

SUMMARY
NTRK1 gene fusions are actionable drivers of numerous human malignancies. Here, we show that expression of the TPR-NTRK1 fusion kinase in immortalized mouse pancreatic ductal epithelial (IMPE) (pancreas) or mouse lung epithelial (MLE-12) cells is sufficient to promote rapidly growing tumors in mice. Both tumor models are exquisitely sensitive to targeted inhibition with entrectinib, a tropomyosin-related kinase A (TRKA) inhibitor. Initial regression of NTRK1-driven tumors is driven by induced expression of BIM, such that BIM silencing leads to a diminished response to entrectinib in vivo. However, the emergence of drug-resistant disease limits the long-term durability of responses. Based on the reactivation of RAF>MEK>ERK signaling observed in entrectinib-treated tumors, we show that the combination of entrectinib plus the MEK1/2 inhibitor cobimetinib dramatically forestalls the onset of drug resistance in vivo. Collectively, these data provide a mechanistic rationale for rapid clinical deployment of combined inhibition of TRKA plus MEK1/2 in NTRK1-driven cancers.
In Brief
Vaishnavi et al. present two novel tissue-of-origin mouse models of transformation and describe a mechanistic rationale for why vertical, combined pathway inhibition of TRKA with MEK inhibition can significantly delay the onset of acquired resistance in numerous mouse models driven by an NTRK1 fusion.
Graphical Abstract

INTRODUCTION
The tropomyosin-related kinase family (TRKA, TRKB, and TRKC) of receptor tyrosine kinases (RTKs) are proto-oncogenes that mediate in their normal guise critical neuronal processes (Smeyne et al., 1994). However, somatic gene fusion events involving NTRK1 (encoding TRKA) encode constitutively active oncoprotein tyrosine kinases that are drivers of numerous human malignancies including lung, colorectal, and pancreatic cancers (O’Reilly and Hechtman, 2019; Pishvaian et al., 2018; Vaishnavi et al., 2015). Moreover, targeted inhibition of TRKA activity in cancers driven by NTRK1 oncogene fusions influence key cancer hallmarks including the cell division cycle and apoptosis (Doebele et al., 2015; Vaishnavi et al., 2013, 2017). The majority (~90%) of pancreatic cancers, and a large percentage (~33%) of lung cancers, are driven by mutationally activated KRAS. Despite improvements in the treatment of KRAS-driven cancers, the 5-year survival rate for such patients remains low (Nevala-Plagemann et al., 2020; Román et al., 2018). The identification of novel, actionable oncoproteins such as TRKA fusions in pancreatic cancer suggests that appropriate pharmacological targeting of driver oncoprotein kinases has the potential to improve patient outcomes in this particularly recalcitrant cancer (Cocco et al., 2019; O’Reilly and Hechtman, 2019; Pishvaian et al., 2018). This may also be true in other cancers since analysis of The Cancer Genome Atlas (TCGA) data suggests that ~0.5% of all human malignancies may be driven by NTRK oncogenes (Okamura et al., 2018; Vaishnavi et al., 2015).
Entrectinib (RXDX-101) is a potent, CNS-penetrant, ATP-competitive tyrosine kinase inhibitor of the TRKA, TRKB, TRKC, anaplastic lymphoma kinase (ALK), and c-ros oncogene 1kinase (ROS1) RTKs (Drilon et al., 2017b). Following clinical trials, the US Food and Drug Administration (FDA) granted entrectinib approval for use in the treatment of ROS1-mutated non-small cell lung cancer (NSCLC) and for agnostic use in NTRK1-driven solid tumors (Drilon et al., 2017b). However, as with most kinase inhibitors, the durability of patient responses is limited by the emergence of drug-resistant disease (Drilon et al., 2017a; Russo et al., 2016). Numerous mechanisms of resistance have been identified in NTRK1-driven cancers, making the identification and treatment of resistant disease after it has emerged challenging (Cocco et al., 2019; Drilon et al., 2017a; Fuse et al., 2017; Okimoto and Bivona, 2016; Russo et al., 2016; Vaishnavi et al., 2015, 2017). Consequently, there is a need to improve our knowledge of how to prevent the emergence of entrectinib resistance in solid tumors driven by TRKA fusions. Here, we describe the development of two new preclinical models of NTRK1-driven solid tumors: the first being pancreatic cancer in which the Translocated Promoter Region-Neurotrophic Receptor Tyrosine Kinase 1 (TPR-NTRK1) oncogene is expressed in conditionally immortalized mouse pancreatic ductal epithelial (IMPE) cells, and the second being a TPR-NTRK1-driven model of lung cancer by oncogene expression in mouse lung epithelial 12 (MLE-12) cells. Both of these models form rapidly growing tumors in mice and are exquisitely sensitive to entrectinib. Moreover, the emergence of drug resistance limits the long-term effectiveness of entrectinib in both models. Importantly, by dissecting the mechanisms by which entrectinib inhibits tumorigenesis, we demonstrate that the addition of a MEK1/2 inhibitor forestalls the emergence of drug resistance in both models. Additionally, this combination approach was superior to single-agent entrectinib in two additional human cancer cell models. Consequently, these data provide an empirical foundation for the clinical deployment of new combination therapy approaches for the treatment of NTRK1-driven malignancies (Cocco et al., 2019).
RESULTS
A TPR-NTRK1-Positive Pancreatic Cancer Is Sensitive to Entrectinib
As described previously (Pishvaian et al., 2018), a 47-year-old man presented with stage IIA (T3N0M0) pancreatic cancer at diagnosis. The patient received neo-adjuvant nanoparticle albumin-bound-paclitaxel and gemcitabine followed by chemoradiation with capecitabine. A restaging computed tomography (CT) scan showed disease progression in the liver. Diagnostic testing of the patient’s primary tumor by next-generation sequencing (FoundationOne) identified a TPR-NTRK1 fusion oncogene. Consequently, the patient was enrolled in the STARTRK-2 clinical trial (NCT02568267) for the TRKA-targeted inhibitor entrectinib at Huntsman Cancer Institute. After 6 months of entrectinib treatment, the primary pancreatic lesion had shrunk by ~38%, as determined by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 criteria (Figures 1A and 1B). In addition, the patient’s carbohydrate antigen 19–9 (CA19–9) level decreased by 94% (Pishvaian et al., 2018). After 8 months of treatment, imaging revealed disease progression at both the primary site and in the liver, and the patient discontinued entrectinib therapy (Pishvaian et al., 2018).
Figure 1. A TPR-NTRK1 Fusion Found in a Pancreatic Cancer Patient Transforms Immortalized Mouse Pancreatic Epithelial (IMPE) Cells In Vitro.
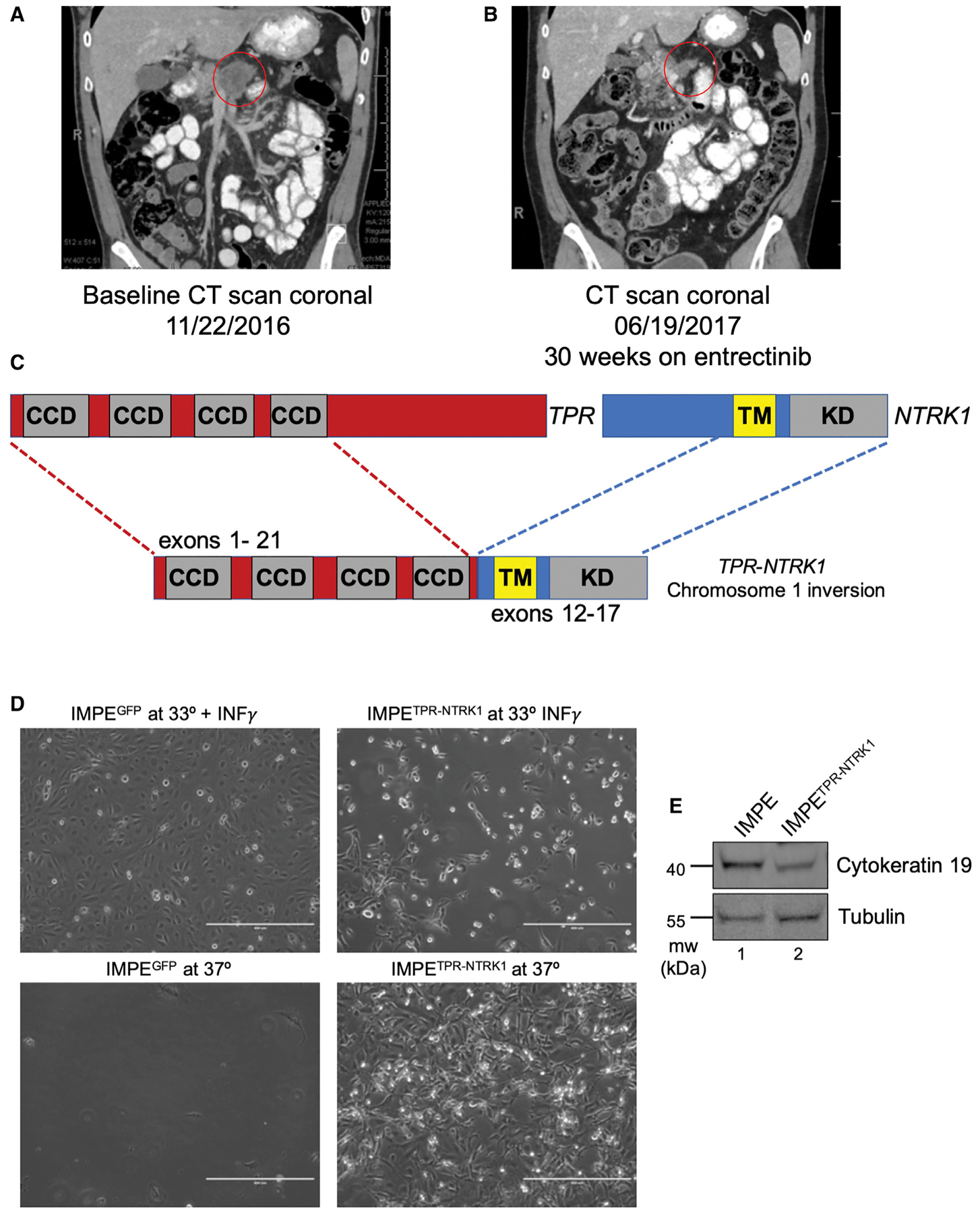
(A) Coronal CT scans of the patient’s tumor at baseline prior to the beginning of treatment with entrectinib. Primary pancreatic adenocarcinoma lesion is circled in red.
(B) Coronal CT scans of the patient’s tumor following prolonged entrectinib treatment, leading to a partial response by RECIST criteria by treatment cycle 7. Residual primary pancreatic tumor is circled in red.
(C) Schematic of the breakpoints of the TPR-NTRK1 fusion oncogene identified through Foundation Medicine analysis. Exons 1–21 of the TPR gene were juxtaposed inframe to exons 12–17 of the NTRK1 gene.
(D) Phase contrast images (10x) of IMPE cells grown under the indicated cell culture conditions. Scale bars indicate 400 μm.
