

Abstract
Purpose:
Targeting non-specific, tumor associated antigens (TAA) with chimeric antigen receptors (CARs) requires specific attention to restrict possible detrimental on-target/off-tumor effects. A reduced affinity may direct CAR-engineered T (CAR-T) cells to tumor cells expressing high TAA levels while sparing low expressing normal tissues. However, decreasing the affinity of the CAR-target binding may compromise the overall anti-tumor effects. Here, we demonstrate the prime importance of the type of intracellular signaling on the function of low affinity CAR-T cells.
Experimental design:
We used a series of single-chain variable fragments (scFvs) with 5 different affinities targeting the same epitope of the multiple myeloma (MM)-associated CD38 antigen. The scFvs were incorporated in three different CAR costimulation-designs and we evaluated the anti-tumor functionality and off-tumor toxicity of the generated CAR-T cells in vitro and in vivo.
Results:
We show that the inferior cytotoxicity and cytokine secretion mediated by CD38 CARs of very low affinity (KD<1.9×10–6 M) bearing a 4–1BB intracellular domain can be significantly improved when a CD28 costimulatory domain is used. Additional 4–1BB signaling mediated by the co-expression of 4–1BBL provided the CD28-based CD38 CAR-T cells with superior proliferative capacity, preservation of a central memory phenotype and significantly improved in vivo anti-tumor function, while preserving their ability to discriminate target antigen density.
Conclusion:
A combinatorial costimulatory design allows the use of very low affinity binding domains (Kd<1μM) for the construction of safe but also optimally effective CAR-T cells. Thus, very-low-affinity scFvs empowered by selected costimulatory elements can enhance the clinical potential of TAA-targeting CARs.
Introduction
Adoptive immunotherapy with genetically engineered T cells bearing tumor-antigen specific chimeric antigen receptors (CAR) holds the potential for effective treatment of hematological malignancies and solid tumors. CARs are synthetic receptors that redirect antigen recognition and mediate T cell activation, in a single molecule, through the fusion of an extracellular antigen-binding moiety, such as a single-chain-variable region (scFv), with an intracellular signaling domain usually derived from the CD3ζ chain1. CARs endow T cells with customizable antigen recognition as scFv domains of different specificity and antigen-binding properties can be interchangeable. These properties confer a broad applicability potential to CAR T cells for a wide range of patients and diseases. Importantly, second- and third-generation CARs provide combined activation and costimulatory signals1,2. The addition of intracellular components from known costimulatory receptors/molecules produces signaling cascades similar to their normal counterparts and enhances T cell activation, expansion and in vivo persistence. Up to date, second generation CAR T cells targeting CD19 have been shown to induce impressive responses in chemotherapy resistant B cell leukemias and lymphomas (80–90% complete remissions in relapsed acute lymphoblastic leukemia) and the majority of clinical studies are performed using CARs containing either CD28 or 4–1BB cytoplasmic domains3–8.
Broadening the applicability of CAR-T cell therapy for various types of tumors remains a challenge since most of the available targets are tumor-associated antigens (TAA), which are not entirely tumor-restricted. In some cases the expression of the target on healthy tissues can be tolerable and clinically manageable, such as the B-cell aplasia caused by CD19 CAR T cells4,9, but in cases where vital tissues are involved off-tumor toxicity can be fatal10–12. Fine-tuning the affinity of the CAR’s binding domain can be a successful and easily applicable strategy to avert “on-target/off-tumor” reactivity of CAR-T cells. CARs of lower affinity targeting Erbb2/Her2, EGFR, CD123 or CD38 showed better discrimination between tumors and normal tissues expressing the same antigen in lower levels13–16. However, decreasing the CAR affinity results in a higher target expression-threshold for T cell activation and, depending on the level of antigen expression on the tumor cells, it may also hamper the efficacy of anti-tumor function17–19. Therefore, it is important to identify the conditions where the best discriminative potential between tumor and healthy tissues can be achieved, by using the lowest possible affinity, in combination with an optimal anti-tumor effect.
The influence of specific costimulatory moieties on the biology and therapeutic efficacy of CAR-T cells has been a subject of many recent studies. CAR-T cells bearing a 4–1BB costimulatory endodomain seem to persist for longer time in patient circulation in comparison to CAR-T cells having a CD28 costimulatory domain3,5,20, due to better maintenance of a memory phenotype and reliance on oxidative metabolism21–24. On the other hand addition of a CD28 endodomain confers a more efficient and rapid cytotoxic ability to CAR-T cells2,23. Moreover, the combination of both synergizing signaling pathways results in even greater CAR-T cell potency, persistence and anti-tumor response25–27. Previous studies evaluating the functionality of CAR-T cells with lower affinities, performed comparisons using the same costimulatory design. It is, therefore, largely unknown whether and how lowering the affinity for the target would affect the costimulatory requirements of CAR-T cells for optimal functionality and persistence.
Here, we hypothesized that the type of CAR mediated costimulatory design is of prime importance for the optimal function of low affinity CAR-T cells. To this end, we investigated a series of scFv’s binding to the same CD38 epitope but possessing 5 different affinities15. We incorporated these scFvs in three different CAR designs and evaluated the anti-tumor functionality, phenotype, and persistence of the generated CAR-T cells in vitro and in vivo. We demonstrate that decreasing the affinity of CARs can, depending on the CAR design, compromise the anti-tumor efficacy of CAR-T cells and that the combinatorial delivery of CD28 and 4–1BB signals potentiates lower affinity CARs and improves their immunotherapeutic potential in vitro and in vivo without increasing on-target/off-tumor toxicity.
Methods
Lower affinity CAR construction
Lower affinity CARs were produced with different germline variable light chains while keeping the variable heavy chain constant (clone 028). Selection and classification of lower affinity antibodies was described previously15. The selected variable heavy and light chains, separated by a G4S linker were PCR amplified with a proofreading Q5-Hotstart polymerase (NEB) using the AfeI containing forward primer 5’ctctgctgctgcctctagcgctgctgctg3’ and the NotI containing reverse primer 5’gttgtgcggccgcgctggacacggtgaccattg 3’. PCR product was purified (Bioké) and cloned into SFG retroviral vector with a T4 ligase (Roche). The scFv was followed by a CD8a transmembrane domain and the 4–1BB and CD3ζ signaling domains or a CD28 transmembrane and intracellular sequence as described in Zhao et al23. The CAR sequences were linked by a P2A sequence44 to a truncated LNGFR, dsRed or 4–1BBL sequence.
Cloning of 4–1BBL
The 4–1BBL sequence was obtained form EBV-LCL cell line 10850, amplified by standard RT-PCR (Thermo Fisher) using a compatible primer pair. cDNA was used as a template to replace the dsRed, separated by a P2A from the CAR-CD28z. The forward primer including a RsrI restriction site 5’atcccggaccgatggaatacgcctctgacg3’ and reverse primers with a SalI restriction site 5’ccgtcgacctattattccgacctcggtgaag 3’ were used to replace the dsRed sequence with the 4–1BBL. The 4–1BBL sequence is separated by the P2A sequences and is therefore expressed separate from the CAR-28z.
Generation of retroviral particles and transduction of T cells
Phoenix-Ampho packaging cells were calcium phosphate transfected with 10 μg CAR constructs. 16 hours post-transfection complete medium (DMEM + 10% FBS) was refreshed, and two and three days after transfection, cell free supernatants containing retroviral particles were collected and directly used for transduction.
Peripheral blood mononuclear cells (PBMCs) from healthy donors (3×106/well) were stimulated with lectin-like phytohemagglutinin (PHA-L) in a 6 well plate (Greiner Bio-One) in culture medium (RPMI-1640, 10% FBS, penicillin;100 U/ml, streptomycin; 100 μg/ml). After 48 hours, 1 ml 3×106/ml of cells were transferred to retronectin coated (15 μg/ml) (Takara) 6-well plates (Falcon). Retroviral transduction was performed by addition of 2 ml virus per well followed by spinoculation (1500g, 1 hour at room temperature) in the presence of 4 μg/ml Polybrene. A second transduction was conducted after 16 hours, replacing 2/3 of the cell supernatant with freshly obtained virus (2 ml). 6–8 hours after the second hit, half of the cell supernatant was replaced by fresh culture RPMI-1640 + 10%FBS and 50 IE/ml rhIL-2 (Proleukin®, Novartis) was added once. 72 hours post-transduction LNGFR, dsRed or 4–1BBL and CD38 expression were measured by flow cytometry to determine transduction efficiency.
Primary cells from MM patients and healthy individuals.
Healthy donor peripheral blood mononuclear cells (PBMCs) from buffy coats (Sanquin blood-bank) or bone marrow mononuclear cells (BM-MNCs) from MM patient’s bone marrow aspirates (~10–40% malignant cells, determined by flow cytometry (CD138+/CD38+)), were isolated from through Ficoll-Paque (GE Healthcare Life Sciences) density centrifugation. Primary apheresis material was thawed and subsequently sorted by EasySep (stem cell technologies) with CD34+ magnetic beads according to manufacturer’s protocol. Isolated cells were directly used in cytotoxicity assays or cryopreserved in liquid nitrogen until use. All primary samples were obtained after informed consent and approval by the institutional medical ethical committee.
