

SUMMARY
CD160 is a signaling molecule that interacts with herpes virus entry mediator (HVEM) and contributes to a wide range of immune responses, including T cell inhibition, natural killer cell activation, and mucosal immunity. GPI-anchored and transmembrane isoforms of CD160 share the same ectodomain responsible for HVEM engagement, which leads to bidirectional signaling. Despite the importance of the CD16Q:HVEM signaling axis and its therapeutic relevance, the structural and mechanistic basis underlying CD160-HVEM engagement has not been described. We report the crystal structures of the human CD160 extracellular domain and its complex with human HVEM. CD160 adopts a unique variation of the immunoglobulin fold and exists as a monomer in solution. The CD160:HVEM assembly exhibits a 1:1 stoichiometry and a binding interface similar to that observed in the BTLA:HVEM complex. Our work reveals the chemical and physical determinants underlying CD160:HVEM recognition and initiation of associated signaling processes.
Graphical Abstract
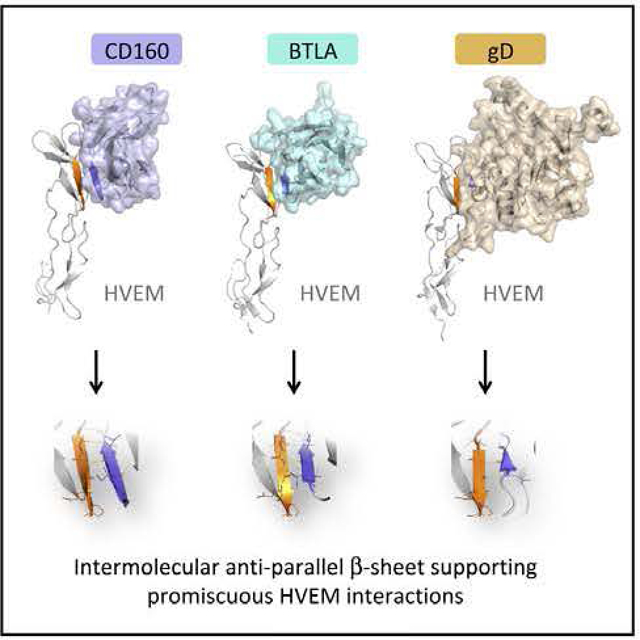
In Brief
Liu et al. determined the structures of human CD160 and its complex with HVEM, revealing the atomic basis for CD160:HVEM recognition. These structures highlight an intermolecular antiparallel β-sheet organization that supports the promiscuous binding properties of HVEM, enabling recognition of CD160, BTLA, and herpes simplex virus glycoprotein D.
INTRODUCTION
CD160 was originally identified as a cell-surface antigen expressed on natural killer (NK) cells, CD8+ cells, a small subset of CD4+ cells, and all intraepithelial lymphocytes (IELs) (Anumanthan et al., 1998; Maiza et al., 1993; Nikolovaetal., 2002).CD160 was initially described as a glycosylphosphatidylinositol (GPI)-anchored receptor with an ectodomain belonging to the immunoglobulin superfamily (IgSF) (Anumanthan et al., 1998; Giustiniani et al., 2007). Subsequently, an alternative isoform of CD160 was reported, which contains a cytoplasmic tail, capable of activating the ERK1/2 signaling pathway (Giustiniani et al., 2009). Both isoforms interact with ligands through the IgSF domain, including modest interactions with classical and non-classical major histocompatibility complex class I molecules, which enhances NK and T cell activity, and promotes tumor angiogenesis (Agrawal et al., 1999; Barakonyi et al., 2004; Fons et al., 2006; Maeda et al.. 2005; Nikolova et al., 2002). A soluble form of CD160, shed from NK cells, has also been reported to inhibit cell-mediated cytotoxicity (Giustiniani et al., 2007).
Herpes virus entry mediator (HVEM) is a prominent member of the tumor necrosis factor (INF) receptor superfamily (TNFRSF), which binds CD160 (Cai et al., 2008; Montgomery et al., 1996) as well as LTα and LIGHT, two members of the TNF ligand super-lamily (TNFSF) (Ward-Kavanagh et al., 2016), Engagement of HEVM with LIGHT expressed on CD8+ T cells provides a stimulatory signal that upregulates immune responsiveness and promotes the development of CD8+ T cell memory (Desai et al., 2017; Mauri et al., 1998). HVEM is an unusual TNFRSF member that directly interacts with IgSF members and others, as evidenced by engaging IgSF members BTLA (B and T lymphocyte attenuator) and CD160, as well as herpes simplex virus glycoprotein D (HSV gD) and neuron-specific SALM5 (Cai and Freeman, 2009; Krummenacher et al., 2005; Zhu et al., 2016). Similar to other TNFRSF members, the HVEM ectodomain is composed of multiple tandem cysteine-rich domains (CRDs). Mutagenesis and structural studies show that LIGHT binds to CRD2 and CRD3 of HVEM, whereas BTLA and CD160 compete for binding on CRD1 of HVEM (Cai et al., 2008; Compaan et al., 2005). In contrast to the costimulatory function of the LIGHT:HVEM trans interaction, BTLA engagement by HVEM delivers a coinhibitory signal to attenuate CD4 T cell activation similar to the CTLA-4 and PD-1 receptor systems (Sedy et al., 2005, 2017; Watanabe et al., 2003) (Figure 1).
Figure 1. The cis and trans Interaction Network of HVEM Hub.
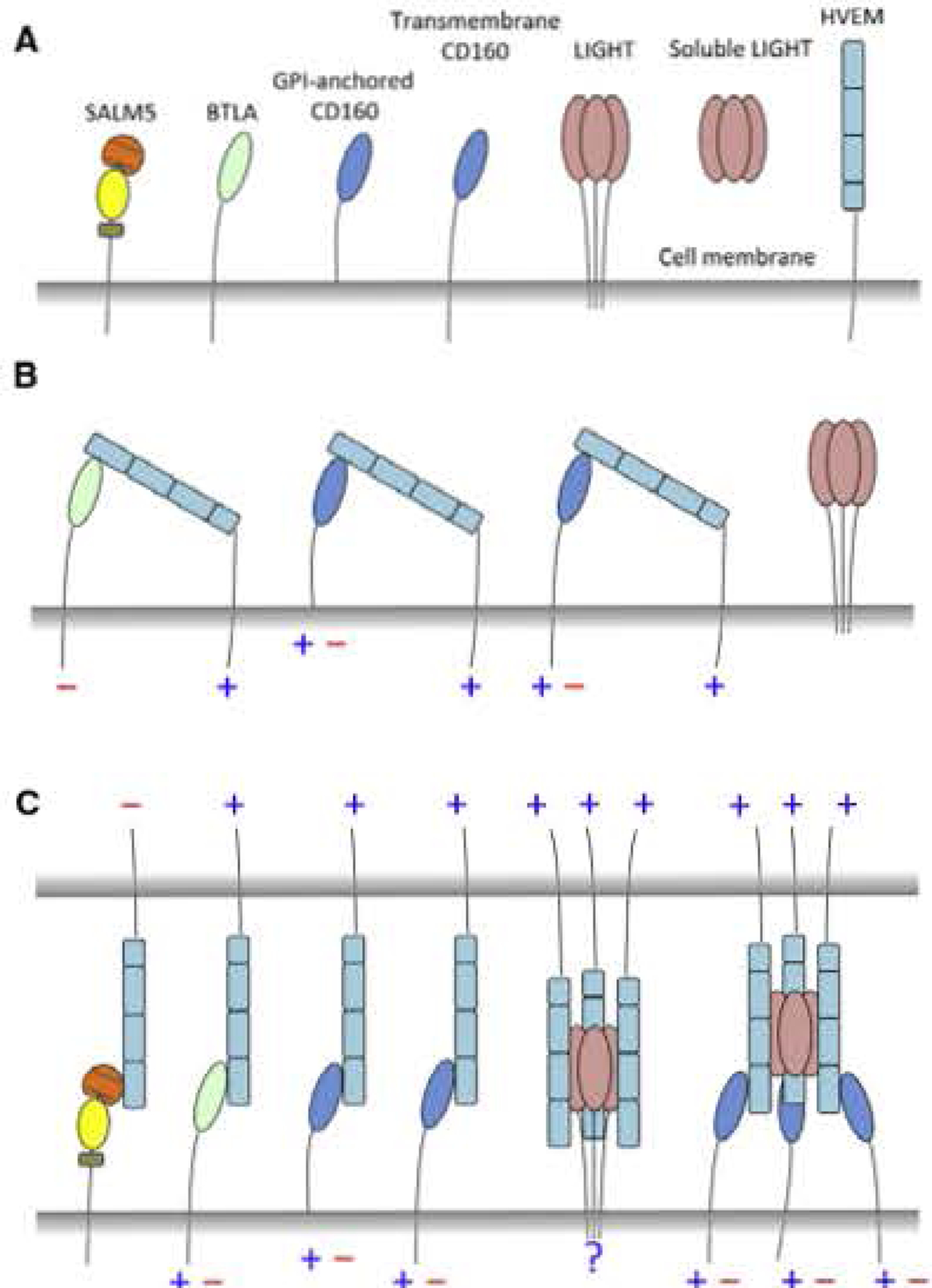
(A) Schematic representation of HVEM and its binding partners.
(B) The cis interactions of HVEM with its receptors. Blue plus symbol (+) indicates stimulatory signaling, while red minus symbol (−) indicates inhibitory signaling.
(C) The trans interactions of HVEM.
The CD160:HVEM interaction elicits distinct functions on different cell types. For example, in T lymphocytes, trans engagement of HVEM and CD160 results in a coinhibitory signal that suppresses CD4+ T cell proliferation and interferon-γ (IFN-γ) production (Cai et al., 2008). In contrast, in NK cells, CD160:HVEM engagement delivers costimulatory signals to boost cytokine production and promote lytic activity, possibly via phosphorylation of AKT and ERK1/2 (Sedy et al., 2013; Tu et al., 2015). In the mucosal system, CD160 on innate-like intraepithelial lymphocytes interacts with epithelial HVEM to activate the Stat3-Reg3 pathways, which enhances the mucosal innate response and provides host defense against bacterial infection (Shui etal., 2012).
CD 160 and HVEM are important regulators, exhibiting multiple functional outcomes, which are sensitive to a number of variables, including the competing engagement of different ligands, expression patterns on different cell types, cis versus trans interactions, and the context-dependent direction of the signaling (Figure 1) (Ward-Kavanagh et al.. 2016). Therapeutic targeting of the CD160:HVEM axis for the treatment of autoimmunity, infectious diseases, and cancer has generated intense interest (Boice et al.. 2016; del Rio et al., 2010; Pasero et al., 2012: Tang et al„ 2016; Ward-Kavanagh et al., 2016); however, despite the numerous opportunities, progress has been hampered by the complexity of the interaction network and the paucity of structural information for CD160. Here we report the crystal structure of CD160 and the CD160:HVEM complex, which reveals a 1:1 stoichiometry and a binding interface similar to that observed in the BTLA:HVEM complex. This structure defines the chemical and physical determinants responsible for recognition and provides a foundation for dissecting biochemical functions important for potential therapeutic applications. CD160 was predicted to belong to the IgSF (Maeda et al., 2005), and early studies suggested that it may form multimeric assemblies (Anumanthan et al., 1998; Giustiniani et al., 2009). Our structures revealed that the ectodomain of CD160 adopts a unique variation of the immunoglobulin (Ig) C2 structure. In addition, the only unpaired cysteine in the CD160 ectodomain is solvent inaccessible and thus unavailable for covalent dimerization, consistent with current solution studies that support a physiological monomer for CD160.