(E) Immunoblot analysis of IMPE and IMPETPR-NTRK1 cell lysates probed for the pancreatic lineage marker cytokeratin (CK)-19 antibody or for tubulin.
Establishing a Pancreatic Epithelial Cell Model of Transformation
The patient’s pancreatic tumor expressed a TPR-NTRK1 fusion oncogene that encompassed exons 1–21 of the TPR gene fused in-frame to exons 12–19 of NTRK1 generated by virtue of an inversion of chromosome 1 (Figure 1C) similar to a fusion oncoprotein identified in thyroid cancer (Greco et al., 1997). Expression of TPR-NTRK1 indicated that the TPR-TRKA fusion protein is a constitutively activated oncoprotein kinase that is autophos-phorylated on tyrosine, leading to activation of critical downstream signaling pathways (Figure S1A). Consistent with this, expression of TPR-NTRK1 in the interleukin-3 (IL-3)-dependent mouse pro-B cell line Ba/F3 rendered the cells capable of cytokine-independent growth (Figure S1B). To establish a model of TPR-TRKA-driven pancreatic cancer, TPR-NTRK1 (or a GFP control) was expressed in conditionally immortalized IMPE cells. IMPE cells were established from the “immortomouse” in which expression of a temperature-sensitive (tsA58) form of a simian virus 40 (SV40) large T antigen (Tag) is controlled by the interferon-γ (IFNγ)-regulated H-2Kb promoter (Jat et al., 1991). Consequently, IMPE cells proliferate in vitro in the presence of exogenously added IFNγ at 33°C under conditions where SV40 large tag (LT) inhibits the function of both TP53 and the RB family of tumor suppressors. Indeed, when IMPE cells are cultured at 37°C in the absence of IFNγ, they undergo growth arrest and then cell death (Koizumi et al., 2004). Strikingly, ectopic expression of TPR-NTRK1, but not GFP, in IMPE cells resulted in immediate morphological changes and rapid cell growth at both 33°C and 37°C Figure 1D). Moreover, these TPR-NTRK1-transformed IMPE cells maintained pancreatic identity, evidenced by expression ofthe pancreatic epithelial marker cytokeratin-19 (Figure 1E) (Bouwens, 1998). Interestingly, TPR-NTRK1 expression drove transformation of IMPE cells even at the non-permissive temperature for Large TtsA58 without additional genetic alterations. Surprisingly, mutationally activated epidermal growth factor receptor (EGFR)L858R, which is frequently detected in lung cancer and is also capable of converting Ba/F3 cells to cytokine-independent proliferation, was unable to transform IMPE cells (Figure S2) (Jiang et al., 2005). Together, these data indicate that TPR-NTRK1 is a driver of oncogenic transformation of both mouse and human pancreatic cells.
IMPETPR-NTRK1 Cells Are Sensitive to Entrectinib Both In Vitro and In Vivo
Having established IMPETPR-NTRK1 cells, we determined their tumorigenic potential as well as their response to entrectinib both in vitro and in vivo. As expected, both TPR-NTRK1-expressing IMPE and Ba/F3 cells displayed activation of the RAS-regulated RAF>MEK>ERK mitogen-activated protein (MAP) kinase signaling pathway (Figure 2A; Figures S1C and S1D). Treatment of these cells with escalating concentrations of entrectinib elicited a dose-dependent decrease of both phospho-TRKA (pTRKA) and also in the magnitude of RAF>MEK>ERK signaling (Figure 2A; Figure S1C). This response was specific to treatment of IMPETPR-NTRK1 cells with entrectinib and was not observed in parental IMPE cells or if the IMPETPR-NTRK1 cells were treated with gefitinib, an EGFR inhibitor (Figure 2A; Figure S1D). We failed to detect TPR-TRKA-mediated activation of the phosphatidylinositol-3 kinase>AKT pathway in IMPETPR-NTRK1 cells, nor was pAKT altered following entrectinib treatment (data not shown). Moreover, both IMPETPR-NTRK1 and Ba/F3TPR-NTRK1 cells displayed sensitivity to the anti-proliferative effects of entrectinib (Figure 2B; Figure S1E). Finally, the proliferation of parental IMPE or Ba/F3 cells was insensitive to entrectinib or gefitinib, demonstrating the expression of the TPR-TRKA oncoprotein is specifically what renders these cells entrectinib sensitive (Figure 2B; Figure S1F). These data indicate that TPR-TRKA signals through the RAF>MEK>ERK pathway in IMPE cells and that the IMPETPR-NTRK1 cells are sensitive to the anti-proliferative effects of entrectinib.
Figure 2. IMPETPR-NTRK1 Cells Are Sensitive to Entrectinib In Vitro and as Tumors In Vivo, but Drug-Resistant Tumors Eventually Emerge.

(A) Immunoblot analysis of lysates of IMPETPR-NTRK1 cells treated for 2 h with DMSO (vehicle control) or the indicated doses of gefitinib orentrectinib and probed for the abundance of the proteins indicated.
(B) Dose-response curve of IMPETPR-NTRK1 cell proliferation following treatment with DMSO, entrectinib, or gefitinib. Calculated half-maximal inhibitory concentration (IC50) values are listed in nM. Error bars indicate the standard error of the mean (SEM).
(C) Quantification of subcutaneous tumor formation in non-obese diabetic-severe combined immunodeficiency (NOD-SCID) mice implanted with either 106 IMPE cells engineered using lentiviral gene transduction to express either GFP (IMPEGFP) or TPR-NTRK1 (IMPETPR-NTRK1) measured over 2–8 weeks as indicated.
(D) Quantification of tumor volume in a subcutaneous model of IMPETPR-NTRK1 tumorigenesis in response to once-daily (q.d.) or twice-daily (b.i.d.) treatment with 60 mg/kg entrectinib. White arrow on x axis and dark black arrow in graph indicate day when drug treatment was started and finished, respectively. Error bars indicate the SEM. Statistical analysis was performed using a one-way ANOVA, where **p < 0.01. n = 40 mice.
(E) Bioluminescent images of IMPETPR-NTRK1 cells engineered to express firefly luciferase and then orthotopically implanted into the pancreata of immune-compromised mice (n = 18). Pancreatic tumor burden was imaged 7 days after cell implantation and then again 7 days following treatment with either vehicle control or entrectinib (60 mg/kg, q.d.) as indicated.
(F) Quantification of luminoscore of bioluminescent images using LivingImage software of “luciferized” IMPETPR-NTRK1 cell-derived pancreatic lesions either from vehicle- or drug-treated groups over time. Entrectinib-treated mice were dosed every day until entrectinib resistance emerged. Error bars indicate the SEM. Statistical analysis was performed using a two-tailed t test, where ****p < 0.0001.
(G) Kaplan-Meier survival curve tracking survival of mice treated as described in (C) and (D). Statistical analysis was performed using a log rank Mantel-Cox test, where ****p < 0.0001.
To determine the tumorigenic potential of IMPETPR-NTRK1, they were engineered to express a firefly luciferase reporter and then injected either subcutaneously or orthotopically into the pancreata of immunocompromised mice. Whereas parental IMPE cells failed to form tumors in mice, IMPETPR-NTRK1 cells formed rapidly growing tumors both subcutaneously and orthotopically (Figures 2C–2F). In both models, treatment of mice bearing IMPETPR-NTRK1-derived tumors displayed rapid, dramatic responses to entrectinib even when using different clinically relevant dosing regimens (Figures 2D–2F). In the subcutaneous model, we consistently observed rapid and complete shrinkage of even large tumors (≥500 mm3) within 4–5 days (Figure 2D). Consistent with observations in humans, IMPETPR-NTRK1 tumor-bearing mice were not cured, as cessation of drug treatment resulted in tumor re-growth within 1 week (Figure 2D, see days 14–26). Similarly, in the orthotopic pancreatic cancer model, the luciferase signal in entrectinib-treated mice was significantly reduced within 7 days (two-tailed t test, p < 0.0001) (Figure 2F). Moreover, entrectinib treatment significantly prolonged survival compared with vehicle treatment by ≥70 days (log rank Mantel-Cox test, p < 0.0001) (Figure 2G). Collectively, these data indicate that this model of TPR-NTRK1-driven pancreatic cancer is exquisitely sensitive to targeted inhibition of TRKA kinase activity.
The Pro-apoptotic BCL2 Family Protein BIM Mediates Sensitivity of IMPETPR-NTRK1 Cells to Targeted TRKA Inhibition
Intrigued by the dramatic tumor regression observed in this model of pancreatic cancer, we set out to elucidate the molecular basis of the response to entrectinib. Twenty-four hours following entrectinib addition, we noted readily detectable, dose-dependent expression of the longest isoform of BIM (BIM-EL), a pro-apoptotic member of the BCL2 family (Figure 3A). Since there is prior evidence indicating the importance of BIM induction in cancer cell response to targeted therapy (Cartlidge et al., 2008; Deng et al., 2007; Gong et al., 2007; Ng et al., 2012), we evaluated the kinetics of BIM induction and the expression of other known markers or mediators of apoptosis following entrectinib treatment. Indeed, 12 h after drug addition, BIM expression had reached peak levels, correlating with proteolysis of poly(ADP-ribose) polymerase (PARP) (Figure 3B). However, we noted that at 18 or 24 h after drug addition, there was clear evidence of reactivation of RAF>MEK>ERK signaling as evidenced by increased pERK (Figure 3B), suggesting that single-agent TRKA inhibition was insufficient to maintain durable inhibition of this pathway. Importantly, pTRKA remained completely inhibited at 18 and 24 h, indicating that entrectinib was still effectively inhibiting its direct target (Figure 3B).
Figure 3. BIM Mediates the Sensitivity of IMPETPR-NTRK1 Cell-Derived Pancreatic Tumors to Entrectinib.

(A) Immunoblot analysis of BIM or tubulin expression in lysates of IMPETPR-NTRK1 cells treated with DMSO control (0) or with 1 or 100 nM entrectinib for 24 h.
(B) Immunoblot analysis of protein expression or modification in lysates of IMPETPR-NTRK1 cells treated with 10 nM entrectinib for 0–24 h.
(C) Immunoblot analysis of lysates of BIM-proficient (sgNT) or BIM-deficient (BIMNull) IMPETPR-NTRK1 that were isolated from cells either untreated or treated with 10 nM or 1 μM entrectinib for 24 h as indicated.
(D) Quantification of tumor volume changes in sgNT or BIMNull IMPETPR-NTRK1 cell-derived tumors that developed subcutaneously in immunocompromised mice in response to vehicle or 30 mg/kg entrectinib daily. Error bars represent the SEM. White arrow at the x axis and black solid arrow at the top of the graph indicate the start of drug treatment. Statistical analysis was performed using a one-way ANOVA, where ****p < 0.0001. n = 25 mice.
(E) Dose-response curve of the proliferation of sgNT or BIMNull IMPETPR-NTRK1 cells following treatment with a dose range of entrectinib alone, entrectinib with a fixed dose of 250 nM cobimetinib, or entrectinib with a fixed dose of 1 μM gefitinib, compared with vehicle control. Calculated IC50 values are listed. Error bars represent the SEM.
