

Abstract
Background and aims:
We recently showed that miR-223–3p on high-density lipoproteins (HDL) is exported to endothelial cells, where it inhibits inflammation. However, the origin of miR-2233p on HDL is unknown. We hypothesize that HDL-associated miR-223–3p originates in myeloid cells and is exported to HDL in a scavenger receptor BI (SR-BI)-dependent manner.
Methods:
Polymorphonuclear neutrophils (PMNs) and human monocyte derived macrophages (HMDMs) were incubated with native HDL (nHDL) or discoidal reconstituted HDL (rHDL). Total RNA was isolated before and after incubation. Mature and primary miR-223–3p (pri-mir-223–3p) levels were quantified by real-time PCR.
Results:
Incubation with nHDL and rHDL increased miR-223–3p export from PMNs and HMDMs. In PMNs, nHDL but not rHDL, increased mature and pri-mir-223–3p. Incubation with HDL also increased Dicer mRNA, a critical regulator of miRNA biogenesis. Incubation of HMDMs with nHDL did not increase cellular levels of mature miR-223–3p, but significantly increased pri-mir-223 levels. Incubation with rHDL had no effect on either mature or pri-mir-223–3p levels.
Activated PMNs increased miR-223–3p export to HDL and production of reactive oxygen species and activated protein kinase C. Blocking HDL binding to SR-BI increased miR-223–3p export to HDL in both PMNs and HMDMs, but did not affect mature and primary miR-223–3p levels. Chemical inhibition of cholesterol flux by Block Lipid Transport (BLT)-1 inhibited HDL-induced pri-mir-223 expression in PMNs.
Conclusions:
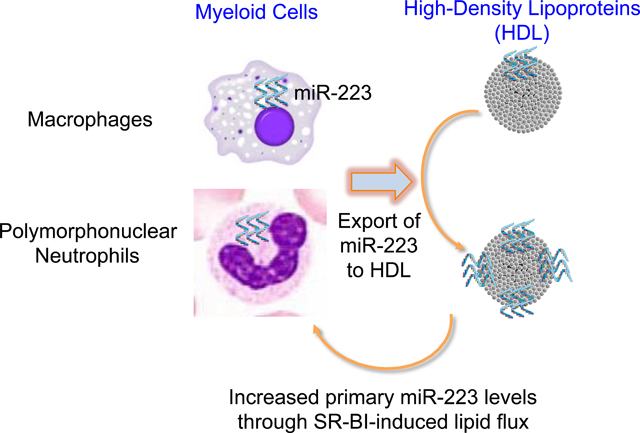
HDL-associated miR-223–3p originates in PMNs and macrophages. HDL stimulates miR-223–3p biogenesis in PMNs in a process that is regulated by SR-BI-mediated lipid flux.
Keywords: Polymorphonuclear neutrophils, High-Density Lipoproteins, microRNAs export, Inflammation-induced atherosclerosis
Graphical Abstract
Introduction
Increased plasma levels of myeloid cells, e.g. polymorphonuclear neutrophils (PMNs) and macrophages/monocytes, are associated with unstable coronary artery disease and increase in cardiovascular events(1–3). For example, PMNs and macrophages are present in atherosclerotic lesions after 4 weeks of high-fat diet in hypercholesterolemic mice, e.g. apolipoprotein E-null (Apoe−/−) mice(3, 4).
High-density lipoproteins (HDL) have many beneficial functions, including inhibition of inflammation and PMN activation(5–8). Infusions of HDL inhibited PMNs adhesion to vascular endothelium and prevented infiltration into the sub-intimal space(9). Activated PMNs are a key source of myeloperoxidase and hydrogen peroxide, which can modify HDL-associated lipids and apolipoproteins, including apolipoprotein A-I (apoA-I)(10). Although the role(s) of PMNs in plaque development are unclear, mutual repressive networks have emerged between HDLs and PMNs; HDL suppress PMN activation and PMNs promote HDL modifications causing HDL dysfunction(10).
Multiple reports support key roles for 22 nucleotide-long non-coding RNAs, namely, microRNAs (miRNAs), in reducing inflammation-induced atherosclerosis(11–13). miRNAs are transcribed as long primary transcripts, which are cleaved by Drosha, bound by its regulatory subunit DGCR8, to produce precursor miRNAs (pre-miRNAs) of ~60–70 nucleotides. Pre- miRNAs undergo a series of processing steps, including cleavage by the endoribonuclease Dicer, to produce single-stranded, mature miRNAs(13). We have recently shown that miRNAs are present in HDL and HDL-associated miRNAs decrease neutrophil adhesion to human arterial endothelial cells in vitro(14). miRNAs have proven to be key regulators of inflammatory cell phenotypes and functions. For example, PMNs, eosinophils, monocytes and macrophages highly express miR-223–3p, which regulates PMN progenitor development, hyperactivity, and recruitment during infection(15–18). We previously demonstrated that miR-223–3p is one of the most abundant miRNAs on HDL and is exported from J774 mouse macrophages to HDL(19). Furthermore, we found that HDL transfers miR-223–3p to recipient cells, including human coronary artery endothelial cells (HCAECs), human umbilical vein endothelial cells (HUVECs) and Huh7 hepatoma cells, and this transfer is dependent on the HDL receptor, scavenger receptor class B type I (SR-BI)(14, 19, 20). The transfer of HDL-miR-223–3p to HCAECs inhibited endothelial inflammatory genes such as intercellular adhesion molecule 1 (ICAM-1) (14). Multiple studies have shown that the up-regulation of ICAM-1 in endothelial cells is associated with sites of lesion formation (21). Taken together, these findings suggest that HDL-miR-223–3p plays an important role in the regression of atherosclerosis. Nonetheless, the mechanisms that govern cellular miR-223–3p export to HDL are largely unknown.
