

Abstract
A common feature of cancer cells is the alteration of kinases and biochemical signalling pathways enabling transformed growth on soft matrices, whereas cytoskeletal protein alterations are thought to be a secondary issue. However, we report here that cancer cells from different tissues can be toggled between transformed and rigidity-dependent growth states by the absence or presence of mechanosensory modules, respectively. In various cancer lines from different tissues, cells had over tenfold fewer rigidity-sensing contractions compared with normal cells from the same tissues. Restoring normal levels of cytoskeletal proteins, including tropomyosins, restored rigidity sensing and rigidity-dependent growth. Further depletion of other rigidity sensor proteins, including myosin IIA, restored transformed growth and blocked sensing. In addition, restoration of rigidity sensing to cancer cells inhibited tumour formation and changed expression patterns. Thus, the depletion of rigidity-sensing modules through alterations in cytoskeletal protein levels enables cancer cell growth on soft surfaces, which is an enabling factor for cancer progression.
For normal cell growth, complex cellular mechanosensing functions are needed to develop the proper growth signals. Mechanical parameters of the micro-environment, as measured by the cells, dictate whether they survive, grow or die. Matrix rigidity is one of the most critical aspects of the micro-environment for normal development and regeneration. However, transformed cancer cells normally bypass the context-dependent matrix rigidity sensing and develop aberrant growth signals. One classic example is the anchorage-independent growth exemplified by cancer cell proliferation on soft agar, which is a hallmark of cancer cells and highlights their capacity for colony formation1. This feature has also been coined transformed growth or ‘anoikis resistance’2.
We recently described rigidity-sensing modules as cytoskeletal protein complexes that contract matrix to a fixed distance. If, during these contractions, the force level exceeds about 25 pN, the matrix is considered rigid3. This is just one of a number of modular machines that perform important tasks in cells, including, for example, the clathrin-dependent endocytosis complex4. Such modular machines typically assemble rapidly from mobile components, perform the desired task and disassemble in a matter of seconds to minutes. They are activated by one set of signals and are designed to generate another set. The cell rigidity-sensing complex is a 2–3-μm-sized modular machine that forms at the cell periphery during early contact with matrix well before formation of stress fibres or other later cytoskeletal structures3,5–8. It is powered by sarcomere-like contractile units (CUs) that contain myosin IIA, actin filaments, tropomyosin 2.1 (Tpm 2.1), α-actinin 4 and other cytoskeletal proteins7. The correct length and duration of contractions are controlled by receptor tyrosine kinases (RTKs) through interactions with cytoskeletal proteins6. Furthermore, the number of CUs is dependent on EGFR or HER2 activity as well as on substrate rigidity8. On rigid surfaces, CUs stimulate the formation of mature adhesions often leading to growth. However, on soft surfaces, contractions are very short-lived with rapidly disassembly of adhesions, leading to cell death by anoikis3,7. The failure of cancer cells to activate anoikis pathways on soft matrices prompted us to postulate that the absence of rigidity-sensing CUs in cancer cells enables anchorage-independent growth.
Cytoskeletal proteins are integrated into many complex cellular functions, and their roles are well studied in normal cells9. However, the role of cytoskeletal proteins, and particularly CU components, in cell transformation and cancer development is still not clear. Mutations and abnormal expression of various cytoskeletal or cytoskeletal-associated proteins have been reported in many cancer studies10: myosin IIA has been identified as a tumour suppressor in multiple carcinomas11,12; the expression level of Tpm 2.1 is highly suppressed in a variety of cancer cell lines13; and Tpm 3 (including Tpm 3.1 and Tpm 3.2) is commonly overexpressed in primary tumours and tumour cell lines14. However, it is still unclear whether these cytoskeletal proteins act as tumour suppressors or activators. For example, α-actinin 4 is reported to be a tumour suppressor in certain cases15,16 but an activator in others17. These proteins are all necessary components of rigidity-sensing modules.
There is a potential relation between malignant transformation and loss of the ability of cells to form active rigidity-sensing modules because of altered cytoskeletal protein levels. In our recent studies we found that rigidity-sensing activity was missing in MDA-MB-231 breast cancer cells but was preserved in normal MCF 10A mammary epithelial cells, as defined by local contractions of submicrometre pillars3. In contrast, both cell lines developed actin flow-driven traction forces on the substrates. The rigidity sensing of MDA-MB-231 cells could be restored following re-expression of Tpm 2.1 (ref.3). Previous studies have shown that restoration of normal levels of Tpm 2.1 blocked transformed growth13,18 and invasion19 of MDA-MB-231. Conversely, depletion of Tpm 2.1 from the normal MCF 10A cells caused both transformation and loss of rigidity sensing3,13. Furthermore, removal of another CU component, α-actinin 4, in mouse embryonic fibroblasts (MEF) disrupted rigidity sensors and enabled rapid growth on soft matrices7. These findings indicated that not only were altered cytoskeletal protein levels involved in transformation, but also that there may be a strong correlation between the loss of rigidity sensing and transformation. The wide diversity of cancer cells from many different tissues are thought to function through many different growth pathways, but may have the common feature that they lack the rigidity-sensing modules that would normally suppress growth on soft surfaces.
Here we report that various types of cancer cells lack rigidity sensing because of altered cytoskeletal protein levels, and can be restored to a rigidity-dependent growth state by restoring normal levels of rigidity-sensing components. In correlation with the restoration of rigidity sensing, cells undergo a reversible cell state change, including changes in contractile behaviour as well as adhesion and cytoskeletal organization, plus marked changes in expression patterns. This indicates that alterations of specific cytoskeletal proteins are sufficient to switch reversibly between a transformed state and a normal state across different cancer cell types. It also highlights the importance of cell mechanosensing in the control of normal growth.
Fundamental differences between normal and cancer cells
To understand the nature of the differences between normal cells and transformed cancer cells, we analysed the forces developed by normal and cancer cells on discrete adhesion sites. To that end, we used 500-nm-diameter polydimethylsiloxane (PDMS) pillars coated with fibronectin, which support the formation of cell adhesions similar to those formed on continuous surfaces5. In the case of human foreskin fibroblast (HFF) cells, these formed 2–3-μm-sized CU) (Fig. 1a, blue vectors) at the cell edge that tested substrate rigidity. A typical CU contained a pair of pillars that were contracted toward each other by an average of 60 nm, with a half-deflection time of 30 s (Fig. 1b). More than 25% of the pillars pulled by HFF cells contributed to CU formation during early spreading on fibronectin matrices (Fig. 1e). When pillars were part of contractile pairs, they had a narrower distribution of displacement than those pulled centripetally by cells (Fig. 1c). Furthermore, the average displacement of contractile pairs was significantly greater than the radial contractions (Fig. 1d). In contrast, HT1080 cancer cells (a fibrosarcoma line from an untreated patient carrying an IDH1 mutation20) had very few contractile pairs and the radial contractions were significantly greater in magnitude than those in the HFFs (Fig. 1h–j). There was no significant difference between the total cell-spreading areas of HFF and HT1080 cells 20 min after seeding on pillars (Fig. 1f,g). Thus, overall patterns of contractility were markedly different between normal fibroblasts and HT1080 fibrosarcoma cells, indicating that they were in fundamentally different physical cellular states.
Fig. 1 |. Fundamental differences between normal and transformed cells during initial spreading.
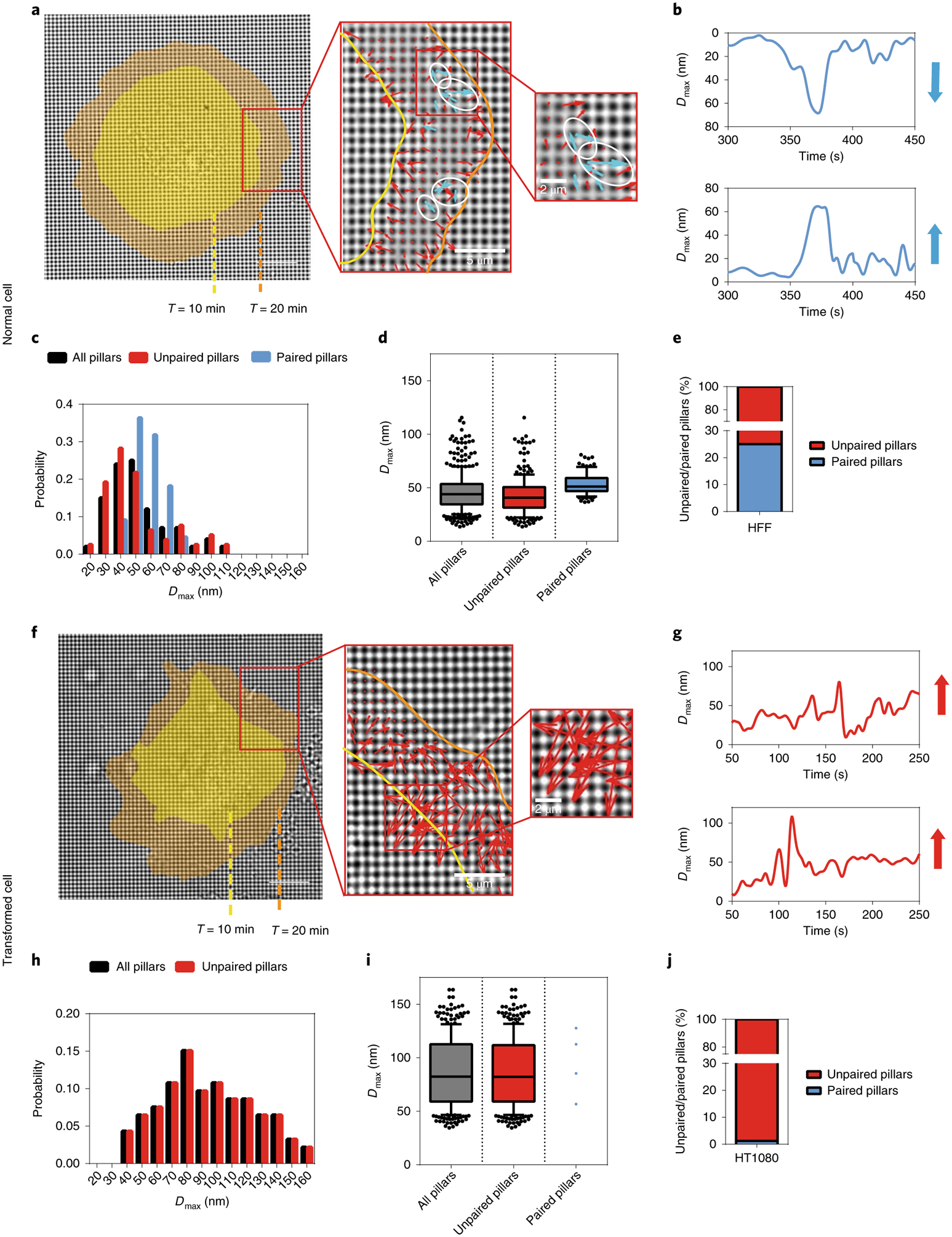
a, Left: actual CUs observed at the outward extending edge of a HFF (normal) cell spreading on FN-coated pillars. Cell edges are marked in yellow or orange corresponding to time points 10 or 20 min after seeding, respectively (scale bar, 10μm). Middle and right: enlarged images of boxed areas showing detected CUs. Arrows represented pillar movements: blue, detected CUs; red, non-CUs. White circles highlight several examples of identified CUs. b, Displacement versus time plots of two adjacent pillars highlighted by white circles in a. Blue vectors represent the relative displacement directions and magnitude of those two pillars. The neighbouring pillars were moving directly towards each other and were highly correlated in time in HFF cells. c, Histogram distribution of maximum pillar displacement (Dmax) from one HFF cell. Black denotes all tracked pillars, red denotes those that did not form any CUs during imaging (unpaired pillars) while blue denotes those that formed CUs (paired pillars). d, Box-and-whisker plots of pillar Dmax for three HFF cells. e, Bar graph of unpaired/paired pillar percentages of 400 pillars from three cells. f, Left: actual CUs observed in a HT1080 (transformed) cell (scale bar, 10μm). Middle and right: enlarged images of boxed areas. g, Displacement versus time plots of two adjacent pillars in f. h, Histogram distribution of pillar Dmax for one HT1080 cell. i, Box-and-whisker plots of pillar Dmax summary for three HT1080 cells. j, Bar graph of unpaired/paired pillar percentages of 400 pillars from three cells.