Based on these data, and previously published work, we hypothesized that BIM may be necessary for the response of IMPETPR-NTRK1-derived tumors to entrectinib. To test this, we used CRISPR-Cas9-mediated genome editing to silence BIM expression in IMPETPR-NTRK1 cells. A clonal population of IMPETPR-NTRK1 cells that do not express BIM (BIMNull) was identified and confirmed by immunoblotting (Figure 3C). To determine the effects of BIM silencing on the sensitivity of IMPETPR-NTRK1-derived tumors to entrectinib in vivo, both control (sgNT) and BIMNull cells were implanted subcutaneously into immunocompromised mice and treated daily once the tumor volume was ≥250 mm3 (Figure 3D). As expected, the sgNT IMPETPR-NTRK1 tumors responded to entrectinib, showing a complete tumor response within 7 days of treatment (Figure 3D). By contrast, entrectinib treatment of BIMNull IMPETPR-NTRK1 tumors resulted in disease stabilization for ~4 days, followed by tumor growth thereafter (Figure 3D). These experiments revealed that BIMNull IMPETPR-NTRK1 tumors were significantly less sensitive to entrectinib than the relevant controls (ANOVA, p < 0.0001) and provide strong evidence that induced BIM expression is required for the response of IMPETPR-NTRK1 tumors to entrectinib (Figure 3D).
To determine what was promoting the proliferation of the BIMNull IMPETPR-NTRK1 cells in the presence of entrectinib, we performed a mouse RTK phospho-array, to identify whether a new RTK was activated in the presence of entrectinib and the absence of BIM (Figure S3). The phospho-array revealed several phosphorylation sites that were elevated, including sites on EGFR, HER2, MET, AXL, and EPH-A2 (Figure S3C). We further validated this finding of BIMNull drug resistance through in vitro proliferation of the BIMNull cells, which displayed an ~15-fold increase in the half-maximal inhibitory concentration (IC50) to entrectinib (Figure 3E). Importantly, we were able to validate the importance of EGFR activation by combining entrectinib with the EGFR inhibitor gefitinib that restored the IMPETPR-NTRK1 cells’ proliferative sensitivity back to below the baseline of the BIM-proficient IMPETPR-NTRK1 cells; reducing the IC50 for proliferation by 37-fold down to 83 nM (Figure 3E). These data suggest that, in the absence of BIM, EGFR is important for the proliferation of IMPETPR-NTRK1 cells. These data further suggest that entrectinib-induced BIM expression is essential for the anti-tumor effects of entrectinib in IMPETPR-NTRK1-derived tumors.
Combined Inhibition of Both TPR-TRKA plus MEK1 Forestalls the Onset of Acquired Drug Resistance in IMPETPR-NTRK1-Driven Pancreatic Cells
Mice bearing orthotopic pancreatic IMPETPR-NTRK1 tumors, which had responded to single-agent entrectinib (Figures 2D–2F), were dosed continuously until the emergence of entrectinib-resistant NTRK1+ IMPE (ER-NI) tumors ~78 days post-initiation of drug treatment. As drug-resistant tumors emerged, mice were euthanized and ER-NI cell lines were generated for further analysis. As expected, all six ER-NI cell lines retained their resistance to the drug in vitro, indicating that the property of drug resistance was largely tumor cell autonomous (Figure S4A). Immunoblotting of ER-NI lysates revealed a trend of higher baseline levels of pERK1/2 compared with that of the parental cells (Figure S4B, compare lanes 2–6 with lane 1). Despite substantial efforts in the analysis of these ER-NI cells (e.g., whole-exome next-generation sequencing and extensive pharmacologic testing), we were unable to identify the mechanism(s) of entrectinib resistance (data not shown). Regardless, we hypothesized that delaying entrectinib resistance with a combination drug regimen would be a superior strategy compared with trying to treat entrectinib resistance after it emerges. Hence, based on the perceived importance of RAF>MEK>ERK signaling downstream of TPR-TRKA, and our previous observation that entrectinib-treated IMPETPR-NTRK1 cells displayed a rebound reactivation of RAF>MEK>ERK signaling 18–24 h after drug addition (Figure 3B), we hypothesized that entrectinib alone would be insufficient to maintain durable inhibition of RAF>MEK>ERK signaling in IMPETPR-NTRK1 cells. Thus, in an effort to forestall the emergence of drug resistance, we reasoned that combined pathway-targeted vertical inhibition of both TPR-TRKA plus RAF>MEK>ERK signaling through the combination of entrectinib plus the MEK1/2 inhibitor cobimetinib would increase the durability of the anti-tumor response in this model. To that end, we treated IMPETPR-NTRK1 cells for either 2 or 24 h with either entrectinib alone, cobimetinib alone, or the combination of both drugs and evaluated effects on downstream signaling by immunoblotting. Treatment of IMPETPR-NTRK1 cells either with entrectinib or cobimetinib alone effectively inhibited pERK at 2 h, but reactivation of pERK was readily detected at 24 h following either single-agent treatment (Figure 4A). By contrast, combined treatment of IMPETPR-NTRK1 cells with both entrectinib plus cobimetinib (E+C) gave profound inhibition of pERK1/2 at both 2- and 24-h time points (Figure 4A, compare lanes 2 and 3 with lanes 6 and 7). Moreover, consistent with the effects on pERK1/2, the E+C combination gave a more rapid and sustained elevation in BIM expression (Figure 4A) compared with the single agents. These data suggest that the E+C combination prevents the reactivation of pERK1/2 signaling observed with entrectinib alone and that by more durably suppressing RAF>MEK>ERK signaling may be more effective than TRKA monotherapy.
Figure 4. Combined Inhibition of TPR-TRKA Plus MEK1/2 Forestalls the Onset of Drug Resistance in IMPETPR-NTRK1 Cell-Derived Tumors.

(A) Immunoblot analysis of lysates of IMPETPR-NTRK1 treated with different combinations of DMSO, entrectinib (100 nM), cobimetinib (250 nM), or the two inhibitors combined at either 2 or 24 h post-drug treatment.
(B) Immunoblot analysis of lysates of IMPETPR-NTRK1 cells treated with different combinations of DMSO, entrectinib (100 nM), gefitinib (1,000 nM), or the two inhibitors combined at either 2 or 24 h after drug treatment.
(C) Dose-response curve of IMPETPR-NTRK1 cell proliferation following treatment with a dose range of entrectinib alone, entrectinib with a fixed dose of 250 nM cobimetinib, or entrectinib with a fixed dose of 1 μM gefitinib, compared with control. Calculated IC50 values for entrectinib in each treatment group are listed. Error bars indicate the SEM.
(D) Quantification of tumor burden in immunocompromised NOD-SCID mice implanted with IMPETPR-NTRK1 cells treated q.d. with vehicle (blue), 5 mg/kg cobimetinib (orange), 30 mg/kg entrectinib (gray), or the combination of cobimetinib plus entrectinib (yellow). Arrows indicate the initiation of drug treatments. Error bars indicate the SEM. Statistical analysis was performed using a one-way ANOVA, where ****p < 0.0001. n = 40 mice.
(E) Kaplan-Meier survival curve comparing the effects of each inhibitor used in (C) on survival of each arm using the same color scheme. Statistical analysis was performed using a log rank Mantel-Cox test, where ****p < 0.0001.
(F) Immunoblot analysis of lysates of IMPETPR-NTRK1 tumors established subcutaneously in NOD-SCID mice. Tumor-bearing mice were treated with one dose of (1) vehicle (corn oil, 0, lanes 1), (2) entrectinib (30 mg/kg), (3) entrectinib (30 mg/kg) plus cobimetinib (5 mg/kg), or (4) entrectinib (30 mg/kg) plus gefitinib (25 mg/kg) for the indicated times (6–48 h) prior to euthanizing the mice for preparation of cell lysates for immunoblot analysis with the indicated antisera. Two unique tumors were pooled for each lysate, and two unique lysates were tested for each drug treatment. The immunoblots shown are representative images of the tumors tested.
These data revealed that the combination of E+C more effectively inhibited RAF>MEK>ERK signaling, but we were curious to know what mediates reactivation of this pathway in the presence of single-agent entrectinib. Our hypothesis was that this may be mediated by EGFR activation, a mechanism previously identified as critical in other NTRK1-driven cancer models as well as in our BIMNull IMPETPR-NTRK1 cell model (Figure 3E) (Vaishnavi et al., 2017). To test this, we repeated the experiment described above (Figure 4A) but replaced cobimetinib with the EGFR inhibitor gefitinib. Strikingly, the combination of entrectinib plus geftinib (E+G) also prevented the reactivation of RAF>MEK>ERK signaling observed 24 h after single-agent entrectinib (Figure 4B, compare lane 7 with lane 3). Hence, these data suggest reactivation of RAF>MEK>ERK signaling at later times after entrectinib is mediated by EGFR. Moreover, the ability of entrectinib to inhibit the proliferation of IMPETPR-NTRK1 cells in vitro was greatly potentiated by the addition of a fixed dose of either cobimetinib or gefitinib (8- and 13-fold increased sensitivity, respectively; Figure 4C). Activation of EGFR signaling detected at 24 h reveals the molecular basis for why the IC50 (Figure 2A, <10 nM) for the inhibition of pTRKA did not correlate with the IC50 of inhibition of cell proliferation in response to entrectinib monotherapy on IMPETPR-NTRK1 cells (Figure 2B, 202 nM). Taken together, these data provide evidence that inhibition of TRKA in IMPETPR-NTRK1 cells leads to an initial blockade of RAF>MEK>ERK signaling that then rebounds due to activation by EGFR.
To determine whether the E+C drug combination provided more durable anti-tumor control in vivo, mice bearing IMPETPR-NTRK1-driven tumors were treated once daily with either entrectinib, cobimetinib, or the combination of both agents (Figure 4D). IMPETPR-NTRK1-driven tumors were largely resistant to treatment with cobimetinib alone. As described above, IMPETPR-NTRK1-driven tumors were exquisitely sensitive to the anti-tumor effects of entrectinib initially, but all mice developed drug-resistant tumors by ~35 days of drug treatment (Figure 4D). Strikingly, the E+C combination significantly forestalled the emergence of drug resistance in this model (ANOVA, p < 0.0001) such that all E+C-treated mice remained in response after 45 days of treatment, at which time all other groups of mice had been euthanized due to tumor burden (Figures 4D and 4E). Consistent with this, the E+C combination also significantly prolonged survival compared with entrectinib alone (log rank Mantel-Cox test, p < 0.0001), where 100% of E+C-treated mice were alive at the time that 100% of entrectinib-treated mice had been euthanized (Figure 4E). These data validate our in vitro findings and reveal that preventing reactivation of RAF>MEK>ERK signaling using a MEK1/2 inhibitor greatly potentiates the durability of responses to entrectinib.