RESULTS
The CD160 Ectodomain Is Monomeric in Solution
Multiple constructs of the human CD160 ectodomain were expressed in Escherichia coli as inclusion bodies and subjected to oxidative refolding (Figures 2 and S1). Two constructs (residues I27-K144 and I27-L158) of CD160 were successfully refolded (Figure 2). Removal of K144 from the C terminus prevented productive refolding under the conditions examined (Figure 2). Size-exclusion chromatography (SEC) indicates that refolded CD160 exists as a single monodisperse species with a molecular weight of ~1 5 kDa (Figure 2). Fractions across the entire peak were subjected to SDS-PAGE analysis under reducing and non-reducing conditions. All fractions migrated with an approximate molecular weight of 13 kDa, the predicted molecular weight of monomeric CD160, suggesting that CD160 exists as a monomer and does not form intermolecular disulfide bonds (Figure 2).
Figure 2. CD160 Is a Monomer in Solution and Adopts a Unique Ig Structure.
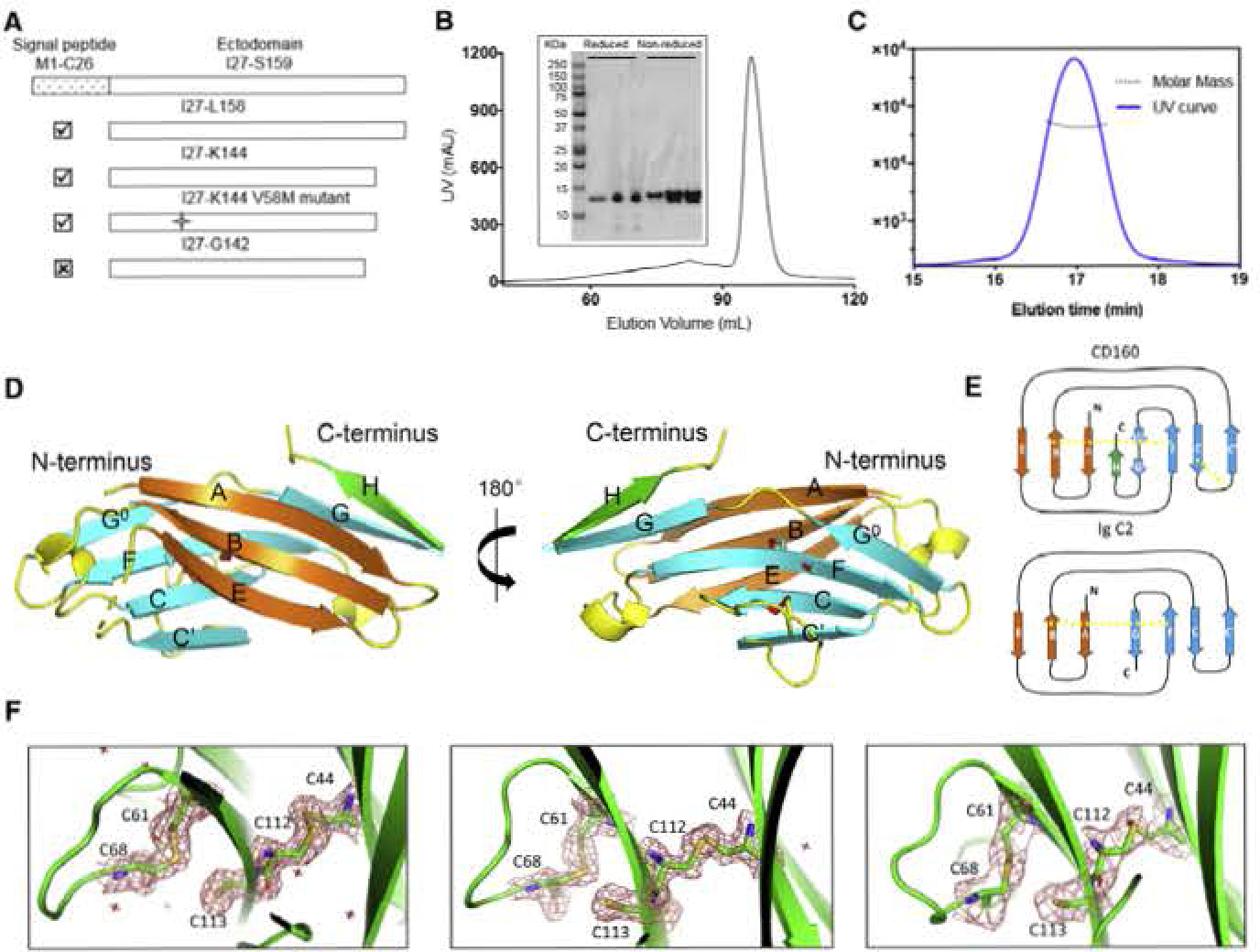
(A) Different construct designs of human CD160. Top panel shows the organization of the human GD160 ectodomain. Cross mark (☒) indicates that this construct was not successfully refolded, whereas a checkmark (☑) indicates successful refolding of the construct.
(B) Size-exclusion chromatography (SEC) demonstrates that refolded CD160 ((27-K144) runs as a single monodisperse peak. Proteins from different SEC fractions were subjected to reducing or non-reducing SDS-RAGE analysis.
(C) SEC-MALS analysis of the refolded CDI60 demonstrates that the molecular weight of CDI60 is 13.2 ± 0.6 kDa. corresponding to the calculated molecular weight of monomeric CD160. The mean value and standard deviation are from three independent experiments
(D) The back β-strand sheet is colored orange and the front β-strand sheet is colored cyan; the unique H strand is colored green; the connecting loops are colored yellow. Two disulfide bonds formed between the B and F strands, and the C and C′ strands are shown as slicks. The cysteine sulfur atoms are colored red. The unpaired C113 residue on strand F is also shown as a stick. Dashed lines indicate loops missing from the structure.
(E) Schematic presentation of the differences between CD160 and the canonical Ig C2 fold. The intramolecular disulfide bonds are indicated by yellow dashed lines.
(F) Clear electron densities for C113 and the adjacent disultide bond formed by the C44-C112 and C61-C68 pairs in the structures of CD160 (left), SeMet V58M GD160 (middle), and sc-CD16Q:HVEM (right). 2Fo-FC electron density maps were contoured at 1.5σ and shown as red mesh.
See also Figure S1.
To confirm that E. coli-expressed and refolded CD160 reflects the behavior of native material, we expressed the human CD160 ectodomain (I27-L158) in Drosophila S2 cells, which also predominately exist as a monomer in solution (Figure S1). The refolded CD1S0 and insect cell-produced CD160 were each subjected to SEC with multiangle light scattering (SEC-MALS) analysis at a concentration of 5 mg/mL. These studies demonstrated that the two CD160 constructs have molecular weights of 13.2 ± 0.6 kDa and 15.1 ± 0.9 kDa (due to glycosylation), corresponding to the molecular weights of monomeric GD160 (Figures 2 and S1). These findings are consistent with the behavior of the entire extracellular domain of CD160 expressed in HEK293S cells, which was also reported to be monomeric in solution (Kojima et al., 2011). Together, these results support a physiological monomer for the CD160 ectodomain.
Crystal Structure of CD160 Reveals a Unique Ig Folding
Refolded CD160 (residues I27-K144) yielded needle-like crystals exhibiting diffraction consistent with the orthorhombic space group date P2,2121, which extended to a resolution of 1.95 A (Table 1). Incorporation of selenomethionine (SeMet) did not generate sufficient anomalous signals for phasing (this construct only possesses two Met residues, one of which is the N-terminal residue). To enhance the anomalous signal, we designed two separate mutants (T103M and V58M) to increase the SeMet content of the protein (Figures 2 and S1). TheT103M mutant failed to refold, whereas V58M refolded well and produced needle-like crystals similar to wild-type protein and diffracted to 1.95 A (Table 1). Phasing based on diffraction data from the SeMet-V58M mutant readily supported structure determination, revealing two independent molecules of CD160 in the asymmetric unit (Figure S1). Superimposition of the two chains shows similar structures with a root-mean-square deviation (RMSD) of 0.4 Å for 107 aligned Cα atom s (Figure S1). All subsequent discussions are based on the B chain.
Table 1.
Data Collection and Refinement Statistics
| SeMet V58M | |||
|---|---|---|---|
| CD160 | CD160 | sc_CD160:HVEM | |
| Data Collection | |||
| Wavelength used (Å) | 0.9786 | 0.97931 | 0.97931 |
| Resolution range (Å) | 1.95–50.00 | 1.95–50.00 | 2.83–50.0 |
| (1.95–1.99) | (1.95–1.98) | (2.88–2.93) | |
| Space group | P212121 | P212121 | 14122 |
| Unit cell (Å) | a = 44.6, | a = 44.7, | a = b = 87.9, |
| b = 51.4, | b = 54.1, | c = 157.7 | |
| c = 95.1 | c = 92.6 | ||
| Unique reflections (n) | 16,516 | 17,092 | 7,332 |
| Redundancy | 10.4 (10.1) | 5.0 (5.3) | 14.1 (13.0) |
| Completeness | 99.8(100) | 99.6(100) | 99.9 (100) |
| I/σ | 26.0 (3.0) | 18.9 (4.2) | 14.5 (2.0) |
| Rmerge | 0.108 (0.936) | 0.106 (0.433) | 0.178 (1.552) |
| Refinement | |||
| Resolution range (Å) | 1.95–47.71 | 1.95–46.69 | 2.88–20.0 |
| (1.95–2.01) | (1.95–2.00) | (2.88–2.96) | |
| Rwork | 0.202 (0.236) | 0.217 (0.214) | 0.234 (0.293) |
| Rfree | 0.253 (0.323) | 0.269 (0.243) | 0.274 (0.358) |
| Average B factor (Å2) | 35.1 | 24.4 | 72.0 |
| RMS band (Å) | 0.018 | 0.019 | 0.013 |
| RMS angles (°) | 1.950 | 2.006 | 1.913 |
| PDB ID | 6NG9 | 6NGG | 6NG3 |
Rmerge = ΣhklΣi|li(hkl) – <l(hkl)>/ΣhklΣili(hkl).
Rwork = Σ|Fc – Fo|/ΣFo.
Parentheses indicate statistics tor the highest-resolution bin.