Transformed cells lack local contractions
To further examine whether transformed cells generally lack rigidity-sensing CU formation, we tested three additional cancer lines and a non-cancerous transformed cell line. The bases of transformation for these cells were all different: Cos7 cells were derived from African green monkey kidney fibroblast cells by SV40 transformation; MDA-MB-231 is a human breast cancer line that forms tumours in nude mice21,22; SKOV3 is a human ovarian adenocarcinoma line with an epithelial-like morphology23; LLC is a lung carcinoma line from a C57B L mouse24. We measured CU activity as previously described8 and normalized it to cell-spreading area. Consistent with previous publications, HFF cells generated 39 CUs per 100 μm2 on rigid pillars (k = 8.4 pN nm−1) and 24 CUs per 100 μm2 on soft pillars (k = 1.6 pN nm−1) per 10 min during initial spreading (Fig. 2a,b). In contrast, all transformed cells produced <2 CUs per 100 μm2 per 10 min on the two pillar types (Fig. 2a,b). To further demonstrate the extent of this phenomenon, four more highly contractile and metastatic human cancer cell lines were examined for CU activity: A375P, human skin malignant melanoma line25; A2058, a highly metastatic human skin melanoma cell line26; PANC1, a highly contractile human pancreatic cancer line27; and U251, a human glioblastoma line28. All four cell lines had <5 CUs per 100 μm2 on soft and rigid pillars (Supplementary Fig. 2c,d). Since transformation was also caused by v-Src transfection29, we tested whether HFF cells transfected with v-Src were able to sense substrate rigidity. The v-Src-expressing HFFs s howed a >50% decrease in the number of CUs formed on both rigid and soft pillars compared with non-transfected pillars (Supplementary Fig. 2j). Thus, in various transformed backgrounds, cells had low levels of rigidity sensing.
Fig. 2 |. Transformed cells lack rigidity sensing and rigidity sensor proteins.
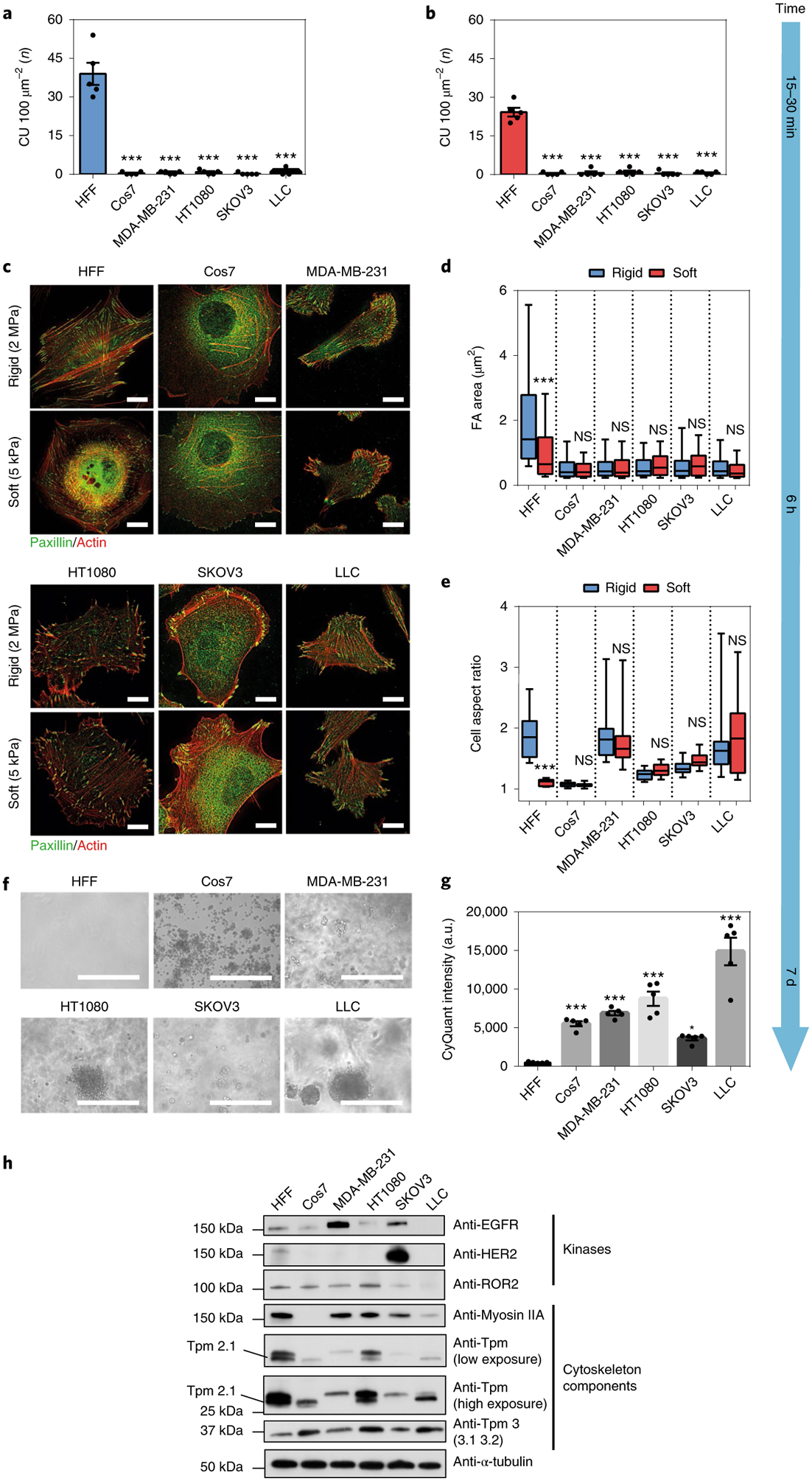
a,b, Average number of CUs per100 μm2 generated by HFF or various transformed cell lines on rigid (a, k = 8.4 pN nm−1) and soft (b, k = 1.6 pN nm−1) pillars per 10 min, respectively (n = 5 cells in each case; ***P < 0.001). c, Staining for actin (red) and paxillin (green) in HFFs and other transformed cells on hard (2 MPa) and soft (5 kPa) PDMS surfaces after plating for 6 h (scale bars, 10 μm). d,e, Box-and-whisker plots of single focal adhesion area (FA) (d) and cell aspect ratio (e) of HFFs and various transformed cells after plating for 6 h on soft (red) or rigid (blue) PDMS surfaces (>150 adhesions from n ≥ 10 cells were analysed in each case; ***P < 0.001; NS, not significant). f,g, Soft agar assay showing growth of various transformed cells, but not HFF cells, after 7-d culture (scale bars, 400 μm; n = 5 in each case; error bars represents s.e.m.; ***P < 0.001; a.u., arbitrary units).
To determine whether cells were able to distinguish between rigid and soft surfaces at a later spreading stage, we plated all ten cell lines on stiff (2 MPa) and compliant (5 kPa) fibronectin-coated PDMS surfaces for 6 h. Cells were then fixed and stained for paxillin and actin as markers for adhesion formation and morphological change, respectively (Fig. 2c and Supplementary Fig. 2e–h). As previously described30, HFF cells polarized on rigid PDMS and spread less on soft PDMS in a rounded shape. However, all nine transformed cell lines showed no significant difference in cell polarization level or adhesion size on the surfaces, with a 400-fold difference in rigidity (Fig. 2d,e and Supplementary Fig. 2i). In addition, to further investigate how ‘normal’ and ‘transformed’ lines would behave on substrates even softer than 5 kPa, four selected cell lines (HFF, Cos7, MDA-MB-231 and HT1080) were cultured on 4-, 2- or 1-kPa soft hydrogel-coated or bare glass surfaces overnight. Notably, the mean cell spread area of HFF cells was ~25-fold larger on glass substrates than on 1-kPa surfaces. However, for all three transformed lines, there was no significant change in cell spread area under all conditions tested (Supplementary Fig. 2a,b). Thus, transformed cells were less sensitive to substrate rigidity compared to normal cells.
