

Abstract
Increasing rates of drug-resistant Gram-negative (GN) infections, combined with a lack of new GN-effective antibiotic classes, is driving the need for the discovery of new agents. Bacterial metabolism represents an underutilized mechanism of action in current antimicrobial therapies. Therefore, we sought to identify novel antimetabolites that disrupt key metabolic pathways and explore the specific impacts of these agents on bacterial metabolism. This study describes the successful application of this approach to discover a new series of chemical probes, N-(phenyl) thioacetamide-linked 1,2,3-triazoles (TAT) that target cysteine synthase A (CysK), an enzyme unique to bacteria that is positioned at a key juncture between several fundamental pathways. The TAT class was identified using a high-throughput screen against Escherichia coli designed to identify modulators of pathways related to folate biosynthesis. TAT analog synthesis demonstrated a clear structure-activity relationship, and activity was confirmed against GN antifolate-resistant clinical isolates. Spontaneous TAT resistance mutations were tracked to CysK, and mode of action studies led to the identification of a false product formation mechanism between the CysK substrate O-acetyl-L-serine and the TATs. Global transcriptional responses to TAT treatment revealed that these antimetabolites impose substantial disruption of key metabolic networks beyond cysteine biosynthesis. This study highlights the potential of antimetabolite drug discovery as a promising approach to the discovery of novel GN antibiotics, and the pharmacological promise of TAT CysK probes.
Keywords: antimetabolite; Gram-negative drug discovery; antifolate; 1,2,3-triazole; cysteine synthase; high-throughput screen
Graphical Abstract
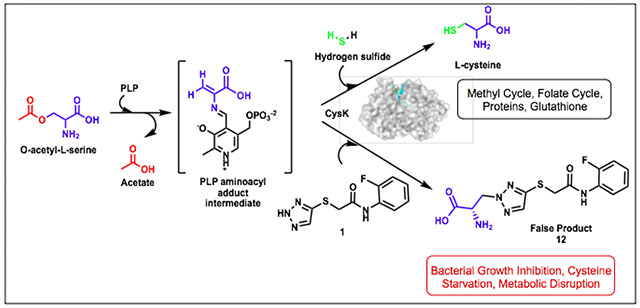
Incidences of multidrug-resistant bacterial infections are increasing at a formidable pace and account for significant morbidity, mortality, and financial burden worldwide.1, 2 The output of new antibiotics for clinical use with efficacy against Gram-negative (GN) bacteria has been especially low over the past several decades, with a general lack of innovative mechanisms of action (MOA).3 The failure to discover new classes of antibiotics effective against GN bacteria in the last 50 years calls for adaptation of our drug discovery approaches to identify antibiotic targets that may have been passed over in favor of targets that are either previously validated or easier to identify through traditional screening methods. In recent years, there has been a resurgence in interest towards bacterial metabolism as a source of new antimicrobials. The number of existing clinical therapeutics targeting bacterial metabolism represents less than 10% of FDA approved antibacterial agents.4, 5 Therefore, we sought to identify antimetabolites that disrupt key metabolic pathways and utilize these compounds to explore essential mechanisms in bacterial metabolic pathways.
In this study, we present the discovery of thioacetamide-linked 1,2,3-triazoles (TATs), which act against the GN pathogen Escherichia coli through disruption of cysteine biosynthesis. These molecules were discovered using a conditional high-throughput screen for antimetabolites inhibiting E. coli growth by engaging pathways related to tetrahydrofolate. The folate biosynthesis pathway was selected as it exemplifies the therapeutic utility of selectively targeting bacterial metabolism. This pathway has been a historically useful antibiotic target for over eighty years since the discovery of the sulfonamides.6, 7 The efficacy and selectivity of antimicrobial antifolates can be attributed to the absence of most of the de novo folate biosynthetic machinery in humans and because folates are essential in bacteria for the biosynthesis of a diverse array of metabolites including purines, thymidine, pantothenate, and amino acids.8
The TATs identified from this screen were found to target a pathway adjacent to folate biosynthesis, the cysteine biosynthetic pathway (Figure 1). Homocysteine, derived from cysteine, feeds into both the folate cycle and the methyl cycle producing methionine and S-adenosylmethionine.9 In prokaryotes and lower eukaryotes, cysteine synthase A (CysK) catalyzes the production of L-cysteine from O-acetyl-L-serine (OALS) and sulfide (Figure 1).10 Most microorganisms have an additional isoenzyme, cysteine synthase B (CysM), which is capable of the same function, but understood to be more active under anaerobic conditions.11 Mammalian systems lack an enzyme homologous to CysK and acquire cysteine either through the diet or the transsulfuration pathway.12 In bacteria, L-cysteine acts as a precursor for several metabolites necessary for redox defense, virulence, and basic cell functions.10 Cysteine provides the thiol group present in several molecules including glutathione, iron-sulfur-clusters, thiamine, and methionine. Importantly, CysK is positioned at the juncture of cysteine biosynthesis and the reductive sulfate assimilation pathway (RSAP) and consequently mediates the primary route of sulfur acquisition for several microbial species.13 CysK is therefore central to multiple key metabolic processes (Figure 1) and represents an attractive bacterial target due to its absence in humans.
Figure 1. Pathways of interest probed in the antimetabolite screen.
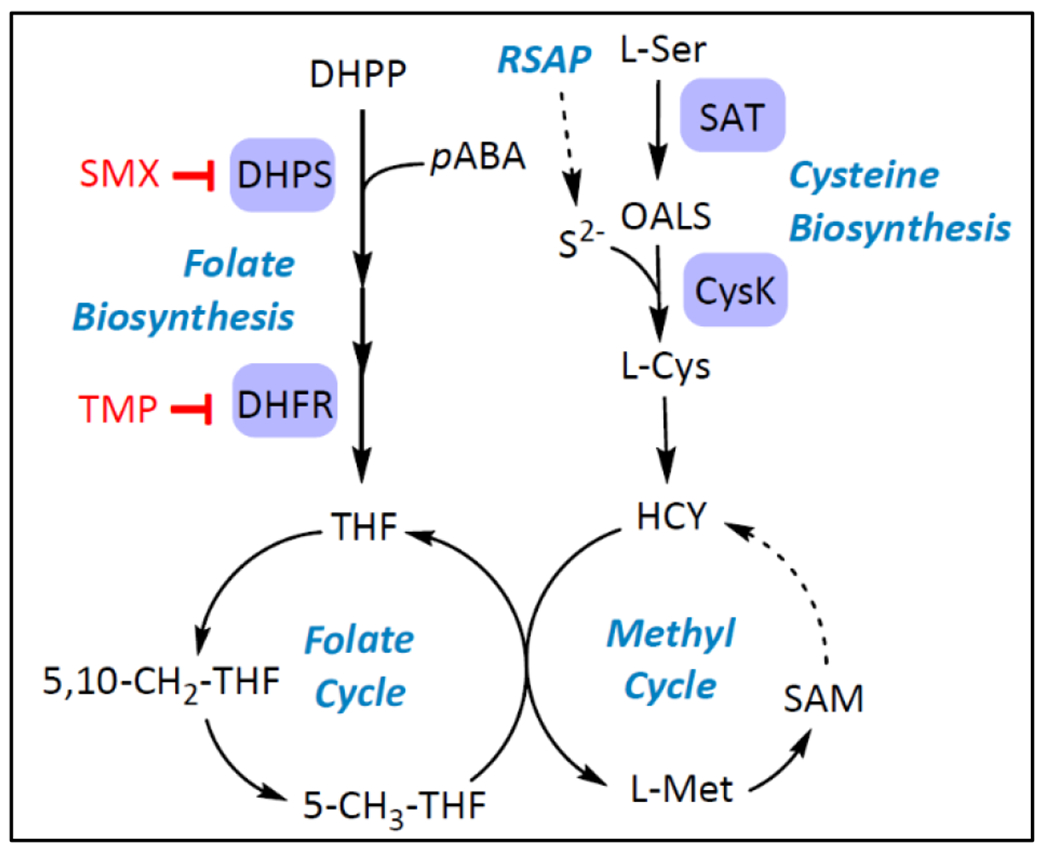
Products of folate and cysteine biosynthesis pathways feed into folate and methyl cycles. Pathway representations are simplified. RSAP = Reductive sulfate assimilation pathway; DHFR = Dihydrofolate reductase; SMX=Sulfamethoxazole; TMP = Trimethoprim; THF = Tetrahydrofolate; HCY = Homocysteine; SAM = S-adenosylmethionine.
There have already been several efforts towards the development of CysK as an antimicrobial target. The CysK enzymes of several bacterial, mycobacterial, and protozoal species have been characterized and explored for inhibitor development.12, 14–23 Cysteine production is allosterically regulated by the upstream enzyme serine acetyltransferase (SAT), which forms an inhibitory complex through the C-terminal tail of SAT with CysK in the absence of the OALS substrate.24 The majority of these recently described CysK inhibitors are peptides which mimic the C-terminal tail of SAT.22, 25
Triazoles are well known pharmacophores for a diverse range of biological and pharmacological activity. An early study on S. Typhimurium CysK showed that 1,2,4-triazole with weak antimicrobial effects is an inhibitor of CysK which mediates the formation of a false product between OALS and the triazole.14 In this study, we demonstrate that the mode of action of the TATs is also through a CysK-mediated formation of a false product between OALS and the TATs. However, the TATs are greater than 64-fold more potent than 1,2,4-triazole. The TATs are amenable to modification, and their antimicrobial activity displays a distinct structure-activity relationship. Interestingly, RNA-seq studies on TAT-treated E. coli revealed that the TATs impart widespread effects on multiple biosynthetic pathways beyond cysteine biosynthesis including sulfur assimilation, aromatic amino acid biosynthesis, and nucleic acid biosynthesis. These studies highlight the potential of antimetabolites to impose substantial disruption of key metabolic networks in pathogenic microbes as a strategy to discover novel GN-effective antibiotics.