Cell lines
The human MM cell line, UM9 (unmodified or luciferase (Luc-GFP)-transduced) was cultured in RPMI-1640 (Thermo Fisher) + 10% FBS (Invitrogen) + antibiotics (penicillin;100 U/ml, streptomycin; 100 μg/ml). The mouse fibroblast cell line NIH/3T3 cell line was obtained from ATCC and transduced with a lentivirus to express human CD38. NIH/3T3 (modified or CD38-transduced) cells and Phoenix Ampho cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Thermo Fisher) + GlutaMAX.10% FBS (Invitrogen) and penicillin (100 U/ml) and streptomycin (100 μg/ml).
Flow cytometry
Flow cytometry assays were performed on BD LSRFortessa. Viable cells were determined with live/dead cell marker (LIVE/DEAD® Fixable Near-IR; Life Technologies L10119). Transduction efficiency and associated CAR expression was measured with an APC conjugated antibody towards NGFR (CD271) (Biolegend) for CAR-4–1BBz-LNGFR and APC antibody 4–1BBL (CD137L) (Biolegend) for CAR-28z-41BBL. CAR-28z-dsRed were measured in the PE-CF594 channel to detect dsRed. Additional antibodies were used for weekly differentation phenotype: CD3, CD4, CD8, CD38, (BD Bioscience), CD45RA and CD62L (Biolegend), for exhaustion assays antibodies: PD-1, LAG-3 and TIM-3 (Biolegend) and for cytotoxicity assays: CD3, CD14, CD19, CD38, CD56 and CD138 (BD Bioscience). To distinguish Mock/CAR T cells from target cells, target cell were stained with 0.5 μM Violet tracer (Thermo Fisher) for 25 minutes and washed before cytotoxicity assay co-cultures. Flow cytometry data analysis was performed with FACS Diva 6.1 software.
Proliferation assays
CAR T cells were counted and stimulated weekly with irradiated (50 Gy) CD38+ UM9 cells or (80 Gy) 3T3-CD38. Starting seven days post-transduction, 1×106 CAR+ T cells were seeded in a 24-well plate containing 3×105 UM9 or 3T3-CD38 cells, to a volume of 1–1.5 ml. No additional cytokines were added and when a cell count of ~2×106 cells/ml was exceeded the cell culture was split to a density of 1×106 per well.
Flow cytometry-based cytotoxicity assay
Seven to ten days after transduction serial dilutions (Effector:Target 3:1, 1:1 or 1:3) of CAR T cells were incubated with Violet tracer (Thermo Fisher) labeled BM-MNC or PBMC for 16–24 hours. After addition of Flow-Count™ Fluorospheres (Beckman 7547053) cells were harvested and stained for different CD markers (see section flow cytometry) to distinguish different subsets. Viable cells were then quantitatively analyzed through Flow-Count-equalized measurements. Percentage cell lysis was calculated as followed and only if the analyzed target cell population contained >500 viable cells in the untreated samples. % lysis cells = 1 − ((# viable target cells in treated wells/# of beads) / (#viable target cells in untreated wells/# of beads)) × 100%.
Bioluminescent Imaging based cytotoxicity assay
Seven to ten days after transduction serial dilutions (effector:target 10:1, 3:1, 1:1, 1:3 or 1:10) of CAR T cells were incubated with Luc-GFP-transduced human MM cell line UM9. The luciferase signal produced by surviving UM9 cells was determined after 16–24 hours with a GloMax® 96 Microplate Luminometer (Promega) within 15 minutes after the addition of 125 μg/mL beetle luciferin (Promega). % lysis cells = 1 − (BLI signal in treated wells / BLI signal in untreated wells) × 100%.
Cytokine measurements
To determine cytokine production by CAR T cells, cell supernatants were harvested 24 hours after co-culture with target cells (UM9, 3T3 or MM-BM). To measure cytokines we used Cytokine Bead Array (CBA) Human Th1/Th2/Th17 cytokine kit (BD) according to manufacturer protocol. In brief, a mixture of capture beads (IL-2, IL-4, IL-6, IL-10, IL17A, TNF and IFN-γ), PE-detection reagent and cell supernatant were incubated for 3 hours. Beads were washed and analyzed by a BD standardized flow cytometry assay.
In vivo xenograft studies
RAG2−/−γc−/− mice used in this study were bred and maintained at the Amsterdam Animal Research Center. We used an in vivo model, in which a humanized bone marrow-like environment is created in mice to allow the growth of human MM tumors or normal CD34+ cells in their natural niche. Briefly, hybrid scaffolds consisting of three 2- to 3-mm3 triphasic calcium phosphate particles were coated in vitro with human bone marrow mesenchymal stromal cells (BM-MSC)(2×105 cells/scaffold). The scaffolds were implanted subcutaneously into the mice29. Eight to twelve weeks after implantation, for the anti-tumor model 10×106 luciferase-transduced MM cells (UM9) were injected i.v.. Or in the separate off-tumor experiment, 1×106 fluorescent (FarRed) labeled healthy CD34+ cells were injected transcutaneously into the scaffold. After one week, when the tumor or CD34+ cells became detectable by bioluminescence imaging (BLI) or Fluorescence live imaging (FLI) respectively, mice were divided in equal groups. Mice received CD38-CAR-BBz, 28z, 28z-BBL or mock-transduced T cells (5×106 cells/mice), by i.v. injection in the tail vein. Tumor growth or CD34+ cell persistence was monitored by weekly BLI/FLI measurements. Postmortem, bone marrow, spleen and scaffolds were harvested from each mouse, bone marrow was flushed and spleen and scaffolds were dissociated. Flushed or dissociated tissues were filtered through a 70 μm filter and single cell suspensions were counted, stained and measured by flow cytometry.
Hematopoietic progenitor cell growth inhibition assay
A total of 2000 CD34+ EasySep sorted (Stem cell technologies) cells from MM patient apheresis material were mixed with effector CD38-CAR T cells at a CART:BM cell ratio of 1:1 in 0.2 mL of RPMI + FBS culture medium. After culturing for 4 hours in this small volume, the cells were resuspended to a final volume of 2 mL with semisolid Methocult (Stem cell technologies, H4534),then plated in 6cm dishes and incubated at 37°C in 5% CO2. Between 14–21 days, the number of colony-forming unit-granulocytes (CFU-G), and CFU-monocytes (CFU-M), were scored under a microscope.
Statistical analysis
Statistical analyses were performed using Graphpad Prism software version 7.0. For normal distributions parametric student’s t-tests were used. In analyses where multiple groups were compared, either a parametric ANOVA with bonferroni posthoc test or nonparametric Kruskal-Wallis test were used with subsequent multiple comparison. Two-tailed statistical tests were always used. A p value <0.05 was considered significant.
Results
CD38-CAR constructs combining different affinities and costimulation design.
We used CD38 as a model for a TAA since it is highly and uniformly expressed on MM cells but is also present at lower levels on subsets of healthy hematopoietic cells. We generated 15 different CAR constructs through the combination of 5 scFv domains of variable affinity targeting the same CD38 epitope with 3 described CAR structural designs (Figure 1A). Affinity was ranked from high to very low as depicted in Figure 1B. The scFvs were cloned into vectors encoding the components of the two most popular second-generation CAR structures providing the 4–1BB (BBz) or the CD28 (28z) modalities (Figure 1A). In order to deliver both 4–1BB and CD28 signaling we chose to use a design recently shown to provide the optimal combination of these two signals, where a second generation CD28z CAR is co-expressed with 4–1BBL (28zBBL) (Figure 1A)23,28. The CAR transgenes were linked to functionally irrelevant markers whose expression levels was used as surrogate marker of CAR expression on transduced T cells, as previously validated for these constructs (16). All CAR constructs were well expressed upon transduction on human T lymphocytes without significant difference in expression levels (Figure 1C–D and Figure S1A–B). In contrast to high affinity CAR028 cells, lower affinity CD38-CAR-T cells showed no significant decrease of CD38 expression compared to mock-transduced T cells, indicating reduced CD38-directed cytotoxicity against targets with intermediate CD38 expression (Figure 1E and Figure S1C). After acquiring a decreased CD38 expression on the cell surface, high affinity CD38-CAR T cells were eventually able to expand (see also Drent et al. Haematologica). Therefore, CAR T cells were used at least after one week of culture so that fratricide would not bias the absolute number of CAR T cells.
Figure 1. CAR constructs and expression.
