

 A series of indole compounds were designed and synthesized as CB2 agonist with high efficacy and selectivity.
A series of indole compounds were designed and synthesized as CB2 agonist with high efficacy and selectivity.
Abstract
The CB2 receptor plays a crucial role in analgesia and anti-inflammation. To develop novel CB2 agonists with high efficacy and selectivity, a series of indole derivatives with N-ethyl morpholine moieties (compounds 1–56) were designed, synthesized and biologically evaluated. Compounds 1, 2, 3, 46 and 53 exhibited high CB2 receptor affinity at low nanomolar concentrations and good receptor selectivity (EC50(CB1)/EC50(CB2) greater than 1000). The most active compound, compound 2, was more potent than the standard drug GW405833 for in vitro agonistic action on the CB2 receptor. More importantly, in a rat model for CFA-induced inflammatory hyperalgesia, compound 2 had a potent anti-inflammatory pain effect within 12 hours after administration. At the 1 h time point, compound 2 had a dose-dependent reversal for hyperalgesia with an estimated ED50 value of 1.097 mg kg–1. Moreover, compound 2 significantly suppressed the pro-inflammatory cytokines (IL-1β, IL-6 and TNF-α) in CFA-induced lesions. These protective effects of compound 2 on inflammatory pain were superior to those of GW405833, suggesting that compound 2 may be a promising therapeutic drug that needs further validation.
1. Introduction
Cannabinoid (CB) receptors are a series of membrane receptors that respond to cannabinoids.1 There are several subtypes of cannabinoid receptors, of which, CB1 and CB2 are the definitive cannabinoid receptor subtypes.2,3 The orphan receptor GPR55 and peroxisomal proliferator activated receptors (PPARs) have recently been reported to be novel cannabinoid binding targets.4 Endocannabinoid receptors have been demonstrated to be involved in multiple physiological and pathological functions and to play significant roles in analgesia.5
The typical cannabinoid receptors, CB1 and CB2, are both G-protein-coupled receptors (GPCRs). They share 44% amino acid sequence homology, while the 7-transmembrane region shares 68% amino acid sequence homology. The protein crystal structure of CB1 has been successfully solved in 2016,6 and that of CB2 has been recently solved.7 The CB1 receptor is mainly expressed in the central nervous system (CNS),8 while the CB2 receptor is primarily distributed in the peripheral immune system9 and in diseased brain cells.10 With the different distribution of the two receptors, the CB2 receptor has unique physiological and pharmacological effects, such as immunosuppression, inhibition of tumor cell growth, bone formation, anti-fibrosis, and anti-neurological injury, while the CB1 receptor is involved in psychotropic or other CNS-related effects.11–16 In terms of pain relief, anti-inflammation, and inhibition of cough,16,17 the CB2 receptor is almost non-addictive and non-tolerable, and hence targeting it will have a significant therapeutic value. A significant amount of research has been dedicated to the development of CB2 receptor ligands due to their high efficacy and high selectivity. However, the high homology between the ligand-binding domains of the two receptors has been a challenge in developing selective CB2 receptor ligands. These ligands should avoid the adverse effects of CB1 receptor-mediated psychotropic effects.18 Although a large number of structurally diverse CB2 receptor ligands have been developed, with some progressing to clinical trials,19 none have been developed into successful clinical drugs. One of the main reasons for failure could be due to low receptor selectivity.
The indole ring is a privileged scaffold in medicinal chemistry,20 and several types of compounds having the indole moiety have been demonstrated to display CB2 activity.21–28 We found that several of these CB2 ligands contain an N-ethyl morpholine ring structure (Fig. 1). WIN-55212-2 was first reported by Sterling-Winthrop as a non-selective cannabinoid receptor agonist with anti-inflammatory activity (Ki(CB1) = 1.9 nM, Ki(CB2) = 0.3 nM), and is regularly used in pharmacological and behavioral research. AM630 is a selective inverse agonist of the CB2 receptor, while GW405833 (L768242) is a selective agonist of the CB2 receptor. A-796260 is another excellent CB2 receptor agonist and has demonstrated good efficacy in various pain models such as postoperative pain and neurological pain. Compound E is the most active compound in a series of novel CB2 receptor selective ligands with indolopyridone as the scaffold.
Fig. 1. (A)–(E): Representative indole-based CB2 ligands with N-ethyl morpholine moieties. N-Ethyl morpholine moiety (in red); indole moiety (in blue).