The overall structure of CD160 generally conforms to the Ig C2 domain fold, with back and front antiparallel β sheets (Bodelon et al., 2013; Chattopadhyay et al., 2009). The back sheet contains the A, B, and E strands and the front sheet contains the G, G°, F, C, and C′ strands as is conventional in Ig C2 domains (Figure 2). However, the CD160 structure is unique in that residues T135-K144 make a turn after the G strand, forming a unique strand (denoted as H) that runs antiparallel to the G strand (Figure 2). The additional H strand is important for the stability of CD160, as shortening this segment prevents refolding (Figures 2 and S1).
Two disulfide bonds are present in the CD160 structure, including the hallmark IgSF disulfide formed between the B (G44) and F (C112) strands on the back and front sheets, respectively, and a non-canonical disulfide bond (C61 and C68) between the C and C′ strands (Figure 2). Notably, an unpaired cysteine, C113, was observed with clear side-chain electron density on the F strand, in the center of the concave front sheet (Figure 2). 0113 is solvent inaccessible and stericaliy restricted from forming an intermolecular disulfide bond, and the C113S mutant refolded similar to wild-type CD160, suggesting that Cl13 is not essential for integrity of the CD160 structure (Figure S1).
Interaction of CD 160 with HVEM
The binding affinity of CD160 with HVEM was measured by surface plasmon resonance (SPR). Human HVEM, expressed in Drosophila S2 cells, was immobilized on a CM5 sensor chip and various concentrations of S2 cell-derived CD160 or E. coli-derived CD160 were used as analyte, and the equilibrium dissociation constants (KD) calculated using a steady-state model. The experimentally determined KD for S2 cell-derived CD160 and E. coli-derived CD160 were 6.7 ± 0.5 μM and 7.1 ± 0.9 μM, respectively (Figures 3 and S2). These results demonstrate that refolded and S2 cell-produced CD160 possess similar affinities for HVEM and that glycosylation of CD160 makes no appreciable contribution to HVEM binding.
Figure 3. CD160 Interacts with HVEM in a Manner Similar to BTLA, but with Weaker Affinity.
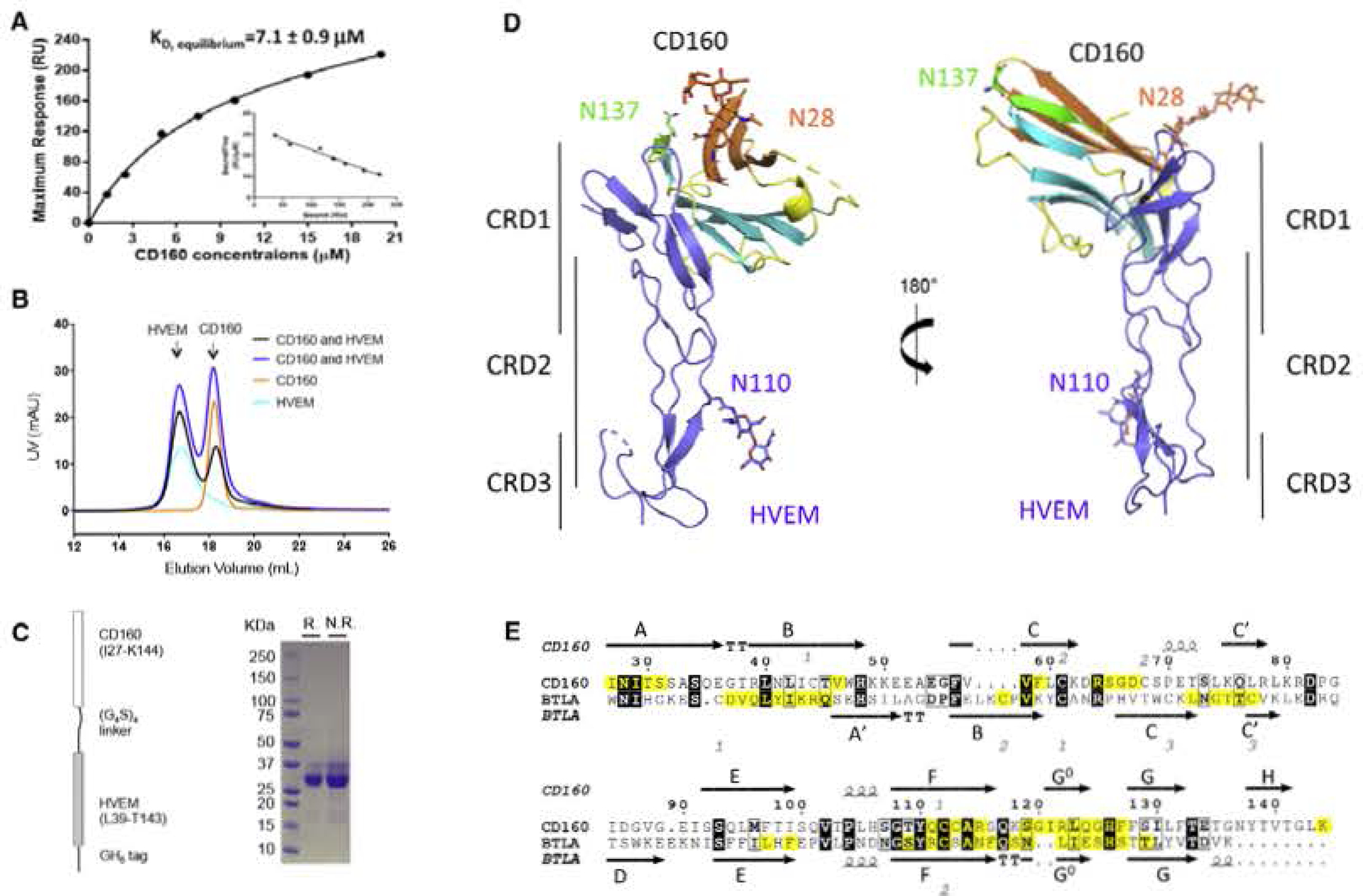
(A) SPR results of injections of E. coli-derived CD160 across immobilized HVEM at various concentrations. The inset shows the linear regression between “Bound/Free” and “Bound” The reported values represent the average of three experiments with the standard deviation.
(B) SEC traces of CD160, HVEM and the mixtures of CD160 and HVEM in different molar ratios. These results indicate that CD 160 and HVEM do not form a stable complex in solution under the conditions examined.
(C) Schematic representation of the single-chain human CD160: HVEM complex as shown on the left panel. Right panel shows the SDS-PAGE result of the purified single-chain protein under reducing (R.) or non-reducing (N.R.) conditions.
(D) Overall structure of the single-chain CD16Q;HVEM complex. CD160 is colored and represented as in Figure 2; HVEM is shown in light blue. The carbohydrate modifications and the associated Asn residues are shown as sticks.
(E) Sequence alignment of human CD160 with human BTLA Ig domains. The secondary structure of CD160 and BTLA is presented (arrows indicate β strands and helical symbols indicate α. helices). Disulfide bonds are denoted by gray numbers near the cysteines. Residues in the binding interface are highlighted yellow. The sequence alignment was performed in the online server CLUSTALW and ESPript 3.0 (Robert and Gouet. 2014; Thompson et al., 1994).
See also Figure S2.
Notably, the measured CD160:HVEM KDs were weaker than the previous reports of BTLA:HVEM KDs (0.97 ± 0.19 μM for mouse BTLA and mouse HVEM) (Nelson et al., 2008; Stiles et al., 2010). As HVEM and BTLA form a stable heterodimeric complex in solution (Figure S2) (Compaan et al., 2005), we tested the solution behavior of the CD160:HVEM assembly. Drosophila S2 cell-derived or E. coli-derived CD160 were mixed together with HVEM in different molar ratios at a final total concentration of 5 mg/mL and the resulting mixtures subjected to SEC analysis. The SEC traces showed no new species forming in the mixtures of CD160 and HVEM (Figures 3 and S2), indicating CD160 and HVEM did not form stable complexes under the conditions examined. As LIGHT has been reported to moderately enhance the binding of CD160 to HVEM (Cai et al., 2008), HVEM and LIGHT were mixed at a final concentration of 5 mg/mL for SEC analysis. Although LIGHT:HVEM formed stable complex in solution, CD160 was not observed to bind the LIGHT:HVEM complex (Figure S2). Taken together, these results demonstrate that CD160 has lower binding affinity than BTLA for HVEM.
Crystal Structure of CD160:HVEM Complex and Epitope Mapping of CD160
All efforts to crystallize the CD160:HVEM complex failed, perhaps due to the modest affinity between these molecules. To circumvent the challenges associated with low affinity, we designed a covalent single-chain CD160-HVEM construct in which the C terminus of CD160 was covalently fused to the N terminus of HVEM via a (GGGGS)4 linker (Figure 3). The single-chain protein was produced using Drosophila S2 cells and purified to homogeneity (Figure 3). This construct yielded rod-like crystals, which exhibited diffraction consistent with the tetragonal space group 14,22 and extended to a resolution of 2.9 A (Table 1), The structure was determined by molecular replacement using CD160 and HVEM (PDB: 4FHQ} as search models.
The asymmetric unit contains one copy of the CD160:HVEM fusion. The CD160 and HVEM domains were clearly defined in the electron density maps, whereas density for the connecting (GGGGS)4 linker was not present (Figures 4 and S3). Residues from multiple segments of CD160 contact CRD1 of HVEM. overlapping with the previously reported BTLA and HSV1 gD binding sites (Carfi et al., 2001; Compaan et al., 2005) (Figures 5 and S3). These residues from CD160 include the CC′ loop (R64-D67), A strand (I27-S31) at the N terminus, G0, G strands (S119-F127), the adjacent F strand (Q111-R115), and the unique C-terminal H strand (K144) (Figures 3 and 4). HVEM residues contributing to the binding interface include K43, D45-E46, E52-P59, Y61, K64-E65, C67-P77, and L87. HVEM residues K43 and D45-E46 make contacts with CC′ loop of CD160, while HVEM residues E52-P59 and L87 contact the G0, F strands, and CC′ loops of CD160 with no observable polar contacts. Furthermore, HVEM residues Y61, K64-E65, and C67-P77 contact the A, G0, and G strands and C-terminal K144 of CD160. Notably, residues T71-E76 of HVEM make the most abundant polar contacts with the G0 strand of CD160 (Figure 4). Most of these polar contacts are main-chain hydrogen bonds configured similarly to the hydrogen bonds observed between the two adjacent antiparallel strands in conventional β-sheet structures. The putative N-linked glycosylation sites (residues N28 and N137) of CD160 are not directly involved in the interface between CD160 and HVEM (Figure 3), consistent with our SPR measurements showing that Drosophila S2- and E. coli-derived CD160 exhibit similar binding affinities for HVEM (Figures 3 and S2).
Figure 4. Binding Interface of the CD160:HVEM Complex and Epitope Mapping of CD160.