Since cell transformation is defined classically as growth on soft agar1, we cultured the various lines in soft agar for 7 d. All five transformed cell lines that were tested formed colonies while HFF cells barely survived (Fig. 2f,g). Thus none of the transformed lines developed a significant number of CUs for rigidity sensing, and this is consistent with their inability to react to differences in matrix rigidity, as well as their ability to grow on soft surfaces.
To establish whether CU formation is a general property of normal cells, we next analysed the behaviour of four further non-transformed cell lines from different origins: a normal African green monkey kidney fibroblast cell line (CV1), a normal human breast epithelial cell line (MCF 10A), a normal ovarian surface epithelial cell line (IOSE 523) and a primary human umbilical vein endothelial cell line (HUVEC). Despite genetic background differences, all four cell lines formed similar numbers of CUs on both rigid and soft pillar surfaces during initial spreading, as did HFFs (Supplementary Fig. 2k,l). Furthermore, similar to HFFs, matrix rigidity had a significant effect on the average single focal adhesion area of all four cell lines after plating for 6 h (Supplementary Fig. 2m–r). Thus, CU formation during initial spreading is a mechanism used universally to test matrix rigidity for non-transformed cells.
Transformed cells lack rigidity sensing due to altered CU component expression levels
To determine whether transformed cells had altered levels of rigidity-sensing components, we performed immunoblot screening of known CU proteins (Supplementary Fig. 2s), including kinases (EGFR, HER2, ROR2 and AXL) and cytoskeletal proteins (Myosin IIA, Tpm 2.1, Tpm 3, FLNA and α-actinin 1 and 4) (Fig . 2i and Supplementary Fig. 2t). Interestingly, the five transformed cell lines all lacked at least one CU component. In addition, the expression levels of selected cytoskeletal proteins followed a similar trend in different transformed backgrounds but not in the non-transformed cell groups. Both myosin IIA and Tpm 2.1 expression levels were suppressed, while that of Tpm 3 (Tpm 3.1 and 3.2) was increased in most cancer lines (Fig. 2i and Supplementary Fig. 2u–w). On the other hand, the pattern of RTK levels varied among cell lines (Fig. 2i and Supplementary Fig. 2t). Taking all the above results into consideration, we observed different patterns of protein depletion in transformed cells, including cytoskeletal components and RTK levels. This raised two issues: (1) CUs may occur after restoration of normal levels of only the altered cytoskeletal components, and (2) alteration of another cytoskeletal protein in restored cells may inhibit CU formation and again cause transformed growth.
Restoration of cytoskeletal protein levels restores rigidity sensing and normal growth while reciprocal depletion reverts cells to transformed growth
To address the above issues, we first tested whether we could block transformed growth by simply restoring the missing cytoskeletal CU components to enable rigidity sensing. We selected Cos7 cells, which lack myosin IIA, as the first candidate. After expression of EGFP-myosin IIA, Cos7 cells produced higher forces on both rigid and soft pillars (Fig. 3a). Also, the density of CUs increased dramatically after myosin IIA expression (Fig. 3b). We next examined whether restoration of CUs during initial spreading would affect cell morphology and cell fate decisions at later stages in the same environment. After spreading for 6 h, Cos7 cells usually formed small focal adhesions of similar size on both soft (0.52 ± 0.03 μm2) and rigid (0.53 ± 0.04 μm2) flat PDMS surfaces (Fig. 3d,f). In contrast, Cos7-IIA (myosin IIA-expressing Cos7 cells) generated larger focal adhesions (1.01 ± 0.05 μm2) on rigid, and smaller adhesions (0.65 ± 0.03 μm2) on soft, surfaces as commonly observed for normal fibroblast cell lines (Fig. 3c,f). In the colony growth assay on soft agar, Cos7-IIA cells did not survive after 7 d in culture whereas control Cos7 cells proliferated and formed colonies (Fig. 3g,h). To further examine whether this suppression of cell survival in a soft environment was due to an overall suppression of growth, we analysed cell proliferation rates on both soft (2 kPa) and rigid (plastic) surfaces during culture for 4 d. Cos7-IIA cells maintained a similar proliferation rate on rigid, but not on soft, surfaces compared with control Cos7 cells (Supplementary Fig. 4d,e). Thus, restoration of rigidity-sensing activity in Cos7 cells caused selective death in a soft environment.
Fig. 3 |. Myosin IIA in Cos7 cells enables normal growth, but Tpm 2.1 silencing restores transformation.
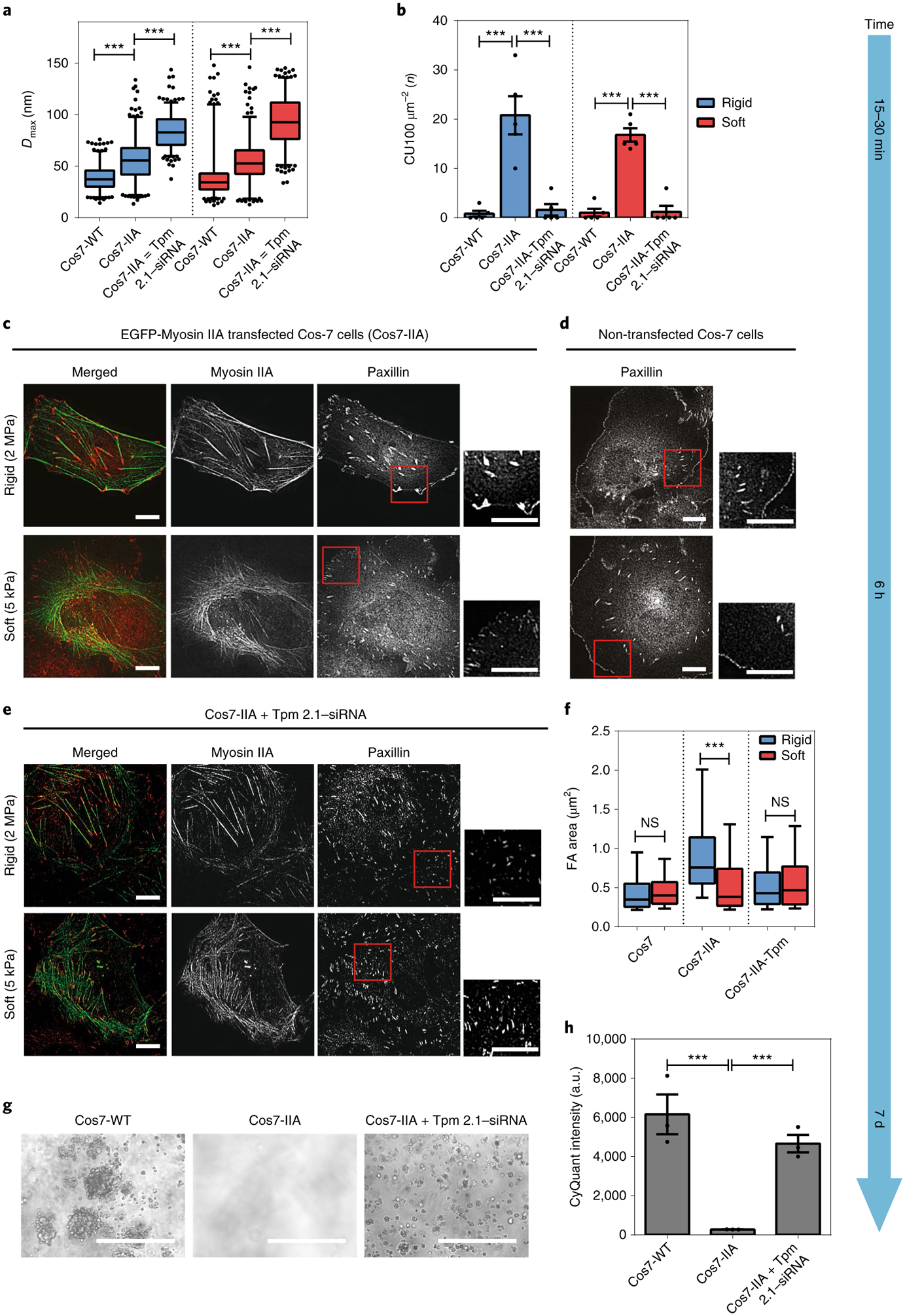
a, Box-and-whisker plots of pillar Dmax for Cos7-WT, Cos7-IIA and Tpm-siRNA-transfected Cos7-IIA cells on rigid (blue) and soft (red) pillars. Silencing of Tpm 2.1 in Cos7-IIA cells increased the average force level on both types of pillar. b, Bar graphs of average CU density per 10 min for Cos7-WT, Cos7-IIA and Tpm 2.1–siRNA-transfected Cos7-IIA cells on rigid (blue) and soft (red) pillars (n = 5 in each case; error bars are s.e.m.; ***P < 0.001). c–e, Paxillin images of EGFP-myosin IIA-transfected Cos7 cells (Cos7-IIA) (c), Cos7 cells (d) and Tpm 2.1-depleted Cos7-IIA cells (e) fixed 6 h following seeding on rigid (2 MPa) or soft (5 kPa) fibronectin-coated PDMS surfaces (scale bars, 10 μm). f, Box-and-whisker plots of single focal adhesion area (FA) of Cos7, Cos7-IIA and Tpm 2.1-silenced Cos7-IIA cells on rigid (blue) and soft (red) PDMS surfaces (>150 adhesions were analysed in each case; ***P < 0.001). g,h, Soft agar assay indicating the growth of Cos7 and Tpm 2.1-knockdown Cos7 IIA cells, but not Cos7-IIA cells, after culture for 7 d (scale bars, 400 μm; n = 3 in each case; error bars are s.e.m.; ***P < 0.001; a.u., arbitrary units).
To test whether restoration of rigidity-sensing activity in Cos7 cells was caused by myosin IIA and not by increasing the total amount of myosin in the cells, we next expressed myosin IIB in Cos7 cells. Cos7-myosin IIB cells generated very few CUs on pillar surfaces during initial spreading (Supplementary Fig. 3a,b) and they formed focal adhesions of similar size on both rigid (0.31 ± 0.05 μm2) and soft (0.35 ± 0.03 μm2) fibronectin-coated PDMS 6 h after plating (Supplementary Fig. 3c,d), which is consistent with a previous study31. Thus, the re-expression of myosin IIA but not myosin IIB in Cos7 cells restored rigidity-sensing activity and blocked transformed growth.
