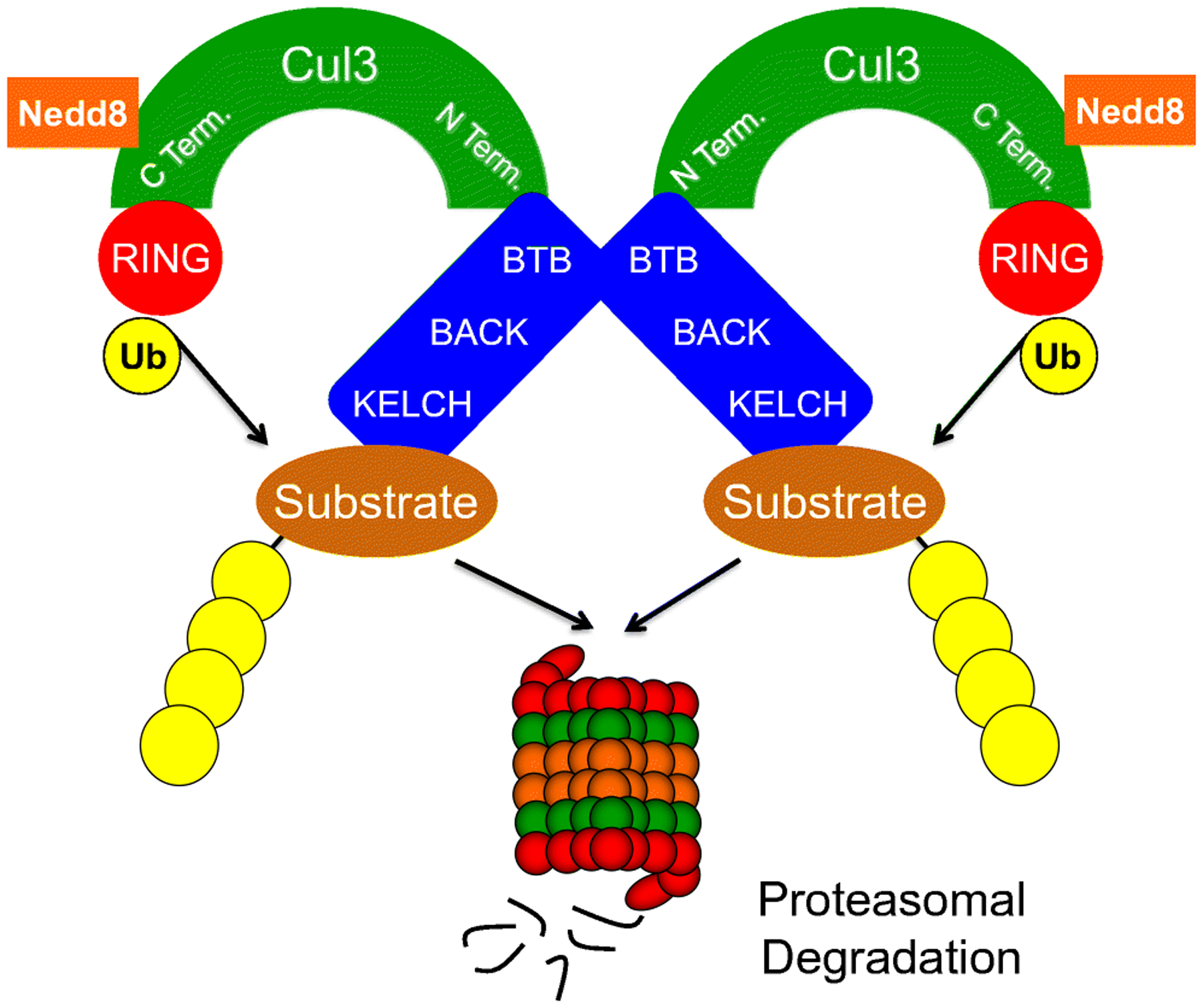
The C-terminus of CUL3 binds the E3 ubiquitin ligase to form the catalytic core while its N-terminus interacts with adaptor proteins delivering substrates for ubiquitylation. Neddylation of CUL3 near the C-terminus (covalent adduction of an 8.6 kDa Nedd8 molecule) is required for its activation. The BTB domain of substrate adaptors interacts with CUL3 N-terminus and supports the homodimerization of CUL3 complexes. The KELCH domain of substrate adaptors recruits target proteins, which are often poly-ubiquitylated and subsequently degraded in the proteasome.
The interactions among CUL3, substrate adaptors and its substrates are highly complex. Quantitative proteomics reveal that CUL3 can potentially pair with hundreds of different substrate adaptors [17]. Given that some adaptors are known to recruit more than one substrate adds a layer of complexity to the molecular organization and target spectrum of CRL3. For example, CUL3 utilizes KLHL12 (Kelch-like 12) to recruit two different substrates, Dishevelled in the Wnt-β catenin pathway in Xenopus and zebrafish embryos and the D4 dopamine receptor in human embryonic kidney (HEK) 293 cells [18, 19]. Similarly, CUL3-KLHL15 (Kelch-like 15) complex ubiquitylates both the CtIP protein in DNA repair and the B’β subunit of protein phosphatase 2A in the brain [20, 21]. Indeed, the full range of substrates delivered by BTB-domain containing substrates to the CRL3 complex remains undefined. The versatility of substrate adaptors implies that CRL3-mediated protein ubiquitylation may be temporally and spatially specific.
1.2. Mechanisms Regulating CRL3 Activity
How different CUL3-complexes are activated is not fully understood. Cullins are cycled between active and inactive states, and this dynamic cycling is essential for efficient ubiquitylation activity of the CRL complex [22]. Neddylation, which is catalyzed by CULLIN-RBX1 itself and a Nedd8-E3 ligase, covalently adducts a ubiquitin-like molecule Nedd8 to a conserved C-terminal lysine residue [22, 23]. This modification is required for CUL3 activation because 1) it alters the structural flexibility of the CUL3 scaffold, allowing the C-terminal CUL3-RBX1 to interact and engage the E3 ubiquitin ligase [24]; and 2) it causes re-orientation of CUL3-RBX1 subdomains and eliminates a binding site for CAND1, a Cullin regulator that selectively binds to unneddylated Cullins to facilitate the exchange of substrate adaptors [25]. While neddylation regulates Cullin activation, deneddylation, the removal of Nedd8 by COP9 signalosome (CSN) is also required for ubiquitylation activity [22]. Cullin activation can be pharmacologically inhibited by blocking neddylation with a small molecule MLN4924 but it should be noted that MLN4924 inhibits all Cullins and other enzyme activities which require Neddylation by the Nedd8-E3 ligase.