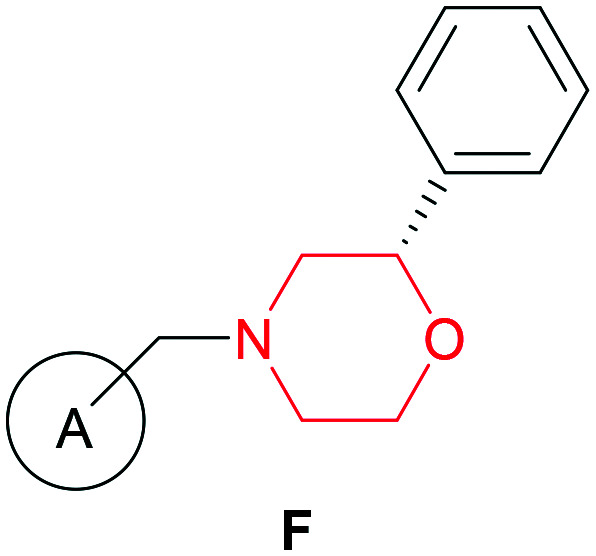
It is well known that several selective CB2 receptor ligands could be developed by modifying the morpholine ring alone as a structural scaffold. For example, a series of CB2 receptor agonists having the morpholine ring have been reported by Zindell et al. (EC50(CB2) = 10–15 000 nM) (Fig. 2).29
Fig. 2. (F): Representative structures of morpholine-based CB2 ligands. A: Substituted aromatic rings. Morpholine moiety (in red).
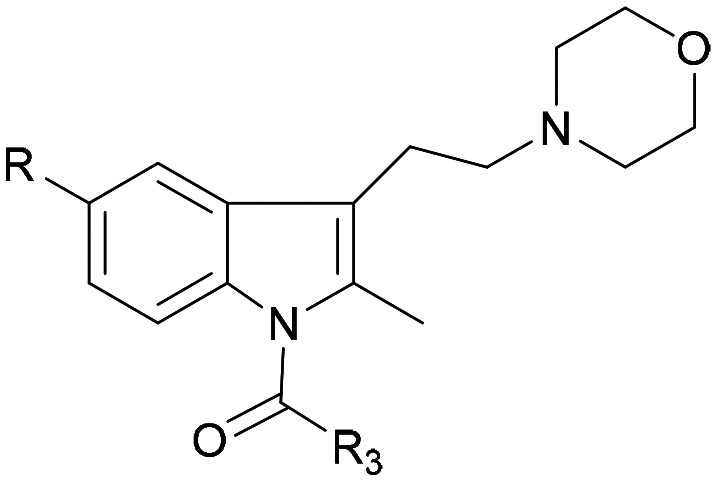
The activity data for CB2 ligands with morpholine or indole moieties, especially when both structures were present simultaneously, were likely to have high affinity and selectivity. The purpose of this study was to synthesize a series of new indole derivatives with N-ethyl morpholine moieties and evaluate their activity and selectivity as CB2 agonists, and then analyze their structure–activity relationship. Potential compounds were screened and assessed for anti-inflammatory properties for pain using in vivo and in vitro studies (Fig. 3).
Fig. 3. Synthesized compounds.

2. Results and discussion
2.1. Chemistry
Compounds 1–7 were synthesized as summarized in Scheme 1. The substituted indole compounds (2a–2d) were obtained through a Fischer indole synthesis reaction of the corresponding phenylhydrazine hydrochloride and ethyl levulinate. Ester hydrolysis was used to derive compounds 3a–3d. Subsequently, the carboxyl group at the 3-position of the indole was condensed with the morpholine ring to obtain compounds 4a–4d. LiAlH4 was used to reduce compounds 4a–4d to derive compounds 5a–5d. For the last step, N at the 1-position of the indole ring was reacted to substituted acid chloride to derive compounds 1–7.
Scheme 1. Reagents and conditions: (a) CH3COONa, CH3COOH, ethyl levulinate, 70 °C, 5–10 min; (b) H2SO4, C2H5OH, reflux 3–4 h; (c) 6 N NaOH, EtOH, room temperature, 3 h; (d) DMF, EDC·HCl, DMAP, 0 °C, 20–30 min; (e) morpholine, room temperature, overnight; (f) Ar2, LiAlH4, THF, 0 °C to room temperature, 2 days; (g) Ar2, HMPA, KHMDS, THF, –78 °C; (h) Ar2, –22 °C, 30 min; (i) Ar2, room temperature, 2–3 h.

Compounds 8–35 were synthesized as summarized in Scheme 2. First, compounds 5a–5d were reacted with tert-butyl bromoacetate to derive compounds 6a–6d. They were then hydrolyzed to derive compounds 7a–7d. Second, compounds 7a–7d were condensed with nitrogenous compounds to derive compounds 8–35.
Scheme 2. Reagents and conditions: (a) Ar2, NaH, DMF, 0 °C, 30–50 min; (b) BrCH2COOBu-t, 50 °C, 12 h; (c) CF3COOH, CH2Cl2, room temperature, 16 h; (d) DMF, EDC·HCl, DMAP, 0 °C, 30 min; (e) nitrogenous compounds, room temperature, overnight.