We next asked whether reciprocal depletion of another CU component could reverse transformed growth of Cos7-IIA cells. Previously, depletion of Tpm 2.1 caused transformed growth in MCF 10A cells3. Following small interfering RNA depletion of endogenous Tpm 2.1 (Supplementary Fig. 4a), CU formation in Cos7-IIA cells was significantly suppressed (1.6 CUs per 100 μm2 on rigid and 1.2 CUs per 100 μm2 on soft pillars) (Fig. 3b). Despite the lack of CUs, Tpm 2.1-depleted Cos7-IIA cells produced larger displacement (force) on both soft and rigid pillars, indicating that myosin IIA was still active (Fig. 3a). Moreover, consistent with previous studies on MEF cells3, depletion of Tpm 2.1 in Cos7-IIA cells caused a significant decrease in FA size on PDMS surfaces but stress fibres were still evident (Fig. 3e,f). Furthermore, Tpm 2.1-depleted Cos7-IIA cells survived on 2.3-kPa polyacrylamide gels without activating caspase-3 cleavage after 3 d in culture (Supplementary Fig. 4b,c). Different from Cos7-IIA cells and similar to wild type (WT) Cos7 cells, Tpm 2.1-depleted Cos7-IIA cells exhibited an increase in cell proliferation on both soft and rigid surfaces during culture for 4 d (Supplementary Fig. 4f). In the soft agar assay, visible colonies of Tpm 2.1-depleted Cos7-IIA cells formed after 7 d (Fig. 3g,h). Thus, depletion of Tpm 2.1 in Cos7-IIA cells blocked CU formation and restored transformed growth.
To investigate whether this connection between CU formation and transformed growth was cell line dependent, we examined the MDA-MB-231 cell line21. Consistent with previous studies3, restoration of Tpm 2.1 in MDA-MB-231 cells decreased pillar displacement (Fig. 4a) and restored CUs on both soft and rigid matrices (21.6 CUs per 100 μm2 on rigid pillars and 14.8 CUs per 100 μm2 on soft pillars) (Fig. 4b). Tpm 2.1-transfected MDA-MB-231 cells (231-Tpm) distinguished between soft and rigid PDMS by generating larger focal adhesions and spreading to wider areas on rigid surfaces (Fig. 4c,f). In the growth assay, control MDA-MB-231 but not 231-Tpm cells formed colonies in soft agar culture after 7 d (Fig. 4g,h), although both maintained a similar proliferation rate on rigid surfaces (Supplementary Fig. 5d,e).
Fig. 4 |. Tpm 2.1 in MDA-MB-231 enables normal growth but myosin IIA depletion restores transformation.
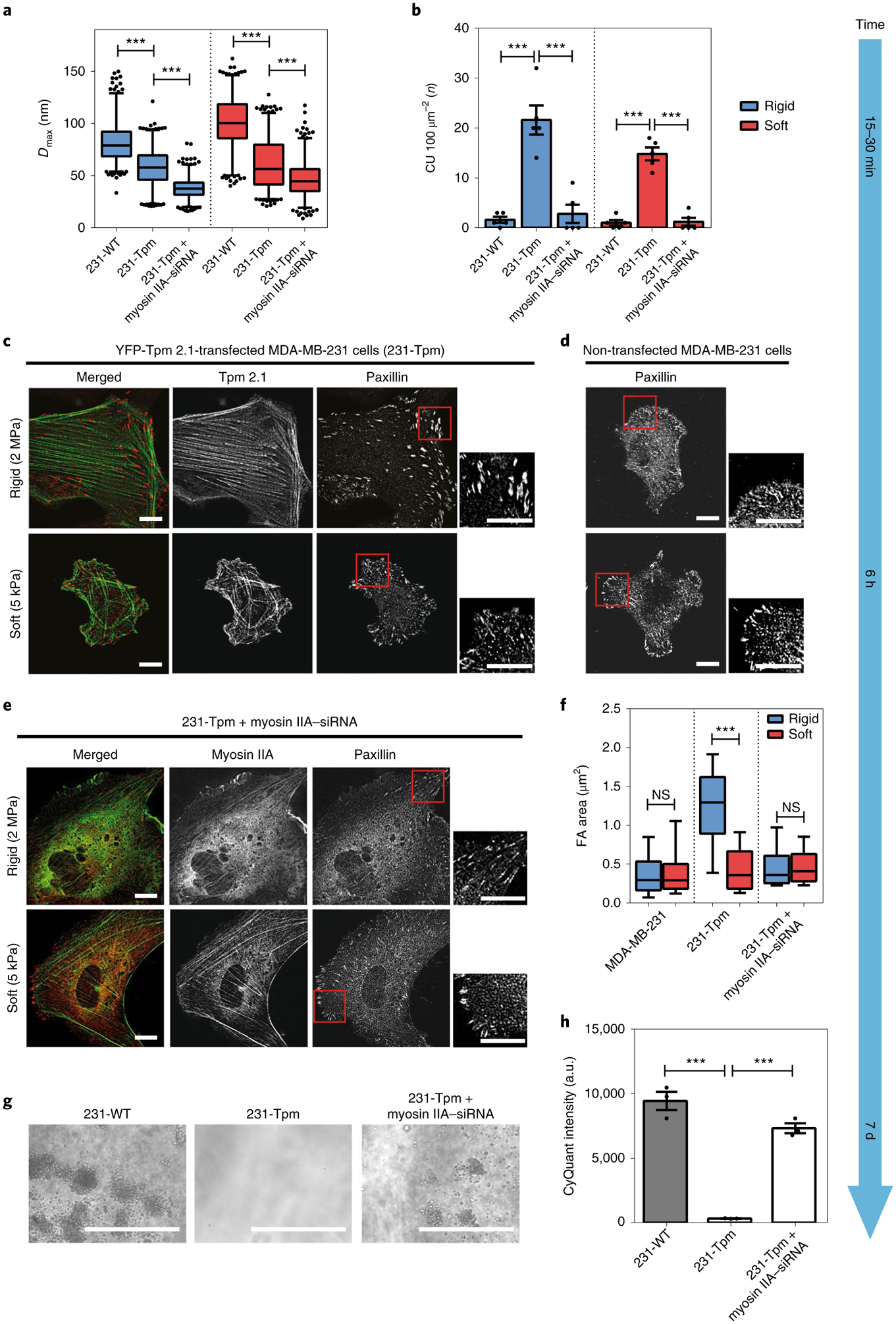
a, Box-and-whisker plots of pillar Dmax by 231-WT, 231-Tpm and myosin IIA–siRNA-transfected 231-Tpm cells on rigid (blue) and soft (red) pillars. Silencing of myosin IIA in 231-Tpm cells decreased the average force level on both types of pillar. b, Bar graphs of average CU density per 10 min for different cells (n = 5 in each case; error bars are s.e.m.; ***P < 0.001). c–e, Paxillin images of YFP-Tpm 2.1-transfected MBA-MB-231 cells (231-Tpm) (c), MDA-MB-231 cells (d) and Myosin IIA-depleted 231-Tpm cells (e) fixed at 6 h following seeding on rigid (2 MPa) or soft (5 kPa) fibronectin-coated PDMS surfaces (scale bars, 10 μm). f, Box-and-whisker plots of single focal adhesion area (FA) for MDA-MB-231, 231-Tpm and Myosin IIA-silenced 231-Tpm cells on rigid (blue) and soft (red) PDMS surfaces (>150 adhesions were analysed in each case; ***P < 0.001). g,h, Soft agar assay showing growth of MDA-MB-231 and Myosin IIA-silenced 231-Tpm cells, but not 231-Tpm cells, after culture for 7 d (scale bars, 400 μm; n = 3 in each case; error bars are s.e.m.; ***P < 0.001; a.u., arbitrary units).
In a reciprocal manner, depletion of endogenous myosin IIA in 231-Tpm cells (Supplementary Fig. 5a) inhibited CU formation on both rigid and soft pillars by fivefold (Fig. 4b) and decreased overall contractility (Fig. 4a). As expected, myosin IIA-depleted 231-Tpm cells showed disruption of stress fibres as well as a significant reduction in focal adhesion size on PDMS surfaces (Fig. 4e,f). Furthermore, myosin IIA-depleted 231-Tpm cells grew as well as WT 231 cells on both soft and rigid surfaces, and the level of cleaved caspase-3 on soft surfaces also decreased twofold (Supplementary Fig. 5b–f). Additionally, after myosin IIA knockdown, 231-Tpm cells formed colonies in soft agar similar to MDA-MB-231 cells after 7 d (Fig. 4g,h). Thus, in the restored cell lines, depletion of other cytoskeletal components that blocked CU formation induced transformed cell growth (Supplementary Table 1). This indicated that rigidity-sensing CU formation blocked anchorage-independent growth, and that loss of rigidity sensing enabled transformed growth in different cell backgrounds with different alterations in signalling pathways.
A low-molecular-weight tropomyosin, Tpm 3, suppresses CU formation
Careful examination of the immunoblot screening data (Fig. 2i) raised a question about why HT1080 cells failed to generate CUs for rigidity sensing despite the necessary CU components having been expressed. Notably, these cells also had high levels of another tropomyosin isoform, Tpm 3 (including Tpm 3.1 and Tpm 3.2), which is highly expressed in many cancers14. To determine whether Tpm 3 would suppress CU formation, we silenced endogenous Tpm 3 in HT1080 cells by siRNA (Fig. 5a). As shown in Fig. 5b, after Tpm 3 knockdown pillar displacement was significantly lower than in the control group on both rigid and soft pillars, which is consistent with the observation that Tpm 3.1/2 are the tropomyosin isoforms that activate Myosin IIA ATPase in vitro32. In addition, Tpm 3-depleted HT1080 cells generated 14.4 CUs per 100 μm2 on rigid pillars and 10.8 CUs per 100 μm2 on soft pillars, approximately fivefold higher than control cells (Fig. 5c). Additionally, Tpm 3-depleted HT1080 cells formed larger focal adhesions (1.44 ± 0.07 μm2) on rigid PDMS and smaller adhesions (0.62 ± 0.03 μm2) on soft PDMS (Fig. 5d,e,g). Moreover, the depletion of Tpm 3 or inhibition of Tpm 3 assembly on actin filaments by TR100 (ref.14) or ATM 3507 (ref.33) decreased the size and number of colonies formed by HT1080 cells in soft agar after culture for 7 d (Fig. 5f,h). Thus, these results support the idea that the high level of Tpm 3 protein in HT1080 cells suppressed CU formation and thereby stimulated transformed growth.