Since SR-BI is (i) an HDL receptor, (ii) a bidirectional transporter of cholesterol, and (iii) a critical regulator of HDL-associated miRNA uptake, we hypothesized that SR-BI contributes to miR-223–3p cellular export to HDL. In this study, we aimed to determine if PMNs and human monocyte-derived macrophages (HMDMs) export miR-223–3p to HDL and whether this process is dependent on SR-BI. Furthermore, we sought to determine if HDL induce miR-223–3p transcription in PMNs and if the export of miR-223–3p from PMNs impacts cellular miR-223–3p levels. The impact of PMN activation on HDL-miR-223–3p export was also investigated. Results from this study demonstrate that PMNs and HMDMs export miR-223–3p to HDL, and this is linked to HDL-induced primary-miR-223 (pri-mir-223) transcription. We also show that HDL-induced miR-223 transcription is regulated by SR-BI-induced lipid flux.
Materials and methods
HDL isolation:
Healthy volunteer blood samples (n=10) were collected into EDTA tubes. Plasma was isolated by centrifugation (3,000 × g for 10 min at 4 °C) and native HDL (nHDL) were isolated from plasma by density-gradient ultracentrifugation (DGUC) in the 1.063<d<1.25 g/ml density range and/or fast-protein liquid chromatography (FPLC), as previously described(22, 23). HDL fractions isolated from FPLC were determine by a cholesterol assay (Raichem, CLINIQA Corporation, USA), combined and concentrated. The total protein concentrations of isolated HDL samples were quantified using the bicinchoninic acid (BCA) assay (Thermo Scientific, USA) (PMID: 3843705). All isolated HDL samples were used individually and not pooled. All participants provided written consent, which was approved by the Human Research Ethics Committee of the University of New South Wales Australia (HC-13174).
Human primary cell isolation:
Human neutrophils were isolated from peripheral blood of healthy volunteers (n=10) using polymorphprep (AXIS, Olso, Norway)(7). Contaminating erythrocytes were lysed with red blood cell lysis incubation for 15 min at room temperature. Isolated neutrophils were suspended in RPMI 1640 containing 10% (v/v) FBS. All procedures were conducted at room temperature to avoid activating neutrophils (24, 25). Human monocytes were isolated from healthy donor buffy coat preparations (n=10) (New South Wales Red Cross) by density gradient centrifugation. The monocytes were differentiated into HMDMs for 7–9 days, as previously described(26). The miR-223–3p copy numbers were calculated using the following tools: https://www.thermofisher.com/au/en/home/references/ambion-tech-support/rna-tools-and-calculators/dna-and-rna-molecular-weights-and-conversions.html and http://scienceprimer.com/copy-number-calculator-for-realtime-pcr.
Tissue culture:
Freshly isolated human PMNs and HMDMs were cultured at a density of 1×106 cells/well (12-well plates) in serum-free RPMI 1640 medium. For each experiment, a total of 6 wells were used for each experiment (each well contained a final HDL protein concentration of 0.2–1 mg/ml). PMNs or HMDMs were incubated with phosphate buffered saline (PBS), HDL (0.5–4 h for PMNs or 16 h for HMDMs) or discoidal reconstituted HDL (rHDL) for 4 h. After incubation, cells were harvested and medium from the 6 wells was pooled for HDL or rHDL isolation. PMNs were activated with 1 μM N-formyl-L-methionyl-L-leucyl-phenylalanine (fMLP, ab141806, Abcam) for 45 min at 37 °C, washed with PBS and then incubated for 4 h with HDL (final protein concentration 1 mg/ml). To evaluate SR-BI involvement, cells were pre-incubated (1 h at 37 °C) with either 10 μM Block Lipid Transport-1 (BLT-1, SML0059, Sigma-Aldrich) or DMSO (vehicle); rabbit anti-SR-BI blocking antibody (1:200 dilution, NB400–113, Novus Biological) or rabbit IgG (vehicle). After pre-incubations, PMNs or HMDMs were washed with PBS and incubated for 4 h (PMNs) or 16 h (HMDMs) with HDLs (final protein concentration 1 mg/ml). All experiments were repeated ≥3 times using multiple independent donors of human neutrophils and HMDMs.
Transcriptomics:
Total RNA was isolated using Qiazol miRNAEasy kits (Qiagen), as previously described(14) and quantified by spectrophotometry (Nanovue). For HDL and rHDL samples, total RNA was isolated from approximately 100 μg of total protein from DGUC-HDL or sequential DGUC-HDL followed by FPLC-HDL, and 20 μg of FPLC-HDL pre and post cell treatment. For HDL and rHDL samples, Caenorhabditis elegans miR-39 was spiked in after the Qiazol step for normalization. Total RNA was reverse transcribed using TaqMan microRNA reverse transcription kit (Applied Biosystems- Catalogue number 4366596) and high capacity RNA-to- cDNA kit (Applied Biosystems- Catalogue number 4387406) according to manufacturer’s protocol for mature miRNA and primary miRNA/mRNA, respectively. For real-time PCR quantification, reverse transcription product was used with TaqMan miRNA assays (Applied Biosystems) and TaqMan pri-miRNA assays (Applied Biosystems) for quantification of mature miR-223 (Assay ID: 002295) and primary miRNA (Assay ID: Hs03303017_pri), respectively. All real-time PCR values expressed as 2-(CT[miR−223]-CT[control]) were normalized to appropriate controls (Applied Biosystems): U6 (for cellular miR-223 quantification; Assay ID: 001093), β-actin (for pri-miR-223 quantification; Catalogue number: 4333762F), and cel-miR-39 or arbitrary 32 (for HDL and rHDL miR-223 quantification; Assay ID: 000200). For HDL or rHDL miRNA expression, the real-time PCR was normalized to the HDL total protein concentration determined by BCA assay.
Statistics:
Data were presented as mean±SEM. Groups were compared using one-way ANOVA or unpaired two-tailed Student’s t test as appropriate (Prism). Bonferroni multiple comparison tests were used to compensate for multiple testing. A p value of <0.05 was considered to be significant.