Compounds 36–56 were synthesized as summarized in Scheme 3. Compounds 2a–2c were reacted with N-(2-chloroethyl) morpholine under a strong base to derive compounds 8a–8c, and then hydrolyzed and condensed to produce compounds 36–56.
Scheme 3. Reagents and conditions: (a) Ar2, NaH, DMF, 0 °C, 30–40 min; (b) N-(2-chloroethyl) morpholine, 45 °C,4–6 h; (c) EtOH, 6 N NaOH, room temperature, 3 h; (d) DMF, EDC·HCl, DMAP, 0 °C, 30 min; (e) nitrogen-containing compound, room temperature, overnight.

2.2. In vitro agonistic activity and SAR
The synthesized compounds (1–56) were evaluated for the agonistic activity and selectivity for the CB2 receptor. EC50 values were determined via fluorometric measurements using CHO-HACB2- Gα16 cell lines (Chinese hamster ovarian cancer cells overexpressing CB2 and Gα) and CHO-HACB1- Gα16 cell lines (Chinese hamster ovarian cancer cells overexpressing CB1 and Gα).30,31 GW405833 was used as the positive control for activity assays. The results are presented in Table 1.
Table 1. EC50 values for compounds 1–56.
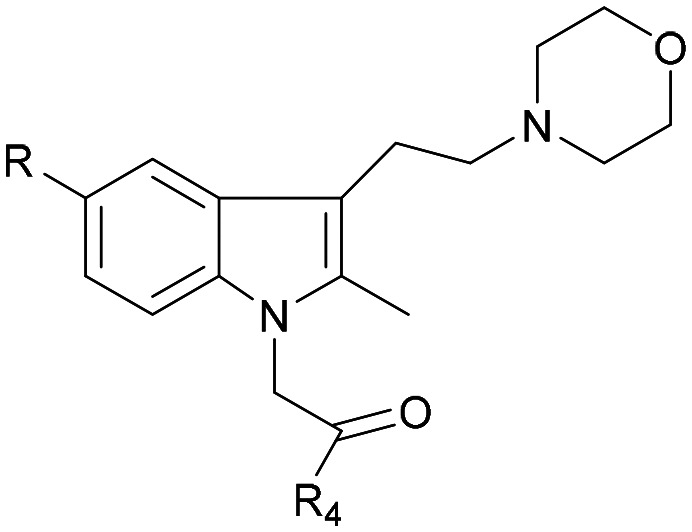
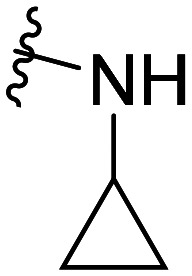
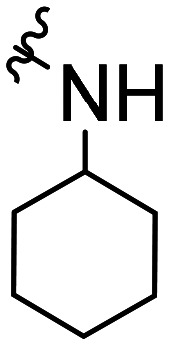
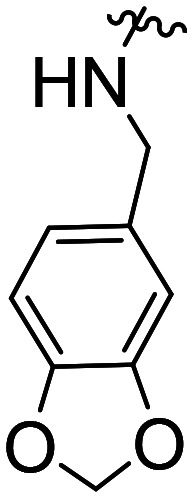
| Comp. | Structural skeleton | R | R2 | R3 | R4 | EC50 (CB2, nM) | EC50 (CB1, nM) |
| GW405833 |
|
CH3O |
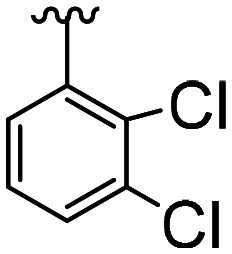
|
50.7 ± 14.1 | 16.1 × 103 ± 5.9 × 103 | ||
| 1 | Cl | 116.6 ± 41.6 | 79.8 × 103 ± 17.3 × 103 | ||||
| 2 | F | 18.9 ± 4.5* | 29.2 × 103 ± 14.4 × 103 | ||||
| 3 | Br | 52.3 ± 6.2 | >100 × 103 | ||||
| 4 | CH3O |
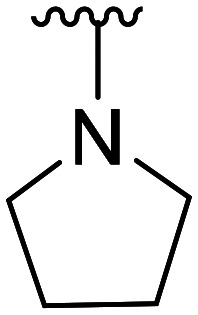
|
26.4 × 103 ± 16.1 × 103 | NA | |||
| 5 | Cl | NA | NA | ||||
| 6 | F | NA | NA | ||||
| 7 | Br | NA | NA | ||||
| 8 |
|
CH3O |
|
NA | NA | ||
| 9 | Cl | 5.3 × 103 ± 3.0 × 103 | NA | ||||
| 10 | F | 3.9 × 103 ± 1.6 × 103 | NA | ||||
| 11 | Br | 8.2 × 103 ± 3.2 × 103 | NA | ||||
| 12 | CH3O |
|
11.6 × 103 ± 0.4 × 103 | NA | |||
| 13 | Cl | NA | NA | ||||
| 14 | F | NA | NA | ||||
| 15 | Br | NA | NA | ||||
| 16 | CH3O |
|
28.1 × 103 ± 19.7 × 103 | NA | |||
| 17 | Cl | NA | NA | ||||
| 18 | F | NA | NA | ||||
| 19 | Br | NA | NA | ||||
| 20 | CH3O |
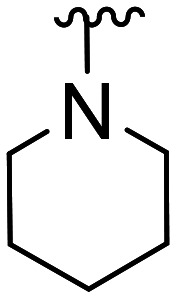
|
41.9 × 103 ± 22.0 × 103 | NA | |||
| 21 | Cl | NA | NA | ||||
| 22 | F | NA | NA | ||||
| 23 | Br | NA | NA | ||||
| 24 | CH3O |
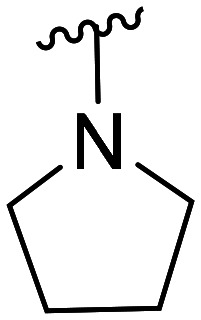
|
>100 × 103 | NA | |||
| 25 | Cl | NA | NA | ||||
| 26 | F | NA | NA | ||||