RESULTS AND DISCUSSION
A high-throughput antimetabolite screen yields discovery of thioacetamide-linked 1,2,3-triazoles
To identify antimetabolites that engage folate biosynthesis and related pathways in GN bacteria, nearly 13,000 compounds were screened for growth inhibition of E. coli K12 MG1655 at 50 µM in the nutrient-restricted M9 minimal media. This media lacks metabolites known to mask the growth inhibitory action of clinically established and experimental antifolates.26, 27 Four libraries were screened, including: a “Lead-Like” set of pharmacologically tractable small molecules (lipophilicity cLogP<3, Mol wt <300); a Fragment collection (98% of compounds have a Mol wt < 200); a collection of FDA-approved Bioactives with diverse therapeutic indications; and an in-house collection of experimental antifolates. Compounds were considered primary hits if they exhibited at least 50% E. coli growth inhibition. 136 primary hits were obtained, resulting in a primary hit rate of 1.1% (Figure 2A and 2B).
Figure 2. Screen for E. coli growth inhibition in M9 yielding N-(phenyl) thioacetamide-linked 1,2,3-triazoles.
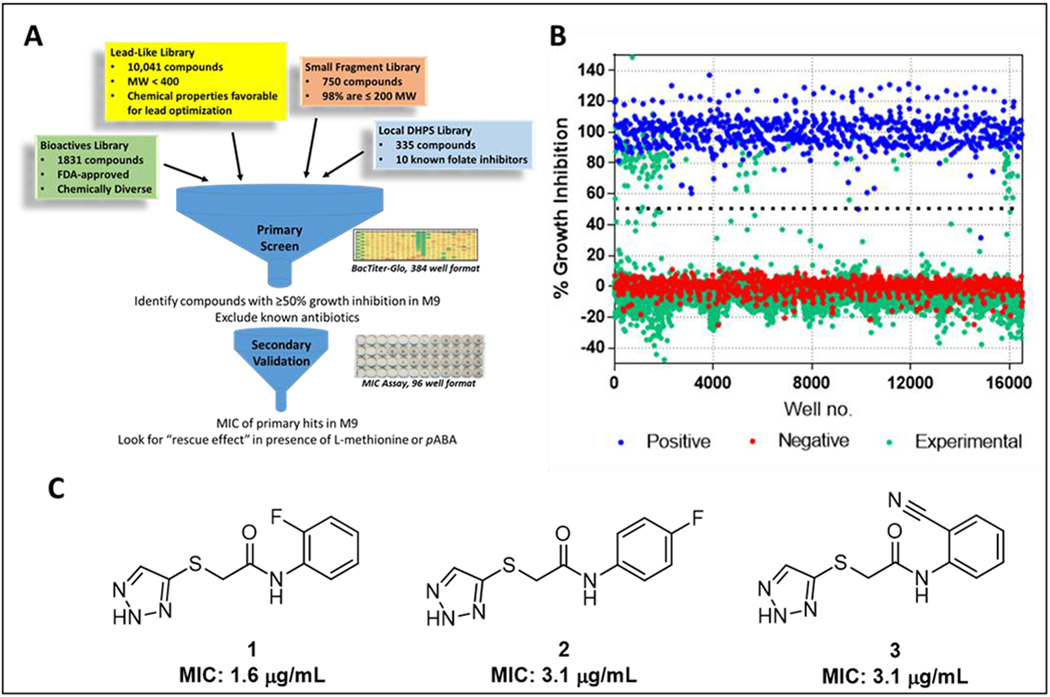
(A) Overall schematic of screen. Testing for antagonization by pABA and L-methionine during secondary validation indicates interaction with pathways related to folate biosynthesis. (B) Scatter plot of all compounds tested. Negative controls (red dots) represent 0.47 % DMSO carrier while positive controls (blue dots) represent 50 μM treatment with chloramphenicol. (C) Chemical structures of three TAT hits from the Lead-Like Library and MIC against E. coli K12 MG1655.
Primary hits were evaluated for interaction with metabolic pathways of interest using a secondary validation assay in which the growth inhibition phenotype is masked in the presence of methionine, a folate derivative, or para-aminobenzoic acid (pABA), a substrate of the folate pathway enzyme dihydropteroate synthase (DHPS).26, 27 First, primary hits from the Bioactives collection that were known to belong to antibiotic classes, including the antifolates, were evaluated. The sulfonamide antifolates, which compete with pABA to form a DHPS-mediated false product with the other substrate dihydropterin pyrophosphate, provided and internal validation standard. Growth inhibition with the sulfonamides was fully masked by pABA while methionine rescued growth inhibition to a more moderate extent. The remaining non-antibiotic primary hits were subsequently evaluated (Figure 2A, Supplementary Figure 1). Twelve compounds from the Bioactives collection with known therapeutic indications were identified. MICs ranged from 0.21 to 42 μM and included chemotherapies targeting thymidylate synthase and DNA synthesis (Supplementary Table 1). Most of the remaining validated hits originated from the Lead-Like library. Among these, three of the most potent compounds shared a 2-((1H-1,2,3-triazol-5-yl)thio)-N-(phenyl)acetamide scaffold (hereafter referred to as thioacetamide-linked 1,2,3-triazoles or TATs, compounds 1-3) (Figure 2C). Bacteriostatic growth inhibition of these compounds could be rescued by methionine but not pABA, indicating the compounds disrupt pathways related to methionine biosynthesis by a mechanism that is independent of direct DHPS interaction (Supplementary Table 1). Therefore, this scaffold was chosen for further evaluation.
Structure-activity relationship of TAT analogs
To explore the structure-activity relationship (SAR) of the TATs, a focused set of analogs was synthesized and tested for E. coli growth inhibition (Figure 3, Supplementary Table 2). The N-phenyl ring was tolerant to substitutions with small effects on potency, as represented by compounds 4-6. N-methylation of the triazole, compound 7 abolished activity, and substitution of the 1,2,3 triazole with a 1,2,4 triazole, compound 8, reduced efficacy four-fold (Figure 3). Modifications to the central portion of the molecule including isosteric replacement of the thioether with a ether or amine (10-11), greatly reduced the potency, and the reduction of the amide also reduced overall potency (9). These data demonstrate that the TAT series is amenable to optimization by medicinal chemistry and presents a clear SAR with a requirement for the 1,2,3-triazole to retain optimal activity.
Figure 3. SAR summary of the TAT series.
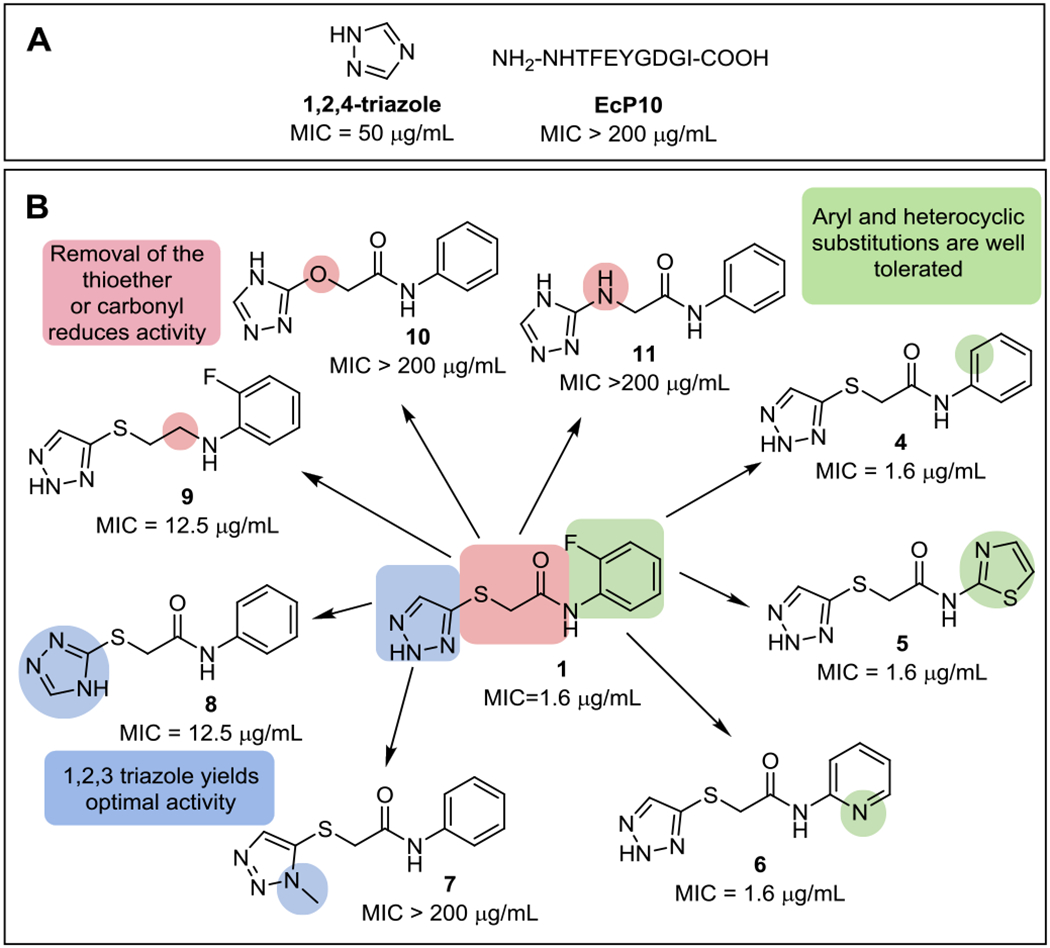
(A) 1,2,4-triazole and EcP10, representing the last ten amino acids at the C-terminal tail of E. coli SAT, are previously described CysK-targeted molecules. (B) Original TAT hit 1 and analogs. Color coding highlights regions of interest relative to the original hit 1. The E. coli MG1655 K12 MIC is shown for all compounds. Complete MIC data is listed in the Supplementary Table 2.