At this point, a critical question remained: to what extent does EGFR signaling promote reactivation of RAF>MEK>ERK signaling in entrectinib-treated IMPETPR-NTRK1 tumors in vivo? To address this, IMPETPR-NTRK1 -derived tumors were established, and mice were treated as follows: (1) a single dose of entrectinib (30 mg/kg, E); (2) combination of E+C (30 mg/kg + 5 mg/kg, E+C); or (3) combination of E+G (30 mg/kg + 25 mg/kg, E+G) for 6, 12, 24, or 48 h at which time tumor lysates were prepared for immunoblot analysis. This analysis revealed that single-agent entrectinib inhibited pTRKA for ≥24 h, with reactivation of the TPR-TRKA kinase detected at 48 h. However, despite ongoing inhibition of pTRKA, RAF>MEK>ERK signaling was fully reactivated by 24 h after addition of entrectinib (Figure 4F). Although the addition of either cobimetinib (Figure 4F) or geftinib (Figure 4F) had no effect on the kinetics of decrease or re-accumulation of pTRKA, both agents provided more durable inhibition of RAF>MEK>ERK signaling (≥48 h) and more robust expression of BIM compared with single-agent entrectinib. These results reinforce the molecular basis for why combination therapy is superior to single-agent entrectinib because of the more durable inhibition of RAF>MEK>ERK signaling achieved.
BIM expression can be regulated transcriptionally as well as post-translationally by ERK1/2-mediated phosphorylation of serine 69 that, in turn, elicits β-transducin SV40 repeat-containing protein (β-TRCP)-mediated ubiquitination of BIM, leading to its proteasomal degradation (Cartlidge et al., 2008; Dehan et al., 2009; Luciano et al., 2003; Sunters et al., 2003). To understand the effects of the various drug treatments on BIM expression, IMPETPR-NTRK1 cells were treated with entrectinib or the E+C or E+G drug combinations with a period of drug removal from 1 to 6 h, with BIM phosphorylation or expression monitored by immunoblotting. Following single-agent entrectinib, we observed the pERK rebound signal at 24 h, and persistent levels of pS69-BIM, paired with transient expression of total BIM (Figure S5A). Both the E+C and E+G combination therapies revealed no restoration of pBIM and a more stable induction of BIM following drug removal (Figures S5B and S5C). These data provide additional evidence that the E+C or E+G drug combinations are superior in inducing higher, more stable levels of BIM expression than monotherapy and are consistent with a superior anti-tumor response.
Establishing a Model of TPR-NTRK-Driven Lung Cancer
Since NTRK1 gene fusions are observed in lung cancer, we generated TPR-NTRK1-expressing MLE-12 cells (Vaishnavi et al., 2013). MLE-12 cells are a weakly tumorigenic cell line derived from a pulmonary tumor in mice with Large T expression under the control of the surfactant protein C (SPC) promoter (Wikenheiseret al., 1992). When re-implanted into immunocompromised mice, MLE-12 cells generate tumors over 6–9 months (Wikenheiser et al., 1992). MLE-12 cells were engineered to stably express TPR-TRKA (MLE-12TPR-NTRK1) or GFP control (MLE-12GFP) in a manner similar to that described for IMPE cells. Phase contrast images of MLE-12TPR-NTRK1 or MLE-12GFP cells revealed no distinct morphological differences, except a modest increase in the refractory appearance of MLE-12TPR-NTRK1 compared with control (Figure 5A). Subcutaneous implantation of MLE-12TPR-NTRK1 cells into immunocompromised mice resulted in rapid tumorigenesis in 19 of 20 mice. By contrast, 0 of 10 mice injected with MLE-12EGFP cells formed tumors by 8 weeks post-implantation (Figure 5B). To determine the effects of entrectinib, cobimetinib, or gefitinib, either alone or in combination, MLE-12TPR-NTRK1 tumor-bearing mice were treated with these agents as described for IMPETPR-NTRK1 cells (Figure 4). Interestingly, this new TPR-NTRK1-driven lung cancer model revealed similar trends as the IMPE pancreatic cancer model. Treatment with entrectinib led to inhibition of pTRKA at 2 and 24 h, but RAF>MEK>ERK signaling was only inhibited at 2 h and was fully reactivated at 24 h (Figure 5C). Cobimetinib inhibited RAF>MEK>ERK signaling at 2 h, but pathway activity was restored by 24 h. However, combined treatment with E+C suppressed RAF>MEK>ERK signaling at both 2 and 24 h. Moreover, the most robust induction of BIM-EL expression was observed with the E+C combination (Figure 5C, E+C). The rebound of RAF>MEK>ERK signaling observed in MLE-12TPR-NTRK1 cells after 24 h of entrectinib treatment was mediated by EGFR, as discerned from the combined effect of E+G, which suppressed RAF>MEK>ERK pathway reactivation at 24 h (Figure 5D). Similarly, the most robust induction of BIM expression was observed with the E+G combination. Consistent with these findings, while the MLE-12TPR-NTRK1 cells are more sensitive to entrectinib monotherapy than the IMPETPR-NTRK1 cells, the addition of a low fixed dose of gefitinib, but not cobimetinib, resulted in a strong shift in sensitivity to inhibition of proliferation in vitro (Figure 5E).
Figure 5. Generation of a MLE-12 Cell Line Model of Lung Cancer Driven by the TPR-NTRK1 Fusion Kinase.
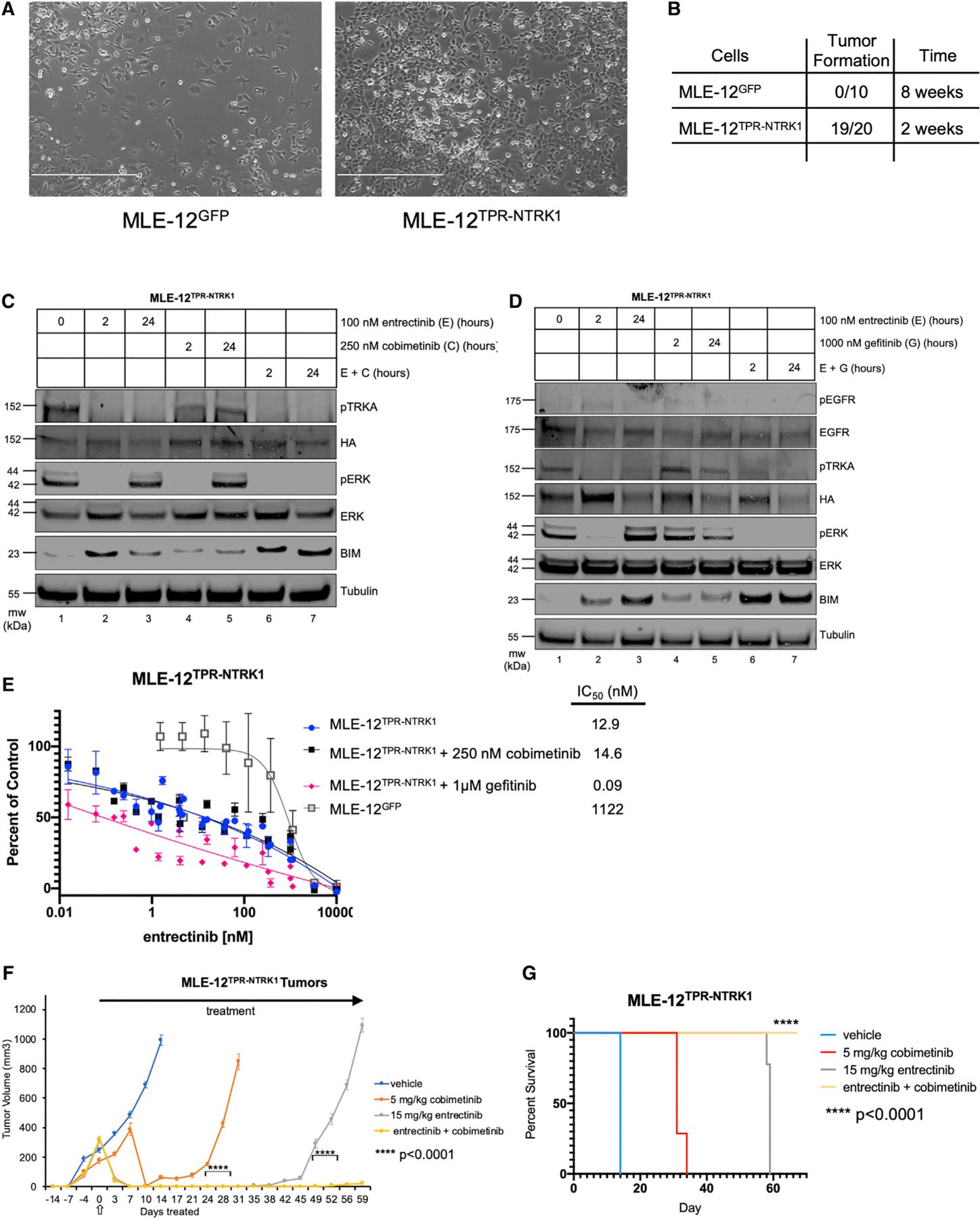
(A) Phase contrast images (10x) of MLE-12TPR-NTRK1 or control MLE-12GFP cells grown under standard tissue culture conditions. Scale bars represent 400 μm.
(B) Quantification of subcutaneous tumor formation in NOD-SCID mice implanted with either 106 MLE-12TPR-NTRK1 or control MLE-12GFP cells assessed at either 2 or 8 weeks post-cell implantation.
(C) Immunoblot analysis of lysates of MLE-12TPR-NTRK1 cells treated with DMSO (0), entrectinib (100 nM), cobimetinib (250 nM), or entrectinib plus cobimetinib for either 2 or 24 h as indicated. Blots were probed with the indicated antibodies to assess the effects of the various treatments as indicated.
(D) Immunoblot analysis of lysates of MLE-12TPR-NTRK1 treated with DMSO, entrectinib (100 nM), gefitinib (1 μM), or the combination of entrectinib plus gefitinib for either 2 or 24 h as indicated. Blots were probed with the indicated antibodies to assess the effects of the various treatments as indicated.
(E) Curves of dose response of MLE-12TPR-NTRK1 or MLE-12GFP cells to treatment with a dose range of entrectinib either alone or in combination with a fixed dose of cobimetinib (250 nM) or a fixed dose of gefitinib (1 μM). Calculated IC50 values are listed. Error bars represent the SEM.
(F) Quantification of tumor burden overtime in immunocompromised NOD-SCID mice implanted with MLE-12TPR-NTRK1 cells treated q.d. with vehicle (blue), 5 mg/kg cobimetinib (orange), 30 mg/kg entrectinib (gray), or the combination of cobimetinib plus entrectinib (yellow). Arrows indicate the initiation and duration of drug treatments. Error bars represent the SEM. Statistical analysis was performed using a one-way ANOVA, where ****p < 0.0001. n = 40 mice.
(G) Kaplan-Meier survival curve comparing the effects of each inhibitor used in (F) on survival of each arm using the same color scheme. Statistical analysis was performed using a log rank Mantel-Cox test, where ****p < 0.0001.