Results
HDL induces miR-223 transcription and export in PMNs
miR-223–3p is highly expressed in PMNs, with 1.0×104 ±1.1×102 copies per cell for freshly isolated PMNs from healthy donors. Incubation of nHDL with PMNs for 4 h significantly increased HDL-miR-223–3p levels at nHDL concentrations of 0.5 mg/ml (p<0.05) and 1 mg/ml (p<0.0001) (Fig. 1A). Time dependent experiments showed that miR-223–3p levels in nHDL were increased after 4 h of incubation with PMNs (Fig. 1B). To demonstrate that the DGUC- purified nHDL were not contaminated with extracellular vesicles (EV) such as exosomes, immunoblotting for known EV markers was performed on isolated nHDL with whole plasma and cell lysates as controls. Although EVs have been reported with similar density to HDL, we did not observe EV markers, including ALIX, TSG101, Flotilin-1, and HSP70, in isolated nHDL (Supplementary Fig.1). Furthermore, incubations of HDL isolated from plasma by FPLC or DGUC followed by FPLC (DGUC>FPLC) with PMNs for 4 h significantly increased HDL-miR-223–3p levels (Supplementary Fig.2). Similar to nHDL, rHDL incubations with PMNs for 4 h significantly increased HDL-miR-223–3p levels (p<0.001) (Fig. 1B). To determine if the export of miR- 223–3p to HDL changed cellular miR-223–3p levels, miR-223–3p was quantified in PMNs at each time point. HDL at 1 mg/ml protein concentration significantly increased cellular miR-223–3p levels (Fig. 1C, p<0.0001) and miR-223–3p levels were increased by 2.0±0.1-fold after 4 h of incubation (Fig. 1D, p<0.0001). To assess if the increase in cellular miR-223–3p levels is linked to HDL-induced changes to pri-mir-223 levels, PMNs were incubated with HDL (0.2–1 mg/ml) at multiple time-points. At a concentration of 1 mg/ml, nHDL significantly increased pri- mir-223 levels at each time-point e.g. 2.8±0.2-fold at 0.5 h (p<0.0001) and 2.4±0.2-fold at 4 h (p<0.0001) (Fig. 1E and F). It appears that the simple contact of HDL with PMNs can lead to the export of miR-223–3p from PMNs to HDL independently of HDL concentration. However, higher HDL protein concentrations (1 mg/ml) may lead to increased mobilization and export of miR-223–3p pool in PMNs, and therefore increased miR-223–3p transcription. The up-regulation of pri-mir-223–3p with a high HDL protein concentration (1 mg/ml) is associated with increased mature miR-223–3p levels in PMNs. Taken together, these results suggest that HDL increases cellular miR-223–3p biogenesis and export from PMNs. However, rHDL (1 mg/ml) were able to increase mature, but not primary miR-223–3p levels in PMNs (Fig. 1D and F).
Fig. 1.

Real-time PCR quantification.
(A and B) Real-time PCR quantification of HDL or rHDL-associated miR-223 levels pre- and post-incubation with PMNs (rHDL concentration 1 mg/ml; incubation time 4 h) reported as fold-change of relative quantitative values (normalized to cel-miR-39 and HDL or rHDL total protein concentration). (C and D) Real-time PCR quantification of mature miR-223–3p levels in PMNs after 0.5–4 h incubation with PBS, HDL (0.2–1 mg total protein/ml) or rHDL (1 mg/ml for 4 h). (E and F) Primary miR-223 (pri-mir-223) levels in PMNs incubated for 0.5–4 h with PBS, HDL (0.2–1 mg total protein/ml) or rHDL (1 mg/ml for 4 h) quantified by real-time PCR (n≥3). Data are presented as mean ±SEM. *p<0.05, **p<0.01, ****p<0.0001.
PMN activation promotes HDL-miR-223–3p export
Studies have shown that activated PMNs, through the production of reactive oxygen species and activation of protein kinase C (PKC), contribute to atherosclerosis development(27). To confirm that fMLP increases both superoxide anion production and PKC activation in PMNs, cells were incubated with fMLP or PBS and superoxide anion production and PKC activation were measured. Both superoxide anion generation and PKC activity were significantly increased in fMLP-activated PMNs (Fig.s 2A and B). To determine if PMN activation impacts miR-223–3p export to HDL, HDL-miR-223–3p levels were measured before and after HDL incubation with fMLP-activated PMNs. Strikingly, HDL-miR-223–3p export was significantly increased >2-fold with fMLP activation (13.1±3.4-fold, p<0.0001) compared to PBS-treated non-activated PMNs (6.3±1.0-fold, p<0.05) (Fig. 2C). The increased miR-223–3p export to HDL from fMLP-activated cells was accompanied by a significant reduction in cellular mature miR-223–3p levels compared to the PBS-treated non-activated PMNs (Fig. 2D). Furthermore, HDL treatments significantly increased pri-mir-223 levels in both activated and non-activated PMNs with no significant difference between the cell states (Fig. 2E). Activation of PMNs did not alter the ability of HDL to increase the mature miR-223–3p and pri-mir-223 levels (Fig.s 2D and E). Mature miR-223–3p and pri-mir-223 changes in both activated and non-activated PMNs after HDL incubations were accompanied by increases in Dicer mRNA, a critical regulator of miRNA biogenesis (13) (Fig. 2F). These results suggest that miR-223–3p export from PMNs is not directly linked to miR-223–3p biogenesis or cellular mature miR-223–3p levels. Nevertheless, these results show that activated PMNs are more likely to export miR-223 to HDL compared to non-activated PMNs.
Fig. 2.