Fig. 5 |. High levels of Tpm€3 (3.1 and 3.2) inhibit cell rigidity sensing.
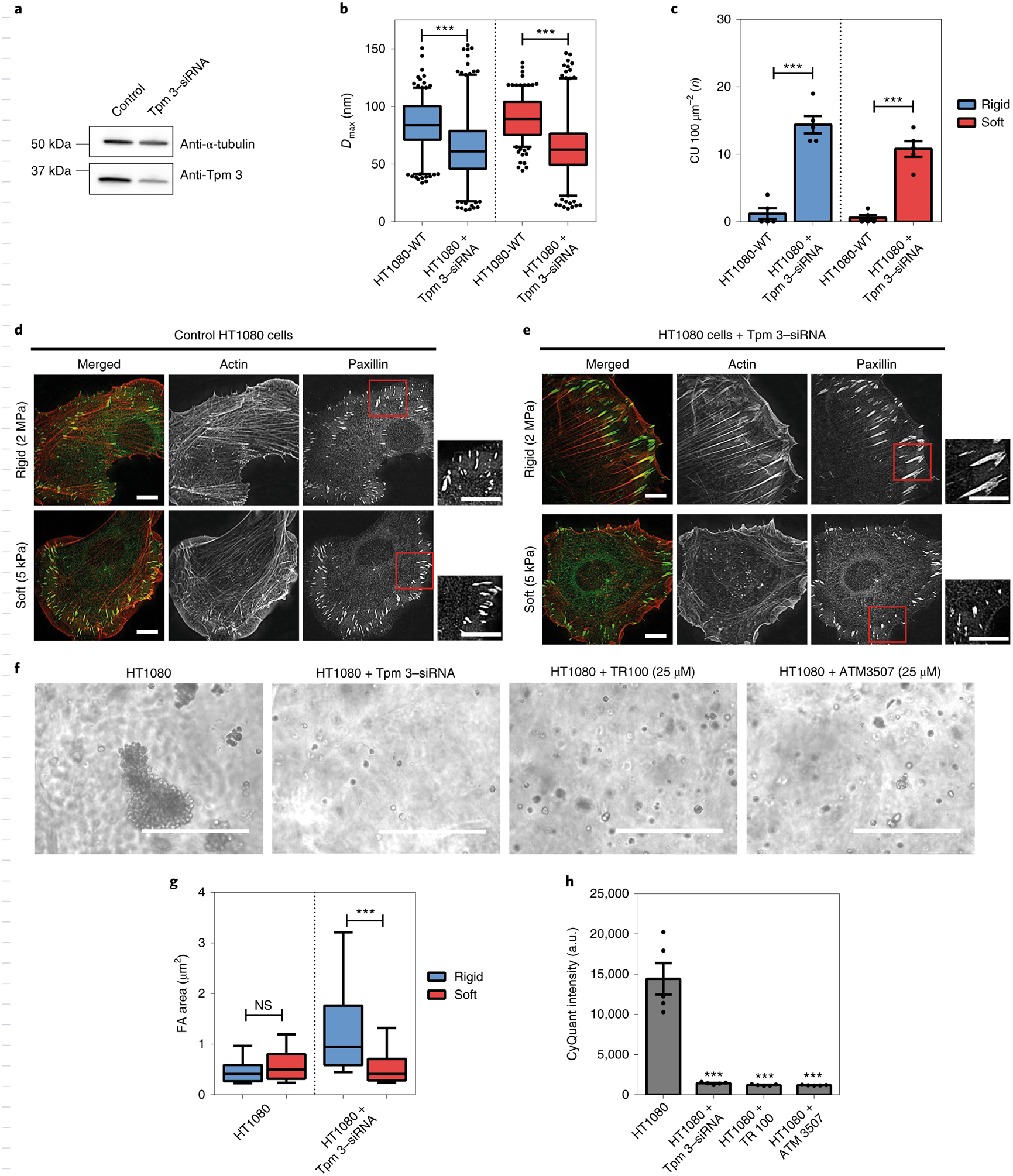
a, Immunoblot showing Tpm 3 (Tpm 3.1 and 3.2) levels in HT1080 cells treated with scramble or anti-Tpm 3–siRNA. b, Box-and-whisker plots of pillar Dmax for control or Tpm 3-depleted HT1080 cells on rigid (blue) and soft (red) pillars. c, Average CU density per 10 min for control and Tpm 3-depleted HT1080 cells on two pillar substrates of differing rigidity (n = 5 in each case; error bars are s.e.m.; ***P < 0.001). d,e, Staining of actin (red) and paxillin (green) in control (d) or Tpm 3-depleted HT1080 cells (e) on hard (2 MPa) and soft (5 kPa) PDMS surfaces after plating for 6 h (scale bars, 10 μm). f, Bright field images of cell colony growth after culture for 7 in soft agar (scale bars, 400 μm). g, Box-and-whisker plots of single focal adhesion area (FA) for control and Tpm 3-depleted HT1080 cells on rigid (blue) and soft (red) PDMS surfaces (>250 adhesions were analysed in each case; ***P < 0.001; NS, not significant). h, Soft agar assay showing growth of control HT1080 cells, but not of Tpm 3-depleted cells or those treated with Tpm 3 inhibitors (TR100 and ATM 3507) (n = 5 in each case; error bars are s.e.m.; ***P < 0.001; a.u., arbitrary units).
To determine whether Tpm 3 depletion would activate CU formation even in the absence of Tpm 2.1, we knocked down Tpm 3 by siRNA in MD A-MB-231 cells lacking endogenous Tpm 2.1 (Supplementary Fig. 7a). Although Tpm 3 depletion caused a decreased level of force in MDA-MB-231s (Supplementary Fig. 7b), it failed to increase CU density and rigidity-dependent adhesion formation on both rigid and soft surfaces (Supplementary Fig. 7c–f). This indicated that Tpm 2.1 expression is necessary for CU formation and cell rigidity sensing.
Reduced expression of high-molecular-weight tropomyosins (including Tpm 2.1) and increased expression of low-molecular-weight tropomyosins (including Tpm 3.1 and Tpm 3.2) were reported in cells transformed by various oncogenes, carcinogens and viruses34. In addition, Tpm 2.1 co-polymerized on the same actin filaments with Tpm 3.1 and Tpm 3.2 but not with other Tpm isoforms in vitro32. The fact that silencing of Tpm 3 in Tpm 2.1-expressing transformed cells increased the number of CUs during early spreading raised the possibility that there was competition between Tpm 2.1 and Tpm 3 (Tpm 3.1 and Tpm 3.2) during CU formation. To test this hypothesis, we asked whether overexpression of Tpm 3 (Tpm 3.1 or Tpm 3.2) in a normal fibroblast cell line would suppress cell rigidity sensing. To that end, HFF cells expressing high levels of NeonGreen-Tpm 3.1 or EYFP-Tpm 3.2 were analysed on pillar surfaces. Forces produced by these cells were significantly higher than by WT HFF cells on both rigid and soft surfaces (Supplementary Fig. 8a). Conversely, CU density was decreased (Supplementary Fig. 8b) and Tpm 3-overexpressed HFF cells formed adhesions of similar size on both rigid and soft PDMS surfaces after plating for 6 h (Supplementary Fig. 8c–e). This indicated that high expression levels of Tpm 3.1 or Tpm 3.2 had inhibited CU formation as well as rigidity sensing, potentially through competition with Tpm 2.1.
To further examine the behaviour of Tpm 2.1 in Tpm 3.1-over-expressing cells, we fixed and stained endogenous Tpm 2.1 in HFFs with or without NeonGreen-Tpm 3.1 overexpression after 15-min spreading on pillars. Consistent with previous observations3, Tpm 2.1 concentrated at cell edges in control HFFs. However, this peripheral localization of Tpm 2.1 disappeared in T pm 3.1-over-expressing cells (Supplementary Fig. 8f–i). Thus, we suggest that high levels of Tpm 3 inhibited cell rigidity-sensing CU formation by competion with endogenous Tpm 2.1.
Restoration of cell rigidity sensing in transformed cells suppressed tumour formation in vivo
Since the restoration of CUs in MDA-MB-231 cells blocked colony formation in soft agar, we hypothesized that restoration of rigidity-sensing activity might suppress tumour growth in vivo. To test this hypothesis, we embedded the same number of stably EGFP-N3-transfected MDA-MB-231 (231) cells or YFP-Tpm 2.1-transfected 231-Tpm cells in Matrigel and placed the cell plug in a chick chorioallantoic membrane (CAM) model35 (Fig. 6a). Consistent with the in vitro experiments, the average weight of tumours formed by 231-Tpm cells was 39.5% lower than for 231 cells (Fig. 6b–d). In addition, tumour volume increased by 51% on average with 231 cells after 4 d but, in contrast, that of 231-Tpm cells decreased by 20% during the same time period (Fig. 6e). In the hematoxylin and eosin staining assay, tumour cell density was significantly lower in the 231-Tpm group than in the 231-control group (Supplementary Fig. 9a).
Fig. 6 |. Restoration of rigidity sensing in transformed cells blocks tumour formation in vivo.