Based on these results, we tested the effects of the E+C combination on the response of MLE-12TPR-NTRK1 tumors in immunocompromised mice. MLE-12TPR-NTRK1 tumor-bearing mice were randomized into four treatment arms (10 mice per arm) to receive once-daily dosing of vehicle, cobimetinib, entrectinib, or the combination of E+C. As with the IMPETPR-NTRK1 cells, single-agent entrectinib promoted striking tumor regression, but drug-resistant tumors started to appear ~49 days post-initiation of therapy (Figure 5F). In contrast to IMPETPR-NTRK1 tumors, MLE-12TPR-NTRK1 tumors were sensitive to single-agent cobimetinib, but drug-resistant tumors started to appear ~28 days post-initiation of therapy. The E+C combination did not accelerate the initial reduction in tumor size observed with entrectinib alone but dramatically enhanced the durability of response compared with either of the single agents (ANOVA, p < 0.0001) (Figure 5F). Overall, the E+C-treated mice displayed a striking and significant survival advantage compared with all of the other treatment groups (log rank Mantel-Cox test, p < 0.0001) (Figures 5F and 5G). Hence, these data from a model of TPR-NTRK1-driven lung cancer are consistent with the data from our model of TPR-NTRK1-driven pancreatic cancer and firmly reinforce the ability of cobimetinib to forestall the onset of resistance to entrectinib in both models.
As a final test of these observations, we used two human cancer cell lines that express a mutationally activated NTRK1 fusion kinase. CUTO-3.29 cells were derived from a human lung cancer cell line isolated from the index lung cancer patient that harbored an MPRIP-NTRK1 fusion (Vaishnavi et al., 2013). KM12 cells are a colorectal cancer-derived cell line that expresses a TPM3-NTRK1 fusion (Vaishnavi et al., 2013, 2017). Although previous studies have identified an important role for EGFR signaling in these cell lines, the studies have not tested the potential utility of vertical inhibition of TRKA plus MEK1/2 inhibition to forestall drug resistance. To that end, CUTO-3.29 or KM12 cells were treated with (1) entrectinib, (2) cobimetinib, (3) combination E+C, or (4) combination E+G for 2 or 24 h at which time cell lysates were analyzed by immunoblotting. As observed in IMPETPR-NTRK1 and MLE-12TPR-NTRK1 cells, treatment with entrectinib inhibited RAF>MEK>ERK signaling (pERK) at 2 h, but the pathway was reactivated by 24 h post-drug addition, despite the fact that pTRKA remained inhibited (compare lanes 1–3 in Figures 6A and 6B). However, addition of either cobimetinib or gefitinib prevented reactivation RAF>MEK>ERK signaling at 24 h post-drug addition (compare lanes 3 and 7 in Figures 6A and 6B). Moreover, drug-induced expression of BIM was most robust with the E+C or E+G combinations.
Figure 6. Combined Inhibition of MEK or EGFR Is Superior to TRKA Monotherapy in Patient-Derived Lung and Colorectal Cancer Models Driven by an NTRK1 Fusion.
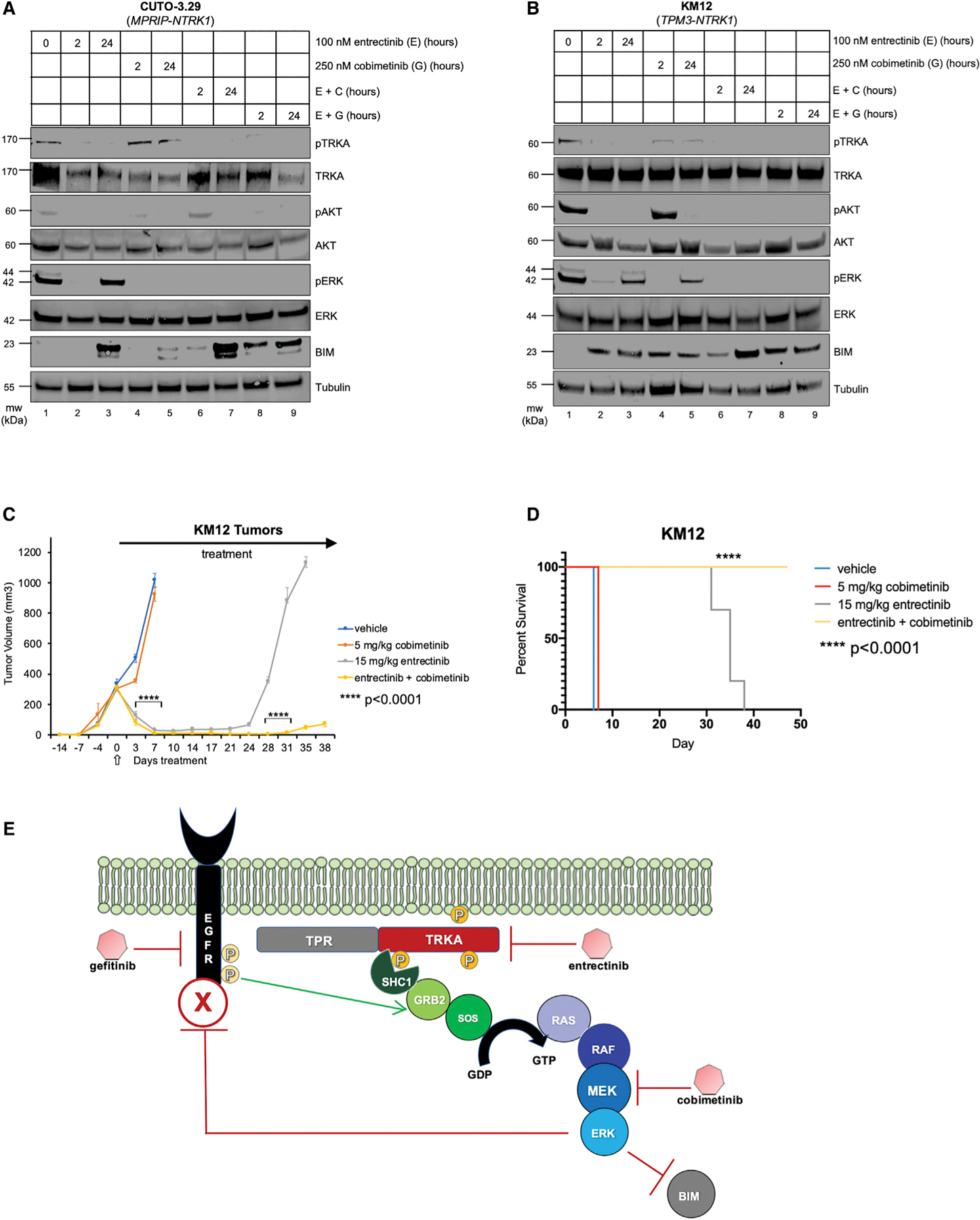
(A) Immunoblot analysis of MPRIP-TRKA-expressing CUTO-3.29 lung cancer lysates treated with DMSO, entrectinib (100 nM), cobimetinib (250 nM), gefitinib (1,000 nM), or the two inhibitors combined over 2- or 24-h time points.(B) Immunoblot analysis of TPM3-TRKA+ KM12 colorectal cancer lysates treated with DMSO, entrectinib (100 nM), cobimetinib (250 nM), gefitinib (1,000 nM), or the two inhibitors combined over short (2-h) or long (24-h) time points.
(C) Quantification of tumor volume in immunocompromised NOD-SCID mice implanted with KM12 cells treated q.d. with vehicle, 5 mg/kg cobimetinib, 15 mg/kg entrectinib, or the combination of cobimetinib plus entrectinib. Arrows at the x axis or in the graph indicate the start of drug treatments. Error bars indicate the SEM. Statistical analysis was performed using a one-way ANOVA, where ****p < 0.0001. n = 40 mice.
(D) Kaplan-Meier survival curve comparing the effects of each inhibitor used in (C) on survival of each arm. Statistical analysis was performed using a log rank Mantel-Cox test, where ****p < 0.0001.
(E) Schematic of the proposed model of the response of NTRK1-mutated tumors to entrectinib. Prior to manipulation, the TRKA fusion oncoprotein kinase signals through the RAF>MEK>ERK pathway, leading to enhanced transit through the cell division cycle, suppression of apoptosis, but also feedback inhibition of other RTKs including EGFR. Entrectinib-mediated inhibition of TRKA activity results in reduced feedback inhibition, leading to increased signaling by EGFR that is capable of sustaining RAF>MEK>ERK signaling in the cells. Consequently, the addition of an inhibitor of either RAF>MEK>ERK signaling (cobimetinib) or of EGFR (gefitinib) prevents (re)activation of RAF>MEK>ERK signaling, leading to enhanced suppression of the pathway and more robust induction of BIM. Consequently, the combination of entrectinib plus cobimetinib forestalls the onset of drug resistance in multiple preclinical models of NTRK1-driven cancer.
To determine whether the E+C combination would provide more durable tumor control, KM12 cells were injected into immunocompromised mice and when tumors were ~250 mm3, mice were treated once daily with vehicle, cobimetinib, entrectinib, or the combination of E+C as described above. Again, the E+C combination significantly delayed the onset of acquired resistance compared with entrectinib alone at 28 days post-initiation of drug treatment and also significantly extended the survival of KM12 tumor-bearing mice (ANOVA and log rank Mantel-Cox text, p < 0.0001) (Figures 6C and 6D). These data reinforce the findings made with the IMPETPR-NTRK1 and MLE-12TPR-NTRK1 cells and extend the potential utility of this approach to colorectal cancers driven by mutationally activated NTRK1+ fusion oncogenes.
DISCUSSION
Over the past 20 years, a deeper understanding of subsets of malignancies defined by predictive biomarkers of proto-oncogene activation, in combination with development of pharmacological inhibitors of oncoprotein kinases such as EGFR, BRAF, ALK, ROS1, and TRKA, has had a dramatic impact in the treatment of diseases such as melanoma and lung cancer (Doebele et al., 2015; Drilon et al., 2018; Kwak et al., 2010; Lynch et al., 2004; Shaw et al., 2014; Subbiah et al., 2020; Vaishnavi et al., 2013). Unfortunately, the emergence of lethal drug-resistant disease continues to reduce the durability of patient responses. In BRAF-mutated melanoma and lung cancer, vertical pathway inhibition with the combination of inhibitors of both BRAFV600E plus MEK1/2 has served to enhance the depth and durability of the initial response and has also diminished some of the toxicities associated with monotherapy (Subbiah et al., 2020). Here, we extend the vertical inhibition paradigm to cancers driven by NTRK1 fusion oncogenes, even in malignancies that are noted for their therapeutic recalcitrance such as pancreatic cancer (Cocco et al., 2019). Initiated by the response of a patient whose pancreatic tumor was driven by a TPR-NTRK1 fusion oncogene and who responded to entrectinib, we generated new models of pancreatic cancer or lung cancer driven by the TPR-TRKA oncoprotein kinase. It is noteworthy that the TPR-TRKA oncoprotein kinase was able to transform IMPE cells cultured at 37°C in the absence of IFNγ, conditions under which both the TP53 and the RB tumor suppressor pathways should be reactivated (Koizumi et al., 2004). This contrasts with the inability of KRASG12D or EGFRL858R to transform these cells under the same conditions (McMurray et al., 2008). Furthermore, we noted that EGFRL858R, frequently detected in lung cancer, was also insufficient to transform IMPE cells, despite the fact that it can readily convert Ba/F3 cells to IL-3-independent growth. The potency of TPR-TRKA to transform IMPE cells could be due to differences in signal pathway activation or could be due to differences in the magnitude of signal pathway activation (Marshall, 1995). Indeed, it has previously been shown that activation of RAF>MEK>ERK signaling at different levels can drive strikingly different biological outcomes, some of which are mediated by engagement of various tumor suppressors and/or regulators of the cell cycle or apoptosis (Collado et al., 2005; Marshall, 1995; Murphy et al., 2008; Sarkisian et al., 2007; Woods et al., 1997; Xu et al., 2013; Zhu et al., 1998). These observations may emphasize the importance of using relevant tissue-specific models of oncogenic transformation when studying the biochemistry and tumor biology of novel oncoproteins.