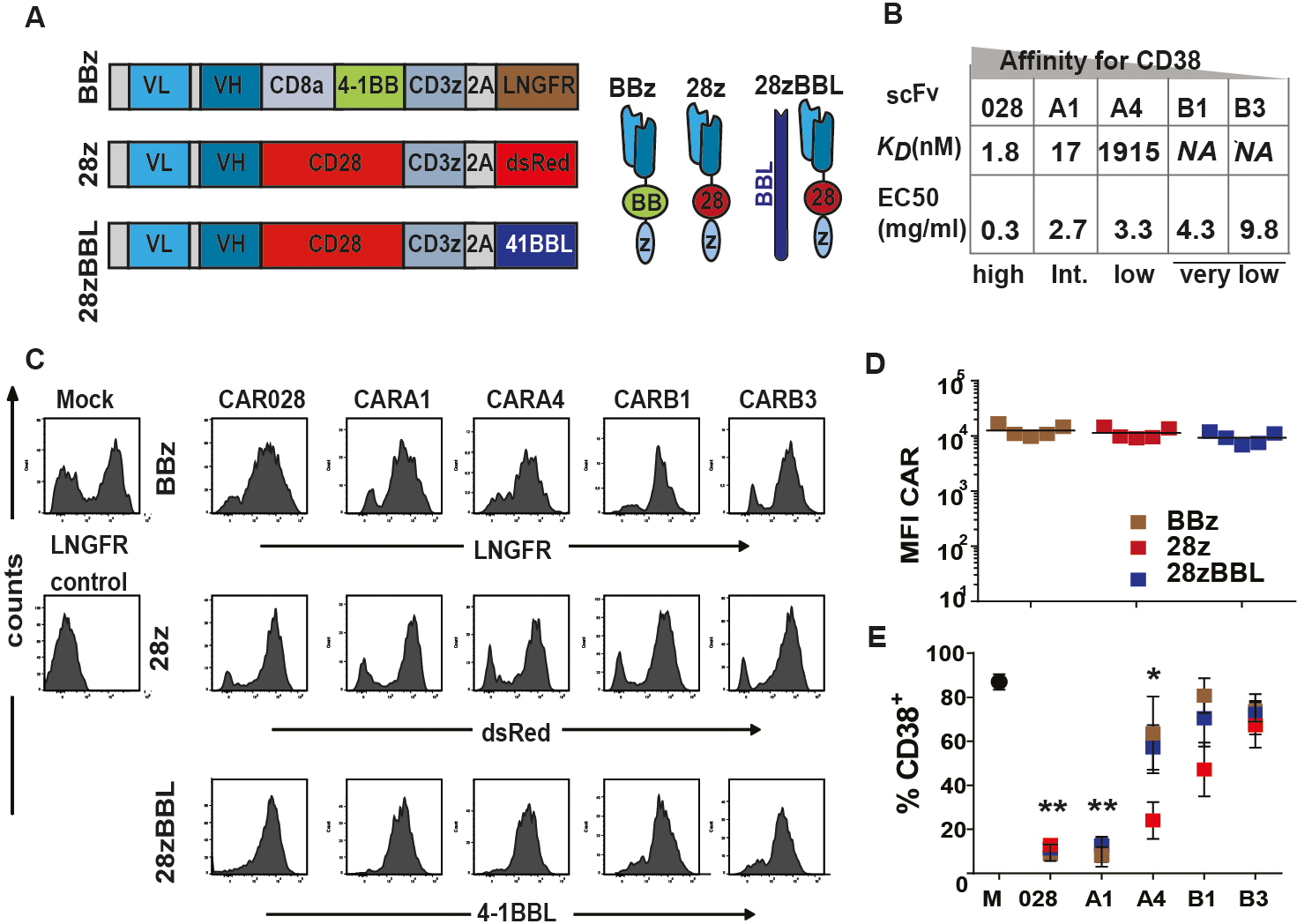
(A) Left panel shows a graphic overview of CAR construct design. The scFv light (VL) and heavy (VH) chains are followed by: 1) the CD8a transmembrane domain, the 4–1BB and CD3ζ signaling domains (BBz) linked by a P2A sequence44 to a truncated LNGFR or 2) the CD28 transmembrane and intracellular sequence and CD3ζ signaling domain (28z) linked to dsRed fluorescent marker or 3) the 28z construct linked to the 4–1BBL coding sequence. (B) Summary of the characteristics of scFvs and the parental antibodies, the surface-plasmon-resonance determined Kd-value (nM) and half-effective concentration (EC50) when titrated on CHO-CD38 cells (μg/ml), described in15. (C) Flow cytometry histogram plots of expression of LNGFR, dsRed and 4–1BBL after transduction of T cells with BBz, 28z or 28z-BBL. (D) Mean fluorescent intensity (MFI) of marker expression. (E) % CD38+ CAR-T cells when transduced with different affinity CD38CARs. NA=not available, * indicates p value <0.05 and ** <0.01 compared to mock, using standard student’s t-test analysis.
CD28 costimulation lowers the affinity threshold for efficient cytotoxicity and cytokine production
To elucidate the potential functional aberrancies caused by lowering the affinity of CD38-CAR-T cells in relation to their costimulatory design, we first studied their lytic capacity against an MM cell line with CD38-expression similar to primary MM cells (>20.000 molecules/cell) (Figure S2). For CD38-CARs carrying the 4–1BB costimulatory domain, lowering the affinity further than Kd=1,9 μM (CARB1 and CARB3) substantially diminished anti-tumor cytotoxicity (Figure 2A). In striking contrast, when a CD28 domain was used in the CAR design, alone or in combination with 4–1BBL, the cytotoxic potential of CAR-T cells was not impacted at all by affinity change. Even CARB1 and CARB3 cells, with >1000-fold lower affinity for CD38 as compared to the high affinity CAR028, displayed no significant decrease in cytotoxicity (Figure 2A).
Figure 2. CD28 signaling improves lytic capacity and cytokine production of affinity-tuned CD38-CAR T cells.
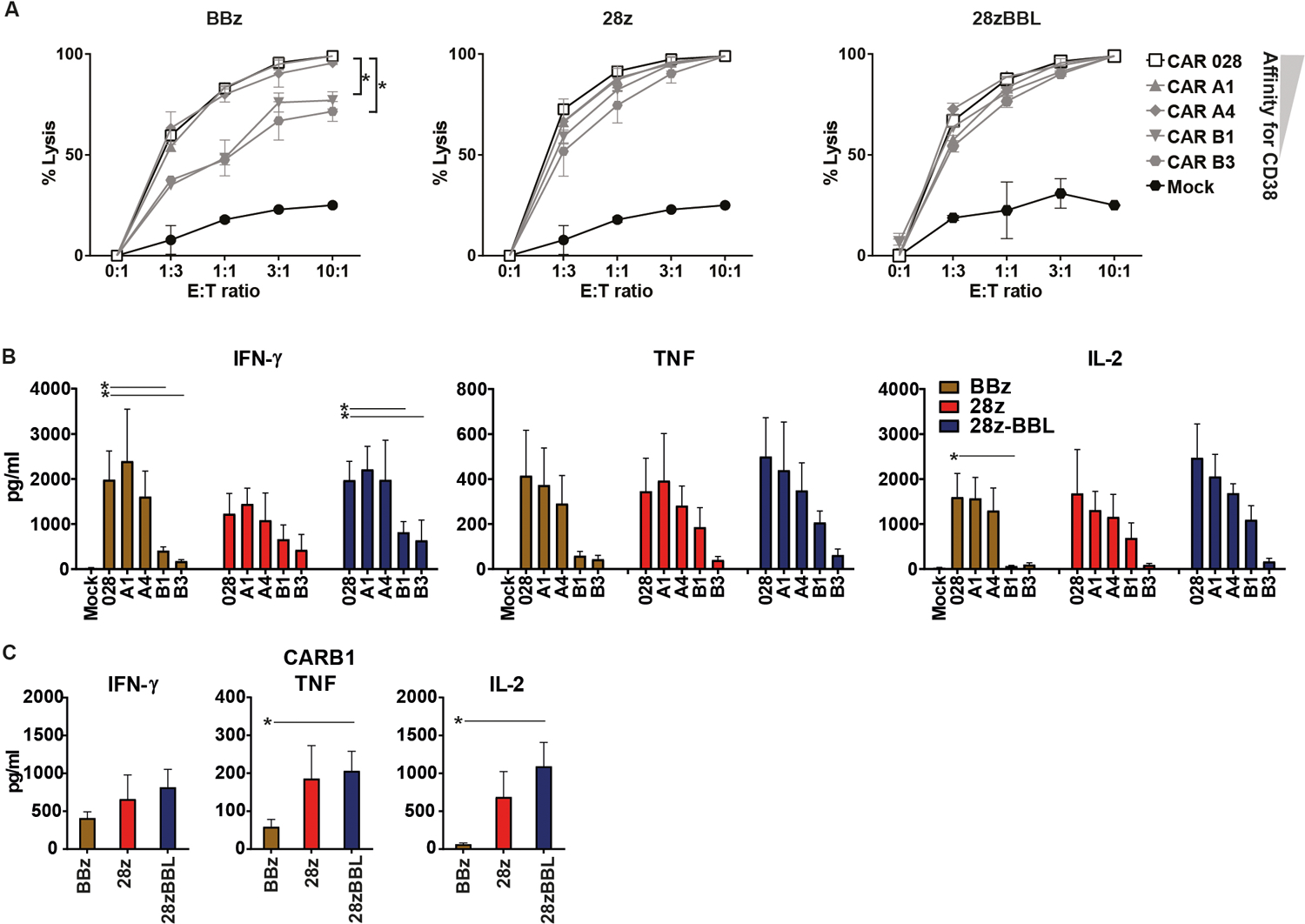
(A) Functional experiments were performed seven to ten days after transduction. The series of high and low affinity CAR T cells were incubated with Firefly-Luciferase-transduced human MM cell line UM9 (n=3 for each condition). The luciferase signal produced by surviving UM9 cells was determined after 16–24 hours within 15 minutes after the addition of 125 μg/mL beetle luciferin. % lysis cells = 1 − (BLI signal in treated wells / BLI signal in untreated wells) × 100%. (B) 24 hours after co-incubation with UM9 (E:T ratio 1:1), cell supernatants were harvested to measure cytokine secretion with a flow cytometry-based assay. Graph shows the secretion of IFN-γ, TNF and IL-2. (C) Comparison of cytokine production of CARB1 with different costimulatory designs. Mean values (+/− SEM) are shown (n=3 each condition). * indicates p value <0.05 ** <0.01 and *** <0.001 using one-way analysis of variance and subsequent multiple comparison, ns: non-significant.