TATs are effective against GN species including clinical antifolate-resistant strains and do not display cytotoxicity
The effect of the TATs on a broad spectrum of bacterial species as well as against human cell lines was evaluated. Growth inhibition was observed against E. cloacae for compound 1 at 5 µg/mL with more moderate yet detectable activity against other GN species (25-200 µg/mL), including Klebsiella pneumoniae, Acinetobacter baumannii, and Pseudomonas aeruginosa (Supplementary Table 3). MICs were not detected for any of the Gram-positive or mycobacterial strains tested, but this absence of activity could be due to interference of nutrients in the media required for the growth of those organisms. Lead compounds 1-3 and 5 were tested against Vero and HepG2 mammalian cell lines and did not show any cytotoxicity (Supplementary Table 4).
To ensure the TATs were effective against antifolate-resistant clinical strains, select TATs were tested against a panel of recent GN clinical isolates from St. Jude Children’s Research Hospital with varying resistance to antifolates (Supplementary Table 5). It was found that the lead inhibitors 1, 4, 5, and 6 maintained efficacy against the collection of E. coli and K. pneumoniae isolates with the exception of one multidrug resistant K. pneumoniae strain. TAT MICs among the panel had a low MIC range for the E. coli isolates ranging between 1.0 and 3.5 μg/mL, and a more varied MIC profile amongst the K. pneumoniae isolates ranging between 0.78 and >100 μg/mL. Resistance to either SMX or TMP did not correspond with increased MICs to the TATs (Supplementary Table 5). The TAT series is therefore not susceptible to currently circulating antifolate resistance mechanisms and the MICs acquired in lab strains are predictive of activity against clinically derived isolates.
Spontaneous mutant generation and cell-based studies reveal CysK as a TAT target
To identify the target of the TAT compounds, spontaneous E. coli K12 mutants were generated on M9 agar containing compounds 1, 2, or 3 at 100 µg/mL. The resulting resistance frequencies for compounds 1, 2, and 3 were 1.3 × 107, 2.7 × 107, and 2.8 × 107, respectively. Genomic sequencing of these mutants revealed nonsynonymous mutations in the cysK gene encoding CysK (Supplementary Table 6). The mutations were modeled onto an existing structure of E. coli CysK (PDB 5J43)28 (Figures 4A and 4B). The affected residues tracked to the active site cleft between the two domains of the CysK fold type II pyridoxal-5’-phosphate (PLP)-dependent enzyme, which contains the binding sites of both the PLP cofactor and the OALS substrate.29 The two mutated sites P37 and R44 were proximal to the invariant K42 necessary for internal aldimine linkage with PLP in the absence of substrate. S70 and G71 are located in a conserved loop that is known to interact with the carboxyl groups of three distinct CysK ligands: the C-terminal isoleucine of SAT, the aminoacrylate reaction intermediate, and the benzoic acid moiety of a thiazolidine inhibitor of M. tuberculosis CysK1.19
Figure 4. CysK target confirmation studies.
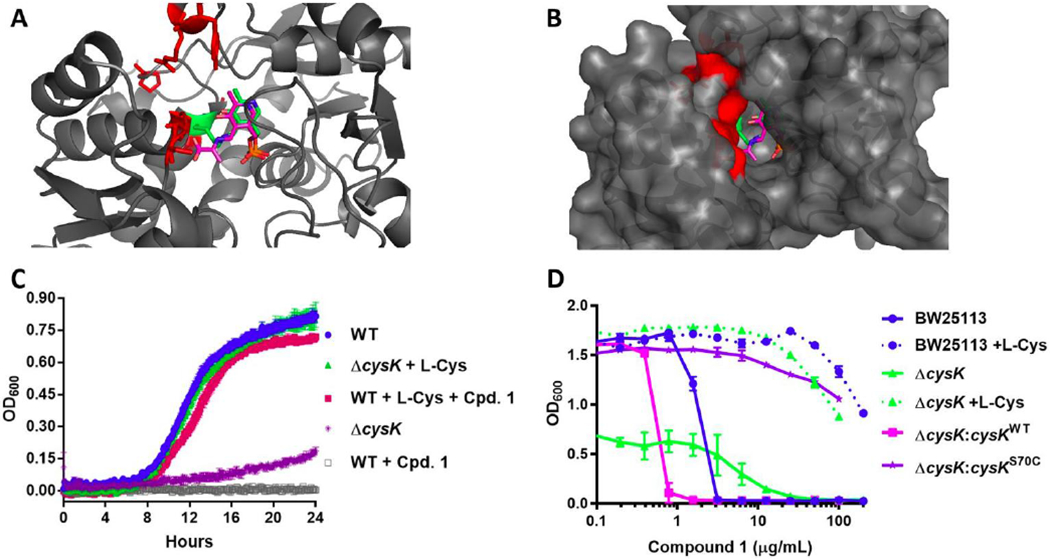
(A) Residues altered by spontaneous mutants induced by compound 1 are highlighted in red on the E. coli CysK structure (PDB 5J43)28. The PLP cofactor is represented in green sticks and the aminoacrylate intermediate from the M. tuberculosis structure (PDB2Q3D) is overlaid in pink sticks. (B) Surface map representation. (C) Survival studies of E. coli BW25113 and ΔcysK in the presence of 16 μg/mL (10x MIC) compound 1 and/or 10 μg/mL L-cysteine. (D) Episomal expression of the cysK gene harboring the spontaneous mutation S70C (Supplementary Table 3) confers resistance to compound 1.
To further confirm CysK as the putative target for the TATs, a panel of CysK isogenic mutants was assembled in the background strains E. coli BW25113 and JW2407 (ΔcysK) (Supplementary Table 3A).30 The cysK knockout had a clear growth defect in M9, which could be restored by supplementation with cysteine (Figure 4C). Addition of cysteine also rescued the WT strain from growth inhibition by compound 1. Episomal expression of the cysK gene harboring the spontaneous mutation S70C conferred resistance to 1 in the ΔcysK background (Figure 4D, Supplementary Table 3A).
Interestingly, overexpression of cysK in the WT E. coli strain by constitutive heterologous cysK expression did not confer resistance to TAT compound 1 (Supplementary Table 3A). A lack of resistance achieved through target overexpression is a hallmark of antimetabolite drugs such as the sulfonamides, which form a DHPS-mediated false product with DHPP.31 When normal metabolic flux is diverted through false product formation, increased abundance of the target does not overcome the growth inhibitory effect. This supported the hypothesis that the TATs may have a false product mechanism similar to the previously described mechanism of 1,2,4-triazole against CysK.
Interactions with cysteine-related metabolic networks
To determine if metabolites related to cysteine metabolism could antagonize the growth inhibition of CysK inhibitors, MIC rescue studies were performed using compound 1 and the 1,2,4-triazole. Supplementation with serine, OALS, and glutathione, which are closely metabolically related to the CysK reaction, increased the MIC of Compound 1 128-fold, and cysteine increased the MIC more than 256-fold (Supplementary Table 7). A similar MIC rescue profile was observed with 1,2,4-triazole, indicating this molecule and the TATs could share CysK as a target. EcP10, a peptidomimetic designed to represent CysK inhibitors mimicking the C-terminal tail of SAT22, had no detectable MIC against E. coli, so metabolite rescues could not be measured (Supplementary Table 7).
To determine if interactions in this metabolic network could result in potentiation effects between antifolates and the TAT series, susceptibility testing was performed with the ΔcysK strain against antifolates and other antibiotic classes (Supplementary Table 8). The ΔcysK phenotype mimics growth inhibition induced by compound 1 (Figure 4C), thus hypersusceptibility to other inhibitors in this genetic background may predict synergistic drug interactions with CysK inhibition. The ΔcysK strain was found to be more sensitive to TMP, the pABA biosynthesis inhibitor MAC173979,26 and two DHPS inhibitors including SMX and a pterin-SMX conjugate (Compound 15 from Zhao et al.27) (Supplementary Table 8). The ΔcysM strain was also tested but showed no significant shift in MIC, likely due to reduced expression of cysM under aerobic conditions.11 Subsequent checkerboard synergy testing did not reveal synergy between compound 5 and the antifolates against the parent E. coli strain, but 5 could potentiate both DHPS inhibitors pterin-SMX and SMX, as represented by an FIC of 0.125 for both DHPS inhibitors in the presence of compound 5 (Supplementary Figure 2). This potentiation effect indicates that inhibition of DHPS is more effective when cysteine biosynthesis is disrupted, possibly through enhanced perturbation of shared metabolic pathways (Figure 1).