Previous research in melanoma, chronic myelogenous leukemia, and lung cancer has emphasized the importance of the regulation of BIM downstream of RAF>MEK>ERK signaling for the response of cancer cells to pathway-targeted therapy (Faber et al., 2011, 2012; Ng et al., 2012). Research reported here further extends this paradigm by the observation that BIM silencing leads to more rapid onset of entrectinib resistance in the impeTPR-NTRK1 model of pancreatic cancer. Our observations suggest that BIM induction is more rapid and robust with the vertical inhibition strategy compared with monotherapy. Our research also indicates a critical role for elevated signaling through EGFR as an adaptive response to entrectinib-mediated inhibition of TRKA-containing fusion oncoproteins that serves to promote tumor cell proliferation in the absence of BIM. Indeed, the importance of BIM in the therapeutic response of different cancers to pathway-targeted therapy might also be exploited by additional strategies such as the use of inhibitors of the anti-apoptotic BCL2 family proteins or of autophagic recycling pathways (Ewings et al., 2007; Hazar-Rethinam et al., 2018; Kinsey et al., 2019). Although mouse models are not always reliable indicators of drug safety and tolerability in humans, it is notable that no toxicity or adverse side effects were observed in tumor-bearing mice treated with the E+C combination. Moreover, while TRKA-directed monotherapy was highly effective for short-term anti-tumor responses, more durable tumor control was achieved using a combination of a TRKA and MEK1/2 inhibitor. These data are consistent with that of others who recently reported that mechanisms of acquired resistance to TRKA inhibitors often occur through reactivation of RAF>MEK>ERK signaling through point mutations in key members of the pathway (Cocco et al., 2019). Specifically, it was recently reported that a different combination of TRKA plus MEK1/2 inhibitors also enhanced the durability of the anti-tumor response following acquired resistance in patient-derived xenograft (PDX) models of lung cancer that had acquired resistance through mutational activation of the ERK1/2 pathway (Cocco et al., 2019). Our research is consistent with these observations and further supports them by adding an additional level of mechanistic understanding as to why this combination is superior (Figure 4F). Although impeTPR-NTRK1 tumors in mice gave rise to drug-resistant variants, the mechanism(s) of entrectinib resistance in this remains unknown but is likely to involve mechanisms that promote reactivation of RAF>MEK>ERK signaling, either alone or in combination with other signaling pathways.
Based on the mechanistic data presented here, we propose the following model (Figure 6E) to explain the rationale for combination therapies involving E+C or E+G. At baseline, the TPR-TRKA oncoprotein kinase signals constitutively through the RAS-regulated RAF>MEK>ERK MAP kinase pathway to (1) promote the cell division cycle; (2) suppress apoptosis; and (3) activate negative feedback mechanisms that suppress the activity of other RTKs such as EGFR, either directly through signal transduction feedback or indirectly through transcriptional induction of negative regulators of RTK signaling as has been described in BRAF-mutated colorectal cancers and in other circumstances (Lito et al., 2012;Prahallad et al., 2012). Inhibition of TPR-TRKA kinase activity with entrectinib inhibits signaling through the RAF>MEK>ERK pathway, leading to elevated expression of BIM but also activation of EGFR signaling. When entrectinib is re-administered (once daily), signaling through the MAP kinase (MAPK) pathway is re-set, likely through heterologous desensitization (allowing the tumor to remain sensitive following drug treatment every 24 h for some period of time [in mice, our data show this can work for roughly 30–85 days, depending on the location of the tumor model and cell type utilized]) (Figure 6E). However, combined inhibition of TPR-TRKA plus MEK1/2 (vertical inhibition) or TPR-TRKA plus EGFR (parallel inhibition) prevents reactivation of RAF>MEK>ERK signaling, leading to more durable suppression of this key proliferative pathway. Feedback inhibition from the MAPK pathway to EGFR following targeted therapy has been previously observed in different tissues and contexts (Lito et al., 2012; Prahallad et al., 2012).
In summary, this research indicates that even with a disease as treatment refractory as pancreatic cancer, the identification of rare actionable mutations in proto-oncogenes such as NTRK1 can lead to a striking short-term therapeutic patient response. Furthermore, these studies also suggest that a combination of FDA-approved targeted therapies against TRKA (e.g., entrectinib, larotrectinib) plus MEK1/2 (bini, cobi-, or trametinib) should be clinically tested for the ability to substantially enhance the durability of response for these patients beyond that achieved with a single-agent TRKA inhibitor.
STAR ★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents or resources should be directed to and will be fulfilled by the Lead Contact, Dr. Martin McMahon.
Materials Availability
All unique/stable reagents generated in this study are available from the lead contact, Dr. Martin McMahon, upon request.
Data and Code Availability
This study did not generate or analyze any datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human patients
Written informed consent was obtained from the patient prior to use of the patient’s tumor sample. The one patient described in this study was a 47 year old male with pancreatic adenocarcinoma. The consent form and protocol was reviewed and approved by the University of Utah Institutional Review Board.
Cultured cell lines
All cells were cultured in RPMI (Roswell Park Memorial Institute) 1640 media supplemented with 10% fetal bovine serum, 1% penicillin plus streptomycin and cultured at 37°C unless otherwise specified. This will herein be referred to as 10% RPM11640. Parental IMPE cells were supplemented with recombinant mouse lnterferon-γ (IFNγ, Peprotech), and cultured in a 33° incubator. Parental Ba/F3 cells were additionally supplemented with 100ng/ml of interleukin-3 (IL-3, Peprotech). HEK293T cells were maintained in DMEM media with 10% fetal bovine serum and 1% penicillin plus streptomycin. All established human cell lines used for these studies have been authenticated by STR profiling and mycoplasma testing is done quarterly using PlasmoTest (Invivogen). Parental IMPE cells and their various derivatives were cultured in 10% RPM11640 in the absence of IFNγ, and transferred to the non-permissive temperature (37°C) to test for oncogenic transformation. Parental MLE-12 cells were purchased from ATCC, and were cultured in HITES medium (DMEM F12/HAM base medium: Insulin 0.005 mg/ml, Tranferrin 0.01 mg/ml, Sodium selenite 30 nM, Hydrocortisone 10 nM, Betaestradiol 10 nM, HEPES10 nM, L-Glutamine2 mM, and 2% FBS), but following tumorigenic transformation by the TPR-NTRK1 fusion the cells were cultured in RPMI. Normal MLE-12 or GFP transduced MLE-12 cells do not grow in RPMI. CUTO-3.29 were kindly provided by Dr. Robert Doebele from the University of Colorado, and KM12 cells were purchased from the National Cancer Institute; both human cell lines were also grown in RPMI media. ER-NI cells were grown in 10% RPM11640 media, and cultured continuously in the presence of entrectinib. All phase contrast images of cultured cells were taken with an EVOS microscope at 10x magnification (Invitrogen), and contain a scale bar of 400 microns in the image.
Mouse models/animals
Animal care and procedures were approved by the Institutional Animal Care and Use Committee Office (IACUC) of the University of Utah under protocol #18–10003. Mice were housed in groups in microisolator cages with bedding enrichment on ventilated racks in an AAALAC approved vivarium. Mice were provided with standard chow and water through a lixit system installed into the housing racks. Mice received two health checks daily by the institute’s animal husbandry staff. Male and female NOD.Cg-Prkdcscid/J mice were obtained from the Preclinical Research Resource breeding colony or purchased from Jackson Laboratories for all mouse experiments used in this study. Males and females were age matched within three weeks of each other, and randomly distributed equally for each experiment once they were at least 6 weeks of age.
METHOD DETAILS
Plasmid cloning and viruses
Based on analysis of the chromosomal breakpoints within the TPR and NTRK1 genes identified in the patient’s pancreatic tumor DNA (Foundation Medicine using reference genome GRCh37/hg19) outlined in Figure 1C, a TPR-NTRK1 gene fusion was chemically synthesized (GenScript) and inserted into the pLenti6/UbC/V5-DEST Gateway lentiviral mammalian expression plasmid (ThermoFisher) using restriction enzyme cloning with 5′ BamHI and 3′ Xhol endonucleases. From the NCBI reference sequence database, human transcript Genbank: NM_002529 was used as the NTRK1 transcript the exon calls were based on using methods previously described (Vaishnavi et al., 2013). Lentiviral supernatants were generated by co-transfection of HEK293T cells with a three vector lentiviral system: using either an empty vector, GFP or pLenti6-TPR-NTRK1 expression vector combined with the lentiviral packaging and envelope plasmids PCMV-Δ8.9 and PCMV-VSVG. pCMV-VSV-G was a gift from Dr. Robert Weinberg (Addgene plasmid # 8454; http://addgene.org/8454; RRID:Addgene_8454).
Blasticidin selection of both IMPE and Ba/F3 cells was performed at 1ug/ml. pFUGW-Pol2-ffLuc2-eGFPforthe expression of both EGFP and firefly luciferase was a generous gift from Glenn Merlino (Addgene plasmid # 71394; http://addgene.org/71394; RRid:Addgene_71394. Retroviral constructs EGFR WT-pBABE and EGFR-L858R were a gift from Matthew Meyerson (Addgene plasmid #11011 and #11012; http://addgene.org/11011; RRID:Addgene_11011 or RRID:Addgene_11012) (Greulich et al., 2005). Retroviral production was performed similarly, using the mouse host cell packaging plasmid pCL-Eco. Puromycin selection was performed for each plasmid, respectively. CRISPR-CAS9-mediated genome silencing of BIM was performed by annealing and cloning both the 5′-GCACAGGAGCTGCGGCGGAT-3′ sequence (sgBIM) and a reverse complement into BsmB1-digested pLenti-CRISPRv2 plasmid (Addgene plasmid #52961 http://addgene.org/52961; RRID:Addgene_52961. Plasmid was a generous gift from Feng Zheng). One BIM sgRNA was tested, using this plasmid. Successful silencing CRISPR-CAS9-mediated silencing of BIM with this sgRNA was verified by Sanger sequencing. From the pooled population, 12 single cell clones were isolated to confirm BIMNull cells by immunoblot analysis. A single cell sgNT clone was also isolated from a pooled population using an identical strategy with 12 single cell clones all retaining BIM protein expression by immunoblot analysis.