We further analyzed the impact of an affinity decrease on a later effector function, such as the cytokine secretion. All CAR constructs in our study showed no significant unspecific cytokine production against a CD38 negative MM cell line (Figure S3). A remarkable decrease of all cytokine production was observed for BBz CARs bearing very low affinity scFvs (Figure 2B). Although 28z and 28z-BBL CAR-T cells also showed an affinity-dependent decrease in cytokine secretion, this effect was not significant or significant only for CARB3 (Figure 2B). Specifically for CARB1 28z and 28z-BBL designs maintained approximately 4-times higher TNF-α and up to 15 times higher IL-2 levels than BBz design (Figure 2C). Therefore, the affinity threshold for effective anti-tumor cytotoxicity and efficient cytokine production seems to be lower for CARs that incorporate a CD28 intracellular domain.
Since cytotoxic function and cytokine secretion are the consequence of antigen engagement and CAR-T cell activation, we investigated whether the decrease of affinity would lead to a reduced intensity of downstream signaling in BBz compared to 28z CD38CARs. However, we found no impact of affinity on the phosphorylation of Zap70, the main mediator of signaling downstream of CD3ζ, on the BBz or the 28zBBL CARs at rest and 24 hours after antigen stimulation (Figure S4A and S4B). For 28z CAR designs we observed a slight decrease of pZap70 when decreasing the affinity, which was statistically significant only for the very low affinity CARs B1 and B3 (Figure S4B). Therefore, the reduction of the cytotoxic potential of lower affinity BBz CD38CARs could not be explained by the intensity of the downstream CD3ζ-mediated CAR signaling in our system.
Combination of CD28 and 4–1BB signaling improves in vitro proliferative capacity of low affinity CAR-T cells.
We, further, weekly stimulated the CD38CAR-T cells in vitro with irradiated NIH-3T3 cells expressing high levels of CD38 (Figure S2A). In this system activation and costimulation signals depends solely on the CAR binding to CD38. In all affinity groups BBz CARs showed a better proliferation potential in comparison to 28z CARs confirming previous reports22–24, although the difference was not statistically significant (Figure 3A). Notably, 28z-BBL CARs showed a stable growth response to repetitive antigen stimulations, which was always consistently higher, compared to the growth rates of both BBz and 28z CARs (Figure 3A). Reduction of this stable proliferative response was only seen with the lowest affinity CARB3 (Figure 3A). Hence, CARB1 sets the threshold below which CD38 CARs of all designs showed a reduced proliferative capacity and most importantly, CD28 and 4–1BB signaling synergize for an optimal and persistent proliferative response irrespective of CAR affinity.
Figure 3. Combination of CD28 and 4–1BB signaling improves in vitro proliferation and delays differentiation of affinity-tuned CD38-CAR T cells.
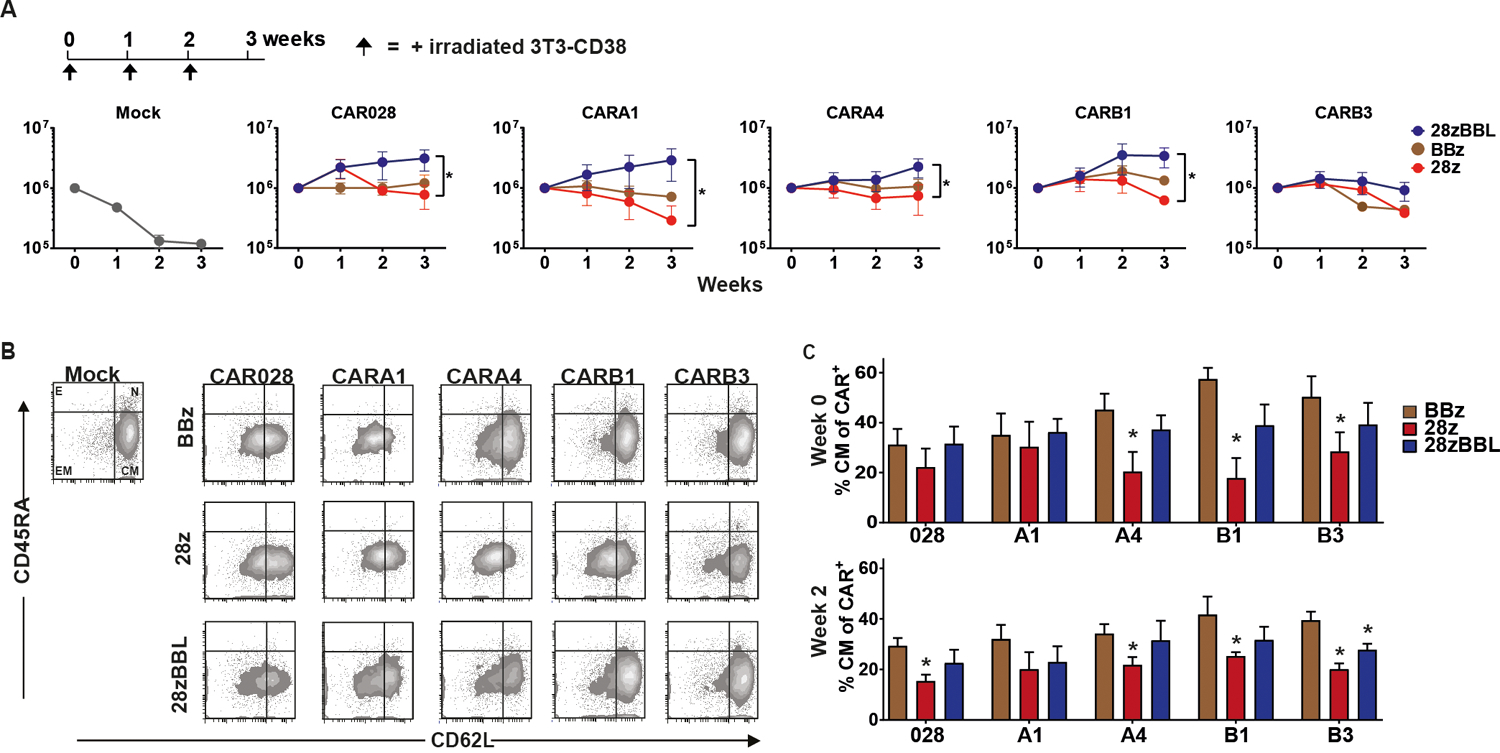
(A) Starting one week after transduction CD38-CAR T cells were co-cultured with mouse fibroblast cells NIH-3T3 transduced with human CD38 at E:T ratio 3:1, and re-stimulated weekly. No cytokines were added to culture. Cells were counted and corrected for % of CAR+ cells as determined by flow cytometry. (B) Flow cytometry density plots of phenotypic profile of each CD38-CAR affinity and costimulation type cell type at week 0, before expansion. Cells have either a naive (N) (CD45RA+/CD62L+) central memory (CM) (CD45RA−/CD62L+), effector memory (EM) (CD45RA−/CD62L−) or effector phenotype (CD45RA+/CD62L−). (C) Percentage of CAR+ cells that have a CM phenotype. Graphs depict mean +/− SEM (n=3 for each condition). Statistical analysis was done using one-way analysis of variance and subsequent multiple comparison, * indicates p value <0.05, between BBz and 28z.
4–1BB signaling endows low affinity CAR-T cells with a less exhausted memory phenotype
We further analyzed the differentiation status and the exhaustion level of affinity-tuned CD38-CAR-T cells during expansion on 3T3-CD38 cells. At the end of production (week 0) CD38-CAR-T cells equipped with 4–1BB signaling, either BBz or 28z-BBL, showed a higher percentage of central memory (TCM) cells compared to 28z CAR-T cells, which was significant for low and very low affinity groups (Figure 3B–C). The same pattern was observed even after 2 weeks of expansion (Figure 3C). We then analyzed the expression pattern of inhibitory receptors PD-1, TIM-3 and LAG-3 (Figure 4A) and determined the percentage of cells expressing none, one, two or three of the markers (Figure 4B). Overall, compared to 28z CD38-CAR-T cells, BBz and 28z-BBL CD38-CAR-T cells showed a lower percentage of triple positive cells (expressing all three markers) for the higher affinity CARs and higher percentage of triple negative cells (expressing none of the three markers) in all affinity groups (Figure 4B and Figure S5). Thus, including 4–1BB signaling moieties in the CAR-T cell design results in longer preservation of a central memory phenotype and a delay of the induction of immune-inhibitory receptors on the cell surface. The effect of 4–1BB costimulation in ameliorating exhaustion of CAR-T cells has been previously attributed to modulation of the CAR signaling22. We found that indeed BBz and especially 28zBBL CD38CARs showed less phosphorylation of Zap70 than CARB1–28z at a basal level as well as after antigenic stimulation across all affinity levels (Figure S4C and S4D), indicating a possible role of the 4–1BB-pathway in regulating the CAR signaling strength.
Figure 4. Exhaustion of affinity-tuned CD38-CAR T cells.