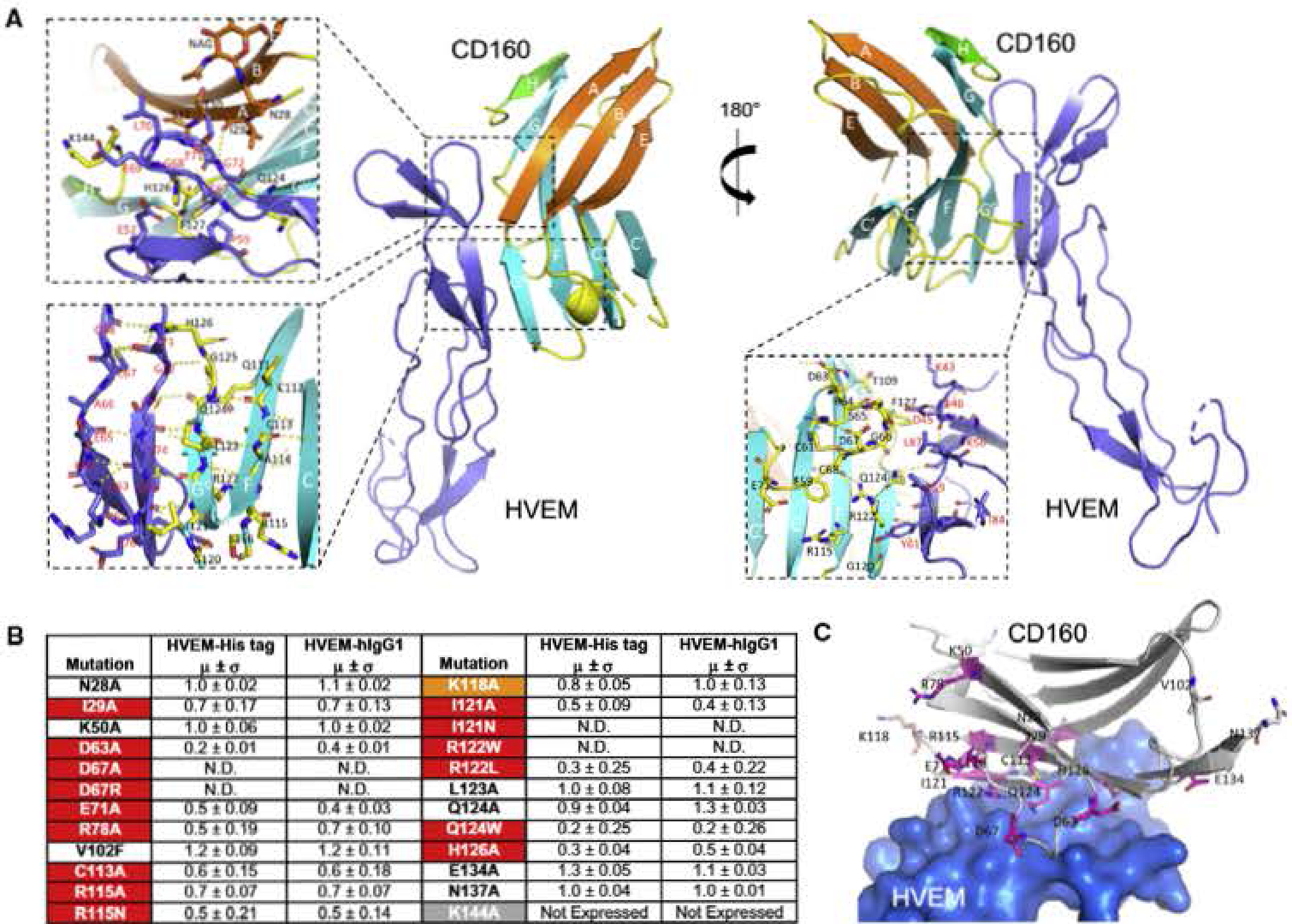
(A) Detailed map of CD16Q:HVEM interaction. The dashed box shows an expanded view of the interactions between human GDI60 and human HVEM. The top left panel emphasizes HVEM residues C67-G72 and their contacts with the A and H strands of CD160. The bottom left panel shows an expanded view of HVEM strands (T71-E7S and the adjacent R62-G68) in contact with CD160 strands (G0 strand composed of G120-G125 and the adjacent F strand composed of Q111-S116). The bottom right panel emphasizes parts of the CD160 CC′ loop (R64-D67) in direct contact with HVEM.
(B) Epitope mapping results of human CD160. The relative binding affinities are presented by the values obtained when normalized to the witd-type CD160 binding affinity of value 1 (standard deviations are results of triplicate measurements). N.D. stands for no detectable binding. The residues are categorized as sensitive residues when more than 20% binding affinity was lost, and are highlighted in red (lost in both assays) and orange (lost in only one assay).
(C) Structural representation of the CD160 epitope mapping results. The residues subjected to epitope mapping are shown as sticks. The sensitive residues with more than 20% compromised binding in both assays are colored purple and residues with more than 20% compromised binding in only one assay are colored orange. HVEM is shown as light-blue surface.
See also Figure S3.
Figure 5. Comparison of CD160:HVEM, BTLA: HVEM, and gD:HVEM Structures.
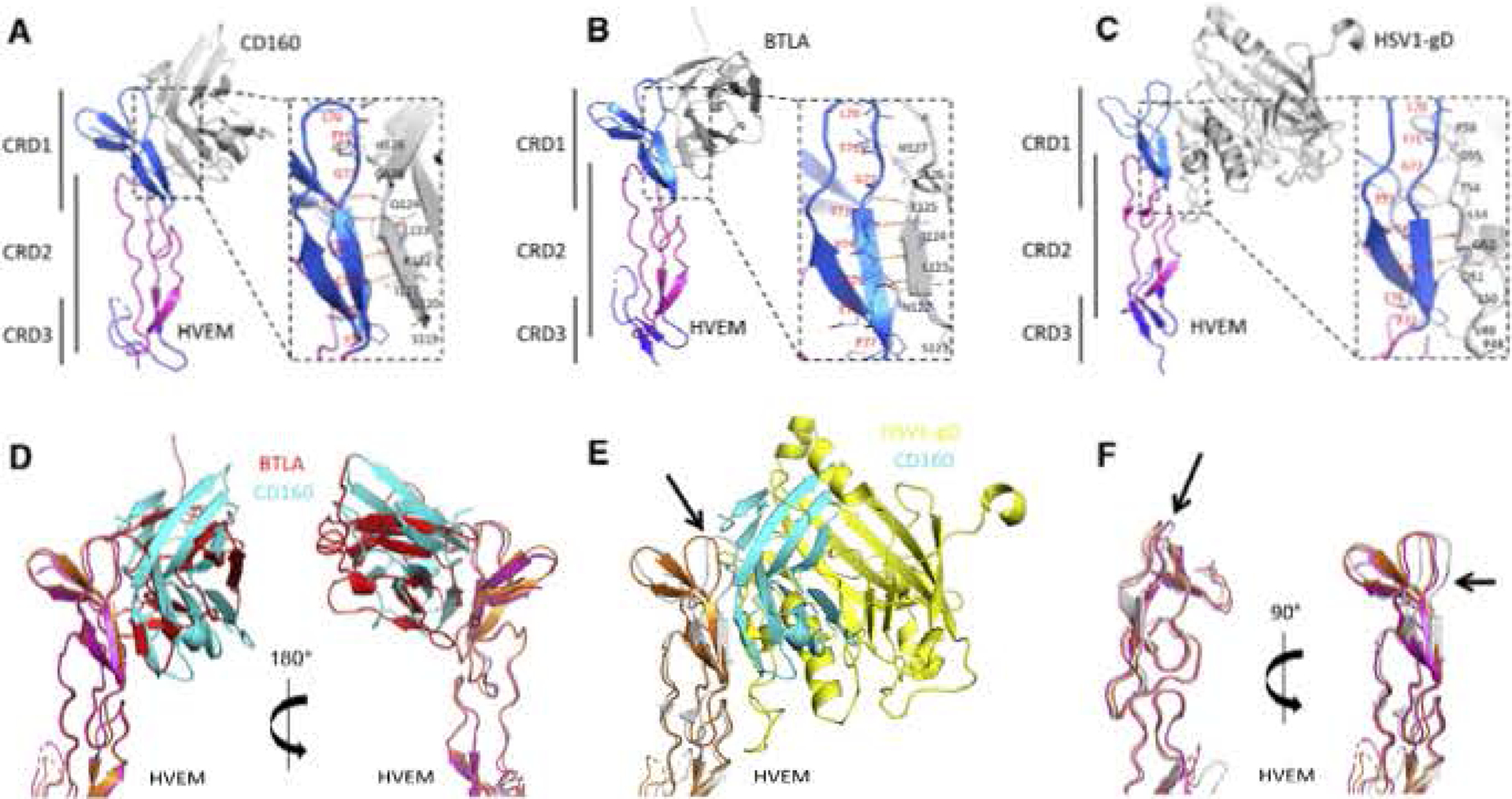
(A-C) Human HVEM structures are colored according to CDR: CRD1 (marine blue), CRD2 (cyan), and CRD3 (blue). Human CD160 (A), human BTLA (B), and HSV1-gD (C) are shown as gray cartoons. Expanded views emphasize the conserved interaction paradigm (intermolecular and antiparallel β sheet) formed by the HVEM strand (T71-E76) contacting with the ligands.
(D-F) Superimposition of the HVEM structures from CD160:HVEM, BTLA:HVEM, and gD:HVEM complexes. CD160 is shown as cyan and HVEM is shown as orange from the CD160:HVEM structure. BTLA is shown as red and HVEM is shown as magenta from the BTLA: HVEM structure. HSV1-gD is shown as yellow and HVEM is shown as gray from the gD:HVEM structure. Superimposition of the HVEM structures from the CD160:HVEM and BTLA:HVEM complexes (D) and GD16Q:HVEM and gD:HVEM complexes (E). Superimposition of HVEM from CD ISO: HVEM, BTLA:HVEM, and gD:HVEM structures (F) Indicates that HVEM from the gD:HVEM structure is most disparate. The significant conformational changes of HVEM residues K64-V74 are indicated by the black arrows.
See also Figure S4.
To further validate the crystallographically observed interface, we utilized an epitope mapping strategy in which wild-type and mutant CD160 proteins were fused to the human PD-L1 transmembrane domain and a cytoplasmic mCherry fluorescent protein, and expressed on the surface of HEK293F (HEK293 freestyle) cells. Cells expressing CDl60-mCherry fusion proteins were challenged with monovalent HVEM-His-tagged protein or bivalent HVEM-hlgGI to determine the consequences of specific mutations on HVEM binding, In general, both HVEM reagents gave similar binding results. The N28A and N137A mutations, which eliminate the putative glycosylation sites on CD160, did not significantly affect HVEM binding, consistent with our SPR data demonstrating that glycosylation of CD160 is not required for the CD160:HVEM interaction (Figure 4). In contrast, CD160 129A, which forms a main-chain-to-main-chain contact with HVEM G72, significantly affected HVEM binding, possibly by altering the local conformation of the CD160 A strand (Figure 4).