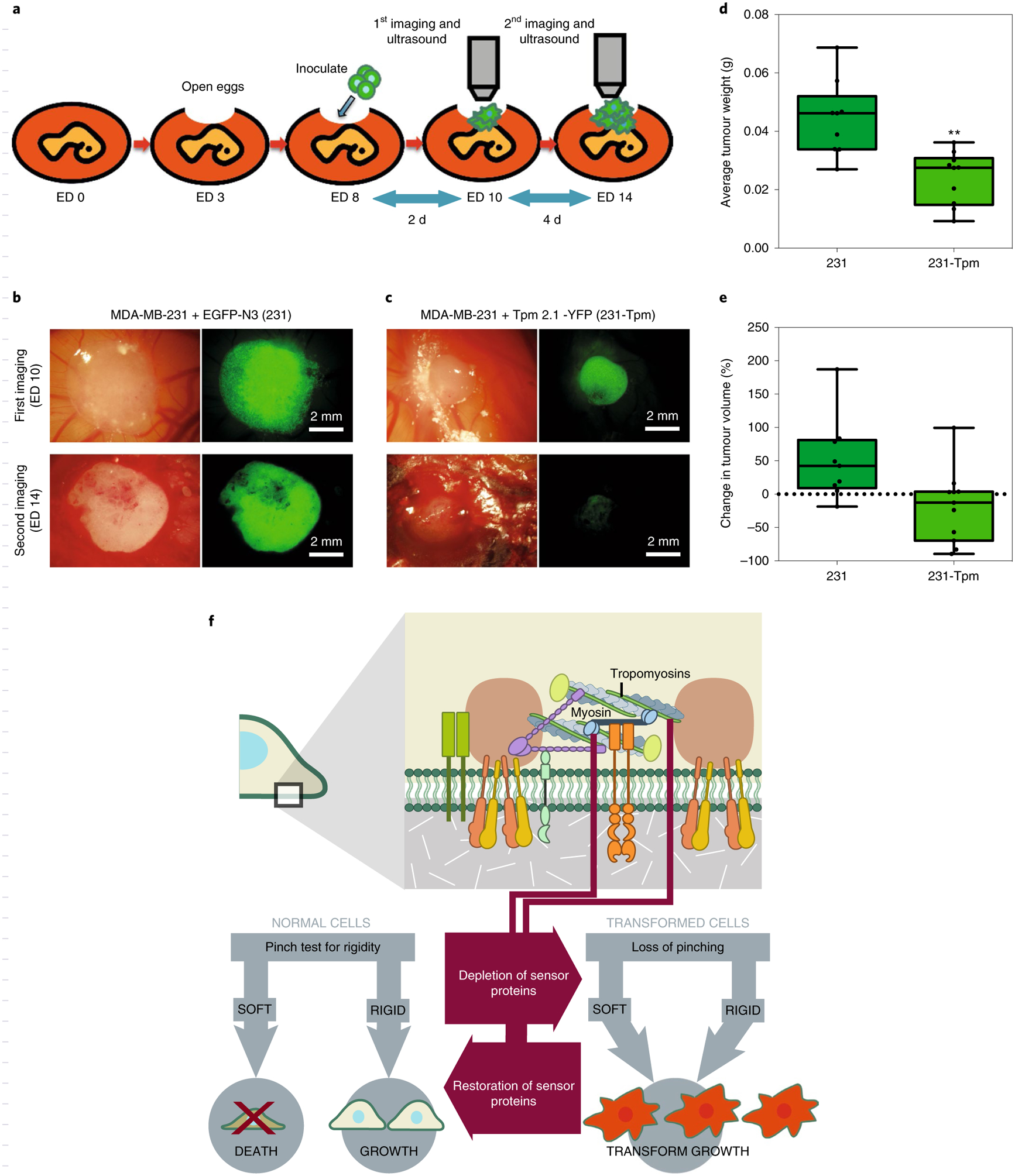
a, Schematic illustration of the CAM assay indicating the critical experimental steps. b,c, Bright field and fluorescence images of 231 (b) and 231-Tpm cells (c) grown in the CAM assay at ED 0 and 14. d, Boxand-whisker plots of average tumour weight of 231 and 231-Tpm cells at ED 14 (n = 9 for 231 group and n = 10 for 231-Tpm group; **P < 0.01). e, Box-and-whisker plots of tumour volume percentage changes in 231 and 231-Tpm cells from ED 10 to 14. f, Schematic illustration of the relationship between cell mechanosensing and transformed growth.
To determine whether the presence of rigidity sensing caused changes in cell expression patterns, we performed RNA-sequencing (RNA-seq) analyses of five cell line pairs that differed only in the expressed levels of Tpm 2.1 or Tpm 3 (Supplementary Table 2). In the case of MDA-MB-231 versus 231-Tpm cells, there were 362 upregulated and 700 downregulated genes; and for Tpm 2.1-depleted HFF versus control HFF cells, there were 244 upregulated and 415 downregulated genes. Overexpression of Tpm 3.1 or Tpm 3.2 in HFFs caused dramatic changes in 1,864 a nd 1,268 gene expression levels, respectively. In HT1080 cells, silencing of Tpm 3 led to up- and downregulation of hundreds of genes. Thus, it seems that there are major changes in cell protein composition when changing from the transformed to the normal state—that is, after restoration of rigidity sensing (Fig. 6f).
Discussion
Based on the current and related findings, transformed growth in cancer cells correlates with their inability to sense substrate rigidity. Across different cancer types, the unbalanced expression of CU-related cytoskeletal proteins decreases rigidity-sensing CU formation, which results in transformed cell growth. When rigidity-sensing CUs are rebuilt in these transformed cells, rigidity-dependent growth is restored and tumour formation in vivo is inhibited. Overexpression of another cytoskeletal protein, Tpm 3 (including Tpm 3.1 and Tpm 3.2), which is overexpressed in many cancers, causes CU disruption. These findings strongly support the hypothesis that transformed growth in cancer cells requires the depletion of rigidity-sensing modules.
One of the major pathways to induction of transformation is the expression of mutant Ras, which is normally activated by a variety of RTKs that participate in cell proliferation, transformation and regulation of differentiation36. For example, MDA-MB-231 cells carry a K-Ras mutation37. In previous studies, mutant Ras isoforms caused a reduction in tropomyosin 2.1, possibly through the upregulation of miR-21 (ref.38). In terms of other ways of transforming cells—for example, virus transformation—certain rigidity-sensing cytoskeleton protein expression levels are affected. SV40 transformation led to the loss of myosin IIA expression in Cos7 cells. In MEF cells, SV40 transformation increased Tpm 3 but, not Tpm 2.1, expression levels39. As we show here, overexpression of Tpm 3 decreased the number of CUs by competing with Tpm 2.1. Thus, classical ways of transformation normally alter different cytoskeletal protein expression levels and therefore cause reduction in CUs. This further supports the relationship between rigidity-sensing CU formation and cell transformation.
An additional connection between cancer and local CUs arises from the role played by tyrosine kinases in rigidity sensing. Previous studies have shown that rigidity sensing requires the action of Src family kinases (SFKs). Knocking out the upstream activator, RPTPα, or the three Src kinases, Src, Yes and Fyn (SYF cells), blocks the ability of those cells to sense substrate rigidity and also enables growth on soft surfaces40,41. This is consistent with our recent study which showed that ErbB family members (EGFR and HER2) are recruited to adhesion sites by SFKs on rigid surfaces and catalyse the formation of CUs in early cell spreading on rigid surfaces8. These findings indicate that RTKs involved in both cancer and epithelial-to-mesenchymal transition also play important roles in rigidity-sensing regulation.
There are marked differences in the level of expression of the various tyrosine kinases among the different transformed cell lines. This is consistent with the belief that many RTKs can contribute to the potential of cancer cells to grow and are involved in rigidity sensing30. Because cells test the rigidity of new micro-environments very early, anoikis, which has been linked to activation of death-associated protein kinase1 (DAPK1)42,43, would normally block growth in many metastatic environments. However, if soft matrices do not activate anoikis because the rigidity sensor is missing, many growth-activation pathways have time to function. Cytoskeletal changes will alter the nature of the mechanical signals collected by cells from their micro-environment, and RTKs can expand on those signals for further cell fate decision-making. Thus, RTKs may facilitate growth in inappropriate environments if growth regulation by mechanosensors of rigidity or other factors is aberrant.
Transformation involves a major change in cell behaviour that can be described as a change in cell state. It is therefore surprising that low levels of Tpm 2.1, myosin IIA or α-actinin 4 can cause transformation in a variety of cell backgrounds. However, recent studies show that rigidity-sensing cadherin contractions in epithelial cells also involve Tpm 2.1, indicating that mechanosensing even in epithelial monolayers involves Tpm 2.1 (ref.44). When myosin IIA is present, transformed cells develop very high forces on pillars. MDA-MB-231 cells show rapid transformed growth without myosin IIA, indicating that the loss of rigidity sensing—but not the level of force at these initial stages—activates transformed growth. Myosin IIs are normally present in most cancer cells, and they then develop very high forces on substrates45, which may enhance growth and metastasis in an in vivo context. In summary, reduction in rigidity-sensing modules is sufficient to cause the transition from a normal to a transformed state.
Severe cancers have many features that are important for tumour growth; however, the depletion of rigidity-sensing modules that causes transformation enables them to escape from the anoikis pathways during metastasis. It is remarkable that reintroduction of the missing CU components in transformed cancer cell lines successfully rebuilds the rigidity-sensing process irrespective of the tissue of origin. Restoring the ability to correctly sense rigidity causes many transformed cancer cells to die on soft surfaces without further manipulations. This provides a different view of blockage of transformed growth—a functional rigidity sensor activates apoptosis pathways on soft surfaces and the depletion of those sensors is often sufficient for cell growth. Although there may be other mechanisms resulting in growth on compliant matrices, the loss of rigidity-sensing modules is a robust mechanism. We therefore propose that transformed growth is, in essence, a mechanobiological phenomenon since restoration of rigidity sensing by cytoskeletal proteins that lack enzymatic activity suppresses growth. In the same way that modular sensory units are required for the stop signal in autopilot cars, modular rigidity-sensing units are needed for cells to sense the wrong matrix micro-environment and provide stop signals for growth.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of code and data availability and associated accession codes are available at https://doi.org/10.1038/s41563–019-0507–0.
Methods
Cell culture and transfection.