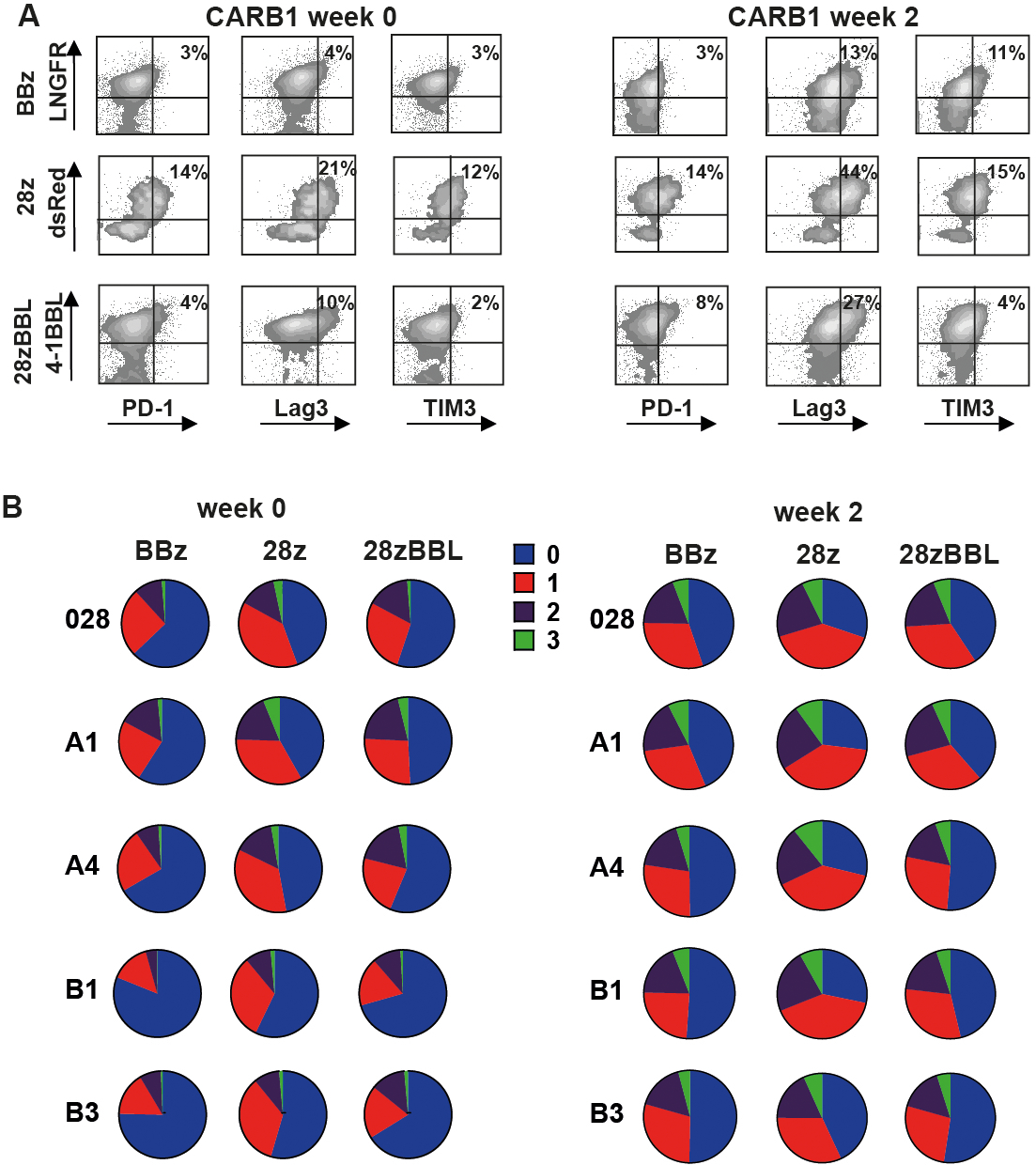
(A) Flow cytometry density plots illustrating expression of inhibitory receptors PD-1, Lag3 and TIM3, 2 weeks after stimulation with 3T3-CD38. Representative figure of 3 independent experiments. (B) Pie charts illustrating the % of cells expressing either 0, 1 (PD-1+, Lag3+ or TIM3+), 2 (PD-1+/Lag3+ or PD-1+/TIM3+ or Lag3+/TIM3+) or 3 (PD-1+, Lag3+ and TIM3+) exhaustion markers gated on live CD3+CAR+ cells. Mean values of n=3 are shown.
Combined costimulation better supports very low-affinity CD38 CAR-T cells to control tumor growth in vivo
CARB1 was the most optimal low-affinity CAR used in our in vitro studies, showing an effective cytotoxic capacity similar to high-affinity CAR028 when coupled to a CD28 domain and the best proliferative response when including a 4–1BB-signaling moiety. Thus, we further investigated the anti-MM effects of CARB1 with either a BBz, 28z or 28zBBL design using a previously described xenograft murine model, in which tumor cells are grown in humanized BM-like niches (Figure 5A)29,30. In this model MM tumor cells preferentially populate the humanized BM scaffolds as revealed by BLI 1 week after injection (Figure S6, week 1). Treatment with CARB1–28z or CARB1–28zBBL T cells resulted in a significant delay of tumor progression compared to mock treated mice while the CARB1-BBz cells failed to control tumor growth (Figure 5B–D, Figure S6). Post-mortem analysis of the scaffold material, 7 weeks after T cell injection, showed significantly lower numbers of (GFP+/CD38+/CD138+) tumor cells in all CAR treated groups compared to mock treated group (Figure 5E). The CARB1–28zBBL group showed the lowest median of tumor cell numbers in the scaffolds compared to both CARB1-BBz and CARB1–28z. When looking specifically at CAR-T cell numbers, CARB1–28z cells initially expanded, reached higher effector:tumor (E:T) ratio than CARB1-BBz T cells and rapidly reduced tumor burden at 3 weeks after injection (Figure S7) but they did not persist as their numbers were significantly reduced after 7 weeks (Figure 5F). On the other hand, BBz CAR-T cells showed a more delayed pattern of expansion at week 3 (Figure S7), but they persisted for longer and eventually achieved control of tumor growth in the scaffolds (Figure 5E–G). These data indicate that CARB1-BBz T cells failed to control tumor spread to secondary sites (e.g. murine bone marrow, skull) (Figure S6). Indeed, although CARB1-BBz T cells infiltrated the murine BM and persisted longer than CARB1–28z cells, they failed to restrict the local tumor cell growth (Figure S8). Importantly, CARB1–28zBBL T cells displayed the best expansion and persistence features compared to the other groups and eventually achieved the best anti-tumor effect as they reached the highest E:T ratios within the scaffolds and murine BM at both an early (3 weeks) and a later (7 weeks) time point (Figure 5G, Figure S7). Previously, using a BBz CAR design we found that CARA4-BBz could elicit a significant anti-MM effect in vivo15. Interestingly, when displaying data from both experiments (Figure 5H), we found that the anti-MM activity of CD38-CAR-T cells is sensitive to affinity reduction from A4 to B1 level when a BBz costimulatory design is used. Most notably, providing both CD28 and 4–1BB signaling can potentiate even very-low-affinity CARB1-T cells to cause significant reduction of MM tumor growth, comparable to that obtained after treatment with a high affinity CAR028-BBz CAR (Figure 5H).
Figure 5. Very low-affinity CD38-CAR-T cells with 28z and 4–1BBL costimulation show better tumor control and persistence in vivo.