Biophysical studies characterize the CysK–TAT interaction
Despite evidence from the cellular studies demonstrating CysK as the TAT target (Figures 4C and 4D), traditional biophysical assays including differential scanning fluorimetry and surface plasmon resonance failed to indicate direct binding interactions between compound 1 and purified E. coli CysK (EcCysK) (data not shown). UV-Vis spectroscopy proved to be a more successful approach in studying interactions between CysK, compound 1, and the OALS substrate. PLP-dependent enzymes such as CysK have a distinct spectroscopic profile reflecting the state of the aldimine intermediates.25 Apo CysK has an absorption maximum around 412 nM. This maximum shifts towards 470 nM upon binding of substrate which reflects a change from the internal aldimine linkage to the external. Additionally, Apo CysK has an emission maximum at 509 nM when excited at either 298 nM or 412 nM. A spectroscopic analysis of compound 1 as well as the mechanistic controls 1,2,4-triazole and EcP10 were performed to profile the binding mode of each ligand.
As expected, EcCysK demonstrated a 410 nm absorption maximum which shifted to 466 nm in the presence of OALS, indicating a PLP shift from the internal K42 aldimine linkage to an external aldimine linkage with OALS (Figure 5A). Addition of the EcP10 peptide to the CysK enzyme alone markedly increased the fluorescence emission intensity at 510 nm upon excitation at 410 nm, which is typical of previously established allosteric CysK inhibitors,25 but not observed with the treatment of 1,2,4-triazole or compound 1 (Figure 5B). Additionally, when either compound 1 or the 1,2,4-triazole molecule were added in combination with OALS, the peak absorbance shift from 410 nm to 466 nm associated with OALS forming the external aldimine was absent for both compounds (Figure 5A). The interactions of OALS with EcCysK as well as that of compound 1 with the OALS-EcCysK complex were both found to be concentration-dependent, indicating a true effect on the E. coli CysK enzyme (Supplementary Figure 3). The diminishment of the substrate-induced spectroscopic changes in the presence of the two triazole-based molecules, the TAT analog 1 and the 1,2,4-triazole molecule, suggests turnover of the external alidimine from CysK, potentially forming a false product with OALS and restoring the cofactor to the internal aldimine linkage. Conversely, the combination of EcP10 and OALS resulted in an overall absorbance profile that is very similar to substrate binding alone, indicating EcP10 had very little interference with substrate adduct formation.
Figure 5. Spectroscopic interaction of compound 1 with CysK.
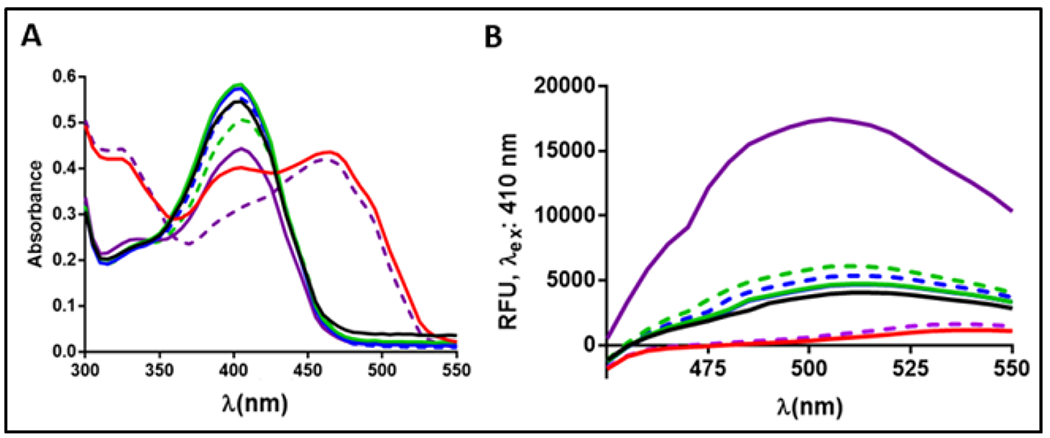
(A) Scanning absorbance and (B) fluorescence spectra of CysK with no ligand (black), OALS (red), 1 (green), 1,2,4-triazole (blue), and EcP10 (purple). CysK-OALS complex and inhibitor combinations are in dashed lines.
MOA studies to explore false product formation
Results demonstrating the dependency of the triazole moiety for activity (Figure 3) and the similarities with 1,2,4-triazole14 within both cellular and spectroscopic studies (Supplementary Table 3A, Figure 5) supported the hypothesis that the TATs are capable of participating in a CysK-mediated false product reaction (Figure 6A). To confirm this MOA more directly, liquid chromatography tandem mass-spectrometry (LC-MS/MS) was employed to detect the false product formed from both the purified CysK enzyme and E. coli cells treated with compound 1. The predicted false product (12) formed between compound 1 and OALS was synthesized as a bioanalytical standard for these experiments (Figure 6A). Compound 12 had no detectable MIC activity, even when tested with outer membrane permeabilizers and a hyperporinated E. coli strains (Supplementary Table 3B), indicating that the TATs do not act as traditional prodrugs that are inactive until a biotransformation event yields a growth-inhibitory form, or that compound 12 is not adequately permeable across the cytoplasmic membrane and must be generated in the cytosol to be toxic to the cell. In vitro incubation of 25 μM CysK, 1 mM OALS, and 1 mM compound 1 recovered both compound 1 (501 ±18μM) and the predicted metabolite compound 12 (404 ±20 μM), whereas reactions lacking CysK did not form detectable false product (Supplementary Table 9). Comparisons of the false-product standard (12) and experimentally derived false product mass spectra fragmentation patterns confirm the structure of the false product (Supplementary Figure 4, Figure 6C).
Figure 6. LC-MS/MS bioanalytical studies of CysK-mediated false product formation.
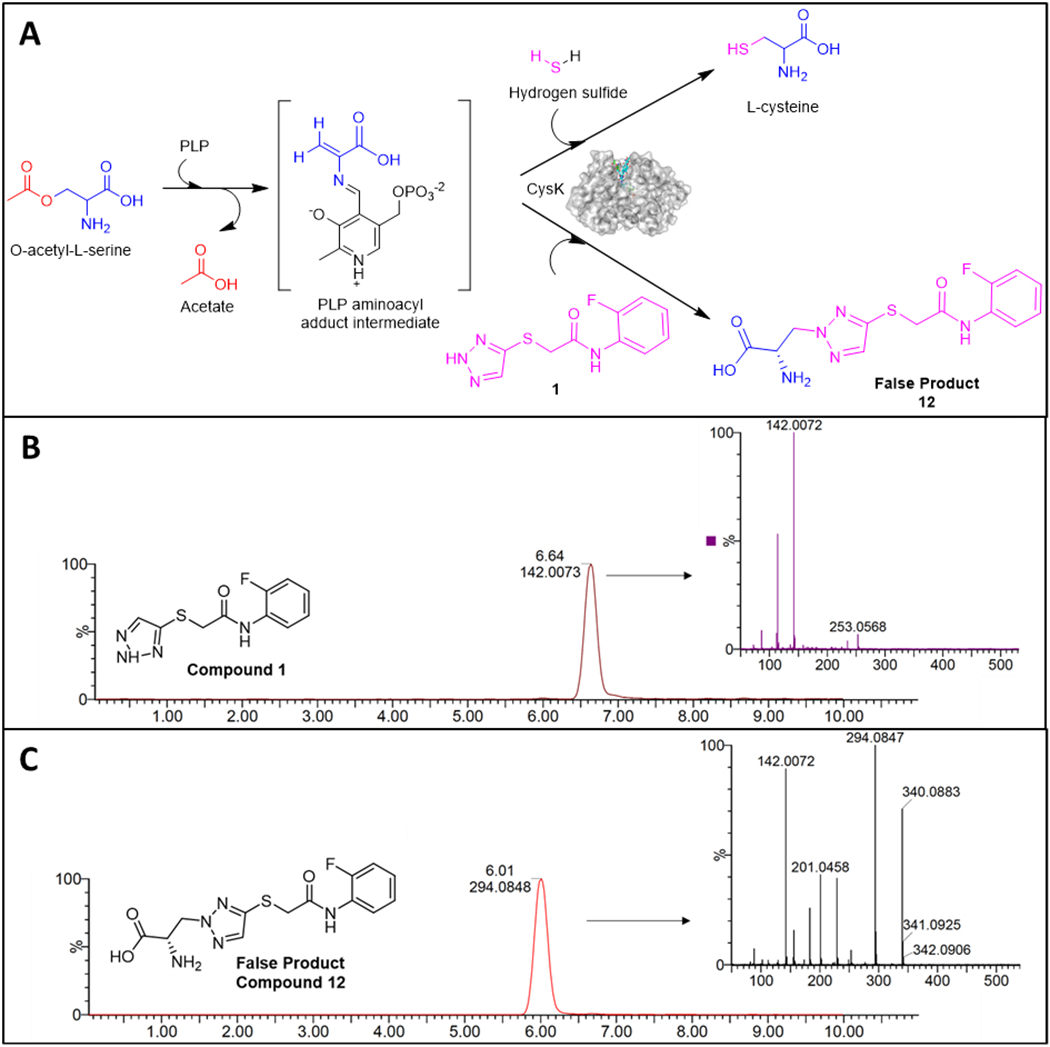
(A) The putative false product reaction mediated by CysK between OALS and compound 1 and LC-MS/MS spectra of E. coli cell extracts after treatment of compound 1 showing LC-MS/MS profile for (B) compound 1 and (C) the predicted false product (12) are shown.