| 27 | Br | NA | NA | ||||
| 28 | CH3O |
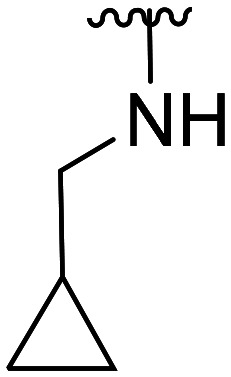
|
>100 × 103 | NA | |||
| 29 | Cl | 14.3 × 103 ± 4.0 × 103 | NA | ||||
| 30 | F | 4.2 × 103 ± 2.7 × 103 | NA | ||||
| 31 | Br | 5.4 × 103 ± 5.1 × 103 | NA | ||||
| 32 | CH3O |
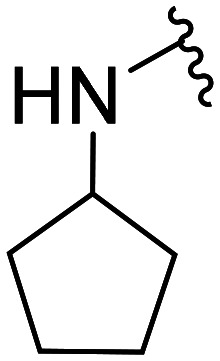
|
5.6 × 103 ± 2.3 × 103 | NA | |||
| 33 | Cl | 16.1 × 103 ± 5.1 × 103 | NA | ||||
| 34 | F | 5.5 × 103 ± 2.3 × 103 | NA | ||||
| 35 | Br | 20.2 × 103 ± 3.6 × 103 | NA | ||||
| 36 |
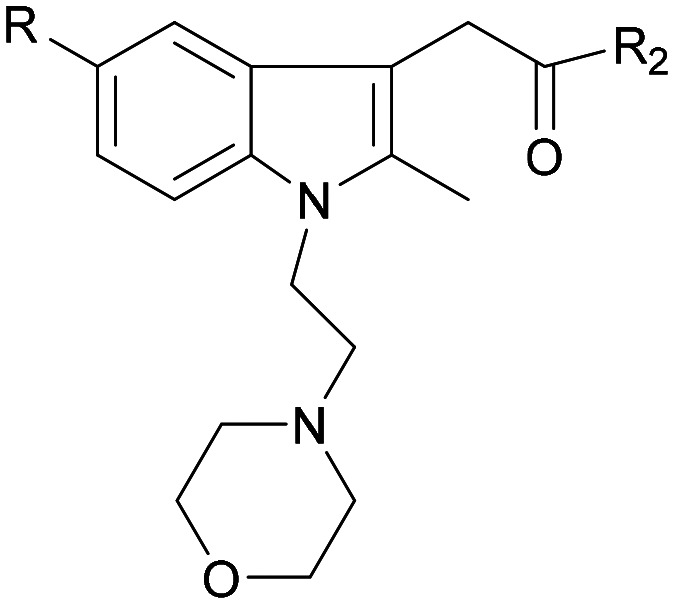
|
CH3O |
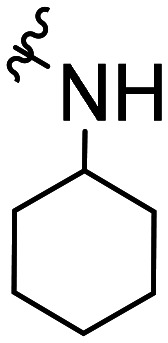
|
>100 × 103 | NA | ||
| 37 | Cl | >100 × 103 | NA | ||||
| 38 | F | >100 × 103 | NA | ||||
| 39 | CH3O |
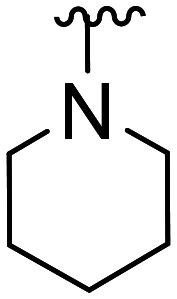
|
>100 × 103 | NA | |||
| 40 | Cl | NA | NA | ||||
| 41 | F | NA | NA | ||||
| 42 | CH3O |
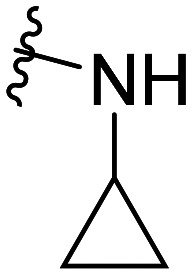
|
NA | NA | |||
| 43 | Cl | 1.2 × 103 ± 0.3 × 103 | NA | ||||
| 44 | F | 4.1 × 103 ± 0.6 × 103 | NA | ||||
| 45 | CH3O |
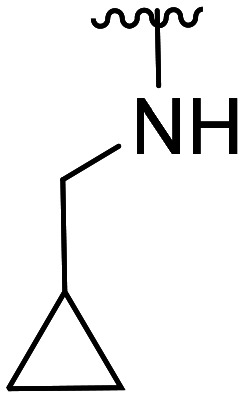
|
NA | NA | |||
| 46 | Cl | 594.0 ± 110.0 | NA | ||||
| 47 | F | 3.1 × 103 ± 2.0 × 103 | NA | ||||
| 48 | CH3O |
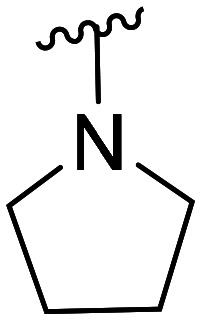
|
NA | NA | |||
| 49 | Cl | NA | NA | ||||
| 50 | F | NA | NA | ||||
| 51 | CH3O |
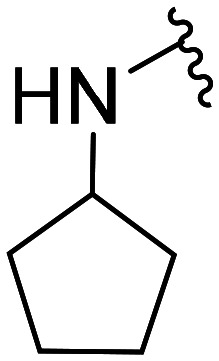
|
NA | NA | |||
| 52 | Cl | 2.7 × 103 ± 0.8 × 103 | NA | ||||
| 53 | F | 476.1 ± 274.8 | NA | ||||
| 54 | CH3O |
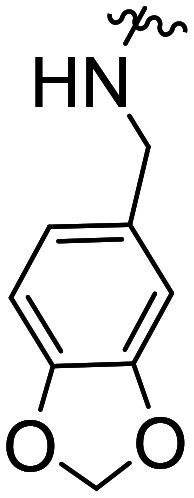
|
NA | NA | |||
| 55 | Cl | NA | NA | ||||
| 56 | F | NA | NA |
GW405833 is known to be a selective agonist of the CB2 receptor, and the activity data obtained from our experiments demonstrated that the EC50(CB1)/EC50(CB2) was greater than 1000. Hence, we figured that any EC50(CB1)/EC50(CB2) greater than 1000 would have good selectivity. From Table 1, 29 compounds had agonistic activity for the CB2 receptor, with 23 of these compounds having μM levels and selectivity greater than 1000. Compounds 1, 2, 3, 46, and 53 had excellent EC50 values at the nM level. Compound 2 had the best EC50(CB2) (18.9 ± 4.5 nM) and was superior to the standard drug GW405833 (EC50(CB2) = 50.7 ± 14.1 nM).