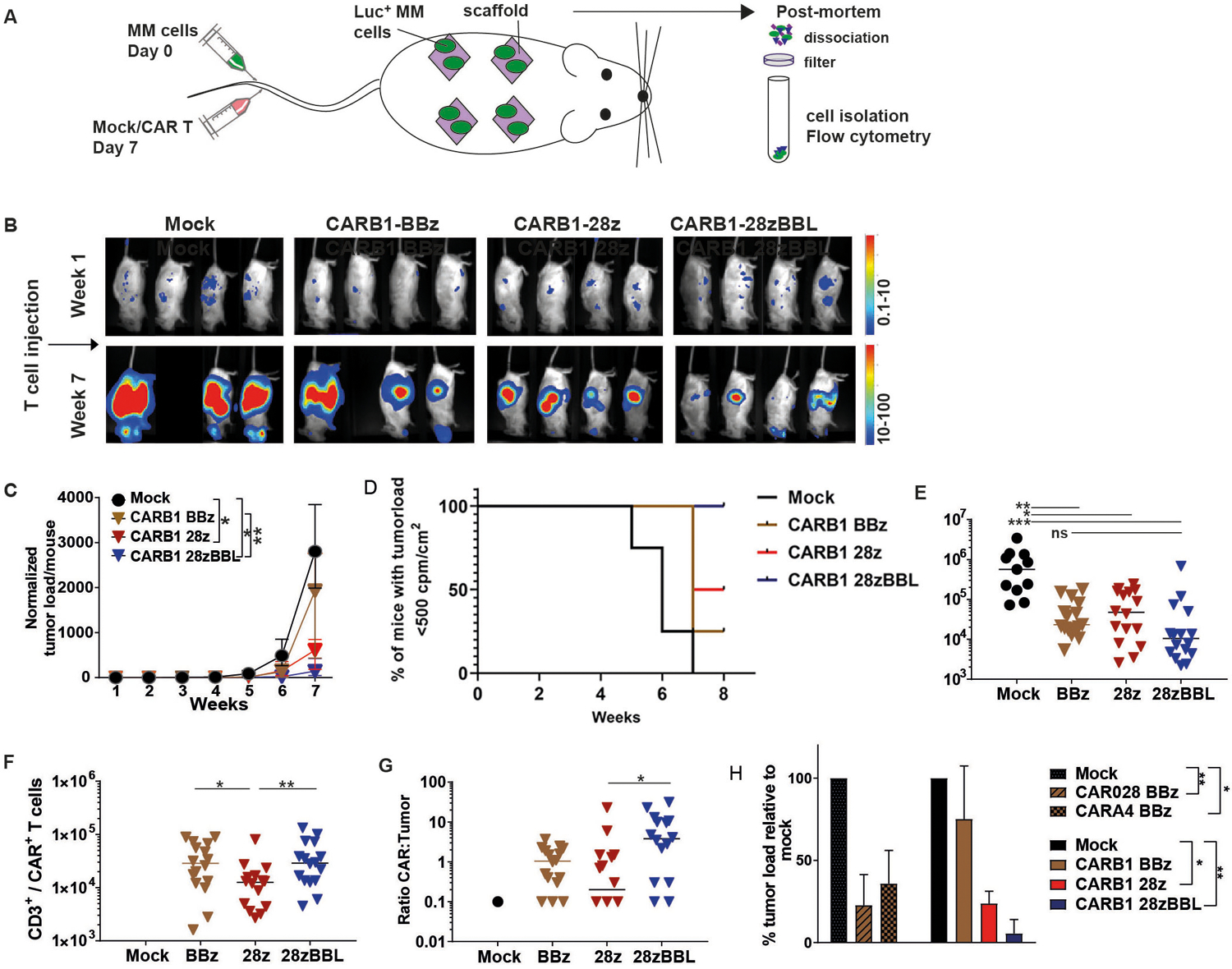
(A) Schematic of in vivo experimental set-up. Scaffolds consisting of three 2- to 3-mm3 triphasic calcium phosphate particles coated in vitro with human mesenchymal stromal cells were implanted subcutaneously in RAG2−/−γc−/− mice. 8–12 weeks later, mice were i.v. injected with 10×106 cells of luciferase-transduced UM9 cells. One week after tumor injection mice were treated with i.v. injections of 5×106 mock, CD38-CARB1 BBz, −28z or −28z-BBL T cells. (B) Representative bioluminescence images (BLI) are shown n=4 per group for week 1 and 7 (all BLI images in Figure S4). (C) Quantification of BLI measurements, normalized to week 1. Each group contained 4 mice and each mouse harbored 4 scaffolds (n=4). Indicated are the median values of normalized tumor load per group +/− range, * indicates p value <0.05, and ** <0.01 using Kruskal-Wallis analysis of variance. (D) Kaplan-Meier curve based on the set up of a maximum BLI measurement. In our model BLI measurement reaches a plateau after between 8 weeks without the tumor being lethal for the mice. Therefore we have chosen week 7 as the end point of our experiments. This plateau roughly corresponds with 500 cpm/m2. Thus we considered this BLI value as the potential human endpoint of the experiment to obtain a Kaplan-Meier curve. (E, F, G) Post-mortem analysis of scaffolds harboring UM9 tumor cells and infiltrated T cells. Scaffolds were dissociated and filtered through a 70μm filter. Single cells were stained for mouse and human CD45, counted and analyzed by flow cytometry. (E) Absolute numbers of MM cells (GFP+/CD138+). (F) Number of CAR-T cells (CD45+/CD3+ and LNGFR+ or dsRed+ or 4–1BBL+). Mock <103 T cells. (G) Ratio between CAR T cells and MM cells. N=4, results are median cell number of 16 scaffolds (4 per mouse) +/− range. (H) Since tumor growth was similar for the mock treated group between experiments and tumor load between treatment groups was equal before CAR T cell injection, we normalized BLI measurements to mock control values and compared data to previous experiment15. Displayed is the median of relative tumor load (quantified BLI measurements) at week 7, relative to mock (set at 100%), n=4 mice. * indicates p value <0.05, ** <0.01, ***<0.001 using Kruskal-Wallis analysis of variance.
Combined costimulation does not increase off-tumor toxicity of low affinity CAR-T cells
Since very-low-affinity CARB1–28zBBL T cells elicited a similar anti-MM response to that of high affinity CAR028–28zBBL in vitro, we further investigated, in a whole bone marrow cytotoxicity assay, if that would be at the expense of increased on-target/off-tumor cytotoxicity against normal hematopoietic cells, which are known to express intermediate-to-low levels of CD38 (200–3000 molecules/cell) (Figure S2). As observed when using UM9 cells, there was a significant decrease in anti-MM cytotoxicity for CARB1- and CARB3-BBz T cells compared to CAR028 (Figure S9A). When the identical scFvs were coupled to the 28z costimulatory domain (+/− 4–1BBL) the overall anti-MM cytotoxicity was improved and was not significantly different from that of CAR028 (Figure S9A). CARA4-, CARB1- and CARB3-BBz T cells resulted in a very limited, non-significant lysis of healthy mononuclear cells (MNC) (Figure S9B and Figure S10) expressing 200–3000 CD38 molecules/cell (Figure S2). Importantly, even in a 28z or 28zBBL format, the very-low-affinity CARB1- and CARB3-T cells did not elicit significant MNC lysis despite their effective anti-MM cytotoxic response (Figure S9B and Figure S10).
We next evaluated the on-target off-tumor toxicity of CARB1 T cells against human FarRed-labeled CD34+ normal hematopoietic progenitor cells in vivo, in a modified version of our murine xenograft model15 (Figure 6A). The FarRed-labeled cells were still detected 14 days after CAR-T cell injection in all treatment groups. Thus, in our system treatment with either high or low affinity CD38-CARs leaves CD34+/CD38− cells intact and does not inhibit overall hematopoiesis (Figure 6A–B). This was also demonstrated in a colony-forming assay where colony-forming capacity of CD34+ cells was not affected whether the CD34+/CD38+ cell subset was eliminated by CAR028–28zBBL cells or left intact by CARA4-BBz and CARB1–28zBBL cells (Figure S11A–D). Post-mortem analysis revealed that, low affinity CARB1–28zBBL T cells caused no significant decrease of the percentage and absolute numbers of CD34+/CD38+ progenitors (2.680 CD38 molecules/cell) and total CD38+ cells (Figure 6C–D and Figure S11E) compared to mock treated controls, in contrast to high affinity CAR028–28zBBL T cells and similar to CARA4-BBz cells, which were previously shown to have reduced off-tumor toxicity15. Therefore, we conclude that although equipping very-low-affinity CD38-CARs with combined CD28 and 4–1BB costimulatory signaling moieties can significantly improve their anti-tumor cytotoxic function, this does not induce off-tumor cytotoxicity of healthy cells expressing lower levels of the target antigen.
Figure 6. Very low-affinity CD38-CAR T cells with 28zBBL design do not lyse healthy CD38+ hematopoietic cells in vivo.
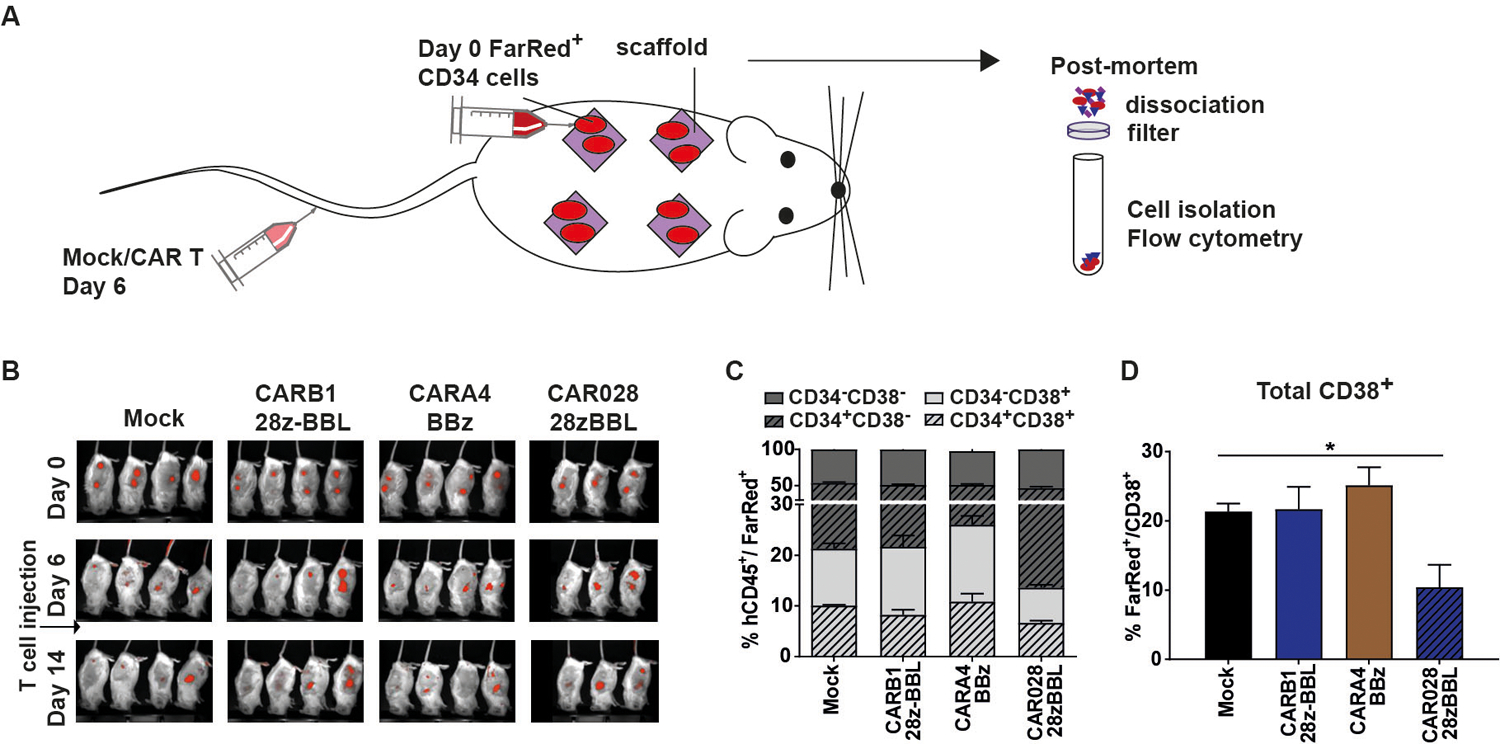
(A) Mice were injected intrascaffold with 1 × 106 fluorescently (FarRed) labeled CD34+ hematopoietic progenitors and treated 6 days later with i.v. injections of 5 × 106 mock, high affinity CAR028–28z-BBL, or low affinity CARA4-BBz or CARB1–28z-BBL T cells. (B) Fluorescence images (FLI) are shown per group at week 1, 2 and 3. (C) Percentages of cells expressing CD34 and/or CD38 and (D) percentage of total CD38+ cells in the total population of FarRed+ cells within the scaffolds. Median values (+/− range) of two scaffolds per mouse and n=4 mice per group. * indicates p value <0.05, using Mann-Whitney test.