Superoxide anion production and PKC activity were assessed in PMNs incubated for 45 min at 37° C with PBS or fMLP (1 μM) (A and B). PMNs were pre-incubated with PBS or fMLP (1 μM) then incubated for 4 h with HDLs (1 mg protein/ml). HDL-associated miR-223 levels pre- and post-incubation (C), intracellular mature miR-223–3p levels (D), pri-miR-223 levels (E), and Dicer mRNA levels (F) were quantified by real-time PCR with relative quantitative values reported as fold-change (N≥3). Data are presented as mean ±SEM (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
SR-BI antagonizes HDL-miR-223–3p export
HDL’s primary receptor, SR-BI, mediates selective uptake of cholesteryl esters (CE) from HDL and free cholesterol efflux to HDL from multiple cell types, including myeloid cells(28–30). We have previously reported that HDL delivery of miR-223–3p to Huh7 hepatoma cells was dependent on SR-BI(19). Nonetheless, it is currently unknown if SR-BI also regulates miRNA export from PMNs to HDL. Using real-time PCR, we showed that SR-BI was expressed in PMNs; however, the PMN SR-BI levels were lower than in HMDMs (Fig. 3A). Agarose gel electrophoresis was used to conform SR-BI primers specificity and purity (Supplementary Fig.3). To determine if PMN HDL-miR-223–3p export is dependent on HDL binding to SR-BI, PMNs were pre-incubated with an SR-BI blocking antibody (31) and HDL-miR-223–3p levels were quantified by real-time PCR before and after incubation with PMN for 4 h. Remarkably, SR-BI blocking antibodies significantly increased PMNs miR-223–3p export to HDL (16.1±4.7fold, p<0.001) over pre-incubation, while the non-immune IgG control only increased HDLmiR-223–3p levels 4.9±1.1-fold compared to pre-incubation (p<0.05) (Fig. 3B). These results suggest that SR-BI represses miRNA export to HDL from PMNs. To determine if HDL induction of PMNs miR-223–3p expression is mediated through SR-BI binding, cellular miR-223–3p and pri-mir-223 levels were quantified by real-time PCR. Most interestingly, inhibition of HDL binding to SR-BI with blocking antibodies failed to attenuate HDL-induced expression of both pri-mir-223 and mature miR-223–3p as compared to IgG control treatments (Fig.s 3C and D). These results suggest that HDL-induced increase in mature and primary miR-223 levels in PMNs is not mediated by HDL binding to SR-BI, SR-BI associated cell signaling or HDL-CE uptake.
Fig. 3.

SR-BI mRNA levels in HMDMs and PMNs were determined by real-time PCR and normalized to β-actin (A). PMNs were pre-incubated for 1 h at 37 °C with IgG or SR-BI blocking antibody (1:200 dilution) then 4 h incubation with HDL (1 mg protein/ml). HDL-associated miR-223 levels (B), intracellular mature miR-223 levels (C) and intracellular pri-mir-223 levels (D) were quantified by real-time PCR and relative quantitative values are reported as fold-change (N≥3). Data are presented as mean ±SEM (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
miR-223–3p export from PMNs to HDL is not dependent on SR-BI-mediated cholesterol flux
HDL binding to SR-BI and SR-BI-mediated bidirectional cholesterol flux are two distinct processes that can be separated by the use of BLT-1, which selectively increases HDL binding to SR-BI and inhibits SR-BI-mediated bidirectional cholesterol flux(30, 32, 33). To determine if miR-223–3p export from PMNs to HDL is dependent on SR-BI-mediated lipid flux, PMNs were pre-incubated with BLT-1 for 1 h prior to incubation with HDL for 4 h. BLT-1 treatment of PMNs significantly increased miR-223 export to HDL compared to pre-treatment with the control vehicle (DMSO) (Fig. 4A). These results suggest that PMNs miRNA export to HDLs is independent from SR-BI-mediated cholesterol efflux and selective uptake of HDL-CE. To determine if HDL-induced expression of mature miR-223–3p is associated with SR-BI-mediated cholesterol flux, real-time PCR was used to quantify mature miR-223 levels in PMNs treated with BLT-1 or vehicle control. HDL-induced miR-223–3p expression significantly increased, not decreased, with chemical inhibition of SR-BI cholesterol transfer (BLT-1) (Fig. 4B). To determine if HDL-induced miR-223 biogenesis in PMNs is dependent on SR-BI-mediated lipid transfer, pri-mir-223 levels were quantified by real-time PCR in HDL-treated PMNs with BLT-1 or vehicle control treatments. BLT-1 treatments attenuated HDL induced pri-mir-223 levels (Fig. 4C). These results suggest that PMN export of miR-223–3p to HDL and HDL-induced increase in mature miR-223–3p levels are not dependent on HDL-binding to SR-BI or SR-BI-mediated cholesterol flux. Nonetheless, these results also suggest that HDL-induced pri-mir-223 expression is linked to SR-BI-mediated cholesterol flux.
Fig. 4.

PMNs were pre-incubated for 1 h at 37 °C with 10 μM BLT-1 or DMSO, then incubated for 4 h with HDLs (1 mg protein/ml). HDL-associated miR-223 levels (A), intracellular mature miR-223 levels (B) and intracellular pri-mir-223 levels (C) were quantified by real-time PCR with relative quantitative values reported as fold-change (N≥3). Data are presented as mean ±SEM (*p<0.05, **p<0.01, ***p<0.001).