CRL3 complexes are regulated by phosphorylation and dephosphorylation. Phosphorylation of adaptor proteins positively or negatively modulates substrate ubiquitylation by altering the binding affinity among CRL3 components [26–28]. In addition, some CUL3 complexes require Ca2+-sensitive co-adaptors for substrate binding and translate transient rises in intracellular Ca2+ concentration into persistent substrate ubiquitylation and consequent physiological processes [29]. Thus, as CRL3 complex regulates protein turnover and relevant biological actions, its own activity is regulated by key intracellular processes such as neddylation, phosphorylation, dephosphorylation, and calcium mobilization.
1.3. Disease-Causing Mutations in CUL3
A role for CUL3 in BP regulation was first revealed when mutations in CUL3 were identified in a severe form of FHHt. Whole exome sequencing of FHHt patients from 41 unrelated families revealed multiple CUL3 mutations located in the splice donor and acceptor sites surrounding exon 9. The mutations were different but they all caused impaired splicing of exon 9 (termed exon skipping) resulting in an in-frame deletion of amino acids 403–459 in a mutated CUL3 protein named CUL3Δ9. Due to altered structural flexibility and molecular binding, CUL3Δ9 exhibits severe impairment in its ability to ubiquitylate targets. FHHt due to mutations in CUL3 is an autosomal dominant disease. At least 5 mechanisms have been reported to contribute to its dominant negative or aberrant activity: 1) CUL3Δ9 supports decreased ubiquitylation activity towards normal substrates [9, 30, 31]; 2) CUL3Δ9 promotes degradation of substrate adaptors through enhanced binding [9, 10, 31]; 3) CUL3Δ9 auto-ubiquitinates and degrades itself [10, 32••]; 4) CUL3Δ9 inhibits wild-type CUL3 (CUL3WT) through the formation of inhibitory and unstable CRL3 heterodimers [31]; and 5) CUL3Δ9 hampers interaction with Cullin regulators CSN and CAND1 [10, 33•].
2. Role of CUL3 in Renal Electrolyte Transport
2.1. The CUL3-KLHL3-WNK pathway
The hypertension and hyperkalemia features of FHHt are reversed by thiazide diuretics, implicating a role of increased sodium reabsorption and enhanced NCC activity in the distal convoluted tubule (DCT) where CUL3 and KLHL3 are both expressed [8]. In the DCT, CUL3-KLHL3-RBX1 complex recruits and ubiquitylates WNK (with-no-lysine) kinases, promoting their degradation through the proteasome (Figure 2). WNK kinases regulate the phosphorylation and activation of the thiazide-sensitive Na+-Cl−-Cotransporter (NCC) through a signaling cascade involving SPAK (SPS-related proline/alanine kinase) and OSR1 (oxidative stress-responsive kinase) [34]. Mutations CUL3, KLHL3 and WNK cause FHHt through a common mechanism involving increased abundance of WNK kinases, excessive activation of SPAK/OSR1, and enhanced phosphorylation of NCC. Increasing NCC activity alone is sufficient to reduce K+ secretion by downstream nephron segments [35], but WNKs also suppress the renal outer medullary K+ channel ROMK (Kir1.1; encoded by KCNJ1) by promoting its internalization from the cell surface [36, 37]. Thus, by controlling the normal turnover of WNK kinases, CUL3-KLHL3 ubiquitin ligase complex regulates the balance between renal Na+/Cl− reabsorption and K+ secretion.
Figure 2. Tissue-specific Actions of CUL3 and CUL3Δ9.
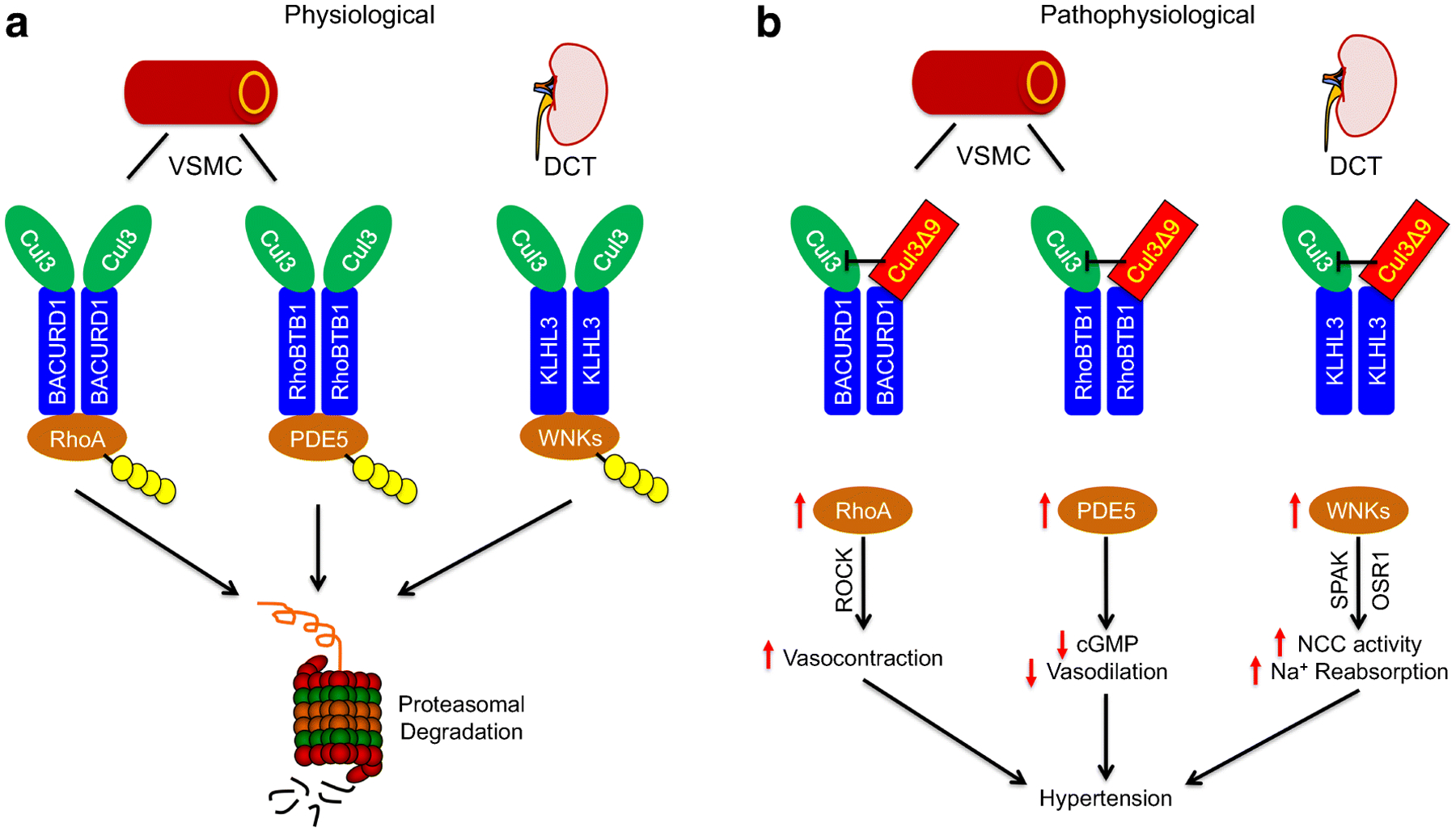
A) CUL3 employs different adaptor proteins to support the physiological turnover of key molecules in different tissues. In the vascular smooth muscle cells (VSMC), CUL3-BACURD1 ubiquitylates RhoA and CUL3-RhoBTB1 ubiquitylates PDE5. In the distal convoluted tubule (DCT), CUL3-KLHL3 ubiquitylates WNK1 and WNK4. Ubiquitylated target proteins are degraded in the proteasome. B) CUL3Δ9 interferes with CUL3WT and impairs substrate ubiquitylation and degradation leading to accumulation of target proteins. In the VSMC, increased RhoA/Rho kinase (ROCK) signaling augments vasocontraction, while enhanced PDE5 decreases cGMP bioavailability and vasodilation. Likewise, increased WNKs in the DCT promote NCC activation and Na+ reabsorption through the SPAK/OSR1 cascade. These pathophysiological mechanisms collectively cause the hypertension phenotype.