The CC′ loop of CD160, which is stabilized by a disulfide bond, proved to be important for HVEM binding. Single mutations (D63A, D67A, D67R, or E71A) in this loop dramatically reduced HVEM binding (Figure 4). GD160 D67 does not form direct contacts with HVEM, but its side chain forms two polar contacts with R64 and S65 of CD160, and D67 mutations may affect binding by altering the conformation of the CC′ loop (Figure 4). CD16Q D63A and E71A, which reside at the N terminus and C terminus of CC′ loop, respectively, do not directly contact HVEM, and may also affect binding by altering the local conformation of the CC′ loop. Notably, CD160 D63 side chain forms intramolecular polar contact with T109, whereas E71 does not form observable hydrogen bond in the structure. R78 is not in direct contact with the interface; the R78A mutant may affect HVEM binding through propagation of local structural effects.
The G0 and G strands reside at the center of CD160:HVEM interaction, and participate in many main-chain-to-main-chain hydrogen bonds, consistent with the observation that “mild” mutations in this region do not significantly alter HVEM binding. For example, the L123A or Q124A single mutations do not significantly affect HVEM binding. However, the Q124W mutant, which introduces a bulkier residue into the interface and is predicted to be sterically incompatible, dramatically lowers HVEM binding (Figures 4 and S4). The I121A mutant decreased HVEM binding, but not as substantially as the I121N mutant. Similarly, R122L decreased HVEM binding but not as dramatically as R122W mutant (Figure 4). H126 of CD160 forms a non-polar side-chain contact with the HVEM C67 side chain. The CD160 H126 side chain also forms van der Waals contacts with HVEM C54-P55 and polar contacts with the main-chain carbonyls of G68 and T71. The H126A mutation greatly diminished HVEM binding (Figure 4). Mutations on the adjacent CD160 F strand also affected HVEM binding, with C113A, R115A, and R115N single mutants exhibiting weaker binding to HVEM, K118A on the FG0 loop had a mild effect on HVEM binding. K50A and V102F, which are distant from the interface, had no measurable impact on HVEM binding (Figure 4).
Comparison of CD160, BTLA, and HSV1-gD Binding with HVEM
The crystal structure of the CD160:HVEM complex revealed a binding mode resembling the structures of the BTLA:HVEM and HSV1 gD:HVEM complexes (Carfi et al., 2001; Compaan et al., 2005). The CD160:HVEM complex buries 759 Å2 of solvent-accessible surface area on CD160 and 686 Å2 on HVEM for a total of 1,445 Å2. The BTLA:HVEM complex (PDB: 2AW2) buries 927 Å2 of solvent-accessible area on BTLA and 898 on HVEM for a total of 1, 825 Å2. The gD:HVEM complex (PDB: 1JMA) buries 852 Å2 of solvent-accessible area on gD and 724 Å2 on HVEM for a total of 1,576 Å2. In general, CD160, BTLA, and gD bind to similar and overlapping surfaces on HVEM.
Examination of the CD160:HVEM, BTLA:HVEM, and gD:HVEM structures reveals a shared interaction paradigm, which underlies the binding promiscuity exhibited by HVEM. The HVEM β strand composed of residues T71-E76 interacts with each ligand so as to form a two-stranded discontinuous antiparatlel sheet (Figure 5). In the CD160:HVEM and BTLA:HVEM complexes, the G0 strands form four pairs of main-chain-to-main-chain hydrogen bonds, while in the gD:HVEM complex three pairs of main-chain hydrogen bonds are formed (Figure 5) (Carfi et al., 2001). These main-chain-to-main-chain hydrogen-bonding interactions reside at the centers of the binding interfaces.
Supeninposition of the HVEM structures from CD160:HVEM and BTLA;HVEM complexes reveals similar overall structures for HVEM with an RMSD of 0.9 Å for 102 aligned Cα atoms, as well as for the HVEM CRD1 with an RMSD of 0.5 Å for 34 aligned Cα, atoms (Figure 5). Although the structures of CD160 and BTLA align well with an RMSD of 2.1 A for 107 aligned Cα, atoms (Figure S2), structural superimposition of their complexes based on HVEM shows a substantial difference in the relative placement of the CD160 and BTLA molecules (Figure 5). Alignment of HVEM from CD160:HVEM BTLA HVEM and gD:HVEM complexes indicates HVEM in the gD:HVEM complex adopts the most disparate conformation in CRD1 (Figures 5 and S3).
Epitope Mapping of HVEM
Constructs of full-length human HVEM fused with eGFP on the C terminus were transfected into HEK293F cells and challenged with wild-type CD160, BTLA, gD, and LIGHT to assess the rotative binding. The HVEM G72D mutant dramatically decreased the binding of CD160, BTLA, and gD, but not LIGHT (Figure 6). Modeling of the G72D mutation suggests that the altered side chain makes an unfavorable steric contact with CD160, resulting in reduced binding affinity (Figure S4). Since G72 is also important for the secondary structure of HVEM (Figure 4), G72D may affect binding through altering the secondary structure of the HVEM contact strand. The HVEM T73A or V74E mutants, which reside in the center of the recognition interface, also significantly reduced the binding of CD160 BTLA HSV1-gD to HVEM, but not LIGHT to HVEM. The V74A mutant was previously reported to compromise binding to both BTLA and CD160, as measured by SPR (Kojima et al., 2011). T71A or E76A, at the periphery of the HVEM contact strand, did not have a significant effect on CD160 and BTLA binding. Although HVEM T71 does not contact gD, the T71A mutant reduced gD binding to HVEM, indicating slightly different binding mechanisms/poses among the three ligands (Figure 6). The side chain of HVEM Y61 makes a polar contact with CD160 G120 (Figure 4), and it has been reported that the Y61A mutant exhibits no binding to cells expressing BTLA or CD160. Interestingly, the HVEM Y61C mutation found in patients with somatic lymphoma has been reported to significantly compromise binding to both BTLA and CD160 (Schmidt et al., 2016; Ware and Sedy, 2018).
Figure 6. Epitope Mapping of HVEM.
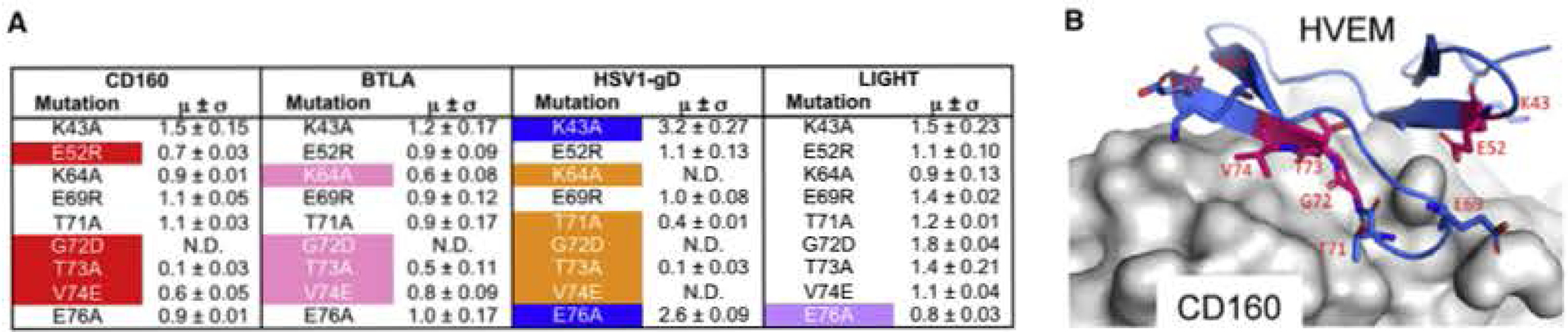
(A) Epitope mapping results of human HVEM, The relative binding affinities are represented by the values when normalized to the wild-type HVEM binding affinity (standard deviations are results of triplicates). N.D. stands for no detectable binding. The residues are categorized as sensitive residues when more than 20% binding affinity was lost. These affected residue’s are highlighted by color. Two residues with more than 2-fold increased affinities are colored blue.
(B) Structural representation of the CD160:HVEM epitope mapping results. The HVEM residues subjected to epitope mapping are shown as sticks. The sensitive residues with more than 20% compromised binding are highlighted by purple color. CD160 is shown as gray surface.
HVEM K43, E52, K64, and E69, outside the primary recognition strand, also contribute to the interface between HVEM and ligands, Mutations at these sites had diverse outcomes on different ligands. The E52R mutant dramatically reduced CD160 binding but not the others. The K64A mutant significantly decreased gD binding and compromised BTLA binding, but did not affect CD160 and LIGHT binding (Figure 6). The E69R mutant, which is located within the binding interface, did not affect binding to any of the ligands. Interestingly, the single K43A and E76A mutants both exhibited 2- to 3-fold enhanced binding to gD (Figure 6). This behavior is likely the consequence of removing nearby repulsive electrostatic interactions between gD and HVEM. Notably, none of the mutants altering CD160, BTLA, or gD binding exhibited an effect on LIGHT binding, or vice versa (Figure 6), consistent with the previous observation that LIGHT does not compete with these ligands (Cai et al., 2008).
DISCUSSION
Early evidence using monoclonal antibody BY55 suggested that CD160 might form a multimer (Anumanthan et al., 1998; Maiza et al., 1993), whereas our biochemical and structural data, as well as a previous report (Kojima et al., 2011), indicates that the CD160 ectodomain is monomeric. The unpaired cysteine C113 is solvent inaccessible and thus unlikely to form an intermolecular disulfide bond. Some Ig-domain proteins undergo strand swapping, which could possibly alter the structure to expose the unpaired cysteine; however, C113 is located on the F strand. The Ig-domain hallmark disulfide bond stabilizes the B and F strands, which prevents the F strand from significant conformational reorganization. Thus, the ectodomain of CD160 by itself is unlikely to form multimers involving disulfide bonds.
The monomeric structure of CD160 resembles the structure of BTLA, though with some unique structural features (Figure S2). This similarity also suggests that analogous to BTLA, CD160 interacts with HVEM in a 1:1 stoichiometry, in contrast to the 3:3 stoichiometry of the LIGHT:HVEM assembly. The CD160:HVEM interaction has been suggested by previous functional studies. For example, both CD160 and BTLA can bind to HVEM in cis form to inhibit T cell activation, whereas LIGHT does not bind to HVEM in the cis form (Cheung et al., 2009). The 3:3 stoichiometry of the LIGHT;HVEM interaction may impose more constraints, preventing membrane-anchored LIGHT and HVEM from adopting the configurations required to form cis interaction. In the cases of CD160 and BTLA, the simple 1:1 stoichiometry does not impose conformational restrictions that would preclude cis interactions (Figure 1).