HMDMs export miR-223 to HDL
To determine if other myeloid lineage cells export miR-223–3p to HDL and if HDL promote miR-223 expression in these cells, HMDMs were tested. miR-223–3p was found to be highly-abundant in HMDMs at 7.3×103 ±7.9×102 miR-223–3p copies per cell. Similar to PMNs, HMDMs were found to export miR-223–3p to HDL, as HDL-miR-223–3p levels significantly increased by 3.5±1.1-fold after 16 h incubation compared to before (p<0.05) (Fig. 5A). Furthermore, HMDMs were also found to export miR-223–3p to rHDL (Fig. 5A). Nevertheless, in HMDMs, HDL treatments did not increase cellular levels of mature miR-223–3p (Fig. 5B). To determine if HDL promotes pri-miR-223 expression in HMDMs, pri-mir-223 levels were quantified by real- time PCR after 16 h HDL or PBS treatments. HDL treatments significantly increased pri-mir-223 levels by 1.3±0.09-fold (p<0.01) (Fig. 5C), but rHDL incubations had no effect on either mature or primary miR-223–3p levels in HMDMs (Fig.s 5B and C). These results suggest that HDL have the capacity to accept miR-223–3p from multiple myeloid cell types, but HDL induced expression of pri-mir-223 and mature miR-223–3p are likely mediated by distinct mechanisms. To determine if miR-223–3p export from HMDMs to HDL is dependent on HDL binding to SR-BI, HMDMs were pre-incubated with a SR-BI blocking antibody and HDL miR-223–3p levels were quantified. HMDMs miR-223–3p export to HDL significantly increased with SR-BI blocking antibody treatment (Fig. 5D). To determine if blocking HDL binding to SR-BI affected cellular levels of mature miR-223–3p and pri-mir-223, mature and primary levels of miR-223–3p were measured in the presence or absence of SR-BI blocking antibody. Blocking HDL binding to SR-BI did not affect mature miR-223–3p or pri-mir-223 levels (Fig.s 5E and F).
Fig. 5.

HMDMs were incubated for 16 h with HDL or rHDL (1 mg total protein/ml). HDL and rHDL-associated miR-223 levels (A), intracellular mature miR-223 levels (B) and intracellular pri-mir-223 levels (C) were quantified by real-time PCR. HMDMs were pre-incubated for 1 h at 37 °C with IgG or SR-BI blocking antibody (1:200 dilution) then incubated for 16 h with HDL (1 mg protein/ml). HDL-associated miR-223 levels (D), intracellular mature miR-223 levels (E) and intracellular pri-mir-223 levels (F) were quantified by real-time PCR with relative quantitative values reported as fold-change (N≥3). Data are presented as mean ±SEM (*p<0.05, **p<0.01, ***p<0.001).
Discussion
miRNAs are present in all extracellular fluids, including plasma, and protected from degradation by packaging into apoptotic bodies, microvesicles, exosomes, lipoproteins (including HDL), and ribonucleoproteins(34). The role of extracellular miRNAs in atherosclerosis development has garnered much attention in recent years (35, 36). Interestingly, HDL may play important roles in these processes as HDL transfer miR-223–3p to HCAECs where they inhibit inflammation (14). Nevertheless, the cellular sources of miR-223–3p on circulating HDL are not fully understood. Although mature miR-223–3p is present in non-myeloid cell types, such as endothelial cells (14, 37), it is characterized as a myeloid-enriched miRNA and is highly expressed in PMNs, monocytes and macrophages (15). We have previously reported that mouse J774 macrophages export miR-223–3p to HDL in vitro (19); however, myeloid cell miR-223–3p export to HDL warrants further investigation. In the present study, we report that PMNs and HMDMs export miR-223–3p to native HDL and RNA-free rHDL, and thus support myeloid cells’ contribution to extracellular miRNAs on HDL. Most interestingly, activated PMN had increased miR-223–3p export to HDL, which may compliment their established role in atherosclerosis of releasing damaging reactive oxygen species and activating inflammatory processes (27, 38).
One limitation of this study is the lack of evidence on how SR-BI antagonizes miR-223–3p export to HDL, however, this study indicates that cholesterol and miRNA flux with HDL are inversely related. Both BLT-1 and SR-BI blocking antibody treatments increased miR-223–3p export from PMNs to HDL, while both treatments inhibit cholesterol flux. Interestingly, BLT-1 treatment increases HDL binding to the cell surface while inhibiting cholesterol flux through the receptor (33) and SR-BI blocking antibodies inhibit both HDL binding and the bidirectional cholesterol flux. This suggests potential of a secondary transporter or receptor independent of SR-BI or two distinct pathways for miR-223–3p export. For example, inhibition of miR-223–3p export could be linked to SR-BI-mediated signaling and transcriptional regulation of factors that modulate export. Nevertheless, both BLT-1 and SR-BI blocking antibody treatments significantly increased miR-223–3p export to HDL from PMNs (Fig.s 3B and 4B). Previously, we reported that J774 macrophage export of miR-223–3p is regulated by neutral sphingomyelinase 2 (nSMase2) or ceramide, as chemical inhibition of nSMase2 (GW4869) significantly increased miR-223–3p export to HDL (19). Furthermore, the export of miR-223–3p from macrophages to HDL is not likely regulated by ABCA1, as inducing ABCA1 expression with liver-X-receptor (LXR) activation failed to change miR-223–3p export (19). Therefore, the observed negative regulation of miR-223–3p export from neutrophils and macrophages by SR-BI suggests that the mechanism is likely distinct from other cholesterol transporters. Current evidence indicates that miRNA export to HDL is selective; however, it is not known how this miRNA selection occurs. This is an important point as multiple cell types may secrete specific miRNAs to HDL through processes that are regulated by undetermined mechanisms. Another limitation of the study is the biochemical mechanism of miR-223–3p exported including the physical transport across the plasma membrane. Nevertheless, results from this study provide insights into potential cellular sources of miR-223–3p on circulating HDL and the negative role of SR-BI in this process.