Population studies find that more than 60% of pedigrees of previously unexplained FHHt carry disease-causing variants in CUL3 or KLHL3, supporting a causal role of these mutations in this monogenetic form of hypertension [38]. However, although mutations in both genes cause the same disease, subjects with mutations in CUL3 display a more severe phenotype as evidenced by earlier onset and the degree of hypertension and electrolyte abnormalities [8]. Boyden et al first showed that mutations in both CUL3 and KLHL3 cause FHHt in humans, suggesting that the loss of CUL3-KLHL3 activity may be causal [8]. While KLHL3 mutations are either dominant or recessive, disease-causing CUL3 mutations are all dominant and predominantly de novo, resulting in skipping of exon 9 and the production of CUL3Δ9 mutant protein.
The structural rigidity of CUL3 is important for protein-protein interactions within the CRL3 complex. The deletion of 57 amino acids in the region between the C- and N-termini likely increases the flexibility of CUL3 backbone, which alters the relative orientation and position of CRL3 components and the overall catalytic activity [10]. The precise mechanism by which CUL3Δ9 impairs the ubiquitylation of WNK kinases is controversial, but multiple models have been postulated each supported by experimental evidence.
CUL3Δ9 itself cannot support WNK4 degradation:
Wakabayashi et al. initially showed that when expressed in cells, CUL3Δ9 results in increased WNK4 abundance, suggesting that this mutation caused FHHt as a result of loss of function in CUL3-mediated ubiquitylation and degradation of WNK4 [39]. Indeed, in the absence of CUL3WT, CUL3Δ9 protein itself cannot degrade WNK4 even with KLHL3 expressed at normal levels [30]. Conversely, when CUL3WT was present, WNK4 ubiquitylation and degradation was restored. These observations indicate that deletion of exon 9 results in defective ubiquitin ligase activity and suggests that CUL3Δ9 mutations cause FHHt phenotypes through a loss of function in WNK degradation.
CUL3Δ9 degrades substrate adaptor KLHL3:
Despite impaired ability to degrade its normal substrate, CUL3Δ9 causes striking increases in KLHL3 ubiquitylation and promotes KLHL3 degradation [9]. Because the deletion of 57 residues (AA403–459) occurs in the elongated region outside of the C- and N-terminus, CUL3Δ9 mutant protein retains the ability to bind RBX and KLHL3 [10]. This allows CUL3Δ9 to form CRL complexes but the structural alterations result in increased ubiquitylation of its substrate adaptor, in this case KLHL3. However, the enhanced degradation of KLHL3 is not completely proteasomal-dependent, as autophagic degradation of KLHL3 is also increased by this CUL3 mutant [30]. Because KLHL3 is required for WNK kinase ubiquitylation, CUL3Δ9 may cause FHHt through a gain of function by depleting the substrate adaptor KLHL3 [9, 40]. Importantly, the DCT may be exquisitely sensitive to the effects of CUL3Δ9 since KLHL3 is expressed at very high levels in this segment [9], and in a mouse model of FHHt, CUL3Δ9 did not alter expression of several other CUL3 adaptors in the kidney [40].
CUL3Δ9 exhibits enhanced auto-ubiquitylation and is dominant negative:
CUL3Δ9 also exhibits markedly increased auto-ubiquitylation than CUL3WT [10]. This is because the structural alterations in CUL3Δ9 exposes lysine residues inaccessible to CUL3WT. Auto-ubiquitylation can lead to self-destruction of CUL3Δ9 and what was thought to be apparent haploinsufficiency of CUL3WT [10]. To determine whether the FHHt phenotypes are primarily driven by CUL3 haploinsufficiency, Ferdaus et al. studied Cul3 heterozygous mice (CUL3-Het) and Cul3 heterozygous mice also expressing CUL3Δ9 in the renal epithelium (CUL3-Het/Δ9) [32••]. Consistent with auto-ubiquitylation, CUL3Δ9 protein was undetectable in CUL3-Het/Δ9 mice. However, while both models preserved 50% of endogenous CUL3WT, only CUL3-Het/Δ9 mice exhibited increased activation of WNK4/NCC, hyperkalemia and hypertension. These results reject the haploinsufficiency hypothesis and support the concept that the dominant effects of CUL3Δ9 are required in the pathogenesis of FHHt.
CUL3Δ9 is hyper-neddylated and dissociates from CUL3 regulators:
CUL3Δ9 is also the target of increased Nedd8-ligase activity and is unable to interact with the CSN deneddylase. Schumacher et al showed that while CUL3WT is mono-neddylated, CUL3Δ9 is modified by up to three Nedd8 molecules (8.6 kDa) [10]. The poly-neddylation of CUL3Δ9 decreases its mobility on western blot resulting in multiple CUL3Δ9 bands [10]. When neddylation is blocked by MLN4924, CUL3Δ9 remains neddylated for a longer duration than CUL3WT, consistent with decreased rates of deneddylation. Thus, the hyper-neddylation of CUL3Δ9 may result from both increased neddylation and decreased de-neddylation [10, 9]. A consequence of this is that it impairs the normal cycling between neddylated and de-neddylated states that is necessary to maintain CRL3 ubiquitylation activity. In support of this, genetic deletion of the CSN catalytic subunit Jab1 in the nephron results in increased CUL3 neddylation, decreased KLHL3 and activation of the WNK4-NCC pathway [41••]. Similar effects may be achieved by pharmacological inhibition of CSN [33•]. These phenotypes recapitulate the dominant effects of CUL3Δ9 and confirm the contribution of deficient CSN binding to CUL3Δ9 in FHHt pathogenesis. Of note, structural alterations in CUL3Δ9 also prevents the interaction with the substrate-adaptor exchange factor CAND1, another important regulator of CRL activity [10]. Thus, disruption of CUL3 regulatory mechanisms also contributes to the impairment of CRL3 activity.