Discussion
The applicability of CAR-T cell therapy beyond B cell malignancies is in part impeded by safety concerns about the on-target/off-tumor effect when targeting TAAs. Adjusting the affinity of CAR-T cells to target antigen density is one popular strategy to avert “on-target/off-tumor” toxicity and confer specificity to the tumor13–15,31. For a given affinity, CAR-T cell activation and cytotoxic response is relative to the level of expression of the target and lowering the affinity increases the target-expression threshold for effective CAR-T cell activation16–19,32. This effect favors the ability of CAR-T cells to distinguish high and low target-antigen expression but on the other hand may impede the anti-tumor effectiveness, depending on the level of target-expression on tumor cells. However, CAR-T cell activation and function is a multifactorial process, which is affected not only by affinity but also by costimulation. In this study, we assessed the antitumor function of CAR T cells of different affinities and costimulatory designs and evaluated whether efficient tumor-selectivity achieved by a low affinity CAR could be combined with optimal immunotherapeutic properties delivered by a specific costimulatory design.
CD38 was used as a paradigm of tumor-associated antigen (TAA) and a panel of 5 CD38-targeting scFv domains spanning a wide range of affinities for the target were used15. Importantly, our scFv panel not only covered high and low affinity levels similar to the ones used in other studies14,18,31,32 (Kd ranging between 1.8×10−6 M-1.9×10−9 M), but also included scFvs from antibodies with unmeasurable Kd, whose binding to the target was only measurable in cell-binding assays. It is well demonstrated that costimulatory moieties derived from CD28 or 4–1BB or a combination of the two provide different biological and metabolic characteristics to CAR-T cells leading to differences in anti-tumor lytic capacity, differentiation and persistence2,23,24,33. The CD38-scFv domains were coupled to CD28 or 4–1BB intracellular signaling domains in order to construct the most commonly used second generation CARs. Additionally, based on a study by Zhao et al.23 we coexpressed a 28z-based second generation CAR with 4–1BBL as an effective conformation to provide CAR T cells with both CD28 and 4–1BB signaling.
Our data revealed that the different cell programming, mediated by costimulation, affects also the efficiency of both early and later CAR-T cell responses when scFvs of lower affinity are used. When using affinities in the micromolar range (Kd <1.9 × 10−6 M), 4–1BB-based CD38 CAR-T cells began to lose their capacity for anti-MM lysis and cytokine production (such as IL-2 and TNF-α). Previous studies evaluating targeting of ErbB2 and EGFRvIII positive tumors using low affinity CARs with a 4–1BB intracellular domain reported no significant loss of cytotoxicity or cytokine production against tumor cells. However, Kd values of 1.1×10−9 M and 1.01×10−7 M for ErbB2 and EGFRvIII respectively were the lowest affinities tested in these studies14,31. Interestingly, we found that inclusion of CD28 signaling reduced the affinity threshold for efficient activation after antigen encounter and rescued cytotoxicity and cytokine production in very-low-affinity CD38-CAR-T cells. This indicated that 4–1BB-based CAR designs are more sensitive to lowering the affinity to the antigen than 28z-based CARs. However, the decreased cytotoxicity of very low affinity BBz CD38CAR T cells could not be attributed to a weaker intracellular CAR signaling since we observed no significant difference in Zap70 phosphorylation between BBz CARs of all affinities before or after antigenic stimulation and there was no difference of pZap70 between BBz and 28zBBL CARs. CD28 can lower the threshold for effective TCR activation and can enhance responses in cases of low antigen availability (low avidity)34,35 but its effect in cases of lower TCR affinity is not known. Our results are in contrast with Chmielewski et al.32, reporting an affinity threshold of 10−8, same affinity level as CARA1, below which CD28 signaling did not improve activation of ErbB2-CAR T cells. This discrepancy could be explained by the different dissociation rate described in Chmielewski et al.32, which was 10-fold lower than that of our low affinity CARs. It seems that affinity fine-tuning by keeping a low dissociation rate results in longer interaction of the CAR with the antigen and ensures efficient and potent CAR activation.
Irrespective to the CAR affinity, our data confirmed previous studies showing different biological properties between CAR-T cells bearing CD28 or 4–1BB costimulatory signaling domains22–24,27. For all different affinities in our study 4–1BB-based CAR-T cells showed less rapid differentiation, less exhaustion and better proliferative capacity in vitro than CD28-based CAR T cells. We found that both BBz and 28z CD38-CAR-T cells restricted similarly MM cell growth within the scaffolds in vivo, although they did so by following different kinetics. However, CD28-based CAR-T cells resulted in better control of total tumor growth than BBz CARs and they showed more rapid and efficient tumor elimination even in lower E:T ratio. It should be noted that the 28z and BBz CAR designs of our study contain different transmembrane domains (CD28 and CD8α respectively) which, although not expected, could be a cofounder influencing their functional differences. Recently, Salter et al.36 showed, using high affinity CARs, that 28z design results in significantly stronger intracellular signaling which is responsible for the effector phenotype, decreased persistence and worse anti-tumor performance of 28z compared to BBz CAR T cells. Our data, using also a high affinity CAR, are in line with this observation, showing significantly higher pZap70 in 28z than in BBz CAR028. Interpreting the results from Salter et al. and our study, we have reason to believe that for high affinity CARs, where cytotoxicity between 28z and BBz CARs does not differ, the signaling strength plays a crucial role in determining the persistence and therefore the long-term anti-tumor function of CAR T cells. On the other hand, when using low affinity, the BBz CARs lose their cytotoxic capacity but this is not connected to a decrease or increase of signaling strength, as discussed above. It seems that the cytotoxicity and cytokine production mechanism is not related to the CAR activation level but possibly on another mechanism.
Supplying full CAR costimulation triggering both pathways results in a more balanced T cell stimulation23,27,37,38. We demonstrate that the CD28z+4–1BBL configuration is the optimal design to provide low affinity CD38 CAR-T cells with enhanced anti-tumor cytotoxic potential (through CD28 signaling) and ameliorated proliferative capacity, retention of a memory phenotype and reduced exhaustion (through 4–1BB/4–1BBL signaling) in vitro. Along with other proposed mechanisms23,27 for this combinatorial effect, we found that addition of 4–1BBL expression to a 28z-based CAR restricted the downstream Zap70 phosphorylation in our system. We observed this effect not only at rest but also after antigenic stimulation and for all affinities used.
Furthermore, in our in vivo xenograft model, very-low affinity CAR-T cells (CARB1) having a 28zBBL design resulted in superior restriction of tumor growth and better expansion and persistence as compared to any of the second generation CD38-CAR designs. Most notably, comparative analysis with previous in vivo data revealed that a 28zBBL design could potentiate even very-low affinity CAR-T cells to elicit anti-tumor responses comparable to that obtained by CAR-T cells with >1000 times higher affinity for the target. Clinically most relevant is the fact that this potentiation of anti-tumor function did not compromise the safety of very-low affinity CARs when tested against healthy CD38+ hematopoietic cells in vitro as well as in vivo. Especially in the whole primary BM cytotoxicity assay the CARB1–28zBBL cells were able to successfully eliminate primary MM tumor cells while sparing surrounding healthy hematopoietic cells. Although optimal affinity levels for different individual target molecules cannot be compared, to our knowledge this is the lowest affinity for a TAA-targeting CAR that has been shown to be both efficient and safe. Our findings are informative for the design of CAR constructs and suggest that even scFvs with practically unmeasurable Kd values can be effective when used for CAR-T cell therapy if optimal costimulation is provided.