HFF cells (ATCC), Cos7 cells (ATCC), MDAMB-231 cells (ATCC), HT1080 (ATCC), SKOV3 cells (ATCC), CV1 (ATCC), PANC1 (from Y. Shaked, Technion), U251 (from P. Monzo, IFOM), A2058 (from P. Koeffl , CSI, NUS), A375P (from C. Simpson, ICR) and LLC cells (ATCC) were cultured in DMEM with high glucose supplemented with 10% FBS and 1 mM sodium pyruvate. IOSE 523 cells were cultured in RPMI 1640 with 10% FBS. MCF 10A cells were cultured in DMEM supplemented with 20 ng ml−1 epidermal growth factor, 10 ng ml−1 bovine insulin, 500 ng ml−1 hydrocortisone and 5% horse serum albumin. HUVEC cells (from N. Rafi , MBI) were cultured in Endothelial Cell Growth Medium (Sigma). Cells were transfected with DNA plasmids either using lipofectamine 2000 (Invitrogen) or by the Neon electroporator system (Life Technologies), according to the manufacturer’s instructions. Expression vectors encoding the following fluorescent fusion proteins were used: Tpm 2.1-YFP (also named hTm1), NeonGreen-Tpm 3.1 (also named Tm 5NM1, C-terminal tagged), EYFP-Tpm 3.2 (also named Tm 5NM2) (gifts rom P. Gunnning), Myosin IIAEGFP and Emerald-myosin IIB (gift rom M. W. Davidson group). Two Tpm 3 inhibitors, TR100 and ATM 3507 (gifts rom P. Gunning), were synthesized as previously described14,33. Each of those compounds was dissolved in dimethyl sulfoxide (DMSO, Sigma) at 50 mM as stock concentration and then diluted to the appropriate working concentration for further experiments.
Transfection of siRNA and immunoblotting.
Cells were seeded into a six-well dish on embryonic Day 0 and transfected with 25 μM myosin IIA siRNA (Dharmacon), Tpm 2.1 siRNA (Qiagen) or Tpm 3 siRNA (Dharmacon) using lipofectamine RNAiMAX (Invitrogen) on Day 1. Control cells were transfected with scrambled control siRNA (Dharmacon). Transfected cells were lysed in RIPA buffer (Sigma), and proteins extracted were separated by 4–20% SDS-polyacrylamide gel (Bio-rad) and transferred to PVDF membranes (Bio-rad) at 75 V for 2 h. Membranes were incubated with appropriate primary antibodies at 4 °C overnight: anti-myosin IIA (Sigma, dilution 1:1,000), anti-TM311 (Sigma, dilution 1:1,000), anti-TM γ9d (a gift from P. Gunning, dilution 1:1,000), anti-EGFR (CST, dilution 1:1,000), anti-HER2 (CST, dilution 1:1,000), anti-ROR2 (CST, dilution 1:1,000), anti-FLNA (abcam, dilution 1:1,000), anti-AXL (CST, dilution 1:1,000), anti-α-actinin (Sigma, dilution 1:1,000) and anti-α-tubulin (Sigma, dilution 1:3,000). Primary antibody binding was processed for ECL detection (Thermo Fisher Scientific) with appropriate horseradish peroxidase-conjugated secondary antibodies (Bio-rad). Uncropped immunoblot images are included in Supplementary Fig. 10.
Pillar fabrication, video microscopy and force traction measurements.
Moulds for PDMS pillars were fabricated as described previously6,8,46. PDMS (0.1 g, mixed at 10:1, Sylgard 184; Dow Corning) was poured onto the silicon mould and then flipped onto a plasma-cleaned glass-bottom dish (ibdi). The sample was pressed by an 8-g weight, cured at 80 °C for 3 h to reach a Young’s modulus of 2 MPa and was then de-moulded while immersed in 99.5% isopropanol. Pillars were washed with PBS five times before coating with 10 μg ml−1 fibronectin (Roche) for cell seeding. Time-lapse imaging and traction force measurements were performed as previously described6,8.
Soft and rigid PDMS surface preparation.
Preparation of PDMS surfaces of varying stiffness was performed as previously described30. PDMS substrates were prepared using a Sylgard 184 silicone elastomer kit. Elastomer/curing agent ratios of 10:1 and 75:1 corresponded to Young’s moduli of 2 MPa and 5 kPa, respectively. The mixed components were de-gased and spin-coated at 2,000 r.p.m for 2 min with a Spin Processor on 25-mm microscope coverslips (Marienfeld Superior). Crosslinking of the elastomer was carried out at 80 °C for 4 h. PDMS surfaces were coated with 10 μg ml−1 fibronectin (Roche) overnight at 4 °C. Before cell plating, PDMS surfaces were washed with PBS and growth medium.
Polyacrylamide (PAM) gel preparation.
Glass-bottom dishes (Iwaki) were silanized using 1.2% 3-methacryloxypropyltrimethoxysilane (Shin-Etsu Chemical, Tokyo, Japan) in 100% methanol for 1 h at room temperature, then 2.3 kPa acrylamide gel was prepared as previously described47. Gel surfaces were treated with sulfo-SANPAN (Thermo Fisher Scientific) and exposed under UV light for 5 m before coating with 10 μg ml−1 fibronectin for cell culture.
Fluorescence microscopy and analysis.
Cells were fixed with 4% paraformaldehyde in PBS at 37 °C for 15 min and permeabilized with 0.2% TX-100 for 10 min at room temperature. Samples were blocked with 1% bovine serum albumin in PBS for 1 hat room temperature, incubated with primary antibodies for paxillin (BD, 1:200), cleaved caspase-3 (CST, 1:200), myosin IIA (Sigma, 1:200) or Tropomyosin 2.1 (Sigma, 1:200) at 4 °C overnight and then incubated with secondary antibodies (Molecular Probes) for 1 h at room temperature. Fluorescence images were acquired using a spinning-disc confocal microscope (PerkinElmer Ultraview VoX) attached to an Olympus IX81 inverted microscope body.
Quantifications of single focal adhesion size and cell aspect ratio were performed as previously described3,30. For single adhesion analysis, images of paxillin-stained cells were analysed using Analyze Particles plugin after applying a threshold for background subtraction. The projected area and best-fit ellipse aspect ratio of phalloidin-stained cells were also calculated using ImageJ.
Cell spreading assay on hydrogel surfaces.
Easy Coat Hydrogel surfaces of varying rigidity (1, 2 and 4 kPa) were purchased from Matrigen (no. SV3510-EC), coated with 10 μg ml−1 fibronectin (Roche) for 1 h at 37 °C and washed with PBS and cell culture medium before cell seeding. HFF, MDA-MB-231, Cos7 and HT1080 cells were seeded on hydrogel or glass surfaces overnight (16 h), fixed and stained as described above (fluorescence microscopy and analysis). Fluorescence images were acquired using a Delta Vision System attached to an Olympus IX71 inverted microscope body.
Soft agar assay.
Soft agar assay and CyQuant analysis were performed using CytoSelect 96-Well Cell Transformation Assay (Soft Agar Colony Formation Kit) from Cell Biolabs according to the manufacturer’s instructions. In particular, the final cell culture agar layer solution was 0.4%, corresponding to a Young’s modulus of <2 kPa48. In the inhibitor-treated experiments, 100 μl of cell culture medium or medium containing inhibitors was added to each well. Fresh medium or medium containing fresh inhibitors was changed every 24 h during incubation.
Cell proliferation assay.
Cells (1,000 per well) were plated in fibronectin-coated 2-kPa Hydrogel (Matrigen) or plastic 96-well plates. Cell proliferation measurements were performed using the CyQuant Cell Proliferation Assay Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions (Adherent Cells).
CAM model.
The CAM model was prepared as previously described35. Fertilized chicken eggs were incubated at 37 °C with 60% relative humidity starting from the day of collection, embryonic day 0 (ED 0). On ED 3, a window was made on every egg to prevent the CAM from sticking to the shell during development. Under sterile conditions, a small incision was made at the narrow end of the egg using a pointed surgical rod or needle, and 4–5 ml of albumin was removed through the hole to introduce the CAM. The shell was cut to form a window of diameter ~1 cm, exposing the CAM. This window was sealed with Tegaderm transparent film dressing (3 M). The eggs were further incubated to ED 7 or 8, when the membranes were ready for grafting of cancer cells. MDA-MB-231 cells transfected with GFP-control or YFP-Tpm 2.1 were selected by the antibiotic G418 (Thermo Fisher Scientific) for 2 weeks. For grafting onto CAM, cancer cells were trypsinized, washed by PBS and re-suspended in pre-chilled Matrigel (BD Biosciences). For each egg, the shell was cut to form a larger window. By gently touching the upper periderm layer of the CAM with an autoclaved glass rod, a slightly bruised area was formed and 9 × 105 cells in 50 μl of Matrigel were inoculated dropwise onto this CAM area. After inoculation, the window was sealed again with film dressing. On ED 10 or 11, tumours that had formed were imaged using an Olympus SZX Microscope (optical and fluorescence). For ultrasound imaging, a thin plastic wrap was carefully placed to cover the eggs, directly touching the top of the tumours. Warmed ultrasound transducer gel (Aquasonic 100) was placed over the plastic wrap and a MS550D transducer connected to a three-dimensional (3D) Acquisition Motor was used to image tumours using VisualSonics Vevo 2100 Imaging System in 3D Power Doppler mode. Multiple cross-sectional images were analysed and reconstructed using Vevo Lab v.1.7.0. Imaging was performed again on ED 14 to compare tumour growth. Only tumours with viable embryos were included in data analysis. Following completion of final imaging, tumours were harvested and weighed and the chick embryos were sacrificed.
Gene expression profile.
Transcriptomes of MDA-MB-231, Tpm 2.1-YFP-transfected MDA-MB-231, HFF, Tpm 2.1-depleted HFF, Tpm 3.1/Tpm 3.2-overexpressed HFF, HT1080 and Tpm 3-depleted HT1080 cells were mapped, quantified and indicated as fragments per kilobase million using the RNA-Seq technology49. Briefly, the total RNA of all cell lysate groups was extracted using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. The extracted RNA was processed and analysed by BGI Tech Solutions Co., as previously described50.
Statistical analysis.