Collectively, previous studies have provided compelling evidence supporting that CUL3Δ9 acts in a dominant manner to cause enhanced sodium reabsorption and electrolyte abnormalities through dual gain and loss of function.
While the rare disease FHHt revealed a role for CUL3-KLHL3 dysregulation in hypertension, more recent data suggest modification of the complex may be important for regulating NCC physiologically. For instance, phosphorylation at serine 433 of the adaptor protein KLHL3 (Kelch-like 3) disrupts its substrate interaction with with-no-lysine kinase 4 (WNK4) [26]. Angiotensin II induces phosphorylation at KLHL3-S433 through protein kinase C in the distal convoluted tubule, leading to decreased WNK4 ubiquitylation, enhanced WNK4 levels, increased NCC activity and impaired K+ secretion. Conversely, increases in extracellular K+ induce calcineurin-mediated dephosphorylation at KLHL3-S433. This promotes WNK4 ubiquitylation and degradation via CUL3-KLHL3 complex, resulting in decreased levels of WNK4 protein, NCC activity and sodium reabsorption [27].
2.2. Additional CUL3 mechanisms in the kidney
The pathogenesis of hyperkalemia and hypertension in FHHt is complex and involves multifaceted actions of CRL3. For example, FHHt-causing CUL3 mutations also increase the degradation of KLHL2, a homolog of KLHL3 capable of facilitating WNK ubiquitylation [42]. Moreover, CUL3-KLHL3 also regulates the turnover of claudin-8 in the collecting duct [43]. In transfected cells, dominant KLHL3 mutations impairs the ubiquitylation and degradation of claudin 8 resulting in accumulation of claudin 8. Because claudin 8 facilitates paracellular chloride reabsorption (also known as the chloride shunt), an increase in claudin 8 may decrease chloride concentration in the lumen which diminishes the lumen negative potential necessary to drive K+ secretion. In keeping with this, enhanced paracellular chloride transport has been proposed as an alternative mechanism of hyperkalemia in FHHt [44], although thiazidereversibility does not support this.
Intriguingly, kidney-specific deletion of Cul3 gene in mice results in salt-dependent hypotension, not hypertension, despite increased levels of WNK kinases, NCC protein and NCC phosphorylation [9]. The hypotension is associated with marked renal dysfunction, including depletion of sodium potassium cotransporter 2 (NKCC2) and aquaporin 2, hypochloremic alkalosis and diabetes insipidus. Thus, perturbation in nephron segments other than the DCT may override the increased WNK-NCC pathway to cause salt wasting and polyuria rather than an FHHt phenotype. Moreover, disruption of Cul3 in the renal epithelia causes proximal tubule injury that progresses to fibrosis [45••]. The widespread renal toxicity is also associated with increased cyclin E, a cell cycle regulator important for normal cellular physiology [9, 45••]. Unlike the heterozygous CUL3Δ9 mutation which spares residual CUL3 activity, nephron-specific CUL3 deletion completely abolishes its actions in the kidney. This suggests that basal levels of CUL3 activity is indispensable for the maintenance of renal structure and function.
3. CUL3 Regulation of Vascular Tone
Mounting evidence support that CUL3 also regulates blood pressure through its control of vascular function. In a mouse model expressing a human hypertension-causing mutation in peroxisome proliferator activated receptor γ (PPARγP467L, termed S-P467L), decreased levels of CUL3 and neddylated CUL3 lead to increased RhoA, a substrate of CUL3-mediated ubiquitylation [4]. As a result, S-P467L mice display enhanced agonist-induced vasoconstriction and increased myogenic tone in resistance vessels, and develop hypertension [4, 46, 47]. Supporting a role of CUL3 in human hypertension, small interfering RNA knockdown or pharmacological inhibition of CUL3 increases RhoA protein in human aortic smooth muscle cells. In keeping with this, inhibition of Cullin ubiquitin ligase activity by MLN4924 markedly enhances RhoA-dependent vasoconstriction in normal aortic segments and elevated arterial pressure in C57BL/6 mice. Molecular studies reveal that CUL3 interacts with an adaptor protein BACURD1 to target RhoA to the CRL3 complex for ubiquitylation [31]. These studies provided the first evidence supporting a causal role of decreased CRL3 activity in vascular dysfunction and hypertension.
The contribution of vascular Cul3Δ9 mutations to the hypertension in FHHt has been established by several lines of evidence. First, expression of Cul3Δ9 transgene selectively in vascular smooth muscle (S-Cul3Δ9) recapitulates the hypertension phenotype of FHHt without any measurable electrolyte abnormalities, suggesting the CUL3 mutations cause human hypertension in part through a vascular smooth muscle mechanism [5, 13••]. S-Cul3Δ9 mice exhibit decreased abundance and function of endogenous CUL3WT in blood vessels, resulting in impaired ubiquitylation and degradation of RhoA, increased RhoA activity and enhanced activation of the RhoA/Rho kinase signaling (Figure 2). This markedly increases vasoconstriction in resistance vessels and elevates arterial pressure under baseline conditions. Of note, the hypertension in S-Cul3Δ9 was associated with arterial stiffening in capacitance vessels [5]. Consistent with this, knock-in mice carrying one allele of wildtype Cul3 and one allele of Cul3Δ9 (CUL3WT/Δ9) not only develop hyperkalemic hypertension as observed in FHHt humans, but also exhibit enhanced medial thickness and increased arterial stiffness [10]. Whether the arterial stiffening is secondary to the hypertension or a direct consequence of vascular Cul3Δ9 awaits further investigation.
CUL3 also regulates nitric oxide (NO)-induced vasodilation by promoting degradation of PDE5, a negative regulator of the NO-soluble guanylyl cyclase (sGC)-cGMP pathway [6••, 7••]. For this reason, CUL3 deficiency causes impaired PDE5 ubiquitylation, increased levels of PDE5 protein and enzymatic activity, and decreased bioavailability of cGMP (Figure 2) [6••]. While Cul3Δ9 mutation and CUL3 deletion (S-CUL3KO) in vascular smooth muscle both impair vasodilator function, the latter results in more severe phenotypes [5, 6]. S-CUL3KO mice display progressive decline in smooth muscle NO responsiveness and develop severe hypertension and arterial stiffening. Interestingly, cGMP production is also decreased in mice lacking CUL3 in the vascular smooth muscle, suggesting that CUL3 also controls cGMP biosynthesis through mechanisms yet to be investigated [6••].