The activity results clearly indicated that when the other structures were unmodified to examine the effect of the 5-position substitution of the indole ring on the agonistic activity of the CB2 receptor, the halogen atom F was superior to Br and to Cl, while the methoxy substitution was inconclusive. We hypothesized that the methoxy containing oxygen atoms could form a hydrogen bond with the amino acid in the protein. We confirmed that the 5-position substitution of the indole ring could indeed reduce the binding strength to CB1. Secondly, we found that when the 5-position substitution in the indole skeleton was similar, regardless of whether the N-ethyl morpholine group was located at the 1-position or the 3-position of the indole skeleton, and as long as the amide nitrogen atom of the compound contained an active hydrogen, most of the compounds had agonistic activity for CB2 and were inactive for CB1. There was no CB2 agonistic activity if the compound had no active hydrogen on the amide nitrogen atom. We hypothesized that the active hydrogen on the amide nitrogen atom could form a hydrogen bond with an amino acid in the CB2 receptor protein, but not in the CB1 receptor. That is, the presence of active hydrogen on the amide nitrogen atom enhances the interaction of the ligand with the CB2 receptor and enhances the agonistic activity. Simultaneously, we found that as the size of the ring of the substituted cycloalkyl group on the amide increased, the agonistic activity of the compound on CB2 decreased until no activity was observed. Hence, a cyclopropyl-substituted compound had better CB2 agonistic activity compared to a cyclopentyl-substituted compound, and a cyclopentyl-substituted compound was superior to a cyclohexyl-substituted compound. We speculate that there are two possibilities. One is that the side chains containing the cycloalkyl groups of the compounds were originally embedded in the cavity of the CB2 receptor. With the increase of the size of the ring, the side chains could not enter the cavity, resulting in the decreased binding strength between the compounds and CB2 receptor, and thus, resulting in decreased agonistic activity. Another possible explanation is that the side chain is always in the cavity, but as the size of the ring increases, the relative position of the compound in the cavity changes, causing the amino acid that interacts with the ligand to change. The interaction between the protein and the ligand is reduced or decreased, resulting in decreased activity. Additionally, we found that the N-ethyl morpholine moiety located at the 1 position of the indole skeleton has similar agonistic activity for CB2 to the N-ethyl morpholine moiety located at the 3 position of the indole skeleton.