Our structural and biochemical data indicate that BTLA possesses a higher affinity than CD160 for HVEM, and that BTLA and CD160 compete for overlapping binding sites on HVEM. These findings are consistent with previous reports that BTLA-Ig outcompetes CD160-lg for binding cell membrane-anchored HVEM (Cai et al., 2008). However, the binding of BTLA and CD 160 with HVEM could be affected by many context-dependent factors. Different cellular subsets may express only a single ligand, and even when both BTLA and CD160 are expressed on the same cell surface they could be spatially segregated, which may allow one to functionally dominate over the other regardless of the binding affinity. For example, confocal microscopy showed that in activated CD4+ T cells, CD160 was present in discrete patches and did not colocalize with BTLA (Cai et al., 2008).
Crystal structures of CD160:HVEM, BTLAHVEM, and gD:HVEM reveal a conserved interaction pattern, which emphasizes the β-sheet-like antiparallel main-chain-to-main-chain hydrogen bonds formed between HVEM and these multiple ligands (Figure 5). These main-chain-to-main-chain hydrogen bonds are critical for the interactions of HVEM with CD160, BTLA, and gD. Three HVEM residues (T73, V74, and C75) participating in these main-chain-to-main-chain hydrogen bonds are conserved among different species (Figure S4). Notably, HVEM residue G72 is also conserved among different species but is not present in other TNF receptors (Mukai et al., 2010; Qian et al., 2015; Sudhamsu et al., 2013; Yu et al., 2018). Modeling of HVEM G72D mutant suggests that bulky side chains at this position will sterically interfere with ligand binding. Super imposition of HVEM with other TNF receptors indicates that the G72 analogous residues all present steric effects similar to those of the HVEM G72D mutant (Figure S4); thus, the G72 analogous residues in other TNF receptors may function as antideterminants to prevent interaction promiscuity with members of the IgSF. In contrast, the unique HVEM G72 residue may contribute to the broad promiscuity of HVEM by minimizing steric interactions and conferring maximum structural flexibility (Figure S4).
A similar strategy involving invariant main-chain interactions was also observed in the DcR3 interaction network. The DE loops of TNF ligands TL1A, LIGHT, and FasL all engage DcR3 by recognizing backbone atoms and invariant residue side chains (Liu et al., 2014, 2016: Zhan et al., 2011). Furthermore, in the structures of DcR3 complexes, DcR3 residue Y90 is proposed to function as an antideterminant to preclude binding of other TNFSF members (Liu et al., 2016; Zhan et al., 2011). These shared strategies of common recognition elements and antideterminants define the repertoire of TNF ligands and other interaction partners recognized by DcR3 and HVEM.
STAR ★ METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Steven C. Almo (steve.almo@einstein.yu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HEK293 freestyle cells (LifeTechnologies) were cultured and expanded at 37°C, 5% CO2 in Freestyle media (LifeTechnologies). Drosophila S2 cells were cultured and expanded at 28 °C in Express Five Serum Free Media (LifeTechnologies). E. coli methionine auxotroph B834 (DE3) was cultured at 37°C in M9 medium supplemented with essential elements and Seleno-L-methionine. E.coli BL21(DE3)pLysS strain was cultured and expanded at 37°C in LB media.
METHOD DETAILS
Molecular Cloning and Mutagenesis
The DNA tem plates of the ectodomains of homo sapiens BTLA, CD160 and single chain CD160-HVEM were synthesized by IDT (Integrated DNA Technologies). The genes encoding BTLA (W26-K137) and different length of CD160s were amplified by PCR using primers with endonuclease digestion sites for Ndel and BamHI. The resulting PCR products were digested and ligated into plasmid pET3a for protein production. The human single chain CD160-HVEM was amplified by PCR and then further digested by endonucleases BgIII and Agel. The purified product was ligated into plasmid pMT/Bip/V5-His for His-tag fusion protein production in Drosophila S2 cells.
The gene encoding full length human CD160 was cloned into an engineered pmCherry-N1 vector (Clontech) to be expressed as a protein fused with the human PD-L1 transmembrane segment followed by the fluorophore mCherry at its C-terminus. The gene coding full length human HVEM was cloned into the pGFP-N1 vector (Clontech) for expression as a protein fused with fluorophore eGFP at the C-terminus. All CD160 and HVEM mutants were generated using QuickChange II Site-Directed Mutagenesis Kit (Agilent Technologies). Sequences encoding glycoproteinD from HSV 1 (accession Q05059, residues 25 to 341) were synthesized (gen9), and cloned into a modified version of pIRES-acGFP with the human erythropoietin signal peptide and a C-terminal deca-His tag.
Protein Production and Purification
The pET3a constructs containing coding sequences for human CD160 or BTLA were transformed into E.coli BL21(DE3)pLysS strain, followed by induction with 1 mM IPTG for 5 hrs at 37°C to express protein as inclusion bodies. SeMet-substituted CD160 proteins were expressed in the E. coli methionine auxotroph B834 (DE3) in M9 medium supplemented with essential elements solutions and Seleno-L-methionine (Studier, 2005). The resulting inclusion bodies were purified by lysis of the cell pellets using sonication in buffer composed of 150 mM NaCl, 10 mM DTT, 100 mM Tris-HCI, pH8.0 and 1 mM EDTA. The purified inclusion bodies were washed three times in the buffer composed of 10 mM Tris-HCI, pH8.0, 100 mM NaCl, 10 mM DTT; and 1 mM EDTA supplemented with 0.5% Triton X-100 and one final time in the same buffer but without 0,5% Triton X-100. The washed inclusion bodies were dissolved in buffer composed of 6 M Guanidine-HCI, pH4.6, 10 mM Sodium Formate and 5 mM EDTA, and were further diluted to a concentration of 1 mg/mL in 16 mL of the same buffer. The protein was refolded by adding dissolved inclusion bodies drop-wise to 1000 mL of the refolding-buffer composed of 200 mM Tris-HCI, pH8.5, 0.4 M Arginine-HCI, 2 mM EDTA, 0.5 mM cystamine and 5 mM cysteamine at 4°C with stirring. Five additional aliquots of diluted inclusion bodies (16 mg each) were added every 8 hrs. The refolded protein was concentrated and buffer-exchanged into 10 mM Tris-HCI, pH8.5, 150 mM NaCl with a 350 mL Amicon stirred cell (Millipore) using 5 KDa molecular weight cutoff ultrafiltration membranes (Millipore). The protein solution was clarified by centrifugation for at 6000 × g for 5 min at 4°C. The supernatant was passed through a 0.22 μM filter to further remove aggregates. The resulting solution was passed through an S75 size exclusion column (Amersham) in the HEPES buffer of 20 mM HEPES, 150 mM NaCl, pH7.0 and 1 mM EDTA to obtain the final working protein fractions.
Human LIGHT, HVEM, CD160 and single chain CD160-HVEM proteins expressed in insect cells were purified as described previously (Liu et al., 2014, 2015). Briefly, stable Drosophila S2 cell lines encoding LIGHT (L83-V240), HVEM (L39-C162), CD160 (I27-L158) and single chain CD160 HVEM were induced with copper sulfate (500 μM final concentration) to express the proteins. The expressed proteins from filtered culture supernatant were purified by Ni-NTA column (QIAGEN) and size exclusion chromatography (HiLoad Superdex 75; Amersham).
For HSV-1 gD production, a 500 mL suspension culture of HEK293 freestyle cells (LifeTechnologies) in Freestyle media were transfected with the HSV-1 gD expression plasmid using linear PEI; valproic acid was added to 3 mM 24 hours post-transfection, and culture supernatant was harvested 6 days post-transfection. Protein was affinity purified with nickel resin (HIS60, Clontech) and through gel filtration (Superdex S200 26/60, GE) in PBS.
Crystallization and Structure Determination
Refolded human CD160 (I27-K144) and S2 cell derived single chain CD160-HVEM proteins were concentrated to 5 mg/mL in a HEPES-based buffer of 20 mM HEPES. pH7.0, 150 mM NaCl and 1 mM EDTA. The wild type CD160 (I27-K144). SeMet V58M CD160 (I27-K144) mutant and single chain CD160-HVEM were crystalized by sitting drop vapor diffusion at 277 K. Briefly, 0.3 μL of CD160 in HEPES buffer was mixed with 0.3 μL of buffer composed of 0.2 M Sodium Formate, 0.1 M Tris, pH8.5 and 30% (W/V) PEG4000. Single chain CD160-HVEM protein in 0.3 μL HEPES buffer was mixed with 0.3 μL of solution composed of 0.1 M HEPES pH 7.0, 30% (V/V) Jeffamine ED-2001 pH7.0, and 0.1 M sodium citrate tribasic dihydrate. CD160 crystals appeared in 4 to 5 days and the single chain CD160-HVEM crystal appeared in 1 to 2 months. Crystals were mounted in cryo-buffers composed of 20% glycerol and 80% crystal buffer. Diffraction data were collected at NSLS, Brookhaven National Laboratory beamline X29 and Advanced Photon Source Sector 31, Argonne National Laboratory (Table 1).
The diffraction data were integrated and scaled with HKL2000 (Otwinowski and Minor, 1997). Molecular replacement was unsuccessful, as were attempts to phase using SeMet-substituted protein (only two methionines are present, one of which is the N-terminal residue). TwoCD160 mutants (T103M and V58M) were generated to increase the methionine content and the associated anomalous signal. The T103M mutant failed to refold, while the V58M CD160 mutant expressed and refolded, and SeMet-substituted material provided sufficient anomalous signal for phase determination. Diffraction data from SeMet-substituted V58M CD160 crystals were collected at APS. Six selenium sites from two chains of V58M CD160 mutant were located using the SHELX package of programs (Sheldrick, 2010). These results indicated that the initiating methionine was not cleaved in the E.coli BL21 physstrain. Phases were calculated using PHASER and improved by density modification and averaging using the programs DM, SOLOMON and PARROT in the CCP4 package (Winn et al., 2011). The averaged electron density maps were manually interpreted using COOT (Emsley et al., 2010; Winn et al., 2011). Intermediate partial structures were submitted to ARP/wARP in the CCP4 package to improve electron density maps (Winn et al., 2011). Following several cycles of manual building in COOT and refinement in REFMAC5, the Rwork and Rfree converged to 21.7% and 26.9%, respectively (Emsley et al, 2010; Winn et al., 2011). The structures of native CD160 and SeMet CD160 were phased using V58M CD160 structure. The single chain CD160-HVEM crystal was phased with HVEM (PDB: 4FHQ) and our CD160 structure, and the resulting model subjected to iterative cycles of model building and refinement with COOT and REFMAC5, resulting in Rwork and Rfree of 23.4% and 27.4%, respectively (Emsley et al., 2010). Structural analysis benefited from the online server PDBePISA (Krissinel and Henrick, 2007).