RhoBTB1 (Rho related BTB domain containing 1) is a substrate adaptor delivering PDE5 to the CUL3 complex for ubiquitylation. Interestingly, RhoBTB1 is a PPARγ target gene and is downregulated in the vascular smooth muscle of S-P467L mice, resulting in impaired NO-sGC-cGMP signaling and decreased vasodilation [4]. Inducible smooth-muscle specific restoration of RhoBTB1 on the S-P467L background completely abolishes the vasodilation impairment, arterial stiffening and hypertension [7••]. Importantly, genetic complementation of RhoBTB1 does not correct the increased vasoconstriction which is mediated by enhanced RhoA signaling, suggesting that CUL3 controls vasoconstriction and vasodilation through distinct mechanisms. Of note, smooth muscle RhoBTB1 expression is also downregulated by angiotensin II infusion, and restoration of RhoBTB1 ameliorates impaired vasodilation and hypertension induced by angiotensin II despite a preservation of increased vasocontraction. Collectively, these observations indicate that improvement of vasodilator function was sufficient to mediate the protective effects of RhoBTB1 restoration. Interestingly, RhoBTB1 has recently been reported to be phosphorylated by Rho kinase at multiple serine residues [28]. This differential phosphorylation may regulate its binding to CUL3 and thus the turnover of PDE5 and other unknown substrates.
Mechanistically, CUL3Δ9 impairs the normal turnover of proteins regulating vascular function (RhoA and PDE5) through multiple mechanisms. When expressed in a CUL3-deficient cell line generated by CRISPR-Cas9 genome editing (HEK293T-CUL3−/−), CUL3Δ9 exhibits reduced ubiquitin ligase activity toward RhoA compared to equimolar levels of CUL3WT [31]. This indicates that CUL3Δ9 exhibits a loss of function incapable of supporting normal levels of substrate degradation on its own, consistent with impaired WNK4 degradation observed by Cornelius et al [30]. CUL3Δ9 also binds to adaptor proteins (BACURD1 and RhoBTB1) more efficiently than CUL3WT [31], highly reminiscent of enhanced ubiquitylation and degradation of KLHL3 in the kidney [9, 10]. Moreover, CUL3Δ9 forms an unstable heterodimer with CUL3WT and disrupts the CUL3WT homodimers [31]. This results in decreased levels of CUL3WT and its neddylation in human aortic smooth muscle cells [5], supporting the concept of self-degradation mediated by CUL3Δ9 autoubiquitylation.
4. The CUL3-Keap1-Nrf2 Pathway and Its Antioxidant Affects
CUL3-KEAP1 complex mediates the ubiquitylation of Nrf2 (nuclear factor-E2-related factor 2), a well characterized transcription factor regulating cellular redox homeostasis [12]. KEAP1 sequesters Nrf2 in the CRL3 complex and normally suppresses its cellular abundance through proteasomal degradation. Thus normally, Nrf2 is in limiting amounts because it is sequestered and degraded. Oxidant radicals modify specific cysteine residues on KEAP1, the Nrf2 adaptor, causing conformational changes that releases Nrf2 from KEAP1 and the CRL3 complex [48]. Nrf2 is then free to translocate to the nucleus where it interacts with a cofactor MafK to activate transcription by binding to the antioxidant response element (ARE) on the promoter region of target genes [49]. These include, but are not limited to, the antioxidant genes superoxide dismutase 1–3 (SOD1–3), catalase, NAD(P)H quinone oxidoreductase 1 (NQO1), hemooxygenase-1, and glutathione peroxidase.
Oxidative stress mediated by impairment of Nrf2 responses has been associated with decreased NO bioavailability and endothelial dysfunction, an important risk factor for hypertension and other cardiovascular conditions [49]. For example, Nrf2 activation by tertbutylhydroquinone in human renal glomerular endothelial cells increases NO through upregulation of endothelial NO synthase (eNOS) and dimethylarginine dimethylaminohydrolase (DDAH), which degrades asymmetric dimethylarginine (ADMA), an endogenous inhibitor of eNOS. These protective effects are abolished by Nrf2 knockdown [50]. Interestingly, Nrf2 binds to an ARE on the PPARγ gene and activation of Nrf2 increase PPARγ mRNA. In fact, Nrf2 and PPARγ demonstrate a reciprocal positive feedback in gene expression with coordinated mechanisms to enhance endothelial NO generation. In line with this, PPARγ activation in the endothelium protects from endothelial dysfunction induced by high fat diet, angiotensin II, and aging [51–53].
While the cardiovascular protection mediated by Nrf2 activation is well documented, it is not clear whether perturbations in CUL3 impacts blood pressure through changes in Nrf2 [49]. CUL3 expression is decreased in mouse models of acute kidney injury and chronic kidney disease and in fibrotic human kidney tissues [45••]. As mentioned above, renal tubule-specific disruption of CUL3 causes profound kidney damage featuring widespread tubulointerstitial fibrosis and inflammation [9]. Of note, severe renal injury occurs despite accumulation and activation of Nrf2, suggesting that activation of Keap1/Nrf2 in the nephron alone is not sufficient to compensate the deleterious effects of kidney-specific CUL3 deficiency [45••]. In fact, over-activation of Nrf2 gives rise to reductive stress, which may be as harmful as oxidative stress [54].
Several lines of evidence support that Nrf2 also mediates protective effects in hypertension and chronic heart failure through its antioxidant effects in the brain [49, 55]. Gao et al recently showed that selective deletion of Nrf2 gene in the rostral ventrolateral medulla (RVLM) downregulates Nrf2-targeted antioxidant enzymes and causes central oxidative stress in this key brain stem region. This results in enhanced sympathetic nerve activity, impaired baroreflex function and neurogenic hypertension [56•]. Consistent with a role of Nrf2 in neurogenic hypertension, impaired Nrf2 signaling in the RVLM is also associated with a downregulation of antioxidant genes (Nqo1, Sod2), inflammation, and increased sympathetic outflow in obesity-induced hypertension [57]. Conversely, upregulation of Nrf2 through lentiviral overexpression of Nrf2 or targeted deletion of Keap1 in the RVLM increases antioxidant proteins NQO1 and hemooxygenase-1, improves baroreflex function and attenuates sympathoexcitation in chronic heart failure mice [58].
5. Conclusion
Research in the last decade has established CUL3 as novel regulator of blood pressure. While the CRL3 modulates key biological processes in the cardiovascular and renal systems (Figure 2), its own activity is regulated by molecular mechanisms including neddylation, phosphorylation/dephosphorylation and intracellular calcium mobilization. Proteomic studies predict that CUL3 has identified hundreds of potential adaptor proteins, and only a very small percentage of these have been experimentally investigated [17]. Although CUL3 is versatile in regard to the spectrum of substrates ubiquitylated, this posttranslational modification is highly selective at the molecular level since different adaptor proteins are employed to recruit these substrates. Studies in vascular smooth muscle clearly demonstrate that CUL3 may regulate more than one substrate (RhoA and PDE5, Figure 1) in the same cell type, however, little is known about the crosstalk between these downstream pathways upon disruption of CUL3.