Having confirmed the formation of the CysK-mediated false product (12) in vitro, the possibility of this event in bacterial cells was evaluated in E. coli BW25113 following treatment with compound 1. Drugging, lysis, and sample preparation for LC-MS/MS were adapted from previously described methods.32 Both parent compound (1) and false product (12) were recovered in high yields from compound 1-treated cells (Supplementary Table 9, Figures 6B and 6C). These data indicate that compound 1 accumulates in GN cells and is metabolized to the false product 12 in cellular preparations (Figures 6B and 6C). No product was detected in cells treated with the drug carrier DMSO. These experiments provided direct evidence that compound 1 acts as an antimetabolite resulting in the formation of the predicted false product 12 (Figure 6A) which follows previous studies on the CysK-mediated formation of a product between OALS and other nitrogen-containing heterocycles.14, 33
Transcriptional effects of TATs on bacterial metabolism
Antimetabolites can substantially disrupt the transcription of associated metabolic genes.34 The 1,2,4-triazole was previously described to downregulate transcription of genes within the RSAP by depleting cellular concentrations of OALS, which is a known activator of RSAP-associated genes.14, 35 To explore if compound 1 exhibits similar effects, RNA-Seq analyses were performed with E. coli cellular preparations treated with compound 1 in both M9 and M9T (M9 with taurine as the sole sulfur source) media. The resulting data was deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE142164. Comparison with treatment in M9T is required as the transcription of genes associated with sulfur metabolism varies significantly depending on the available sulfur source.36 Three major operons attributed to sulfur metabolic processes are the cys, ssu, and tau operons responsible for the RSAP, aliphatic sulfonate metabolism, and taurine metabolism, respectively. These operons are negatively regulated by adenosine 5’ phosphosulfate (APS), a derivative of inorganic sulfate, so the effect of compound 1 on sulfur metabolic processes may not be detectable in the standard M9 media that has magnesium sulfate as the sulfur source.
In the absence of drug, the expected transcriptional upregulation of genes associated with sulfur metabolism was observed in the M9T environment relative to M9 (Supplementary Figures 5A and 6).36 Treatment of E. coli with compound 1 in the M9T environment resulted in significant downregulation of the cys, ssu and taur operons, reflecting previously described effects of 1,2,4-triazole on S. typhimurium (Figure 7A, Supplementary Figure 5B).14, 35 TAT-induced dysregulation of genes in pathways linked to the biosynthesis of several amino acids, the shikimate pathway, and nucleotide biosynthesis was observed in both medias. The glycine cleavage system, which plays a key role in both serine and glycine catabolism and is highly interconnected with the folate cycle, was upregulated in M9 in response to treatment by compound 1. Notably, aromatic amino acid biosynthetic pathways were substantially downregulated, especially in M9 (Figure 7B, Supplementary Figure 5C). However, the only amino acids capable of antagonizing compound 1-induced growth inhibition are L-cysteine, L-serine, and L-methionine, suggesting the depletion of cysteine accounts for growth inhibition (Supplementary Table 7). In the absence of TAT, transcription of aromatic amino acid metabolic processes appears to be sensitive to the available sulfur source (Supplementary Figure 5A). Further experimentation is required to fully understand the relationship between aromatic amino acid metabolism, available sulfur sources and cysteine biosynthesis as there are multiple biological associations between these pathways as well as folate biosynthesis.37, 38, 39 These findings suggest the effects of dysregulating CysK function may extend beyond cysteine biosynthesis and disrupt several other metabolically significant pathways.
Figure 7. Pathway networks of E. coli RNA-seq analysis of genes transcriptionally altered by compound 1 treatment.
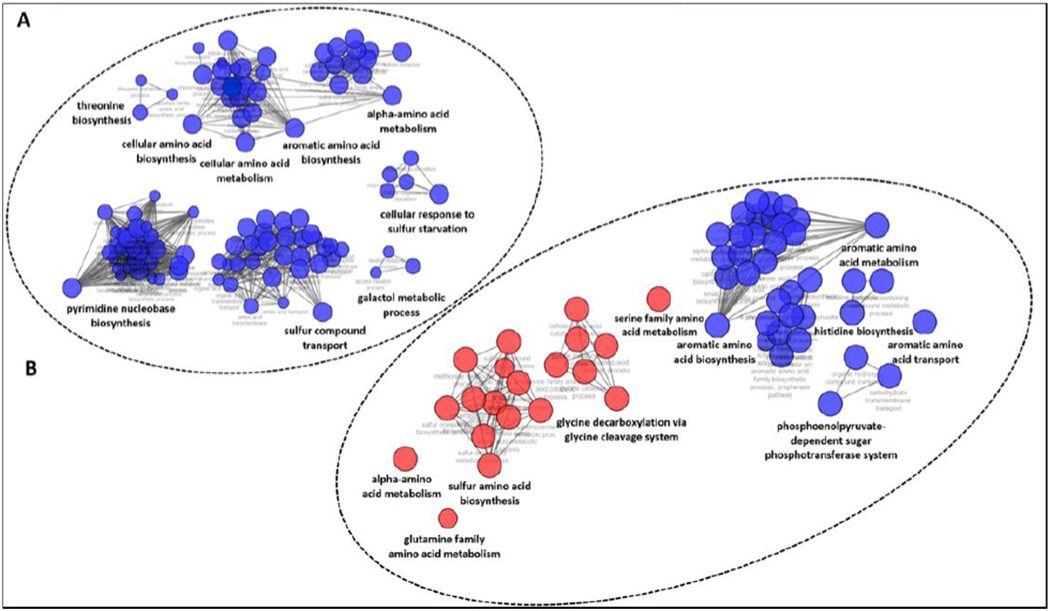
Only genes with at least a 2-fold change in expression and P-value ≤ 0.01 were chosen for the pathway analysis. Blue indicates downregulation and red indicates upregulation. (A) Affected pathway networks after compound 1 treatment in M9T (taurine as the sulfur source). (B) Affected pathway networks after treatment with compound 1 in M9 with magnesium sulfate as the sulfur source. Complete RNA-Seq data are available in NCBI GEO (GSE142164).
CONCLUSION
Bacterial metabolism is a promising avenue for new antibiotic discovery. Currently only a small number of clinically useful antibiotics target bacterial metabolism40, yet antifolates represent one of the most historically successful antibiotic classes and have broad-spectrum utility against a variety of infection types8. We hypothesized that identification of compounds that disrupt key metabolic pathways related to folate biosynthesis could lead to new selective antimetabolites active against Gram negative bacteria. Indeed, within this study a high-throughput screen to identify modulators of pathways related to folate biosynthesis resulted in the identification of previously uncharacterized TAT molecules which target CysK, disrupting cysteine metabolism in Escherichia coli. Multiple orthogonal approaches yielded strong evidence that the growth inhibitory mechanism of TATs is through interaction with the CysK enzyme. This enzyme is at a key juncture between several fundamental metabolic pathways and is absent in humans. Biophysical and biochemical studies demonstrated that the TATs bind to CysK and that the MOA is by false product formation. Rescue of growth inhibition observed with cysteine and glutathione implicates cysteine depletion as the cause of growth inhibition.
Notably, transcriptional analysis of TATs on bacterial metabolism revealed down-regulation of sulfur metabolism genes and disruption of several other key metabolic pathways. False product formation of TAT by CysK may deplete intracellular pools of OALS, which is a plausible explanation for the dramatic transcriptional effects on genes associated with sulfur metabolism. This could have high clinical relevance towards certain pathogens that rely heavily on sulfur to build the thiol group present in key redox defense molecules such as glutathione, mycothiol, and even cysteine itself. Beyond sulfur metabolism, the striking impact of the TATs on transcription of genes associated with aromatic amino acids merits further exploration. Associations between aromatic amino acid biosynthesis and pathways related to CysK have been noted in the existing literature.37–39 Consequently, investigations into the disruption of these pathways could yield highly effective and synergistic inhibitor combinations similar to antifolate combination therapies.
Altogether, this study has produced a new class of selective antimetabolites that act through the CysK enzyme and benefit from favorable drug like properties. The TAT series presents a clear SAR, is amenable to medicinal chemistry optimization, is non-cytotoxic, and is effective against both established E. coli laboratory strains and antifolate drug-resistant clinical isolates. The activity is however limited by antagonism by L-cysteine, which is found in biological matrices, precluding direct antimicrobial development. These efforts highlight the potential of antimetabolite drug discovery as a promising approach towards the discovery of novel Gram-negative antibiotics.
METHODS
Bacterial strains and culture conditions
Strain information and media conditions are listed in Supplementary Table 3A. M9 was made following the recipe for the M9-based minimal media ATCC 2511. For every liter of media, the M9 consisted of 200 mL of 5X M9 salts (Difco 248510), 20 mL of 20% glucose, 2 mL of 1 M MgSO4, 0.1 mL of CaCl2, and 0.1 mL of thiamine. M9AA-CMS consisted of the standard M9 recipe supplemented with all amino acids at concentrations used in Zlitni et al,26 with the exception of L-serine, L-methionine, and L-cysteine. SSM9PR was made as previously described.40
Primary screen
The Bioactives Library was purchased from Selleck in 2014, and the remaining three libraries (Lead-Like, Small Fragment, and Local DHPS Collection) were assembled from compounds available at the Chemical Biology and Therapeutics Department at St. Jude Children’s Research Hospital. Thirteen microliters of M9 media was dispensed into each well of a Nunc 384-well optical bottom plate with PerkinElmer BackSeal applied to the bottom of the plate to accommodate luminescence reads. Source plates containing approximately 10 mM compound were used to stamp 121 nL into the assay plate using a Biomek FXP Automated Liquid Handler. Columns 1, 2, 13, and 14 were reserved for controls, into which chloramphenicol or sulfamethoxazole were added from a control plate. A mid-log culture of E. coli K12 MG1655 was diluted to an OD600 of 0.001 in ATCC medium 2511 before dispensing 13 μL into each well of the drugged assay plates, resulting in 1.3 × 104 CFU per well. The final assay concentration for each compound was 50 µM and the final DMSO concentration was 0.47%.