Prism (GraphPad Software) and Matlab (Math Works) were used for data analysis and graph plotting. Analyses of significant difference levels were carried out using either one-way analysis of variance (for more than two experimental groups), two-tailed Student’s t-test or Mann–Whitney test (in vivo results). No adjustment was used for multiple comparisons. In the box-and-whisker plots, the box spans the interquartile range and the median is denoted by the vertical line with the box. The exact n number for each figure is summarized in Supplementary Tables 3 and 4. Precise P values for all non-significant groups are summarized in Supplementary Tables 5 and 6. All experiments were repeated at least twice.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Data supporting the findings of this study are available within the article and from the corresponding author upon reasonable request;
Supplementary Material
Acknowledgements
The authors thank J.P. Thiery, P. Gunning, N.C. Gauthier and J. Kadrmas for critical reading of the manuscript. We thank D. Lim Gkeok Stzuan (NUHS) and A. Koh Pei Fern (CSI) for their kind help with H&E staining. We thank the members of Sheetz and Bershadsky laboratories for their kind help. This research was supported by funding to the Mechanobiology Institute, National University of Singapore. B.Y. was supported by the NUS grant ‘Activation of Apotosis by Soft Surfaces’ (no. R-714-000-112-133). H.W. is a David and Inez Myers Career Advancement Chair in Life Sciences fellow. M.P.S. is supported by NIH and NUS grants and the Mechanobiology Institute, National University of Singapore.
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41563–019-0507–0.
References
- 1.Hamburger AW & Salmon SE Primary bioassay of human tumor stem cells. Science 197, 461–463 (1977). [DOI] [PubMed] [Google Scholar]
- 2.Frisch SM & S creaton RA Anoikis mechanisms. Curr. Opin. Cell Biol 13, 555–562 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Wolfenson H et al. Tropomyosin controls sarcomere-like contractions for rigidity sensing and suppressing growth on soft matrices. Nat. Cell Biol 18, 33–42 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weinberg J & Drubin DG Clathrin-mediated endocytosis in budding yeast. Trends Cell Biol 22, 1–13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghassemi S et al. Cells test substrate rigidity by local contractions on submicrometer pillars. Proc. Natl Acad. Sci. USA 109, 5328–5333 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang B et al. Mechanosensing controlled directly by tyrosine kinases. Nano Lett 16, 5951–5961 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meacci G et al. α-Actinin links ECM rigidity sensing contractile units with periodic cell edge retractions. Mol. Biol. Cell (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saxena M et al. α-Actinin links ECM rigidity sensing contractile units with periodic cell edge retractions. Nat Mater (2017) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fletcher DA & Mullins RD Cell mechanics and the cytoskeleton. Nature 463, 485–492 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fife CM, McCarroll JA & Kavallaris M Movers and shakers: cell cytoskeleton in cancer metastasis. Br. J. Pharmacol 171, 5507–5523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schramek D et al. Direct in vivo RNAi screen unveils myosin IIa as a tumor suppressor of squamous cell carcinomas. Science 343, 309–313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conti MA et al. Conditional deletion of nonmuscle myosin II-A in mouse tongue epithelium results in squamous cell carcinoma. Sci. Rep 5, 14068 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raval GN et al. Loss of expression of tropomyosin-1, a novel class II tumor suppressor that induces anoikis, in primary breast tumors. Oncogene 22, 6194–6203 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Stehn JR et al. A novel class of anticancer compounds targets the actin cytoskeleton in tumor cells. Cancer Res 73, 5169–5182 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Menez J et al. Mutant alpha-actinin-4 promotes tumorigenicity and regulates cell motility of a human lung carcinoma. Oncogene 23, 2630–2639 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Nikolopoulos SN et al. The human non-muscle alpha-actinin protein encoded by the ACTN4 gene suppresses tumorigenicity of human neuroblastoma cells. Oncogene 19, 380–386 (2000). [DOI] [PubMed] [Google Scholar]
- 17.An HT, Yoo S & Ko J Alpha-actinin-4 induces the epithelial-tomesenchymal transition and tumorigenesis via regulation of snail expression and beta-catenin stabilization in cervical cancer. Oncogene 35, 5893–5904 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Bharadwaj S, Thanawala R, Bon G, Falcioni R & Prasad GL Resensitization of breast cancer cells to anoikis by tropomyosin-1: role of Rho kinase-dependent cytoskeleton and adhesion. Oncogene 24, 8291–8303 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Zhu S et al. MicroRNA-21 targets tumor suppressor genes in invasion and metastasis. Cell Res 18, 350–359 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Rasheed S, Nelson-Rees WA, Toth EM, Arnstein P & Gardner MB Characterization of a newly derived human sarcoma cell line (HT-1080). Cancer 33, 1027–1033 (1974). [DOI] [PubMed] [Google Scholar]
- 21.Freedman VH & Shin SI Cellular tumorigenicity in nude mice: correlation with cell growth in semi-solid medium. Cell 3, 355–359 (1974). [DOI] [PubMed] [Google Scholar]
- 22.Hollestelle A, Elstrodt F, Nagel JH, Kallemeijn WW & Schutte M Phosphatidylinositol-3-OH kinase or RAS pathway mutations in human breast cancer cell lines. Mol. Cancer Res 5, 195–201 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Fogh J, Fogh JM & Or feo T One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J. Natl Cancer Inst 59, 221–226 (1977). [DOI] [PubMed] [Google Scholar]
- 24.Mayo JG Biologic characterization of the subcutaneously implanted lewis lung tumor. Cancer Chemother. Rep 2 3, 325–330 (1972). [PubMed] [Google Scholar]
- 25.Kozlowski JM, Hart IR, Fidler IJ & Hanna N A human melanoma line heterogeneous with respect to metastatic capacity in athymic nude mice. J. Natl Cancer Inst 72, 913–917 (1984). [PubMed] [Google Scholar]
- 26.Fabricant RN, De Larco JE & Todaro GJ Nerve growth factor receptors on human melanoma cells in culture. Proc. Natl Acad. Sci. USA 74, 565–569 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lieber M, Mazzetta J, Nelson-Rees W, Kaplan M & Todaro G Establishment of a continuous tumor-cell line (panc-1) from a human carcinoma of the exocrine pancreas. Int. J. Cancer 15, 741–747 (1975). [DOI] [PubMed] [Google Scholar]
- 28.Bigner DD et al. Heterogeneity of genotypic and phenotypic characteristics of fi een permanent cell lines derived from human gliomas. J. Neuropathol. Exp. Neurol 40, 201–229 (1981). [DOI] [PubMed] [Google Scholar]
- 29.Jove R & Hanafusa H Cell tTransformation by the vViral src. Oncogene 3, 31–56 (1987). [DOI] [PubMed] [Google Scholar]
- 30.Prager-Khoutorsky M et al. Fibroblast polarization is a matrix-rigidity-dependent process controlled by focal adhesion mechanosensing. Nat. Cell Biol 13, 1457–1465 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Shutova MS et al. Self-sorting of nonmuscle myosins IIA and IIB polarizes the cytoskeleton and modulates cell motility. J. Cell Biol 216, 2877–2889 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gateva G et al. Tropomyosin isoforms specify functionally distinct actin filament populations in vitro. Curr. Biol 27, 705–713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Currier MA et al. Identifi ation of cancer-targeted tropomyosin inhibitors and their synergy with microtubule drugs. Mol. Cancer Ther (2017). [DOI] [PubMed] [Google Scholar]
- 34.Helfman DM, Flynn P, Khan P & Saeed A Tropomyosin as a regulator of cancer cell transformation. Adv. Exp. Med. Biol 644, 124–131 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Antony J et al. The GAS6-AXL signaling network is a mesenchymal (Mes) molecular subtype–specific therapeutic target for ovarian cancer. Sci. Signal 9, ra97–ra97 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Yamamoto T, Taya S & Kaibuchi K Ras-induced transformation and signaling pathway. J. Biochem 126, 799–803 (1999). [DOI] [PubMed] [Google Scholar]
- 37.Scholl C et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 137, 821–834 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Frezzetti D et al. Upregulation of miR-21 by ras in vivo and its role in tumor growth. Oncogene 30, 275–286 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Coombes JD et al. Ras transformation overrides a proliferation defect induced by Tpm3.1 knockout. Cell. Mol. Biol. Lett 20, 626–646 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Jiang G, Huang AH, C ai Y, Tanase M & Sheetz MP Rigidity sensing at the leading edge through alphavbeta3 integrins and RPTPalpha. Biophys. J 90, 1804–1809 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sawada Y et al. Force sensing by mechanical extension of the Src family kinase substrate p130Cas. Cell 127, 1015–1026 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kwon T et al. DANGER is involved in high glucose-induced radioresistance through inhibiting DAPK-mediated anoikis in non-small cell lung cancer. Oncotarget 7, 7193–7206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qin R, Wolfenson H, Saxena M & Sheetz M Tumor suppressor DAPK1 catalyzes adhesion assembly on rigid but anoikis on soft matrices Preprint at bioRxiv (2018). [DOI] [PMC free article] [PubMed]
- 44.Yang Y, Nguyen E, Mege R-M, Ladoux B & Sheetz MP Local contractions test rigidity of E-cadherin adhesions Preprint at bioRxiv (2018). [DOI] [PMC free article] [PubMed]
- 45.Mierke CT, Frey B, Fellner M, Herrmann M & Fabry B Integrin α5β1 facilitates cancer cell invasion through enhanced contractile forces. J. Cell Sci 124, 369–383 (2011).References [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cui Y et al. Cyclic stretching of soft substrates induces spreading and growth. Nat. Commun 6, 6333 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakazawa N, Sathe AR, Shivashankar GV & Sheetz MP Matrix mechanics controls FHL2 movement to the nucleus to activate p21 expression. Proc. Natl Acad. Sci. USA 113, E6813–e6822 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li C, Huang Z & Wang RK Elastic properties of soft tissue-mimicking phantoms assessed by combined use of laser ultrasonics and low coherence interferometry. Opt. Express 19, 10153–10163 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Mortazavi A, Williams BA, McCue K, Schaeffer L & Wold B Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Rafi NishaBteMohd et al. A mechano-signalling network linking microtubules, myosin IIA filaments and integrin-based adhesions. Nat. Mater 18, 638 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting the findings of this study are available within the article and from the corresponding author upon reasonable request;