CUL3-based ubiquitin ligase controls the turnover of distinct substrates important for vascular tone, renal electrolyte transport and redox homeostasis. There seems to be spatial and temporal compartmentation of CRL3 activity, allowing precise control of essential physiological processes in the vasculature, kidney, and brain important for blood pressure regulation. As CUL3 is ubiquitously expressed, we have only begun to understand the tissue-specific functions of CUL3-mediated ubiquitin ligase in physiological and pathological conditions. Recently, single nucleotide polymorphisms in CUL3 gene have been associated with protection against essential hypertension, suggesting that altered CUL3 activity may also contribute to the variation of blood pressure at the population scale [59].
Financial support and sponsorship
This study was supported by research grants from the National Institutes of Health (NIH) to CDS (HL084207, HL144807) and JAM (DK098141 and DK117903).
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of Interest
The authors declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published within past 3 years, have been highlighted as:
• Of importance
•• Of major importance
- 1.Kossatz U, Breuhahn K, Wolf B, Hardtke-Wolenski M, Wilkens L, Steinemann D et al. The cyclin E regulator cullin 3 prevents mouse hepatic progenitor cells from becoming tumor-initiating cells. J Clin Invest. 2010;120(11):3820–33. doi: 10.1172/JCI41959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stavropoulos N, Young MW. insomniac and Cullin-3 regulate sleep and wakefulness in Drosophila. Neuron. 2011;72(6):964–76. doi: 10.1016/j.neuron.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mathew R, Seiler MP, Scanlon ST, Mao AP, Constantinides MG, Bertozzi-Villa C et al. BTBZF factors recruit the E3 ligase cullin 3 to regulate lymphoid effector programs. Nature. 2012;491(7425):618–21. doi: 10.1038/nature11548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pelham CJ, Ketsawatsomkron P, Groh S, Grobe JL, de Lange WJ, Ibeawuchi SR et al. Cullin-3 regulates vascular smooth muscle function and arterial blood pressure via PPARgamma and RhoA/Rho-kinase. Cell Metab. 2012;16(4):462–72. doi: 10.1016/j.cmet.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agbor LN, Ibeawuchi SC, Hu C, Wu J, Davis DR, Keen HL et al. Cullin-3 mutation causes arterial stiffness and hypertension through a vascular smooth muscle mechanism. JCI Insight. 2016;1(19):e91015. doi: 10.1172/jci.insight.91015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.••.Agbor LN, Nair AR, Wu J, Lu KT, Davis DR, Keen HL et al. Conditional deletion of smooth muscle Cullin-3 causes severe progressive hypertension. JCI Insight. 2019;5. doi: 10.1172/jci.insight.129793. [DOI] [PMC free article] [PubMed] [Google Scholar]; Selective deletion of CUL3 in vascular smooth muscle results decreases cGMP bioavailability through impaired biosynthesis and increased degradation by PDE5.
- 7.••.Mukohda M, Fang S, Wu J, Agbor LN, Nair AR, Ibeawuchi SC et al. RhoBTB1 protects against hypertension and arterial stiffness by restraining phosphodiesterase 5 activity. J Clin Invest. 2019;130:2318–32. doi: 10.1172/JCI123462. [DOI] [PMC free article] [PubMed] [Google Scholar]; RhoBTB maintains cGMP bioavailability and smooth muscle NO responsiveness by controlling PDE5 activity through CUL3-mediated ubiquitylation.
- 8.Boyden LM, Choi M, Choate KA, Nelson-Williams CJ, Farhi A, Toka HR et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482(7383):98–102. doi: 10.1038/nature10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCormick JA, Yang CL, Zhang C, Davidge B, Blankenstein KI, Terker AS et al. Hyperkalemic hypertension-associated cullin 3 promotes WNK signaling by degrading KLHL3. J Clin Invest. 2014;124(11):4723–36. doi: 10.1172/JCI76126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schumacher FR, Siew K, Zhang J, Johnson C, Wood N, Cleary SE et al. Characterisation of the Cullin-3 mutation that causes a severe form of familial hypertension and hyperkalaemia. EMBO Mol Med. 2015;7(10):1285–306. doi: 10.15252/emmm.201505444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frindt G, Bertog M, Korbmacher C, Palmer LG. Ubiquitination of renal ENaC subunits in vivo. Am J Physiol Renal Physiol. 2020;318(5):F1113–F21. doi: 10.1152/ajprenal.00609.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24(16):7130–9. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.••.Abdel Khalek W, Rafael C, Loisel-Ferreira I, Kouranti I, Clauser E, Hadchouel J et al. Severe Arterial Hypertension from Cullin 3 Mutations Is Caused by Both Renal and Vascular Effects. J Am Soc Nephrol. 2019;30(5):811–23. doi: 10.1681/ASN.2017121307. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study compared the effects of CUL3Δ9 in distal nephron and vascular smooth muscle, and showed that CUL3 mutations cause hypertension through independent renal and vascular mechanisms.