After 16 hours of incubation at 37° C, 26 μL freshly prepared BacTiter-Glo™ was added to each well. The plates were allowed to shake for 4 minutes followed by a luminescence read within 30 minutes of adding the reagent. Four columns total were committed to controls, with two of these columns containing serial dilutions of both CAM and SMX. Therefore, MICs of these standard drugs could be determined within each plate, and the MIC of SMX was shifted by the addition of pABA and methionine. The other two columns containing alternating wells with either 50 μM CAM or 0.47% DMSO, representing full inhibition or full growth, and were used to calculate the Z-factor of each plate. The high-throughput data was analyzed using the Robust Interpretation of Screening Experiments (RISE) application, which was written with Pipeline Pilot (Accelrys, v. 8.5) using the R program.41
Minimal inhibitory concentration testing
Compound solutions were prepared in DMSO and serially diluted 1:2 in 100 μL/well in 96 well round-bottom plates. Bacteria were grown from a single colony to a mid-log OD600 and frozen in 0.5 mL aliquots. These aliquots were used to directly inoculate the assay plates by diluting to an OD600 of 0.001 and plating 100 μL per well, allowing for 105 CFU to be added to each well. After 16 hours of incubation, the MIC was determined as the lowest concentration to visually inhibit bacterial growth for all strains with the exception of M. tuberculosis, which was allowed to incubate for 7 days. Media conditions for each strain are listed in Supplementary Table 3A.
Spontaneous mutant generation and whole genome sequencing
M9 agar was prepared and compounds 1, 2, or 3 were added to individual preparations to achieve 100 μg/mL of each compound. E. coli K12 MG1655 was grown in M9 broth to 108 CFU/mL from a single colony. One mL of this culture was plated on each of three agar plates for each compound. Colonies were counted to calculate the resistance frequency.
Twelve colonies total were selected for further analyses. Five colonies were selected for each of compounds 2 and 3 while 2 colonies were picked for compound 1. Genomic DNA was extracted from overnight cultures of these mutants using the Qiagen DNeasy Blood and Tissue Kit. HiSeq Illumina sequencing was performed and the reads were mapped to the reference genome for the parent strain (GenBank U00096.3) using CLC Genomics Workbench. Variants were detected using fixed ploidy variant detection.
Generation and characterization of E. coli isogenic mutant panel
E. coli BW25113 (WT) and E. coli JW2407 (ΔcysK) were obtained from the Keio collection (Supplementary Table 3).30 The pPJ131 plasmid was kindly gifted from the laboratory of Dr. Charles Rock in the Infectious Diseases Department at St. Jude Children’s Research Hospital. This plasmid is derived from the Stratagene pBluescript plasmid and has the pPET28a multiple cloning site.42 The genes encoding E. coli K12 CysK WT protein (UniProt P0ABK5) as well as the CysK enzyme with observed spontaneous mutation S70C (cysKS70C) were generated and subcloned into NcoI/XhoI cloning site of the pPJ131 vector. The plasmids were electrotransformed into both E. coli WT and ΔcysK. Growth on carbenicillin indicated successful transformation with pPJ131 plasmids and the ΔcysK strains were cultured on kanamycin which is the selective agent for the Keio knockouts.30 The sequencing primers M13F, M13F43, M13R49, and T7 were used to confirm correct sequence of the constructs.
Growth curve studies were carried out in M9 media (ATCC 2511) and when necessary L-cysteine was supplemented at a final concentration of 10 µg/mL and compound 1 was added at a final concentration of 16 µg/mL. At least 2 replicates were included for each group. Each strain was streaked onto agar containing appropriate selective agent and allowed to grow overnight. Single colonies were resuspended to a mid-log OD600 between 0.3 and 0.7 and further diluted to a final OD600 of 0.001 to inoculate a 96 well round bottom plate. The plate was incubated at 37°C and the OD600 was read every 5 minutes for 24 hours for each well using a Pherastar FS microplate reader. The average OD600 read from at least three wells containing media only (no bacteria) was used to normalize the OD600 reads.
For the whole cell dose-response study with compound 1, the experiment was set up in a 96-well plate using the same methods for MIC determination. When necessary L-cysteine was added at a final concentration of 10 µg/mL. The OD600 was read at 24 hours and an average of three bioreplicates was used for determining the dose-response curves.
Cytotoxicity Assay
Cytotoxicity of compounds were assessed using Vero kidney epithelial cells (ATCC CCL-81) and HepG2 hepatocellular carcinoma cells (ATCC HB-8065). Cells were grown in monolayers, harvested using trypsin-EDTA (ATCC 30-2101) and seeded into white, flat-bottom 96-well plates (Nunc 136101, ThermoFisher) at 5000 cells/well (10-15% confluency). Vero cells were diluted in Dulbecco’s Modified Eagle’s Medium (Hyclone: DMEM/High Glucose) and HepG2 in Eagle’s Minimum Essential Medium (EMEM; ATCC 30-2003) both supplemented with 10% fetal bovine serum (ATCC-30-2020). Plates were incubated for 20 hours at 37 °C in the presence of 5% CO2. Compounds were solubilized and serially diluted 2-fold in DMSO followed by addition to respective media at 2x the final concentration. An equal volume of media containing compound was transferred to plates containing seeded cells using a Biomek FXP liquid handling robot (Beck-man Coulter, CA). Plates were incubated for 72 hours followed by viability measurements using Cell Titer-Glo Luminescent Cell Viability assay (G7572 Promega). Assay plates were measured using a PHERAstar FS Multilabel reader (BMG, Cary, NC) at a peak emission wavelength of 590nm. The raw data was normalized to no drug (DMSO only) wells at 100% viability standard. The data was plotted using a non-linear regression-based fitting of inhibition curves in GraphPad Prism version 7 (GraphPad Software, La Jolla CA). The computed IC50 was obtained from the curve where the concentration of test compound inhibited growth by 50%. Results are the average of two technical replicates and error is reported as SD.
Recombinant CysK Protein Expression and Purification
The cysK gene was cloned into pET28a with an N-terminal 6x histidine tag. This plasmid was transformed into BL21(DE3) E. coli. After growth of this strain to an OD600 of 0.5, the bacteria were induced by addition of 1 mM IPTG and overnight growth at 18° C. The induced culture was pelleted via centrifugation, resuspended in 25 mM Tris-HCl, 250 mM NaCl, 1 mM PLP, and 2.5 mM imidazole, pH 8.0 (Buffer A). Addition of lysozyme followed by sonication allowed for lysis of the cell pellets. Cell debris were removed by high-speed centrifugation and the soluble fraction was passed through a 0.22 µM filter. The clarified lysate was run over a Nickel affinity column, washed with five column volumes of buffer A, and elution occurred through a gradient addition of Buffer B, which had the same components as Buffer A with an increased concentration of imidazole at 250 mM. Fractions containing CysK protein were identified via SDS-PAGE, pooled, and run over a HiPrep 26/60 Sephacryl S-300 HR in 50 mM HEPES, 150 mM NaCl, 1 mM PLP, and 1 mM DTT, pH 7.6. It was later determined that DTT could interfere with the normal functioning of the CysK enzyme, so the protein was equilibrated into buffer lacking DTT before all experiments.
Structure Search
A nearest neighbor structure search was performed on the entire St. Jude compound collection with structures from an initially identified set of active compounds, as queries. The MDL Public Keys,43 implemented in Pipeline Pilot,44 were used as molecular fingerprints for the nearest neighbor search. A Tanimoto Coefficient cutoff of 0.8 was used to limit the number of nearest neighbors identified from the search.
Spectroscopy
To ensure the PLP cofactor was fully incorporated into the CysK protein, CysK was allowed to gently rotate with 0.5 mM PLP in 25 mM HEPES, 150 mM NaCl, pH 7.6 for 30 minutes. Since PLP exhibits its own spectroscopic profile, unbound cofactor was removed by then equilibrating CysK into the same buffer lacking PLP by washing with equivalent volumes three times in a 10,000 MWCO centricon. OALS was prepared in the same buffer, while all inhibitory molecules were prepared in DMSO. Final assay concentrations were 100 µM CysK and 500 µM for both substrate and inhibitors. The DMSO concentration for all samples was 1.25%.
Mass-spectrometry
For the in vitro analysis of false product formation, EcCysK was preincubated in PLP, washed, and filtered similarly to the spectroscopy methods, but NaCl was excluded from the assay buffer. 50 µM EcCysK was then incubated with 2 mM OALS to form the aminoacrylate complex for 5 minutes at room temperature. A solution of assay buffer and compound 1 was then added so that the final concentrations were 25 µM for EcCysK, 1 mM for OALS, and 1 mM for compound 1. The reaction was allowed to proceed for 30 minutes at room temperature with gentle rotation. Trifluoroacetic acid was added to a final percentage of 16.6% and the reactions were transferred to ice to stop the reaction. The reactions were run through a 10k centricon to remove CysK enzyme, and the resulting flow-through was used for LC-MS/MS analysis.