Within myeloid cells, miR-223–3p expression is regulated by multiple transcription factors and differentiation processes, but it is not known if the HDL mediated effects on both pri-mir-223 and mature miR-223–3p is through these pathways. HDL treatments increased the cellular levels of pri-mir-223 in PMNs, which may be due to HDL-induced transcription of primary miR-223, increased stability of pri-mir-223, or decreased activity of DGCR8 cleavage of pri-mir-223 to precursor miR-223 (pre-miR-223) in the nucleus. HDL treatments increased mature miR-223–3p levels and also increased Dicer mRNA levels. These results indicate that increased pri-mir-223 levels were not likely the result of deficient miRNA processing, but represent increased miR-223 transcription and biogenesis. However, the molecular mechanisms underlying these HDL effects are likely distinct as HDL-induced pri-mir-223 levels were dependent upon SR-BI-mediated lipid transfer whereas HDL-induced mature miR-223–3p levels were not dependent upon SR-BI activity. For example, chemical inhibition of cholesterol flux by BLT-1, but not SR-BI blocking antibodies, inhibited HDL-induced pri-mir-223 expression. Furthermore, HDL binding to the cell surface contributes to HDL-induced mature miR-223–3p levels since BLT-1 treatment significantly increased mature miR-223–3p levels irrespective of pri-mir-223 levels. Similar to PMNs, blocking HDL binding to SR-BI in HMDMs increased miR-223–3p export to HDL, but did not affect mature and primary miR-223–3p levels. Moreover, HDL-induced pri-mir-223 transcription may be specific to myeloid cells, as HDL treatments were not found to induce pri-mir-223 transcription in HCAECs or HUVECs, as we have previously shown (14). Our results further suggest that the up-regulation of pri-mir-223 in PMNs by HDL is not directly related to the export of miR-223–3p to HDL, as evidenced by the fact that HDL-induced pri-mir-223 transcription was not changed in activated PMNs, although miR-223–3p export was significantly increased. Many factors may contribute to cellular pri-miR-223 and mature miR-223–3p levels, as well as miR-223–3p export to HDL. While the cellular pri-mir-223 and mature miR-223 levels significantly increased after 30 min of HDL treatment, the exported HDL-miR-223 levels were not significantly increased until 4 h of HDL treatment. These differences could be explained by a longer time being required to accumulate exported miR-223–3p on HDL to achieve statistical significance by PCR. We did observe a >2-fold increase in miR-223–3p levels on post-HDL after 30 min compared to the pre-HDL levels, which was a similar effect size to the cellular pri-mir-223 and mature miR-223–3p levels. However, these data are not statistically significant due to variability within the data. Ultimately, there may be a delay for the exported miR-223–3p levels to accumulate on extracellular HDL. As rHDL are miRNA free, we did not expect to observe an increase in mature miR-223–3p levels after rHDL incubations with PMNs. We hypothesize that the increase in PMNs mature miR-223–3p levels after rHDL incubations is due to the re-delivery of the miRNA back to the donor cells as rHDL failed to increase the levels of pri-mir-223 in PMNs.
Although PMNs and HMDMs are both from myeloid lineage, these cell types had distinct response to HDL-induced regulation of cellular miR-223–3p levels. For example, mature miR- 223–3p levels in PMNs were significantly elevated after 4 h HDL incubations, whereas HMDMs miR-223–3p levels were not altered after 16 h incubations. The reason for this difference is unclear, but it may be associated with a greater depletion of HMDM miR-223–3p levels compared to PMNs since the incubation time is longer. On the contrary, PMN fMLP-induced activation decreased intracellular mature miR-223–3p levels compared to non-activated cells. fMLP, a member of the formyl peptide family, is a classic activator of PMNs, and a potent proinflammatory chemo-attractant that produces reactive oxygen species (39, 40). Cellular changes triggered by fMLP-induced activation could explain why miR-223–3p export is increased and mature miR-223–3p levels are decreased. For example, an increase in cytoplasmic Ca2+ levels occurs early in fMLP-induced activation, which activates a cascade of signaling pathways that increases phosphatidylinositol 3,4,5-triphosphate (PIP3), activates PKC, and contributes to an oxidative burst. These factors could contribute to the observed increase in miR-223–3p export to HDL (41–43). The present study supports that in vascular diseases such as atherosclerosis, where the activation of PMNs is promoted; the amount of miR-223–3p on circulating HDL might be increased. There are no previous reports looking at the levels of HDL-miR-223–3p in coronary artery disease but total circulating levels have been shown to be increased(44).
Collectively, results presented here demonstrate that PMNs and HMDMs export miR-223–3p to HDL. We also demonstrate that HDL increases mature miR-223–3p expression and pri-mir-223 biogenesis. These findings provide novel insights into multiple facets of extracellular miRNAs on HDL, including miRNA export from PMNs and HMDMs, the impact of HDL on miRNA transcription and expression in inflammatory cells, the role of HDL’s receptor SR-BI in these processes.
Supplementary Material
Highlights:
The anti-atherogenic microRNA, miR-223–3p, present on high-density lipoproteins (HDL) originates from polymorphonuclear neutrophils (PMNs) and macrophages.
HDL induce both the export of miR-223–3p from PMNs to HDL and the transcription of primary miR-223–3p (pri-mir-223) in PMNs.
miR-223–3p export to HDL is increased when PMNs are activated.
HDL induction of pri-mir-223 transcription in PMNs is dependent on Scavenger Receptor BI (SR-BI)-mediated lipid transfer and not a consequence of HDL binding to SR-BI.
Acknowledgments
Financial support
F.T. is supported by the Australian National Heart Foundation Future Leader Fellowship (Grant 100090). K.C.V. is supported by grants from the National Institutes of Health, National Heart, Lung and Blood Institute HL128996, HL113039, and HL116263; and the American Heart Association CSA2066001. K.A.R. is supported by the National Health and Medical Research Council of Australia program grant 1037903.