- 14.Canning P, Cooper CD, Krojer T, Murray JW, Pike AC, Chaikuad A et al. Structural basis for Cul3 protein assembly with the BTB-Kelch family of E3 ubiquitin ligases. J Biol Chem. 2013;288(11):7803–14. doi: 10.1074/jbc.M112.437996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schumacher FR, Sorrell FJ, Alessi DR, Bullock AN, Kurz T. Structural and biochemical characterization of the KLHL3-WNK kinase interaction important in blood pressure regulation. Biochem J. 2014;460(2):237–46. doi: 10.1042/BJ20140153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ji AX, Prive GG. Crystal structure of KLHL3 in complex with Cullin3. PLoS One. 2013;8(4):e60445. doi: 10.1371/journal.pone.0060445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bennett EJ, Rush J, Gygi SP, Harper JW. Dynamics of cullin-RING ubiquitin ligase network revealed by systematic quantitative proteomics. Cell. 2010;143(6):951–65. doi: 10.1016/j.cell.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rondou P, Haegeman G, Vanhoenacker P, Van Craenenbroeck K. BTB Protein KLHL12 targets the dopamine D4 receptor for ubiquitination by a Cul3-based E3 ligase. J Biol Chem. 2008;283(17):11083–96. doi: 10.1074/jbc.M708473200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Angers S, Thorpe CJ, Biechele TL, Goldenberg SJ, Zheng N, MacCoss MJ et al. The KLHL12-Cullin-3 ubiquitin ligase negatively regulates the Wnt-beta-catenin pathway by targeting Dishevelled for degradation. Nat Cell Biol. 2006;8(4):348–57. doi: 10.1038/ncb1381. [DOI] [PubMed] [Google Scholar]
- 20.Oberg EA, Nifoussi SK, Gingras AC, Strack S. Selective proteasomal degradation of the B’beta subunit of protein phosphatase 2A by the E3 ubiquitin ligase adaptor Kelch-like 15. J Biol Chem. 2012;287(52):43378–89. doi: 10.1074/jbc.M112.420281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferretti LP, Himmels SF, Trenner A, Walker C, von Aesch C, Eggenschwiler A et al. Cullin3-KLHL15 ubiquitin ligase mediates CtIP protein turnover to fine-tune DNA-end resection. Nat Commun. 2016;7:12628. doi: 10.1038/ncomms12628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pintard L, Kurz T, Glaser S, Willis JH, Peter M, Bowerman B. Neddylation and deneddylation of CUL-3 is required to target MEI-1/Katanin for degradation at the meiosis-tomitosis transition in C. elegans. Curr Biol. 2003;13(11):911–21. doi: 10.1016/s0960-9822(03)00336-1. [DOI] [PubMed] [Google Scholar]
- 23.Rabut G, Le Dez G, Verma R, Makhnevych T, Knebel A, Kurz T et al. The TFIIH subunit Tfb3 regulates cullin neddylation. Mol Cell. 2011;43(3):488–95. doi: 10.1016/j.molcel.2011.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell. 2008;134(6):995–1006. doi: 10.1016/j.cell.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J, Furukawa M, Matsumoto T, Xiong Y. NEDD8 modification of CUL1 dissociates p120(CAND1), an inhibitor of CUL1-SKP1 binding and SCF ligases. Mol Cell. 2002;10(6):1511–8. doi: 10.1016/s1097-2765(02)00783-9. [DOI] [PubMed] [Google Scholar]
- 26.Shibata S, Arroyo JP, Castaneda-Bueno M, Puthumana J, Zhang J, Uchida S et al. Angiotensin II signaling via protein kinase C phosphorylates Kelch-like 3, preventing WNK4 degradation. Proc Natl Acad Sci U S A. 2014;111(43):15556–61. doi: 10.1073/pnas.1418342111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.••.Ishizawa K, Wang Q, Li J, Yamazaki O, Tamura Y, Fujigaki Y et al. Calcineurin dephosphorylates Kelch-like 3, reversing phosphorylation by angiotensin II and regulating renal electrolyte handling. Proc Natl Acad Sci U S A. 2019;116(8):3155–60. doi: 10.1073/pnas.1817281116. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrated that phosphorylation/dephosphorylation of KLHL3 alters WNK4 binding and ubiquitylation in the CUL3 complex. Phosphorylation of substrate adaptors is an important regulatory mechanism for CRL3 activity.
- 28.Haga RB, Garg R, Collu F, Borda D’Agua B, Menendez ST, Colomba A et al. RhoBTB1 interacts with ROCKs and inhibits invasion. Biochem J. 2019;476(17):2499–514. doi: 10.1042/BCJ20190203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McGourty CA, Akopian D, Walsh C, Gorur A, Werner A, Schekman R et al. Regulation of the CUL3 Ubiquitin Ligase by a Calcium-Dependent Co-adaptor. Cell. 2016;167(2):525–38 e14. doi: 10.1016/j.cell.2016.09.026. [DOI] [PubMed] [Google Scholar]
- 30.Cornelius RJ, Zhang C, Erspamer KJ, Agbor LN, Sigmund CD, Singer JD et al. Dual gain and loss of cullin 3 function mediates familial hyperkalemic hypertension. Am J Physiol Renal Physiol. 2018;315(4):F1006–F18. doi: 10.1152/ajprenal.00602.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ibeawuchi SR, Agbor LN, Quelle FW, Sigmund CD. Hypertension-causing Mutations in Cullin3 Protein Impair RhoA Protein Ubiquitination and Augment the Association with Substrate Adaptors. J Biol Chem. 2015;290(31):19208–17. doi: 10.1074/jbc.M115.645358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.••.Ferdaus MZ, Miller LN, Agbor LN, Saritas T, Singer JD, Sigmund CD et al. Mutant Cullin 3 causes familial hyperkalemic hypertension via dominant effects. JCI Insight. 2017;2(24). doi: 10.1172/jci.insight.96700. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study clearly demonstrated that CUL3 haploinsufficiency does not cause FHHt, but the dominant effects of CUL3Δ9 in the nephron are required.
- 33.•.Cornelius RJ, Yang CL, Ellison DH. Hypertension-causing cullin 3 mutations disrupt COP9 signalosome binding. Am J Physiol Renal Physiol. 2020;318(1):F204–F8. doi: 10.1152/ajprenal.00497.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]; CUL3Δ9 exhibits impaired binding with COP9 signalosome the deneddylase. This interferes with the neddylation/deneddylation cycle necessary for normal CUL3 function.
- 34.McCormick JA, Mutig K, Nelson JH, Saritas T, Hoorn EJ, Yang CL et al. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab. 2011;14(3):352–64. doi: 10.1016/j.cmet.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grimm PR, Coleman R, Delpire E, Welling PA. Constitutively Active SPAK Causes Hyperkalemia by Activating NCC and Remodeling Distal Tubules. J Am Soc Nephrol. 2017;28(9):2597–606. doi: 10.1681/ASN.2016090948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kahle KT, Wilson FH, Leng Q, Lalioti MD, O’Connell AD, Dong K et al. WNK4 regulates the balance between renal NaCl reabsorption and K+ secretion. Nat Genet. 2003;35(4):372–6. doi: 10.1038/ng1271. [DOI] [PubMed] [Google Scholar]
- 37.Cope G, Murthy M, Golbang AP, Hamad A, Liu CH, Cuthbert AW et al. WNK1 affects surface expression of the ROMK potassium channel independent of WNK4. J Am Soc Nephrol. 2006;17(7):1867–74. doi: 10.1681/ASN.2005111224. [DOI] [PubMed] [Google Scholar]
- 38.Glover M, Ware JS, Henry A, Wolley M, Walsh R, Wain LV et al. Detection of mutations in KLHL3 and CUL3 in families with FHHt (familial hyperkalaemic hypertension or Gordon’s syndrome). Clin Sci (Lond). 2014;126(10):721–6. doi: 10.1042/CS20130326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wakabayashi M, Mori T, Isobe K, Sohara E, Susa K, Araki Y et al. Impaired KLHL3-mediated ubiquitination of WNK4 causes human hypertension. Cell Rep. 2013;3(3):858–68. doi: 10.1016/j.celrep.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 40.Yoshida S, Araki Y, Mori T, Sasaki E, Kasagi Y, Isobe K et al. Decreased KLHL3 expression is involved in the pathogenesis of pseudohypoaldosteronism type II caused by cullin 3 mutation in vivo. Clin Exp Nephrol. 2018;22(6):1251–7. doi: 10.1007/s10157-018-1593-z. [DOI] [PubMed] [Google Scholar]
- 41.••.Cornelius RJ, Si J, Cuevas CA, Nelson JW, Gratreak BDK, Pardi R et al. Renal COP9 Signalosome Deficiency Alters CUL3-KLHL3-WNK Signaling Pathway. J Am Soc Nephrol. 2018;29(11):2627–40. doi: 10.1681/ASN.2018030333. [DOI] [PMC free article] [PubMed] [Google Scholar]; Deletion of Jab1, the catalytic subunit of COP9 signalosome, in the nephron phenocopies the effects of CUL3Δ9. Deficient deneddylation of CUL3 by COP9 signalosome contributes to FHHt.