For the whole cell analysis of false product formation, E. coli BW25113 was grown to mid-log (OD600 of 0.6-0.8) in M9 media at 37°C in an incubator shaking at 225 RPM. The bacteria were pelleted at 4,500 xg for 10 min at 4 °C. Cells were washed and pelleted in PBS a total of two times. The cells were resuspended in PBS for a final time to 3.5mL per 100mL of cells initially cultured. Cells were allowed to equilibrate at 37°C for 5 minutes. 1mL of E. coli was incubated for 10 minutes with a final concentration of 0.5mM of compound 1 in DMSO shaking at 37°C. 800 μL of treated cells were layered over 700μL of supercooled (−78°C) of AR20 silicone oil and pelleted at 13,000 xg for 2 minutes. Once pelleted, the oil and water were carefully removed via pipette. To lyse cells, the pellets were resuspended in 200μL of HPLC grade water was subjected to three freeze-thaw cycles using liquid nitrogen and a 65°C water bath. Cell debris was separated from lysate by centrifugation at 13,000 xg for 2 minutes. 150 μL of extract was removed and set aside. The pellet was resuspended in the remaining 50 μL of water and once thoroughly resuspended, 100μL of HPLC grade methanol was added. The cells were once again pelleted, and lysates were combined. Lysates were pelleted for 10 minutes at 20,000 xg and then further filtered through a 0.22μm filter before injection on to LC-MS/MS.
Samples from both the in vitro and whole cell reactions were analyzed with system with a tandem Waters Acquity M Class series UPLC system and Xevo G2 QTof tandem MS/MS with Zspray. 0.1μl of extract was separated using a Waters C18 300Å (300μm x 150mm) column with solvent A, 0.1% formic acid in water, and solvent B, 0.1% formic acid in acetonitrile. The inlet method for these samples utilized a flow rate of 4 μL/minute with the following gradient: 0-5min, 99% solvent A, 1% solvent B; 5-7 min, 10% solvent A and 90% solvent B; 7-10 min, 99% solvent A. Tandem mass spectra were acquired with a cone voltage of 20V and a collision energy of 20V. High resolution spectra were calibrated by co-infusion of 2 ng/mL leucine enkephalin (Waters lockspray). Data were quantified using Waters MassLynx software where the AUC was determined by integrating the corresponding daughter peak of the parent compound. Concentrations of the unknown compounds were determined by linear fit of corresponding standards. Concentrations are reported as the average of three biological replicates.
RNA-seq Analysis
Two medias were used for the gene expression analysis. M9 is the standard M9-based media ATCC2511 used in previous experiments, and M9T has the same media composition with taurine as the sulfur source. To make M9T, MgSO4 was omitted and MgCl2 was added at a final concentration of 2 mM. Taurine was added at a final concentration of 0.25 mM.36
A single colony of E. coli K12 MG1655 was picked into 5 mL M9 broth and grown overnight at 37°C, 225 RPM shaking. The next day, two 200 µL samples were taken from the overnight culture and pelleted at 3700xG for 10 minutes. The supernatant was removed, and one sample was washed in 1 mL M9 while another sample was washed in 1 mL M9T. The samples were pelleted as before and supernatant removed. The samples were resuspended in 100 µL of their respective medias and added to 100 mL of M9 or M9T. The cultures were allowed to grow to mid-log at 37°C, 225 RPM shaking. Five mL of mid-log culture was added to 50 mL conical tubes containing 50 µL DMSO or 50 µL of compound 1 at 1.56 mg/mL. The cultures were allowed to drug for 30 minutes at 37°C, 225 RPM shaking. Afterwards, 10 mL RNAprotect Bacteria Reagent was immediately added and mixed with the drugged cultures and pelleted at 5,000XG for 10 minutes. The supernatant was discarded, and the pellets were flash frozen. Each group (drugged and undragged in either M9 or M9T) was prepared in triplicate from three different colonies.
For RNA extraction, pellets of E. coli K12 were re-suspended in 100 µL of 1 mg/mL lysozyme and incubated at 37°C for 10 minutes with 5 seconds vortex every 2 minutes. Subsequent steps were carried out using the RNeasy extraction kit (Qiagen) according to protocol. RNA was eluted in 40-50 µL of supplied elution buffer. Residual genomic DNA contamination was depleted using RNA Clean and Concentrator kit (Zymo Research) following the manufacture’s protocol. Samples were eluted in 50 µL of DNase/Rnase-Free water. Quality of RNA samples were measured using an Agilent 2100 Bioanalyzer and RIN values were all > 7.0.
The ScriptSeq™ v2 RNA-Seq Library Preparation Kit was used to prepare the RNA-Seq library. Total RNA was sequenced as 100 bp paired end reads using Illumina HiSeq technology at the Hartwell Center at St. Jude Children’s Research Hospital. Ribosomal RNA sequences were filtered out using BBDuk44. The resulting reads were aligned to the reference Escherichia coli str. K-12 substr. MG1655 genome (NCBI) using Rockhopper 45 to generate the gene counts. Differential gene expression analysis was analyzed using the DESeq2 analysis package 46 in the R statistical computing environment from the raw gene count data of the protein coding sequences generated from Rockhopper. The raw and processed RNA-Seq data associated with this study has been deposited to NCBI GEO under accession number GSE142164.
Supplementary Material
Acknowledgements
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01AI136803, and by ALSAC, St. Jude Children’s Research Hospital.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Health or other funding agencies.
References
- 1.Prevention, C. f. D. C. a. Antibiotic resistance threats in the United States; U.S. Department of Health and Human Services: 2013. [Google Scholar]
- 2.Cassini A; Högberg LD; Plachouras D; Quattrocchi A; Hoxha A; Simonsen GS; Colomb-Cotinat M; Kretzschmar ME; Devleesschauwer B; Cecchini M; Ouakrim DA; Oliveira TC; Struelens MJ; Suetens C; Monnet DL; Group, B. o. A. C., Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: a population-level modelling analysis. Lancet Infect Dis 2019, 19 (1), 56–66. DOI: 10.1016/S1473-3099(18)30605-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith PA; Koehler MFT; Girgis HS; Yan D; Chen Y; Crawford JJ; Durk MR; Higuchi RI; Kang J; Murray J; Paraselli P; Park S; Phung W; Quinn JG; Roberts TC; Rougé L; Schwarz JB; Skippington E; Wai J; Xu M; Yu Z; Zhang H; Tan MW; Heise CE, Optimized arylomycins are a new class of Gram-negative antibiotics. Nature 2018, 561 (7722), 189–194. DOI: 10.1038/s41586-018-0483-6. [DOI] [PubMed] [Google Scholar]
- 4.Prevention, C. f. D. C. a., Outpatient Antibiotic Prescriptions — United States, 2016. 2016. [Google Scholar]
- 5.Hicks LA; Bartoces MG; Roberts RM; Suda KJ; Hunkler RJ; Taylor TH; Schrag SJ, US outpatient antibiotic prescribing variation according to geography, patient population, and provider specialty in 2011. Clin Infect Dis 2015, 60 (9), 1308–16. DOI: 10.1093/cid/civ076. [DOI] [PubMed] [Google Scholar]
- 6.Domagk G, Chemotherapie der bakteriellen infektionen. Angewandte Chemie 1935, 48, 657–667. DOI: 10.1002/ange.19350484202. [DOI] [Google Scholar]
- 7.Yun MK; Wu Y; Li Z; Zhao Y; Waddell MB; Ferreira AM; Lee RE; Bashford D; White SW, Catalysis and sulfa drug resistance in dihydropteroate synthase. Science 2012, 335 (6072), 1110–4. DOI: 10.1126/science.1214641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bermingham A; Derrick JP, The folic acid biosynthesis pathway in bacteria: evaluation of potential for antibacterial drug discovery. Bioessays 2002, 24 (7), 637–48. DOI: 10.1002/bies.10114. [DOI] [PubMed] [Google Scholar]
- 9.Hondorp ER; Matthews RG, Methionine. EcoSal Plus 2006, 2 (1). DOI: 10.1128/ecosalplus.3.6.1.7. [DOI] [PubMed] [Google Scholar]
- 10.Schnell R; Sriram D; Schneider G, Pyridoxal-phosphate dependent mycobacterial cysteine synthases: Structure, mechanism and potential as drug targets. Biochim Biophys Acta 2015, 1854 (9), 1175–83. DOI: 10.1016/j.bbapap.2014.11.010. [DOI] [PubMed] [Google Scholar]
- 11.Kredich N, Biosynthesis of cysteine. In Escherichia coli and Salmonella (ed. Neidhardt FC). ASM Press: 1996; pp 514–527. [Google Scholar]
- 12.Spyrakis F; Singh R; Cozzini P; Campanini B; Salsi E; Felici P; Raboni S; Benedetti P; Cruciani G; Kellogg GE; Cook PF; Mozzarelli A, Isozyme-specific ligands for O-acetylserine sulfhydrylase, a novel antibiotic target. PLoS One 2013, 8 (10), e77558 DOI: 10.1371/journal.pone.0077558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mozzarelli A; Bettati S; Campanini B; Salsi E; Raboni S; Singh R; Spyrakis F; Kumar VP; Cook PF, The multifaceted pyridoxal 5’-phosphate-dependent O-acetylserine sulfhydrylase. Biochim Biophys Acta 2011, 1814 (11), 1497–510. DOI: 10.1016/j.bbapap.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 14.Kredich NM; Foote LJ; Hulanicka MD, Studies on the mechanism of inhibition of Salmonella typhimurium by 1,2,4-triazole. J Biol Chem 1975, 250 (18), 7324–31. [PubMed] [Google Scholar]