Immunoblotting/Arrays
Cells were treated with drugs at the indicated concentrations and for the time indicated as outlined in each figure. Protein extracts were harvested by lysing cells in RIPA lysis buffer with Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific) and diluted into NuPAGE LDS (lithium dodecyl sulfate, pH 8.4) sample loading buffer (Invitrogen) prior to boiling for 10 minutes at 95°C. For lysates made from tumors, mice were euthanized, tumors were excised, and snap frozen in 70% ethanol and dry ice for storage at −80°C. Later, tumors were homogenized with a glass tissue homogenizer in RIPA buffer, and a BCA assay was performed to determine protein concentration prior to boiling. Proteins were separated using standard SDS-PAGE gel electrophoresis with 4%–12% gradient Bis-Tris polyacrylamide gels, transferred to PVDF membranes for immunoblot analysis using an iBlot2 (Invitrogen), and stained with indicated primary antibodies as indicated in each figure. LI-COR brand Intercept (TBS) blocking buffer and IRDye secondary antibodies 680RD and 800CW were utilized for all immunostaining experiments. Membranes were scanned and analyzed using the Odyssey Imaging System and software (LI-COR). The primary antibodies used for immunoblot analysis are described in the key resource table. Phospho BIM (pSerine 69) was generated and previously described by the McMahon Lab (Cartlidge et al., 2008). N = 3 replicates for each immunoblot shown.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-pY490-TRKA | Cell Signaling | Cat# 9141S; RRID: AB_2298805 |
| Anti-HA 6E2/2367 | Cell Signaling | Cat# C29F4/3324S; RRID: AB_1549585 |
| Anti-total TRK A7H6R | Cell Signaling | Cat# 92991S; RRID: AB_2800196 |
| Anti-Cytokeratin 19 D4G2 XP/12434 | Cell Signaling | Cat# D7F7W/13092S; RRID: AB_2722626 |
| Anti-β-Tubulin 9F3/2128 | Cell Signaling | Cat# D3U1W/86298S; RRID: AB_2715541 |
| Anti- pT202/pY204-ERK1/2 D.13.14.4E | Cell Signaling | Cat# 437OS; RRID: AB_10694057 |
| Anti- total ERK L34F12 | Cell Signaling | Cat# 4696S; RRID: AB_10694988 |
| Anti-BIM Y36 | AbCam | Cat# ab32158; RRID: AB_725697 |
| Anti-PARP | Cell Signaling | Cat# 9542S; RRID: AB_2160739 |
| Anti-PUMA | Cell Signaling | Cat# 4976S; RRID: AB_2064551 |
| Anti-BAX | Cell Signaling | Cat# 2772S; RRID: AB_10695870 |
| Anti-pY1068/pY1173-EGFR 1H12/2236S | Cell Signaling | Cat# 53A5/4407S; RRID: AB_331795 |
| Anti-total EGFR D38B1 | Cell Signaling | Cat# 4267S; RRID: AB_2246311 |
| Anti-pS473-AKT D9E | Cell Signaling | Cat #4060L; RRID: AB_2315049 |
| Anti-total AKT 40D4 | Cell Signaling | Cat# 2920S; RRID: AB_1147620 |
| Anti-BMF G81 | Cell Signaling | Cat# 5889; RRID: AB_10835694 |
| Anti-Actin 8H10D10 | Cell Signaling | Cat# 3700; RRID: AB_2242334 |
| Anti-GAPDH D16H11 | Cell Signaling | Cat# 5174; RRID: AB_10622025 |
| anti-pTyr | Millipore | Cat# 4G10; RRID: AB_916370 |
| Anti-Phospho BIM S69 | McMahon Lab | PMID:18715233 |
| Bacterial and Virus Strains | ||
| Stbl3 Chemically Competent E.coli | Thermo Fisher | C73733 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Entrectinib | Ignyta/Genentech | MTA |
| Cobimetinib | Genentech | MTA |
| Gefitinib | SelleckChem | S1025 |
| IFN Gamma | Peprotech | 315–05 |
| IL-3 | Peprotech | 213–13 |
| D-Luciferin | Gold Bio | LUCK-100 |
| Critical Commercial Assays | ||
| Cell Titer 96 Acqueous One | Promega | G3582 |
| Proteome Profiler Mouse RTK array | R&D | ARY014 |
| Experimental Models: Cell Lines | ||
| IMPE (mouse) | University of Rochester | PMID:18500333 |
| 293-T (human) | ATCC | CRL-3216 |
| MLE-12 (mouse) | ATCC | CRL-2110 |
| Ba/F3 (mouse) | ATCC | N/A currently |
| CUTO-3.29 (human) | University of Colorado (MTA) | PMID: 24162815 |
| KM12 (human) | NCI (MTA) | PMID: 24162815 |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD.Cg-Prkdcscld/J | HCI PRR Colony | internal colony |
| Oligonucleotides | ||
| 5′-GCACAGGAGCTGCGGCGGAT-3′ (sgBIM) | IDT | This study |
| Recombinant DNA | ||
| TPR-NTRK1-pLenti-6 | Genscript | This study |
| pCMV-VSV-G | Addgene | 8454 |
| psPAX2 | Addgene | 12260 |
| pFUGW-Pol2-ffLuc2-eGFP | Addgene | 71394 |
| EGFR WT-pBABE | Addgene | 11011 |
| EGFR-L858R-pBABE | Addgene | 11012 |
| pLenti-CRISPRv2 | Addgene | 52961 |
A Proteome Profiler mouse phospho-RTK array kit was purchased from R&D (#ARY014). Briefly, cell lysates from sgNT and BIMNull IMPETPR-NTRK1 cells were harvested and used according to the kit manufacturer’s instructions. Arrays were developed with ECL and scanned with an AZURE scanner. Dot intensity was quantified using FIJI.
Generation of ER-NI cells
Entrectinib resistant - NTRK1 IMPE cells (ER-NI-1) were generated in vitro by culturing IMPE cells continuously in high doses (1 μM for 2 weeks, then 5 μM for 2 weeks) of entrectinib for one month total time. Cells were grown continuously in entrectinib onward. In addition, we generated five entrectinib resistant cell cultures (ER-NI-2 through −6) from drug resistant orthotopic IMPETPR-NTRK1 mouse pancreatic tumors by collecting such tumors from euthanized mice, mincing tumors with a razor, digesting tumors with collagenase (400 U/ml), dispase (5 U/ml), elastase (4 U/ml), and DNase1 (0.25 mg/ml) in Advanced DMEM:F12 HAM media in a 37°C shaker for 15 minutes. After generating a single cell suspension, cells were plated in vitro.
Cell proliferation assays
All cell proliferation assays were performed in media supplemented with 10%(v/v) FBS as previously described using Cell Titer 96 MTS (Promega) (Vaishnavi et al., 2013). Briefly, cells were seeded at 750 or 1000 cells/well and treated for 72 hours at the drug concentrations indicated. Each assay was performed in triplicate and using at least 3 independent biological replicates. Data was plotted and analyzed using GraphPad Prism software, with the 50% inhibitory concentrations (IC50) values calculated as half maximal response.
Mouse experiments
Animal care and procedures were approved by the Institutional Animal Care and Use Committee Office (IACUC) of the University of Utah under protocol #18–10003. For subcutaneous implantation, 106 cells were resuspended into Matrigel and media and injected into each flank. Prior to orthotopic implantation in the pancreas, IMPETPR-NTRK1 cells were infected with virus generated by transfection of a lentiviral plasmid vector encoding a luciferase-eGFP fusion protein. Infected cells were isolated by flow cytometry for EGFP positive cells (Day et al., 2009). Orthotopic implantation into mouse pancreata was performed by HCI’s Preclinical Research Resource Core via standard pancreatic survival surgery. Bioluminescence imaging was performed using the IVIS Spectrum In Vivo Imaging System (Perkin Elmer). Briefly, mice were injected intraperitoneally with approximately 3mg of D-Luciferin (Goldbio) 10 minutes prior to imaging. For subcutaneous tumorigenesis experiments, once tumors reached an average size of ≥ 250 mm3, mice were randomized into treatment groups based on establishing equal tumor size per group. Tumor size was measured twice weekly using digital calipers and tumor volume was calculated with the ellipsoid formula: (length × width2/2) (Tomayko and Reynolds, 1989). All dosing regimens (daily and BID) were performed 7 days a week for the period of time indicated. Entrectinib was initially generously provided by Ignyta and subsequently purchased from MedChem Express. Cobimetinib was generously provided by Genentech Inc. Both drugs were dissolved in corn oil as the vehicle, and delivered by oral gavage on a daily basis. In conjunction with IACUC policy, mice were euthanized when tumor volume exceeded 1 cm3 or if the animal demonstrated any serious health concerns or signs of suffering. The Ullmann-Cullere Body Conditioning Score (BCS) was used to determine animal health status and to determine when and if appropriate euthanasia endpoints were met (Ullman-Culleré and Foltz, 1999).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters are reported in the figures and figure legends. Data are considered significant if p < 0.05. Data are presented as mean ± SEM for biological replicates. Data was analyzed using a Student t test when comparing two conditions. One-way ANOVA test was performed on comparisons of more than two conditions. Survival analysis was performed using Log-rank (Mantel-Cox) test. Statistical analyses were carried out in GraphPad Prism 8 (version 8.4.1).
Supplementary Material
Highlights.
NTRK1 fusions are potent drivers of transformation in pancreatic and lung tissues
BIM mediates the response of NTRK1 fusion-driven cancer cells to entrectinib
EGFR can reactivate signaling through MAPK following TRKA monotherapy
Vertical inhibition of TRKA and MEK is superior in forestalling lethal drug resistance
ACKNOWLEDGMENTS
The authors thank the members of the McMahon lab, past and present, especially Ms. Kayla O’Toole. The authors acknowledge University of Utah and Huntsman Cancer Institute (P30 CA042014) and its various shared resources: (1) Preclinical Research Resource, which provided assistance with tumor models in immunocompromised mice; (2) DNA Sequencing Core Facility; and (3) Flow Cytometry Core Facility. We acknowledge both Ignyta and Genentech for providing entrectinib and cobimetinib and thank Dr. Mark Merchant for his assistance in providing these reagents. Finally, we thank Five for The Fight and Huntsman Cancer Foundation for generous philanthropic support. This research was supported by CA131261 to M.M., P30 CA250971 CCSG to HCI, CA235964 to A.T., and CA228267 and CA246084 to A.V.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107994.
DECLARATION OF INTERESTS
M.M. served on advisory boards to Genentech. I.G.-L. served on a scientific advisory board for Ignyta Pharmaceuticals.