Footnotes
Conflicts of interest
The authors declared they do not have anything to disclose regarding conflict of interest with respect to this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baetta R, and Corsini A. (2010) Role of polymorphonuclear neutrophils in atherosclerosis: current state and future perspectives. Atherosclerosis 210, 1–13 [DOI] [PubMed] [Google Scholar]
- 2.Sweetnam PM, Thomas HF, Yarnell JW, Baker IA, and Elwood PC (1997) Total and differential leukocyte counts as predictors of ischemic heart disease: the Caerphilly and Speedwell studies. American journal of epidemiology 145, 416–421 [DOI] [PubMed] [Google Scholar]
- 3.Carbone F, Mach F, and Montecucco F. (2015) Update on the role of neutrophils in atherosclerotic plaque vulnerability. Current drug targets 16, 321–333 [DOI] [PubMed] [Google Scholar]
- 4.Drechsler M, Megens RT, van Zandvoort M, Weber C, and Soehnlein O. (2010) Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation 122, 1837–1845 [DOI] [PubMed] [Google Scholar]
- 5.Puranik R, Bao S, Nobecourt E, Nicholls SJ, Dusting GJ, Barter PJ, Celermajer DS, and Rye KA (2008) Low dose apolipoprotein A-I rescues carotid arteries from inflammation in vivo. Atherosclerosis 196, 240–247 [DOI] [PubMed] [Google Scholar]
- 6.Wiernik A, Carlson LA, and Jarstrand C. (1986) High-density lipoproteins inhibit the bacterial lipopolysaccharide mediated increase in oxidative metabolism and lysozyme release by neutrophilic granulocytes in vitro. Journal of clinical & laboratory immunology 21, 131–135 [PubMed] [Google Scholar]
- 7.Tabet F, Remaley AT, Segaliny AI, Millet J, Yan L, Nakhla S, Barter PJ, Rye KA, and Lambert G. (2010) The 5A apolipoprotein A-I mimetic peptide displays antiinflammatory and antioxidant properties in vivo and in vitro. Arteriosclerosis, thrombosis, and vascular biology 30, 246–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tabet F, Lambert G, Cuesta Torres LF, Hou L, Sotirchos I, Touyz RM, Jenkins AJ, Barter PJ, and Rye KA (2011) Lipid-free apolipoprotein A-I and discoidal reconstituted high- density lipoproteins differentially inhibit glucose-induced oxidative stress in human macrophages. Arteriosclerosis, thrombosis, and vascular biology 31, 1192–1200 [DOI] [PubMed] [Google Scholar]
- 9.Nicholls SJ, Dusting GJ, Cutri B, Bao S, Drummond GR, Rye KA, and Barter PJ (2005) Reconstituted high-density lipoproteins inhibit the acute pro-oxidant and proinflammatory vascular changes induced by a periarterial collar in normocholesterolemic rabbits. Circulation 111, 1543–1550 [DOI] [PubMed] [Google Scholar]
- 10.Bergt C, Marsche G, Panzenboeck U, Heinecke JW, Malle E, and Sattler W. (2001) Human neutrophils employ the myeloperoxidase/hydrogen peroxide/chloride system to oxidatively damage apolipoprotein A-I. European journal of biochemistry 268, 3523–3531 [DOI] [PubMed] [Google Scholar]
- 11.Gantier MP (2013) The not-so-neutral role of microRNAs in neutrophil biology. J Leukoc Biol 94, 575–583 [DOI] [PubMed] [Google Scholar]
- 12.Tabet F, and Rye KA (2009) High-density lipoproteins, inflammation and oxidative stress. Clinical science 116, 87–98 [DOI] [PubMed] [Google Scholar]
- 13.Vickers KC, Rye KA, and Tabet F. (2014) MicroRNAs in the onset and development of cardiovascular disease. Clinical science 126, 183–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tabet F, Vickers KC, Cuesta Torres LF, Wiese CB, Shoucri BM, Lambert G, Catherinet C, Prado-Lourenco L, Levin MG, Thacker S, Sethupathy P, Barter PJ, Remaley AT, and Rye KA (2014) HDL-transferred microRNA-223 regulates ICAM-1 expression in endothelial cells. Nat Commun 5, 3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, Brummelkamp TR, Fleming MD, and Camargo FD (2008) Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 451, 1125–1129 [DOI] [PubMed] [Google Scholar]
- 16.Dorhoi A, Iannaccone M, Farinacci M, Fae KC, Schreiber J, Moura-Alves P, Nouailles G, Mollenkopf HJ, Oberbeck-Muller D, Jorg S, Heinemann E, Hahnke K, Lowe D, Del Nonno F, Goletti D, Capparelli R, and Kaufmann SH (2013) MicroRNA-223 controls susceptibility to tuberculosis by regulating lung neutrophil recruitment. The Journal of clinical investigation 123, 4836–4848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ward JR, Heath PR, Catto JW, Whyte MK, Milo M, and Renshaw SA (2011) Regulation of neutrophil senescence by microRNAs. Plos One 6, e15810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C, and Bozzoni I. (2005) A minicircuitry comprised of MicroRNA-223 and transcription factors NFI-A and C/EBP alpha regulates human granulopoiesis. Cell 123, 819–831 [DOI] [PubMed] [Google Scholar]
- 19.Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, and Remaley AT (2011) MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nature cell biology 13, 423–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jian B, de la Llera-Moya M, Ji Y, Wang N, Phillips MC, Swaney JB, Tall AR, and Rothblat GH (1998) Scavenger receptor class B type I as a mediator of cellular cholesterol efflux to lipoproteins and phospholipid acceptors. The Journal of biological chemistry 273, 5599–5606 [DOI] [PubMed] [Google Scholar]
- 21.Nakashima Y, Raines EW, Plump AS, Breslow JL, and Ross R. (1998) Upregulation of VCAM-1 and ICAM-1 at atherosclerosis-prone sites on the endothelium in the ApoE-deficient mouse. Arteriosclerosis, thrombosis, and vascular biology 18, 842–851 [DOI] [PubMed] [Google Scholar]
- 22.Rye KA, Garrety KH, and Barter PJ (1993) Preparation and characterization of spheroidal, reconstituted high-density lipoproteins with apolipoprotein A-I only or with apolipoprotein A-I and A-II. Biochimica et biophysica acta 1167, 316–325 [DOI] [PubMed] [Google Scholar]