2.3. Molecular docking studies and SAR
Docking studies were performed in order to explore the effects of the molecular structure on activity. Docking studies were carried out using a crystal structure of human CB2 (PDB ID: 5TZY) obtained from the Protein Data Bank server (; www.pdb.org).
The docking results showed that Phe117, Phe87, Thr114, Met265, Cys288, etc. on the receptor often interact with ligands. The indole ring and N-ethyl morpholine moiety in ligand molecules are often the sites of interaction. The indole moiety often created arene–H interactions with Phe87. The nitrogen atom of morphine often established a hydrogen bond with Thr114. The methylene hydrogen atom in the N-ethyl morpholine moiety often interacts with the sulfur atom in Met265 or Cys288. These docking results confirmed the rationality of the compounds containing both indole and N-ethyl morpholine moieties. The docking results also showed that the presence of active hydrogen on the nitrogen atoms of the amide would facilitate the binding of the compound to the receptor. This confirms our previous speculation about the reasons for the activity of these compounds. For example, for compound 34 and compound 26, the subtle structural difference between the two is whether there is active hydrogen on the amide nitrogen atom. But the activities of the two are quite different. The docking results showed that active hydrogen on the amide nitrogen atom in compound 34 formed hydrogen bond interaction with Thr114, while compound 26 did not (Fig. 4). The amide moieties of other compounds have similar interactions with the receptor. This confirms our previous speculation about the reasons for the activity of these compounds.
Fig. 4. 3D schematic diagram of the interaction pattern of compound 34 (A) and compound 26 (B) with the human CB2 receptor, respectively.

From the docking results, we can find that methoxy groups sometimes participate in the interaction with the CB2 receptor, and sometimes not. This also explains why there is no regularity in the activity of 5-methoxy substituted compounds. However, the methoxy group involved in the CB2 receptor interaction is not the oxygen atom we initially speculated but the hydrogen atom. For example, the hydrogen atom in the methoxyl group in compound 4 forms an arene–H interaction with Trp258, while compound 48 does not (Fig. 5). At the same time, the methoxy group requires a larger space than the halogen atom, so the position of the 5-position methoxy-substituted compound in the CB2 receptor cavity is changed compared with other compounds. These changes also alter the interaction of other groups in the molecule with the CB2 receptor. This is another reason why the activity of the 5-methoxy substituted compound is irregular. For example, for compound 8, because of the presence of the methoxy group, the position of the molecule in the cavity of the CB2 receptor is changed, so that the amide group in the molecule cannot interact with the CB2 receptor like the amide group in compounds 9, 10 and 11 (Fig. 7).
Fig. 5. 3D schematic diagram of the interaction pattern of compound 4 (A) and compound 48 (B) with the human CB2 receptor, respectively.

Fig. 7. The 2D interaction patterns of compounds 8 (A), 9 (B), 10 (C) and 11 (D) with the human CB2 receptor.

The binding modes of compound 2 and GW405833 to the CB2 receptor are conducive to a deeper understanding of why compound 2 is slightly more active than GW405833. The docking posture shows that the indole ring of Trp258 interacts with the hydrogen atom of the 5-position methoxy group in compound GW405833 to form two arene–H interactions. In addition, the hydrogen of the methoxy group interacts with the phenyl ring of Phe117 to form an arene–H interaction. Thr114 forms hydrogen bonds with the nitrogen atoms of morpholine. For compound 2, its indole rings form arene–H interactions with Phe87 and Leu182, respectively. Phe183 forms an arene–H interaction with the hydrogen atom of methylene on the morpholine moiety, which immobilizes the stereoscopic conformation of morpholine, thus making the nitrogen atom on the morpholine group more prominent to Thr114, making it easier for them to form a hydrogen bond. The docking reports showed that the hydrogen bond distance between compound 2 and Thr114 was 3.10 Å, while that between GW405833 and Thr114 was 3.62 Å (Fig. 6).
Fig. 6. 3D schematic diagram of the interaction pattern of GW405833 (A) and compound 2 (B) with the human CB2 receptor, respectively.