SPR to Test Binding
Human HVEM protein derived from Drosophila S2 cells was immobilized on CM5 sensor chips (GE Life Sciences). For determining the binding affinities of purified E. coli expressed CD160 or Drosophila S2 cell expressed CD160, the analyte CD160 proteins were diluted to various concentrations ranging from 20 μM to 1.25 μM and passed over CM5 sensor chips at 25°C in buffer composed of 25 mM HEPES pH7.4, 150 mM NaCl, 3 mM EDTA and 0.005% TWEEN-20 at a flow rate of 20 μL/min. Background responses of blank reference flow channels were subtracted from each response signal of the experimental channel to give the final response level. Equilibrium dissociation constants were calculated by fitting the data to the relationship: Y=BmaxX/(X+KD) (Y is the averaged maximum response of each experiment cell in RUs. X is the concentration of the analytes, KD is the equilibrium dissociation constant and Bmax is the maximum specific binding). The response level curves were exported and analyzed by Prism 5 (Graphpad Software).
SEC-MALS
Solutions of CD160 (5 mg/mL) in buffer containing 20mM HEPES, 150 mM NaCl, 1 mM EDTA, pH7.5 or pH5.5, were subjected to size exclusion chromatography using a WTC030N5 (Wyatt Technology Corporation) column coupled to a Shimadzu HPLC system. Light scattering measurements were performed downstream, using a miniDawn TREOS instrument connected to the column output, followed by Optilab rEX refractive index analysis (Wyatt Technology Corporation). Control experiments were performed with BSA diluted in the same buffer as the sample. Data from these experiments were collected and interpreted using ASTRA software (version 6.0.3.16).
Epitope Mapping of CD160 and HVEM
For mammalian cell transfection, 500 ng CD150 or HVEM plasmids in 50 μL PBS were mixed with 50μL of 0.04 M PEI (Linear Poly-ethylenimine with molecular weight of 25000; Polysciences Inc.). The mixtures were kept still for 10 mins and then added separately to a 24-well plate with each well containing 1 mL of 1 Million/mL Freestyle HEK293 cells (Thermo fisher). After 2–3 days, the cells were harvested and resuspended in PBS to a final concentration of 1 Million/mL.
For staining the CD160 expressing cells, 100 μL of the diluted transfected cells were incubated with 1 μL of 0.5 μg/μL purified HVEM-His or HVEM-lg (R&D systems) for 20 minutes on ice. Cells were pelleted, washed twice and resuspended in 100 μL PBS buffer containing additional 0.5% BSA. 0.5 μL of APC (allophycocyanin) conjugated anti-His-tag antibody (Abeam) or APC conjugated anti-human Ig antibody (Abeam) was added to each well. After 20 mins, the cells were washed once, resuspended in PBS buffer containing additional 0.5% BSA and subjected to FACS (fluorescence-activated cell sorting) analysis. Cells were gated on mCherry positive cells to ensure CD160 expression within the analyzed population. The gated cells were then analyzed for the percentage of APC positive cells. The average percent bound and standard deviations were calculated from three independent experiments.
For staining the HVEM expressing cells, 100 μL of cells expressing HVEM-eGFP proteins were separately mixed with 0.3 μg CD 160-His tag (R&D systems). BTLA-His tag (R&D systems), HSV-1 gD-His tag and LIGHT-His tag proteins. Subsequently 0.5 μg of PE fluorescent anti-His-tag antibody (Abeam) was added to each well. The cells were incubated for 20 mins on ice and washed once with PBS containing 0.2% BSA (PBS-BSA). The cells were resuspended in 100 μL of PBS buffer containing 0.5% BSA and analyzed by FACS. The average percent bound and standard deviations were calculated from three independent experiments.
QUANTIFICATION AND STATISTICAL ANALYSIS
The statistical analysis and the software used can be found in the relevant sections of the methods and the figure/table legends.
DATA AND SOFTWARE AVAILABILITY
The accession numbers for the coordinates and structure factors for the CD160, SeMet V58M CD160 and sc_CD160:HVEM are PDB: 6NG9, PDB: 6NGG and PDB: 6NG3, respectively.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-6xHis lag antibody APC-conjugated | Abeam | Cat#ab72467; RRID: AB_1267596 |
| Anti-6xHis tag antibody PE-conjugated | Abeam | Cat#ab72579: RRID: AB_1267597 |
| Goat anti-human IgG FC (FITC) | Abeam | Cat#ab97224: RRID: AB_10680849 |
| Bacterial and Virus Strains | ||
| E.coli BL21(DE3) pLysS | Promega | N/A |
| E.coli methionine auxotroph B834(DE3) | Novagen | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PEI, MW 25000 | Polysciences Inc. | Cat#23966–1 |
| Recombinant human HVEM Fc | R&D systems | Cat#356-HV/CF |
| Recombinant human BTLA His-lag | R&D systems | Cat#9235-BT-050 |
| Recombinant human CP160His-tag | R&D systems | Cat#6177-CD-0S0 |
| Critical Commercial Assays | ||
| MCSG crystallization suite | Anatrace | MCSG-1 |
| MCSG crystallization suite | Anatrace | MCSG-2 |
| MCSG crystallization suits | Anatrace | MCSG-3 |
| MCSG crystallization suite | Anatrace | MCSG-4 |
| Deposited Data | ||
| Crystal structure of gD:HVEM complex | (Carfi et al., 2001) | PDB: 1JMA |
| Crystal structure of BLA:HVEM complex | (Compaan et al., 2005) | PDB:2AW2 |
| Crystal structure oi TNF:TNFR2 complex | (Mukai et al., 2010) | PDB:3ALQ |
| Crystal structure of equine lentivirus recpetor 1 | (Qian et al. 2015) | PDB:3WVT |
| Crystal structure of HVEM | (Liu et al., 2015) | PDB:4FHQ |
| Crystal structure of Fab:LTα1β2:LTβR complex | (Sudhamsu et al., 2013) | PDB:4MXW |
| Crystal structure of Fab:CD40 complex | (Yu et al., 2018) | PDB:6FAX |
| Crystal structure of sc_CD160:HVEM | This paper | PDB:6NG3 |
| Crystal structure of CD160 | This paper | PDB:6NG9 |
| Crystal structure of SeMet V58M CD160 | This paper | PDB:6NGG |
| Experimental Models: Cell Lima | ||
| Drosophila S2 | Thermo Fisher | N/A |
| HEK293 | Thermo Fisher | N/A |
| Recombinant UNA | ||
| Human CD160-pET3a | This paper | N/A |
| Human BTLA-pET3a | This paper | N/A |
| Human single chain CD16D-HVEM-pMT-Bip-V5-His | This paper | N/A |
| Human CD160-pMT-Blp-V5-His | This paper | N/A |
| Human CD160-pmCherry-N1 | This paper | N/A |
| Human HVEM pGFP-N1 | This paper | N/A |
| HSV1-gD-plRES-acGFP | This paper | N/A |
| Software and Algorithms | ||
| HHL2000 | (Otwinowski and Minor, 1997) | http://www.hkl-xray.com/ |
| SHELX | (Sheldrick, 2010) | http://shelx.uni-goettingen.de/ |
| CCP4 | (Winn et al., 2011) | https://www.ccp4.ac.uk/ |
| COOT | (Emsley et al., 2010) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| ARP/wARP | (Winn et al., 2011) | http://www.embt-hamburg.de/ARP/ |
| REFMAC5 | (Winn et al., 2011) | http://www.ccp4.ac.uk/html/refmac5.html |
| Pymol | Molecular Graphics System, Schrodinger, LLC | https://pyimol.org/2/ |
| CLUSTALW | (Thompson et al., 1994) | https://www.genome.jp/tools-bin/clustalw |
| ESRript 3.0 | (Robert and Gouet, 2014) | https://espript.ibcp.fr/ESPript/ESPript/ |
| PDBePISA | (Krissinel and Henrick, 2007) | http://wwiw.ebi.ac.uk/pdbe/pisa/ |
| Prism 5 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
Highlights.
CD160 adopts a unique monomeric IgC fold
CD160 exhibits lower affinity than BTLA for HVEM
CD160:HVEM structure is similar to that of BTLA:HVEM
β-Sheet interface is important for HVEM promiscuity
ACKNOWLEDGMENTS
We thank the staff o1 X29A beamlines at the National Synchrotron Light Source. Use of the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the US Department o1 Energy, Office of Science, Office o1 Basic Energy Sciences, under contract no. DE-ACQ2-98CH10886. Financial support comes principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the US Department of Energy (DOE), and from the National Center for Research Resources (P41RRQ12408) and the National Institute of General Medical Sciences (P41GM103473) of the National Institutes of Health. Use of the Advanced Photon Source, an Office of Science User Facility operated for the the DOE Office of Science by Argonne National Laboratory, was supported by the US DOE under contract no. DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility. We also acknowledge support from the Albert Einstein Cancer Center (P30CA01333Q), the Einstein Crystallographic Core X-ray Diffraction Facility supported by NIH Shared Instrumentation grant S10 OD020068 and the Albert Einstein Macromolecular Therapeutics Development Facility. This work, was partially supported by the Price Family Foundation arid contributions to the Albert Einstein Center for Experimental Therapeutics by Pamela and Edward S. Pantzer. We thank Dr. Mitchell Kronenberg for stimulating discussions.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.str.2019.05.010.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Agrawal S. Marquet J, Freeman GJ, Tawab A, Bouteiller PL, Roth P, Bolton W, Ogg G, Boumsell L, and Bensussan A (1999), Cutting edge: MHC class I triggering by a novel cell surface ligand costimulates proliferation of activated human T cells. J. Immunol 162, 1223–1226. [PubMed] [Google Scholar]
- Anumanthan A, Bensussan A, Boumsell L, Christ AD, Blumberg RS, Voss SD, Patel AT, Robertson MJ, Nadler LM, and Freeman GJ, (1998). Cloning of BY55, a novel Ig superfamily member expressed on NK cells, CTL, and intestinal infraepithelial lymphocytes. J. Immunol 161, 2780–2790. [PubMed] [Google Scholar]
- Barakonyi A, Rabot M, Marie-Cardine A, Aguerre-Girr M, Polgar B, Schiavon V, Bensussan A, and Le Bouteiller P (2004). Cutting edge: engagement of CD160 by its HLA-C physiological ligand triggers a unique cytokine profile secretion in the cytotoxic peripheral blood NK cell subset. J. Immunol 173 5349–5354. [DOI] [PubMed] [Google Scholar]
- Bodelón G, Palomino C, and Fernández LÁ, (2013), Immunoglobulin domains in Escherichia coli and other enterobacteria: from pathogenesis to applications in antibody technologies. FEMS Microbiol. Rev 37, 204–250. [DOI] [PubMed] [Google Scholar]
- Boice M, Salloum D, Mourcin F, Sanghvi V, Amin R, Oricchio E, Jiang M, Mottok A, Denis-Lagache N, Ciriello G, et al. (2016). Loss of the HVEM tumor suppressor in lymphoma and restoration by modified CAR-T cells. Cell 167, 405–418.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai G, Anumanthan A, Brown JA, Greenfield EA, Zhu B, and Freeman GJ (2008). CD160 inhibits activation of human CD4+ T cells through interaction with herpesvirus entry mediator. Nat. Immunol 9, 176–185. [DOI] [PubMed] [Google Scholar]
- Cai G, and Freeman GJ (2009). The CD160, BTLA, LIGHT/HVEM. pathway; a bidirectional switch regulating T-cell activation. Immunol. Rev 229,244–258. [DOI] [PubMed] [Google Scholar]