- 42.Zhang C, Meermeier NP, Terker AS, Blankenstein KI, Singer JD, Hadchouel J et al. Degradation by Cullin 3 and effect on WNK kinases suggest a role of KLHL2 in the pathogenesis of Familial Hyperkalemic Hypertension. Biochem Biophys Res Commun. 2016;469(1):44–8. doi: 10.1016/j.bbrc.2015.11.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gong Y, Wang J, Yang J, Gonzales E, Perez R, Hou J. KLHL3 regulates paracellular chloride transport in the kidney by ubiquitination of claudin-8. Proc Natl Acad Sci U S A. 2015;112(14):4340–5. doi: 10.1073/pnas.1421441112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schambelan M, Sebastian A, Rector FC Jr. Mineralocorticoid-resistant renal hyperkalemia without salt wasting (type II pseudohypoaldosteronism): role of increased renal chloride reabsorption. Kidney Int. 1981;19(5):716–27. doi: 10.1038/ki.1981.72. [DOI] [PubMed] [Google Scholar]
- 45.••.Saritas T, Cuevas CA, Ferdaus MZ, Kuppe C, Kramann R, Moeller MJ et al. Disruption of CUL3-mediated ubiquitination causes proximal tubule injury and kidney fibrosis. Sci Rep. 2019;9(1):4596. doi: 10.1038/s41598-019-40795-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; Nephron-specific deletion of CUL3 causes widespread kidney damage despite activation of KEAP/Nrf2 signaling. Basal levels of CUL3 activity are required to mainain kidney structure and function.
- 46.Ketsawatsomkron P, Lorca RA, Keen HL, Weatherford ET, Liu X, Pelham CJ et al. PPARgamma regulates resistance vessel tone through a mechanism involving RGS5-mediated control of protein kinase C and BKCa channel activity. Circ Res. 2012;111(11):1446–58. doi: 10.1161/CIRCRESAHA.112.271577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Halabi CM, Beyer AM, de Lange WJ, Keen HL, Baumbach GL, Faraci FM et al. Interference with PPAR gamma function in smooth muscle causes vascular dysfunction and hypertension. Cell Metab. 2008;7(3):215–26. doi: 10.1016/j.cmet.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Itoh K, Tong KI, Yamamoto M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic Biol Med. 2004;36(10):1208–13. doi: 10.1016/j.freeradbiomed.2004.02.075. [DOI] [PubMed] [Google Scholar]
- 49.Chen B, Lu Y, Chen Y, Cheng J. The role of Nrf2 in oxidative stress-induced endothelial injuries. J Endocrinol. 2015;225(3):R83–99. doi: 10.1530/JOE-14-0662. [DOI] [PubMed] [Google Scholar]
- 50.Luo Z, Aslam S, Welch WJ, Wilcox CS. Activation of nuclear factor erythroid 2-related factor 2 coordinates dimethylarginine dimethylaminohydrolase/PPAR-gamma/endothelial nitric oxide synthase pathways that enhance nitric oxide generation in human glomerular endothelial cells. Hypertension. 2015;65(4):896–902. doi: 10.1161/HYPERTENSIONAHA.114.04760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beyer AM, de Lange WJ, Halabi CM, Modrick ML, Keen HL, Faraci FM et al. Endothelium-specific interference with peroxisome proliferator activated receptor gamma causes cerebral vascular dysfunction in response to a high-fat diet. Circ Res. 2008;103(6):654–61. doi: 10.1161/CIRCRESAHA.108.176339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.De Silva TM, Hu C, Kinzenbaw DA, Modrick ML, Sigmund CD, Faraci FM. Genetic Interference With Endothelial PPAR-gamma (Peroxisome Proliferator-Activated Receptor-gamma) Augments Effects of Angiotensin II While Impairing Responses to Angiotensin 1–7. Hypertension. 2017;70(3):559–65. doi: 10.1161/HYPERTENSIONAHA.117.09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Silva TM, Li Y, Kinzenbaw DA, Sigmund CD, Faraci FM. Endothelial PPARgamma (Peroxisome Proliferator-Activated Receptor-gamma) Is Essential for Preventing Endothelial Dysfunction With Aging. Hypertension. 2018;72(1):227–34. doi: 10.1161/HYPERTENSIONAHA.117.10799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dialynas G, Shrestha OK, Ponce JM, Zwerger M, Thiemann DA, Young GH et al. Myopathic lamin mutations cause reductive stress and activate the nrf2/keap-1 pathway. PLoS Genet. 2015;11(5):e1005231. doi: 10.1371/journal.pgen.1005231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tian C, Gao L, Zimmerman MC, Zucker IH. Myocardial infarction-induced microRNA-enriched exosomes contribute to cardiac Nrf2 dysregulation in chronic heart failure. Am J Physiol Heart Circ Physiol. 2018;314(5):H928–H39. doi: 10.1152/ajpheart.00602.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.•.Gao L, Zimmerman MC, Biswal S, Zucker IH. Selective Nrf2 Gene Deletion in the Rostral Ventrolateral Medulla Evokes Hypertension and Sympathoexcitation in Mice. Hypertension. 2017;69(6):1198–206. doi: 10.1161/HYPERTENSIONAHA.117.09123. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates a protective role of Nrf2-mediated antioxidant response in the RVLM. Disruption of Nrf2 in the RVLM results in neurogenic hypertension.
- 57.Balasubramanian P, Asirvatham-Jeyaraj N, Monteiro R, Sivasubramanian MK, Hall D, Subramanian M. Obesity-induced sympathoexcitation is associated with Nrf2 dysfunction in the rostral ventrolateral medulla. Am J Physiol Regul Integr Comp Physiol. 2020;318(2):R435–R44. doi: 10.1152/ajpregu.00206.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ma A, Hong J, Shanks J, Rudebush T, Yu L, Hackfort BT et al. Upregulating Nrf2 in the RVLM ameliorates sympatho-excitation in mice with chronic heart failure. Free Radic Biol Med. 2019;141:84–92. doi: 10.1016/j.freeradbiomed.2019.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li J, Hu J, Sun R, Zhao Y, Liu H, Li J et al. Association between Cullin-3 Single-Nucleotide Polymorphism rs17479770 and Essential Hypertension in the Male Chinese Han Population. Dis Markers. 2017;2017:3062759. doi: 10.1155/2017/3062759. [DOI] [PMC free article] [PubMed] [Google Scholar]