The present analysis focuses on the well-described and most frequently used 4–1BB and CD28 signaling domains. Nevertheless, there are other costimulatory domains that have been used in the design of CARs. The OX40, like 4–1BB belongs to the TNFR family and primarily signals through the NF-κB pathway39,40. On the other hand, CD40L and ICOS are CD28-like and signal through PI3K pathway40–42. Although 4–1BB and CD28 can be considered representatives of the two receptor-families, a different combination of costimulatory moieties may result in similar findings43. Finally, in our study CD38 is also expressed on T cells resulting in significant fratricide when high-affinity CARs were used. Taking this into account, we performed functional assays after one week of culture when we assured that no further fratricide takes place. Although, a first antigen encounter of high affinity CARs could influence comparisons (especially proliferation and phenotype) between CARs of different affinities, we would not expect any influence on the comparisons between same affinity CARs with different costimulatory designs.
In conclusion, we demonstrate here that the selection of the costimulatory design of CARs is of critical importance when using scFv domains with very low affinity for the target. We show that, if equipped with an optimal combination of CD28 and 4–1BB costimulatory moieties, CARs bearing antigen-binding domains with Kd values even lower than 10−6 M can elicit significant anti-tumor cytotoxic and proliferative response without compromising their safety. The results of this study unlock the use of very-low-affinity CARs in order to target TAAs, potentially increasing the capacity to discriminate between tumor and healthy cells and suggest that careful construction of TAA-targeting CARs will enhance their clinical potential.
Supplementary Material
Statement of translational relevance.
The broader applicability of CAR-T cell therapy is restricted by the lack of tumor specific target-antigens while the most promising targets are tumor-associated antigens (TAAs). Decreasing the affinity of a CAR for the target is a popular strategy to avert on-target/off tumor toxicity when targeting TAAs. Here, we evaluated whether lowering the affinity for the target would affect the costimulatory requirements of CAR design for optimal functionality and persistence. We found that the combination of CD28 and 4–1BB signals potentiates even very low affinity CAR-T cells and improves their immunotherapeutic properties while simultaneously preserving their ability to discriminate target-antigen density. Therefore, a combinatorial costimulatory design allows the use of very low affinity binding domains (Kd<1μM) for the construction of safe but also optimally effective CAR-T cells. These results are relevant and inform for the future design of efficient CAR-T cell therapies to optimally target TAAs for hematological and solid tumors.
Acknowledgments:
The RAG2−/−γc−/− mice used in this study were originally obtained from the Amsterdam Medical Center (AMC, Amsterdam, The Netherlands).
Funding: This work is in part financially supported by ‘Fonds Stimulans’ the Netherlands (E.D.), the European Commission (Marie Curie Individual Fellowship to M.T.) and Stichting VUmc CCA (M.T.).
Footnotes
Conflict of interest:
N.W.C.J.v.d.D. has received research support from Janssen Pharmaceuticals, AMGEN, Celgene, Novartis, and BMS, and serves in advisory boards for Janssen Pharmaceuticals, AMGEN, Celgene, BMS, Novartis, Takeda, Bayer, and Servier. S.Z. has received research support from Celgene, Takeda and Janssen Pharmaceuticals and serves in advisory boards for Celgene, Takeda, Janssen and Amgen. H.Y. and J.d.B. are employed by Kuros Biosciences BV. M.S. is cofounder and stockholder of Juno Therapeutics. H.L. has received research support and serves in advisory boards for Janssen and Genmab. R.W.J.G. has received research support from Janssen Research and Development. T.M has received research support from Janssen Research and Development, Celgene, Onkimmune and Genmab. M.T. serves as a consultant of Covagen. The rest of the authors have no conflict of interest to declare.
References
- 1.Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3(4):388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Stegen SJC, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov 2015;14(7):499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med 2014;6(224):224–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maude SL, Frey N, Shaw P a., et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med 2014;371(16):1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (London, England). 2014;385(9967):517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kochenderfer JN, Dudley ME, Kassim SH, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol 2015;33(6):540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turtle CJ, Hanafi L-A, Berger C, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8 + and CD4 + CD19-specific chimeric antigen receptor–modified T cells. Sci. Transl. Med 2016;8(355):355ra116–355ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schuster SJ, Svoboda J, Chong EA, et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med 2017;377(26):2545–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kochenderfer JN, Dudley ME, Feldman S a, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamers CHJ, Sleijfer S, Vulto AG, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J. Clin. Oncol 2006;24(13):904–912. [DOI] [PubMed] [Google Scholar]
- 11.Morgan RA, Yang JC, Kitano M, et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther 2010;18(4):843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parkhurst MR, Yang JC, Langan RC, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther 2011;19(3):620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caruso HG, Hurton LV., Najjar A, et al. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res. 2015;75(17):3505–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu X, Jiang S, Fang C, et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015;75(17):3596–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drent E, Themeli M, Poels R, et al. A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol. Ther 2017;25(8):1946–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arcangeli S, Rotiroti MC, Bardelli M, et al. Balance of Anti-CD123 Chimeric Antigen Receptor Binding Affinity and Density for the Targeting of Acute Myeloid Leukemia. Mol. Ther 2017;25(8):1933–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chmielewski M, Hombach A, Heuser C, Adams GP, Abken H. T cell activation by antibody-like immunoreceptors: increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J. Immunol 2004;173(12):7647–53. [DOI] [PubMed] [Google Scholar]
- 18.Hudecek M, Lupo-Stanghellini M-T, Kosasih PL, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin. Cancer Res 2013;19(12):3153–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walker AJ, Majzner RG, Zhang L, et al. Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol. Ther 2017;25(9):2189–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Porter DL, Hwang W-T, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med 2015;7(303):303ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Milone MC, Fish JD, Carpenito C, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther 2009;17(8):1453–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Long AH, Haso WM, Shern JF, et al. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med 2015;21(6):581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao Z, Condomines M, van der Stegen SJC, et al. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell. 2015;28(4):415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawalekar OU, O’Connor RS, Fraietta JA, et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity. 2016;44(2):380–90. [DOI] [PubMed] [Google Scholar]
- 25.Melero I, Bach N, Hellström KE, et al. Amplification of tumor immunity by gene transfer of the co-stimulatory 4–1BB ligand: synergy with the CD28 co-stimulatory pathway. Eur. J. Immunol 1998;28(3):1116–21. [DOI] [PubMed] [Google Scholar]
- 26.Rudolf D, Silberzahn T, Walter S, et al. Potent costimulation of human CD8 T cells by anti-4–1BB and anti-CD28 on synthetic artificial antigen presenting cells. Cancer Immunol. Immunother 2008;57(2):175–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong X-S, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4–1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther 2010;18(2):413–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stephan MT, Ponomarev V, Brentjens RJ, et al. T cell-encoded CD80 and 4–1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat. Med 2007;13(12):1440–9. [DOI] [PubMed] [Google Scholar]
- 29.Groen RWJ, Noort W a, Raymakers RA, et al. Reconstructing the human hematopoietic niche in immunodeficient mice: Opportunities for studying primary multiple myeloma. Blood. 2012;120(3):9–16. [DOI] [PubMed] [Google Scholar]
- 30.de Haart SJ, van de Donk NWCJ, Minnema MC, et al. Accessory cells of the microenvironment protect multiple myeloma from T-cell cytotoxicity through cell adhesion-mediated immune resistance. Clin. Cancer Res 2013;19(20):5591–5601. [DOI] [PubMed] [Google Scholar]
- 31.Johnson LA, Scholler J, Ohkuri T, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med 2015;7(275):275ra22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chmielewski M, Hombach AA, Abken H. CD28 cosignalling does not affect the activation threshold in a chimeric antigen receptor-redirected T-cell attack. Gene Ther. 2011;18(1):62–72. [DOI] [PubMed] [Google Scholar]
- 33.Abken H Costimulation Engages the Gear in Driving CARs. Immunity. 2016;44(2):214–216. [DOI] [PubMed] [Google Scholar]
- 34.Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science. 1996;273(5271):104–6. [DOI] [PubMed] [Google Scholar]
- 35.Itoh Y, Germain RN. Single cell analysis reveals regulated hierarchical T cell antigen receptor signaling thresholds and intraclonal heterogeneity for individual cytokine responses of CD4+ T cells. J. Exp. Med 1997;186(5):757–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salter AI, Ivey RG, Kennedy JJ, et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal 2018;11(544):eaat6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carpenito C, Milone MC, Hassan R, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci 2009;106(9):3360–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tammana S, Huang X, Wong M, et al. 4–1BB and CD28 Signaling Plays a Synergistic Role in Redirecting Umbilical Cord Blood T Cells Against B-Cell Malignancies. Hum. Gene Ther 2010;21(1):75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Croft M, So T, Duan W, Soroosh P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol. Rev 2009;229(1):173–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol 2013;13(4):227–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watanabe M, Hara Y, Tanabe K, Toma H, Abe R. A distinct role for ICOS-mediated co-stimulatory signaling in CD4+ and CD8+ T cell subsets. Int. Immunol 2005;17(3):269–278. [DOI] [PubMed] [Google Scholar]
- 42.Elgueta R, Benson MJ, de Vries VC, et al. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev 2009;229(1):152–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guedan S, Posey AD, Shaw C, et al. Enhancing CAR T cell persistence through ICOS and 4–1BB costimulation. JCI insight. 2018;3(1):. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim JH, Lee S-R, Li L-H, et al. High Cleavage Efficiency of a 2A Peptide Derived from Porcine Teschovirus-1 in Human Cell Lines, Zebrafish and Mice. PLoS One. 2011;6(4):e18556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.