2.4. In vivo effect of compound 2 on CFA-induced mechanical hyperalgesia associated with inflammation
Intraplantar injection of CFA into the hind paw of rats was used to develop a rat model for hyperalgesia associated with inflammation. A decreased hind paw withdrawal threshold (PWT) to a noxious mechanical stimulus was observed in our rat models.32 Hyperalgesic assessments were conducted at different time intervals within 24 hours. As shown in Fig. 8, a significant group effect (F = 141.1, P < 0.0001) in terms of the PWT value was observed with time and interaction effect (F = 55.54, P < 0.0001; F = 3.960, P < 0.0001). Rats in the CFA + vehicle group had severe hyperalgesia at every time point tested (P < 0.001, respectively). At 1 h, 3 h, 6 h and 9 h after CFA administration, the PWT values were notably increased in rats treated with GW405833 and compound 2 for the different doses tested. However, compound 2 and GW405833 had no anti-hyperalgesic effects at 24 h post CFA administration. compound 2 had a better anti-hyperalgesic effect compared to GW405833 at a dose of 10 mg kg–1, however this difference was not statistically significant. At the 1 h time point, the dose-dependent reversal of hyperalgesia for compound 2 had an estimated ED50 value of 1.097 mg kg–1. Furthermore, the CB1 antagonist (AM281) and CB2 antagonist (SR144528) were used to investigate the inhibition of compound 2 on CFA-induced mechanical hyperalgesia through the CB1 receptor or CB2 receptor. The result in Fig. 8C found that the CB1 antagonist, AM281, did not change the anti-inflammatory hyperalgesic effect of compound 2, being proved by no statistical difference between the compound 2-treated group and compound 2 + AM281-treated group. As expected, the compound 2-treated group had a great statistical difference with the compound 2 + SR144528-treated group (P < 0.001), showing that SR144528 largely lessened the anti-inflammatory hyperalgesic effect of compound 2. In addition, a significant difference between the compound 2-treated group and compound 2 + SR144528-treated group (P < 0.001), and no statistical difference between the compound 2 + SR144528-treated group and SR144528-treated group respectively confirm these points above. These suggested that compound 2 had an anti-inflammatory hyperalgesic effect through the CB2 receptor, but not the CB1 receptor.
Fig. 8. Compound 2 reduces mechanical hyperalgesia associated with inflammation. Animals received an intraplantar injection of 50% CFA (50 μL diluted in 0.9% saline) or saline into the left hind paw. 24 h post injection, rats in the test compound groups were administered with compound 2, GW405833, AM281, SR144528 or their combination. Post-drug PWTs were determined and recorded versus time. (A) Quantitative analysis of reversal at different times after CFA induction (n = 6). (B) The dose–response curve for the reversal of mechanical hyperalgesia by compound 2 is shown for the 1 h time point (n = 6). (C) Quantitative analysis of reversal after different treatments (n = 6). Data were analyzed using one-way analysis of variance (ANOVA) followed by post hoc Tukey's test: ***P < 0.001, **P < 0.01 and *P < 0.05 vs. CFA-vehicle group (A); ***P < 0.001, No: no statistical difference (C).

2.5. In vivo effect of compound 2 on CFA-induced pro-inflammatory cytokine production
Numerous experimental studies have demonstrated that pro-inflammatory cytokines induce or facilitate inflammatory hyperalgesia.33–35 Levels of three critical pro-inflammatory cytokines (interleukin (IL)-1β, IL-6 and tumor necrosis factor (TNF)-α) were measured from paw skin samples. As shown in Fig. 9, the levels of IL-1β, IL-6 and TNF-α were approximately 2.0, 3.3 and 2.3 times higher in rats in the CFA-vehicle group compared to those in rats in the sal-vehicle group. Administration with compound 2 (10, 1 and 0.1 mg kg–1) decreased these pro-inflammatory cytokines in a dose-dependent manner. The levels of IL-1β, IL-6 and TNF-α significantly decreased up to 1.3, 1.9 and 1.1 times the normal after high doses of compound 2 administration respectively. Although there was no statistical difference, compound 2 at a dose of 10 mg kg–1 decreased the IL-1β, IL-6 and TNF-α levels to much lower levels compared to GW405833 (1.4, 2.5 and 1.6 times the normal respectively). However, although compound 2 at a dose of 0.1 mg kg–1 exhibited great inhibition on inflammatory hyperalgesia, there was no obvious effect on the level of pro-inflammatory cytokines, which needs further investigation. Moreover, results in Fig. 9D showed that the compound 2-treated group had no statistical difference with the compound 2 + AM281-treated group, while there was a remarkable difference between the compound 2 + AM281-treated group and AM281-treated group (P < 0.05), suggesting that the CB1 antagonist, AM281, did not inhibit the anti-inflammatory effect of compound 2. In contrast, the CB2 antagonist, SR144528, significantly weakened the inhibition of compound 2 on the level of IL-1β, being reflected in a significant statistical difference between the compound 2-treated group and compound 2 + SR144528-treated group (P < 0.001), and no statistical difference between the compound 2 + SR144528-treated group and SR144528-treated group. Similar results were obtained from the effects on IL-6 and TNF-α (Fig. 9E and F). These findings suggested that compound 2 exerted suppression on these pro-inflammatory cytokines through the CB2 receptor, but not the CB1 receptor.