- 15.Magalhães J; Franko N; Annunziato G; Pieroni M; Benoni R; Nikitjuka A; Mozzarelli A; Bettati S; Karawajczyk A; Jirgensons A; Campanini B; Costantino G, Refining the structure-activity relationships of 2-phenylcyclopropane carboxylic acids as inhibitors of O-acetylserine sulfhydrylase isoforms. J Enzyme Inhib Med Chem 2019, 34 (1), 31–43. DOI: 10.1080/14756366.2018.1518959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagpal I; Raj I; Subbarao N; Gourinath S, Virtual screening, identification and in vitro testing of novel inhibitors of O-acetyl-L-serine sulfhydrylase of Entamoeba histolytica. PLoS One 2012, 7 (2), e30305 DOI: 10.1371/journal.pone.0030305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mori M; Jeelani G; Masuda Y; Sakai K; Tsukui K; Waluyo D; Tarwadi;Watanabe Y; Nonaka K; Matsumoto A; Ōmura S; Nozaki T; Shiomi K, Identification of natural inhibitors of Entamoeba histolytica cysteine synthase from microbial secondary metabolites. Front Microbiol 2015, 6, 962 DOI: 10.3389/fmicb.2015.00962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jean Kumar VU; Poyraz Ö; Saxena S; Schnell R; Yogeeswari P; Schneider G; Sriram D, Discovery of novel inhibitors targeting the Mycobacterium tuberculosis O-acetylserine sulfhydrylase (CysK1) using virtual high-throughput screening. Bioorg Med Chem Lett 2013, 23 (5), 1182–6. DOI: 10.1016/j.bmcl.2013.01.031. [DOI] [PubMed] [Google Scholar]
- 19.Poyraz O; Jeankumar VU; Saxena S; Schnell R; Haraldsson M; Yogeeswari P; Sriram D; Schneider G, Structure-guided design of novel thiazolidine inhibitors of O-acetyl serine sulfhydrylase from Mycobacterium tuberculosis. J Med Chem 2013, 56 (16), 6457–66. DOI: 10.1021/jm400710k. [DOI] [PubMed] [Google Scholar]
- 20.Singh S; Sablok G; Farmer R; Singh AK; Gautam B; Kumar S, Molecular dynamic simulation and inhibitor prediction of cysteine synthase structured model as a potential drug target for trichomoniasis. Biomed Res Int 2013, 2013, 390920 DOI: 10.1155/2013/390920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Westrop GD; Goodall G; Mottram JC; Coombs GH, Cysteine biosynthesis in Trichomonas vaginalis involves cysteine synthase utilizing O-phosphoserine. J Biol Chem 2006, 281 (35), 25062–75. DOI: 10.1074/jbc.M600688200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salsi E; Bayden AS; Spyrakis F; Amadasi A; Campanini B; Bettati S; Dodatko T; Cozzini P; Kellogg GE; Cook PF; Roderick SL; Mozzarelli A, Design of O-acetylserine sulfhydrylase inhibitors by mimicking nature. J Med Chem 2010, 53 (1), 345–56. DOI: 10.1021/jm901325e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amori L; Kathevica S; Bruno A; Campanini B; Felici P; Mozzarelli A; Constantino G, Design and synthesis of trans-2-substituted-cyclopropane-1-carboxylic acids as the first non-natural small molecule inhibitors of O-acetylserine sulfhydrylase. Med. Chem. Commun: 2012; Vol. 3, pp 1111–1116. DOI: 10.1039/C2MD20100C. [DOI] [Google Scholar]
- 24.Huang B; Vetting MW; Roderick SL, The active site of O-acetylserine sulfhydrylase is the anchor point for bienzyme complex formation with serine acetyltransferase. J Bacteriol 2005, 187 (9), 3201–5. DOI: 10.1128/JB.187.9.3201-3205.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Campanini B; Speroni F; Salsi E; Cook PF; Roderick SL; Huang B; Bettati S; Mozzarelli A, Interaction of serine acetyltransferase with O-acetylserine sulfhydrylase active site: evidence from fluorescence spectroscopy. Protein Sci 2005, 14 (8), 2115–24. DOI: 10.1110/ps.051492805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zlitni S; Ferruccio LF; Brown ED, Metabolic suppression identifies new antibacterial inhibitors under nutrient limitation. Nat Chem Biol 2013, 9 (12), 796–804. DOI: 10.1038/nchembio.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao Y; Shadrick WR; Wallace MJ; Wu Y; Griffith EC; Qi J; Yun MK; White SW; Lee RE, Pterin-sulfa conjugates as dihydropteroate synthase inhibitors and antibacterial agents. Bioorg Med Chem Lett 2016, 26 (16), 3950–4. DOI: 10.1016/j.bmcl.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson PM; Beck CM; Morse RP; Garza-Sánchez F; Low DA; Hayes CS; Goulding CW, Unraveling the essential role of CysK in CDI toxin activation. Proc Natl Acad Sci U S A 2016, 113 (35), 9792–7. DOI: 10.1073/pnas.1607112113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schnell R; Oehlmann W; Singh M; Schneider G, Structural insights into catalysis and inhibition of O-acetylserine sulfhydrylase from Mycobacterium tuberculosis. Crystal structures of the enzyme alpha-aminoacrylate intermediate and an enzyme-inhibitor complex. J Biol Chem 2007, 282 (32), 23473–81. DOI: 10.1074/jbc.M703518200. [DOI] [PubMed] [Google Scholar]
- 30.Baba T; Ara T; Hasegawa M; Takai Y; Okumura Y; Baba M; Datsenko KA; Tomita M; Wanner BL; Mori H, Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2006, 2, 2006.0008 DOI: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palmer AC; Kishony R, Opposing effects of target overexpression reveal drug mechanisms. Nat Commun 2014, 5, 4296 DOI: 10.1038/ncomms5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richter MF; Drown BS; Riley AP; Garcia A; Shirai T; Svec RL; Hergenrother PJ, Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 2017, 545 (7654), 299–304. DOI: 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ikegami F; Kaneko M; Kamiyamalsamu H; Murakoshi I, Purification and characterization of cysteine synthase from Citrullus vulgaris. Phytochemistry: 1988; Vol. 27, pp 697–701. [Google Scholar]
- 34.Nixon MR; Saionz KW; Koo MS; Szymonifka MJ; Jung H; Roberts JP; Nandakumar M; Kumar A; Liao R; Rustad T; Sacchettini JC; Rhee KY; Freundlich JS; Sherman DR, Folate pathway disruption leads to critical disruption of methionine derivatives in Mycobacterium tuberculosis. Chem Biol 2014, 21 (7), 819–30. DOI: 10.1016/j.chembiol.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 35.Turnbull AL; Surette MG, L-Cysteine is required for induced antibiotic resistance in actively swarming Salmonella enterica serovar Typhimurium. Microbiology 2008, 154 (Pt 11), 3410–9. DOI: 10.1099/mic.0.2008/020347-0. [DOI] [PubMed] [Google Scholar]
- 36.Gyaneshwar P; Paliy O; McAuliffe J; Popham DL; Jordan MI; Kustu S, Sulfur and nitrogen limitation in Escherichia coli K-12: specific homeostatic responses. J Bacteriol 2005, 187 (3), 1074–90. DOI: 10.1128/JB.187.3.1074-1090.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dosselaere F; Vanderleyden J, A metabolic node in action: chorismate-utilizing enzymes in microorganisms. Crit Rev Microbiol 2001, 27 (2), 75–131. DOI: 10.1080/20014091096710. [DOI] [PubMed] [Google Scholar]
- 38.Yang J; Ogawa Y; Camakaris H; Shimada T; Ishihama A; Pittard AJ, folA, a new member of the TyrR regulon in Escherichia coli K-12. J Bacteriol 2007, 189 (16), 6080–4. DOI: 10.1128/JB.00482-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nikiforova VJ; Bielecka M; Gakière B; Krueger S; Rinder J; Kempa S; Morcuende R; Scheible WR; Hesse H; Hoefgen R, Effect of sulfur availability on the integrity of amino acid biosynthesis in plants. Amino Acids 2006, 30 (2), 173–83. DOI: 10.1007/s00726-005-0251-4. [DOI] [PubMed] [Google Scholar]
- 40.Haselbeck R; Wall D; Jiang B; Ketela T; Zyskind J; Bussey H; Foulkes JG; Roemer T, Comprehensive essential gene identification as a platform for novel anti-infective drug discovery. Curr Pharm Des 2002, 8 (13), 1155–72. DOI: 10.2174/1381612023394818. [DOI] [PubMed] [Google Scholar]
- 41.Team, R. C., R: a language and environment for statistical computing. R Foundation for Statistical Computing: 2013. [Google Scholar]
- 42.Yao J; Ericson ME; Frank MW; Rock CO, Enoyl-Acyl Carrier Protein Reductase I (FabI) Is Essential for the Intracellular Growth of Listeria monocytogenes. Infect Immun 2016, 84 (12), 3597–3607. DOI: 10.1128/IAI.00647-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Durant JL; Leland BA; Henry DR; Nourse JG, Reoptimization of MDL keys for use in drug discovery. J Chem Inf Comput Sci 2002, 42 (6), 1273–80. [DOI] [PubMed] [Google Scholar]
- 44.Bushnell B BBTools. [Google Scholar]
- 45.McClure R; Balasubramanian D; Sun Y; Bobrovskyy M; Sumby P; Genco CA; Vanderpool CK; Tjaden B, Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res 2013, 41 (14), e140 DOI: 10.1093/nar/gkt444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Love MI; Huber W; Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014, 15 (12), 550 DOI: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.