- 23.Michell DL, Allen RM, Landstreet SR, Zhao S, Toth CL, Sheng Q, and Vickers KC (2016) Isolation of High-density Lipoproteins for Non-coding Small RNA Quantification. Journal of visualized experiments : JoVE [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fearon DT, and Collins LA (1983) Increased expression of C3b receptors on polymorphonuclear leukocytes induced by chemotactic factors and by purification procedures. Journal of immunology 130, 370–375 [PubMed] [Google Scholar]
- 25.Forsyth KD, and Levinsky RJ (1990) Preparative procedures of cooling and re-warming increase leukocyte integrin expression and function on neutrophils. Journal of immunological methods 128, 159–163 [DOI] [PubMed] [Google Scholar]
- 26.Rees D, Sloane T, Jessup W, Dean RT, and Kritharides L. (1999) Apolipoprotein A-I stimulates secretion of apolipoprotein E by foam cell macrophages. The Journal of biological chemistry 274, 27925–27933 [DOI] [PubMed] [Google Scholar]
- 27.Doring Y, Drechsler M, Soehnlein O, and Weber C. (2015) Neutrophils in atherosclerosis: from mice to man. Arteriosclerosis, thrombosis, and vascular biology 35, 288–295 [DOI] [PubMed] [Google Scholar]
- 28.de La Llera-Moya M, Connelly MA, Drazul D, Klein SM, Favari E, Yancey PG, Williams DL, and Rothblat GH (2001) Scavenger receptor class B type I affects cholesterol homeostasis by magnifying cholesterol flux between cells and HDL. Journal of lipid research 42, 1969–1978 [PubMed] [Google Scholar]
- 29.Krieger M. (2001) Scavenger receptor class B type I is a multiligand HDL receptor that influences diverse physiologic systems. The Journal of clinical investigation 108, 793–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ji Y, Jian B, Wang N, Sun Y, Moya ML, Phillips MC, Rothblat GH, Swaney JB, and Tall AR (1997) Scavenger receptor BI promotes high density lipoprotein-mediated cellular cholesterol efflux. The Journal of biological chemistry 272, 20982–20985 [DOI] [PubMed] [Google Scholar]
- 31.Lim HY, Thiam CH, Yeo KP, Bisoendial R, Hii CS, McGrath KC, Tan KW, Heather A, Alexander JS, and Angeli V. (2013) Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SR-BI-mediated transport of HDL. Cell metabolism 17, 671–684 [DOI] [PubMed] [Google Scholar]
- 32.Yu M, Romer KA, Nieland TJ, Xu S, Saenz-Vash V, Penman M, Yesilaltay A, Carr SA, and Krieger M. (2011) Exoplasmic cysteine Cys384 of the HDL receptor SR-BI is critical for its sensitivity to a small-molecule inhibitor and normal lipid transport activity. Proceedings of the National Academy of Sciences of the United States of America 108, 12243–12248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nieland TJ, Penman M, Dori L, Krieger M, and Kirchhausen T. (2002) Discovery of chemical inhibitors of the selective transfer of lipids mediated by the HDL receptor SR-BI. Proc Natl Acad Sci U S A 99, 15422–15427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turchinovich A, Weiz L, and Burwinkel B. (2012) Extracellular miRNAs: the mystery of their origin and function. Trends in biochemical sciences 37, 460–465 [DOI] [PubMed] [Google Scholar]
- 35.Hergenreider E, Heydt S, Treguer K, Boettger T, Horrevoets AJ, Zeiher AM, Scheffer MP, Frangakis AS, Yin X, Mayr M, Braun T, Urbich C, Boon RA, and Dimmeler S. (2012) Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nature cell biology 14, 249–256 [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, Liu D, Chen X, Li J, Li L, Bian Z, Sun F, Lu J, Yin Y, Cai X, Sun Q, Wang K, Ba Y, Wang Q, Wang D, Yang J, Liu P, Xu T, Yan Q, Zhang J, Zen K, and Zhang CY (2010) Secreted monocytic miR-150 enhances targeted endothelial cell migration. Molecular cell 39, 133–144 [DOI] [PubMed] [Google Scholar]
- 37.Shi L, Fisslthaler B, Zippel N, Fromel T, Hu J, Elgheznawy A, Heide H, Popp R, and Fleming I. (2013) MicroRNA-223 antagonizes angiogenesis by targeting beta1 integrin and preventing growth factor signaling in endothelial cells. Circulation research 113, 1320–1330 [DOI] [PubMed] [Google Scholar]
- 38.Pende A, Artom N, Bertolotto M, Montecucco F, and Dallegri F. (2016) Role of neutrophils in atherogenesis: an update. European journal of clinical investigation 46, 252–263 [DOI] [PubMed] [Google Scholar]
- 39.Le Y, Yang Y, Cui Y, Yazawa H, Gong W, Qiu C, and Wang JM (2002) Receptors for chemotactic formyl peptides as pharmacological targets. International immunopharmacology 2, 1–13 [DOI] [PubMed] [Google Scholar]
- 40.Kawamoto H, and Minato N. (2004) Myeloid cells. The international journal of biochemistry & cell biology 36, 1374–1379 [DOI] [PubMed] [Google Scholar]
- 41.Laffafian I, and Hallett MB (1995) Does cytosolic free Ca2+ signal neutrophil chemotaxis in response to formylated chemotactic peptide? Journal of cell science 108 ( Pt 10), 3199–3205 [DOI] [PubMed] [Google Scholar]
- 42.Lazzari KG, Proto PJ, and Simons ER (1986) Simultaneous measurement of stimulus- induced changes in cytoplasmic Ca2+ and in membrane potential of human neutrophils. The Journal of biological chemistry 261, 9710–9713 [PubMed] [Google Scholar]
- 43.Frohlich D, Rothe G, Wittmann S, Schmitz G, Schmid P, Taeger K, and Hobbhahn J. (1998) Nitrous oxide impairs the neutrophil oxidative response. Anesthesiology 88, 1281–1290 [DOI] [PubMed] [Google Scholar]
- 44.Liu W, Ling S, Sun W, Liu T, Li Y, Zhong G, Zhao D, Zhang P, Song J, Jin X, Xu Z, Song H, Li Q, Liu S, Chai M, Dai Q, He Y, Fan Z, Zhou YJ, and Li Y. (2015) Circulating microRNAs correlated with the level of coronary artery calcification in symptomatic patients. Scientific reports 5, 16099 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.