REFERENCES
- Bouwens L (1998). Cytokeratins and cell differentiation in the pancreas. J. Pathol 184, 234–239. [DOI] [PubMed] [Google Scholar]
- Cartlidge RA,Thomas GR,Cagnol S,Jong KA,Molton SA,Finch AJ, and McMahon M (2008). Oncogenic BRAF(V600E) inhibits BIM expression to promote melanoma cell survival. Pigment Cell Melanoma Res. 21, 534–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocco E, Schram AM, Kulick A, Misale S, Won HH, Yaeger R, Razavi P, Ptashkin R, Hechtman JF, Toska E, et al. (2019). Resistance to TRK inhibition mediated by convergent MAPK pathway activation. Nat. Med 25, 1422–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguria A, Zaballos A, Flores JM, Barbacid M, et al. (2005). Tumour biology: senescence in premalignant tumours. Nature 436, 642. [DOI] [PubMed] [Google Scholar]
- Day CP, Carter J, Bonomi C, Esposito D, Crise B, Ortiz-Conde B, Hollingshead M, and Merlino G (2009). Lentivirus-mediated bifunctional cell labeling for in vivo melanoma study. Pigment Cell Melanoma Res. 22, 283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehan E, Bassermann F, Guardavaccaro D, Vasiliver-Shamis G, Cohen M, Lowes KN, Dustin M, Huang DC, Taunton J, and Pagano M (2009). betaTrCP- and Rsk1/2-mediated degradation of BimEL inhibits apoptosis. Mol. Cell 33, 109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Shimamura T, Perera S, Carlson NE, Cai D, Shapiro GI, Wong KK, and Letai A (2007). Proapoptotic BH3-only BCL-2 family protein BIM connects death signaling from epidermal growth factor receptor inhibition to the mitochondrion. Cancer Res. 67, 11867–11875. [DOI] [PubMed] [Google Scholar]
- Doebele RC, Davis LE, Vaishnavi A, Le AT, Estrada-Bernal A, Keysar S, Jimeno A, Varella-Garcia M, Aisner DL, Li Y, et al. (2015). An Oncogenic NTRK Fusion in a Patient with Soft-Tissue Sarcoma with Response to the Tropomyosin-Related Kinase Inhibitor LOXO-101. Cancer Discov. 5, 1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drilon A, Nagasubramanian R, Blake JF, Ku N, Tuch BB, Ebata K, Smith S, Lauriault V, Kolakowski GR, Brandhuber BJ, et al. (2017a). A Next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion-Positive Solid Tumors. Cancer Discov. 7, 963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drilon A, Siena S, Ou SI, Patel M, Ahn MJ, Lee J, Bauer TM, Farago AF, Wheler JJ, Liu SV, et al. (2017b). Safety and Antitumor Activity of the Multitargeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib: Combined Results from Two Phase I Trials (ALKA-372–001 and STARTRK-1). Cancer Discov. 7, 400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, Nathenson M, Doebele RC, Farago AF, Pappo AS, et al. (2018). Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med 378, 731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewings KE, Hadfield-Moorhouse K, Wiggins CM, Wickenden JA, Balmanno K, Gilley R, Degenhardt K, White E, and Cook SJ (2007). ERK1/2-dependent phosphorylation of BimEL promotes its rapid dissociation from Mcl-1 and Bcl-xL. EMBO J. 26, 2856–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber AC, Corcoran RB, Ebi H, Sequist LV, Waltman BA, Chung E, Incio J, Digumarthy SR, Pollack SF, Song Y, et al. (2011). BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov. 1, 352–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber AC, Ebi H, Costa C, and Engelman JA (2012). Apoptosis in targeted therapy responses: the role of BIM. Adv. Pharmacol 65, 519–542. [DOI] [PubMed] [Google Scholar]
- Fuse MJ, Okada K, Oh-Hara T, Ogura H, Fujita N, and Katayama R (2017). Mechanisms of Resistance to NTRK Inhibitors and Therapeutic Strategies in NTRK1-Rearranged Cancers. Mol. Cancer Ther 16, 2130–2143. [DOI] [PubMed] [Google Scholar]
- Gong Y, Somwar R, Politi K, Balak M, Chmielecki J, Jiang X, and Pao W (2007). Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med. 4, e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco A, Miranda C, Pagliardini S, Fusetti L, Bongarzone I, and Pierotti MA (1997). Chromosome 1 rearrangements involving the genes TPR and NTRK1 produce structurally different thyroid-specific TRK oncogenes. Genes Chromosomes Cancer 19, 112–123. [PubMed] [Google Scholar]
- Greulich H, Chen TH, Feng W, Jänne PA, Alvarez JV, Zappaterra M, Bulmer SE, Frank DA, Hahn WC, Sellers WR, and Meyerson M (2005). Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2, e313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazar-Rethinam M, Kleyman M, Han GC, Liu D, Ahronian LG, Shahzade HA, Chen L, Parikh AR, Allen JN, Clark JW, et al. (2018). Convergent Therapeutic Strategies to Overcome the Heterogeneity of Acquired Resistance in BRAFV600E Colorectal Cancer. Cancer Discov. 8, 417–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jat PS, Noble MD, Ataliotis P, Tanaka Y, Yannoutsos N, Larsen L, and Kioussis D (1991). Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc. Natl. Acad. Sci. USA 88, 5096–5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Greulich H, Jänne PA, Sellers WR, Meyerson M, and Griffin JD (2005). Epidermal growth factor-independent transformation of Ba/F3 cells with cancer-derived epidermal growth factor receptor mutants induces gefitinib-sensitive cell cycle progression. Cancer Res. 65, 8968–8974. [DOI] [PubMed] [Google Scholar]
- Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, Schuman SS, Shea JE, Seipp MT,Yap JT, et al. (2019). Protective autophagy elicited by RAF/MEK/ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med 25, 620–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi M, Ito D, Fujimoto K, Toyoda E, Kami K, Mori T, Doi R, White-head R, and Imamura M (2004). Conditional transformation of mouse pancreatic epithelial cells: an in vitro model for analysis of genetic events in pancreatocarcinogenesis. Biochem. Biophys. Res. Commun 319, 612–621. [DOI] [PubMed] [Google Scholar]
- Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Jänne PA, Costa DB, et al. (2010). Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med 363, 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, Huang A, Wong WL, Callahan MK, Merghoub T, et al. (2012). Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 22, 668–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G, and Auberger P (2003). Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapo-ptotic function. Oncogene 22, 6785–6793. [DOI] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al. (2004). Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med 350, 2129–2139. [DOI] [PubMed] [Google Scholar]
- Marshall CJ (1995). Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80, 179–185. [DOI] [PubMed] [Google Scholar]
- McMurray HR, Sampson ER, Compitello G, Kinsey C, Newman L, Smith B, Chen SR, Klebanov L, Salzman P, Yakovlev A, and Land H (2008). Synergistic response to oncogenic mutations defines gene class critical to cancer phenotype. Nature 453, 1112–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA, Brown-Swigart L, Johnson L, and Evan GI (2008). Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell 14, 447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevala-Plagemann C, Hidalgo M, and Garrido-Laguna I (2020). From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat. Rev. Clin. Oncol 17, 108–123. [DOI] [PubMed] [Google Scholar]
- Ng KP, Hillmer AM, Chuah CT, Juan WC, Ko TK, Teo AS, Ariyaratne PN, Takahashi N, Sawada K, Fei Y, et al. (2012). A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat. Med 18, 521–528. [DOI] [PubMed] [Google Scholar]
- Okamura R, Boichard A, Kato S, Sicklick JK, Bazhenova L, and Kurzrock R (2018).Analysis of NTRK Alterations in Pan-Cancer Adult and Pediatric Malignancies: Implications for NTRK-Targeted Therapeutics. JCO Precis. Oncol 2018, 10.1200/PO.18.00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okimoto RA, and Bivona TG (2016). Tracking Down Response and Resistance to TRK Inhibitors. Cancer Discov. 6, 14–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly EM, and Hechtman JF (2019). Tumourresponse toTRK inhibition in a patient with pancreatic adenocarcinoma harbouring an NTRK genefusion. Ann. Oncol 30 (Suppl 8), viii36–viii40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pishvaian MJ, Garrido-Laguna I, Liu SV, Multani PS, Chow-Maneval E, and Rolfo C (2018). Entrectinib in TRK and ROS1 Fusion-Positive Metastatic Pancreatic Cancer. JCO Precis. Oncol 2, 1–7. [DOI] [PubMed] [Google Scholar]
- Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, and Bernards R (2012). Unresponsiveness of colon cancerto BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483, 100–103. [DOI] [PubMed] [Google Scholar]
- Roman M, Baraibar I, Lopez I, Nadal E, Rolfo C, Vicent S, and Gil-Bazo I (2018). KRAS oncogene in non-small cell lung cancer: clinical perspectives on the treatment of an old target. Mol. Cancer 17, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo M, Misale S, Wei G, Siravegna G, Crisafulli G, Lazzari L, Corti G, Rospo G, Novara L, Mussolin B, et al. (2016). Acquired Resistance to the TRK Inhibitor Entrectinib in Colorectal Cancer. Cancer Discov 6, 36–44. [DOI] [PubMed] [Google Scholar]
- Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, and Chodosh LA (2007). Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat. Cell Biol 9, 493–505. [DOI] [PubMed] [Google Scholar]
- Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, Riely GJ, Varella-Garcia M, Shapiro GI, Costa DB, et al. (2014). Crizotinib in ROS1-rearranged non-small-cell lung cancer. N. Engl. J. Med 371, 1963–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, Lira SA, and Barbacid M (1994). Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature 368, 246–249. [DOI] [PubMed] [Google Scholar]
- Subbiah V, Baik C, and Kirkwood JM (2020). Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer. Published online June 12, 2020. 10.1016/j.trecan.2020.05.009. [DOI] [PubMed] [Google Scholar]
- Sunters A, Fernandez de Mattos S, Stahl M, Brosens JJ, Zoumpoulidou G, Saunders CA, Coffer PJ, Medema RH, Coombes RC, and Lam EW (2003). FoxO3a transcriptional regulation of Bim controls apoptosis in paclitaxel-treated breast cancer cell lines. J. Biol. Chem 278, 49795–49805. [DOI] [PubMed] [Google Scholar]
- Tomayko MM, and Reynolds CP (1989). Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharmacol 24, 148–154. [DOI] [PubMed] [Google Scholar]
- Ullman-Cullere MH, and Foltz CJ (1999). Body condition scoring: a rapid and accurate method for assessing health status in mice. Lab. Anim. Sci 49, 319–323. [PubMed] [Google Scholar]
- Vaishnavi A, Capelletti M, Le AT, Kako S, Butaney M, Ercan D, Mahale S, Davies KD, Aisner DL, Pilling AB, et al. (2013). Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat. Med 19, 1469–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnavi A, Le AT, and Doebele RC (2015). TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov. 5, 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnavi A, Schubert L, Rix U, Marek LA, Le AT, Keysar SB, Glogowska MJ, Smith MA, Kako S, Sumi NJ, et al. (2017). EGFR Mediates Responses to Small-Molecule Drugs Targeting Oncogenic Fusion Kinases. Cancer Res. 77, 3551–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikenheiser KA, Clark JC, Linnoila RI, Stahlman MT, and Whitsett JA (1992). Simian virus 40 large T antigen directed by transcriptional elements of the human surfactant protein C gene produces pulmonary adenocarcinomas in transgenic mice. Cancer Res. 52, 5342–5352. [PubMed] [Google Scholar]
- Woods D, Parry D, Cherwinski H, Bosch E, Lees E, and McMahon M (1997). Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol. Cell. Biol 17,5598–5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Haigis KM, Firestone AJ, McNerney ME, Li Q, Davis E, Chen SC, Nakitandwe J, Downing J, Jacks T, et al. (2013). Dominant role of oncogene dosage and absence of tumor suppressor activity in Nras-driven hematopoietic transformation. Cancer Discov. 3, 993–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Woods D, McMahon M, and Bishop JM (1998). Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 12, 2997–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate or analyze any datasets or code.