- Carfi A, Willis SH, Whitbeck JC, Krummenacher C, Cohen GH, Eisenberg RJ, and Wiley DC (2001). Flerpes simplex virus glycoprotein D bound to the human receptor HveA. Mol. Cell 8, 169–179. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay K, Lazar-Molnar E, Yan Q, Rubinstein R, Zhan C, Vigdorovich V, Ramagopal UA, Bonanno J, Nathenson SG, and Almo SC (2009). Sequence, structure, function, immunity: structural genomics of costimulation. Immunol. Rev 229, 356–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung TC, Obome LM, Steinberg MW, Macauley MG, Fukuyama S, Sanjo H, D’Souza C, Norris PS, Pfeffer K, Murphy KM, et al. (2009). T cell intrinsic heterodimerrc complexes between HVEM and BTLA determine receptivity to the surrounding microenvironment. J. Immunol 183, 7286–7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compaan DM, Gonzalez LC, Tom I, Loyet KM, Eaton D, and Hymowitz SG, (2005). Attenuating lymphocyte activity: the crystal structure of the BTLA-HVEM complex. J. Biol. Chetn 280, 39553–39561. [DOI] [PubMed] [Google Scholar]
- del Rio ML, Lucas CL, Buhler L. Rayat G, and Rodriguez-Bartiosa JI (2010). HVEM/LIGF1T/BTLA/CD160 cosignaling pathways as targets for immune regulation. J. Leukoc. Biol 87, 223–235. [DOI] [PubMed] [Google Scholar]
- Desai P, Abboud G, Stanfield J, Thomas PG, Song J, Ware CF, Croft M, and Salek-Ardakani S (2017). HVEM imprints memory potential on effector CD8 T cells required for protective mucosal immunity. J. Immunol 199, 296–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fons P, Chabot S, Cartwright JE, Lentant F, L’Faqihi F, Giustiniani J, Herault JP, Guetjuen G, Bono F, Savi P, et al. (2006). Soluble HLA-G1 inhibits angiogenesis through an apoptotic pathway and by direct binding to CD160 receptor expressed by endothelial cells. Blood 108, 2608–2615. [DOI] [PubMed] [Google Scholar]
- Giustiniani J, Bensussan A, and Marie-Cardine A (2009). Identification and characterization of a transmembrane isoform of GDI60 (CD160-TM), a unique activating receptor selectively expressed upon human NK cell activation. J. Immunol 18263–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giustiniani J, Marie-Cardine A, and Bensussan A (2007). A soluble form of the MHC class I-specific CD160 receptor is released from human activated NK lymphocytes and inhibits cell-mediated cytotoxicity. J. Immunol 178, 1293–1300. [DOI] [PubMed] [Google Scholar]
- Kojima R, Kajikawa M, Shiroisht M, Kuroki K, and Maenaka K (2011). Molecular basis for herpesvirus entry mediator recognition by the human immune inhibitory receptor CD160 and its relationship to the cosignaling molecules BTLA and LIGHT. J. Mol. Biol 473 762–772. [DOI] [PubMed] [Google Scholar]
- Krissinel E, and Henrick K (2007). Inference of macromolecular assemblies from crystalline state. J. Mol. Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- Krummenacher C, Supekar VM, Whitbeck JC, Lazear E, Connolly SA, Eisenberg RJ, Cohen GH, Wiley DC, and Carfi A (2005). Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 24, 4144–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Ramagopal U, Chang H, Bonanno JB, Toro R, Bhosle R, Zhan C, and Almo SC (2016). Crystal structure of the complex of human FasL and its decoy receptor DcR3. Structure 24, 2016–2023. [DOI] [PubMed] [Google Scholar]
- Liu W, Vigdorovich V, Zhan C, Patskovsky Y, Bonanno JB, Nathenson SG, and Almo SC (2015). Increased heterologous protein expression in Drosophila S2 cells for massive production of immune ligands/receptors and structural analysis of human HVEM. Mol. Biotechnol 57, 914–922. [DOI] [PubMed] [Google Scholar]
- Liu W, Zhan C, Cheng H, Kumar PR, Bonanno JB, Nathenson SG, and Almo SC (2014). Mechanistic basis for functional promiscuity in the TNF and TNF receptor superfamilies: structure of the LIGHT; DcR3 assembly. Structure 22, 1252–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda M, Carpenito C, Russell RC, Dasanjh J, Veinotte LL, Ohta H, Yamamura T, Tan R, and Takei F (2005). Murine CD160. Ig-like receptor on NK cells and NKT cells, recognizes classical and nonclassical MHC class I and regulates NK cell activation. J. Immunol 3 75, 4426–4432. [DOI] [PubMed] [Google Scholar]
- Maiza H, Leca G, Mansur IG, Schiavon V, Boumsell L, and Bensussan A (1993). Anovel 80-kD cell surface structure identifies human circulating lymphocytes with natural killer activity. J. Exp. Med 178, 1121–1126, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauri DN, Ebner R, Montgomery RI, Kochel KD, Cheung TC, Yu GL, Ruben S, Murphy M, Eisenberg RJ, Cohen GH, et al. (1998). LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity 821–30. [DOI] [PubMed] [Google Scholar]
- Montgomery RI, Warner MS, Lum BJ, and Spear PG (1996). Herpes simplex virus-1 entry into cells mediated by a novel member of the TINF/NGF receptor family. Cell 87,427–436, [DOI] [PubMed] [Google Scholar]
- Mukai Y, Nakamura T, Yoshrkawa M, Yoshioka Y, Tsunoda S, Nakagawa S, Yamagata Y, and Tsutsumi Y (2010). Solution of the structure of theTNF-TNFR2 complex. Sci. Signal 3, ra83. [DOI] [PubMed] [Google Scholar]
- Nelson CA, Fremont MD, Sedy JR, Norris PS, Ware CF, Murphy KM, and Fremont DH (2008), Structural determinants of herpesvirus entry mediator recognition by murine B and T lymphocyte attenuator. J. Immunol 180940–947. [DOI] [PubMed] [Google Scholar]
- Nikolova M, Marie-Cardine A, Boumsell L, and Bensussan A (2002). BY55/CD160 acts as a co-receptor in TCR signal transduction of a human circulating cytotoxic effector T lymphocyte subset lacking CD28 expression. Int. Immunol 74,445–451. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Pasero C, Speiser DE, Derre L, and Olive D (2012). The HVEM network: new directions in targeting novel costimulatory/coinhibitory molecules for cancer therapy. Curr. Opin. Pharmacol 12, 478–485. [DOI] [PubMed] [Google Scholar]
- Qian L, Han X, and Liu X (2015). Structural insight into equine lentivirus receptor 1. Protein Sci. 24, 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert X, and Gouet P (2014). Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt J, Gong S, Marafioti T, Mankel B, Gonzalez-Farre B, Balague O, Mozos A, Cabecadas J, van der Walt J, Hoehn D, et al. (2016). Genome-wide analysis of pediatric-type follicular lymphoma reveals low genetic complexity and recurrent alterations of TNFRSF14 gene. Blood 128, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedy JR, Balmert MO, Ware BC, Smith W, Nemcovicova I, Norris PS, Miller BR, Aivazian D, and Ware CF (2017). A herpesvirus entry mediator mutein with selective agonist action for the inhibitory receptor B and T lymphocyte attenuator. J. Biol. Chem 292 21060–21070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedy JR, Bjordahl RL, Bekiaris V, Macauley MG, Ware BC, Norris PS, Lurain NS, Benedict CA and Ware CF (2013). CD160 activation by herpesvirus entry mediator augments inflammatory cytokine production and cytolytic function by NK cells. J. Immunol 191828–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedy JR, Gavrieli M, Potter KG, Hurchla MA, Lindsley RC, Hildner K, Scheu S, Pteffer K, Ware CF, Murphy TL, and Murphy KM (2005). B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat. Immunol 6, 90–98. [DOI] [PubMed] [Google Scholar]
- Sheldrick GM (2010). Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr 66, 479–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shui JW, Larange A, Kim G, Vela JL, Zahner S, Cheroutre H, and Kronenberg M (2012). HVEM signalling at mucosal barriers provides host defence against pathogenic bacteria. Nature 488, 222–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles KM, Whitbeck JC, Lou H, Cohen GH, Eisenberg RJ, and Krummenacher C (2010). Herpes simplex virus glycoprotein D interferes with binding of herpesvirus entry mediator to its ligands through down regulation and direct competition. J. Virol 84, 11646–11660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW (2005). Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif 41, 207–234. [DOI] [PubMed] [Google Scholar]
- Sudhamsu J, Yin J, Chiang EY, Starovasnik MA, Grogan JL, and Hymowitz SG (2013). Dimerization of LTbetaR by LTalpha1beta2 is necessary and sufficient for signal transduction. Proc. Nati. Acad. Sci. U S A 110, 19896–19901, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W, Wang J, Wang X, and Fu Y-X (2016). Facilitating T cell infiltration in tumor microenvironment overcomes resistance to PD-L1 blockade. Cancer Cell 29,285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, and Gibson TJ (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penaltiesand weight matrixchoice. Nucleic Acids Res. 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu TC, Brown NK, Kim TJ, Wroblewska J, Yang X, Guo X, Lee SH, Kumar V, Lee KM, and Fu YX (2015). CD160 is essential for NK-mediated IFN-gamma production. J. Exp. Med 212,415–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward-Kavanagh LK, Lin WW, Sedý JR, and Ware CF (2016). The TNF receptor superfamily in co-stimulating and coinhibitory responses. Immunity 44,1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware CF and Sedy J (2018). Compositions and methods for treating auto-immune and inflammatory disorders. (Google Patents).
- Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, Hurchta MA, Zimmerman N, Sim J, Zang X, et al. (2003). BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat. Immunol 4, 670–679. [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR. Keegan RM, Krissinel EB. Leslie AG, and McCoy A (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Chan HTC, Orr CM, Dadas O, Booth SG, Dahal LN, Penfold CA, O’Brien L, Mockridge CI, French RR, et al. (2018). Complex interplay between epitope specificity and isotype dictates the biological activity of anti-human CD40 antibodies. Cancer Cell 33, 664–675.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan C, Patskovsky Y, Yan Q, Li Z, Ramagopal U, Cheng H, Brenowitz M, Hui X, Nathenson SG, and Almo SC (2011). Decoy strategies: the structure of TL1A; DcR3 complex. Structure 19, 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Yao S, Augustine MM, Xu H, Wang J, Sun J, Broadwater M, Ruff W, Luo L, Zhu G, et al. (2016). Neuron-specific SALM5 limits inflammation in the CNS via its interaction with HVEM. Sci. Adv 2, e1500637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the coordinates and structure factors for the CD160, SeMet V58M CD160 and sc_CD160:HVEM are PDB: 6NG9, PDB: 6NGG and PDB: 6NG3, respectively.