Fig. 9. Compound 2 inhibits CFA-induced pro-inflammatory cytokine secretion in the paw skin of hyperalgesic rats. Animals were administered with an intraplantar injection of 50% CFA (50 μL diluted in 0.9% saline) or saline into the left hind paw. 24 h post injection, rats in the test compound groups were administered with compound 2, GW405833, AM281, SR144528 or their combination. Pro-inflammatory cytokines were measured after 1 h post drug administration. Quantitative analysis of expression levels for the pro-inflammatory cytokines (A) and (D) IL-1β, (B) and (E) IL-6, and (C) and (F) TNF-α (n = 6). Data were analyzed using one-way analysis of variance (ANOVA) followed by post hoc Tukey's test: ***P < 0.001, *P < 0.05 and No: no statistical difference. Sal, saline.

3. Conclusions
In summary, we designed and synthesized a series of indole compounds with N-ethyl morpholine moieties as CB2 receptor agonists. In vitro activity and molecular docking studies indicate that: (1) the indole ring and N-ethyl morpholine moiety are beneficial to the interaction between the ligand and CB2 receptor. (2) Active hydrogen on the nitrogen atom of amide promotes the binding of the compound to the CB2 receptor. (3) As the size of the ring of the substituted cycloalkyl group on the amide increased, the agonistic activity of the compound on CB2 decreased until no activity was observed. (4) The effect of the 5-position substituent of the indole ring on the agonistic activity of the CB2 receptor is that F is superior to Br and to Cl. (5) The N-ethyl morpholine moiety located at the 1 position of the indole skeleton has similar agonistic activity for CB2 to the N-ethyl morpholine moiety located at the 3 position of the indole skeleton. The most active, i.e. compound 2, had exceptional CB2 receptor selectivity. Furthermore, compound 2 significantly ameliorated inflammatory hyperalgesia within 12 hour post-administration. In addition, pro-inflammatory cytokines were significantly inhibited after treatment with compound 2 in the CFA-induced lesions. These effects were due to its CB2 receptor agonistic action. Consequently, our methods and results may assist medicinal chemists to synthesize similar compounds that have enhanced CB2 agonistic activity profiles.
4. Experimental section
4.1. Chemistry
4.1.1. General procedure for the synthesis of intermediate compounds 2a–2d (ref. 36)
Sodium acetate (3.61 g, 44 mmol) was dissolved in 1 M acetic acid solution at room temperature, and then 4-substituted phenylhydrazine hydrochloride (40 mmol) was added. The mixture was dissolved on heating at 70 ° C. Afterwards, ethyl levulinate (6 mL, 48 mmol) was added dropwise to the reaction solution, and stirred for 5–10 min. Heat was then removed, and the reaction was cooled to room temperature and then filtered. The filter cake was washed with water and then dried. The dried solid was added to 100 mL of H2SO4/EtOH solution and refluxed for 3–4 h. The reaction was analyzed by TLC. After the reaction was complete, the reaction solution was concentrated to 50%, and the pH was adjusted to 6 with 1 M NaOH at 0 ° C. The mixture was then extracted with CH2Cl2, washed with brine, and then dried with anhydrous sodium sulfate. The product was purified by flash chromatography on silica gel (CH2Cl2/CH3OH, gradient elution).
4.1.2. General procedure for the synthesis of intermediate compounds 3a–3d (ref. 37)
6 N NaOH(aq) (9 mL) was added to 100 mL of ethanol, and then the product from the previous step (39 mmol) was added at room temperature. After stirring for 3 h, 50 mL of water was added to terminate the reaction and then ethanol was vacuum evaporated. The pH was adjusted to 3 with 6 N HCl, then extracted with ethyl acetate, washed with brine, and dried with anhydrous sodium sulfate. The product was purified by flash chromatography on silica gel (CH2Cl2/CH3OH, gradient elution).
4.1.3. General procedure for the synthesis of intermediate compounds 4a–4d (ref. 38 and 39)
Compounds 3a–3d (6.8 mmol) were dissolved in 20 mL of DMF, and EDC·HCl (1.57 g, 8.2 mmol) and a catalytic amount of DMAP were added at 0 °C and stirred at 0 ° C for 20–30 min. Morpholine (7.5 mmol) was then added and the temperature was slowly increased to room temperature while stirring overnight. The reaction was detected using TLC. The reaction was quenched with 40 mL water after completion. The mixture was then extracted with ethyl acetate, washed with 1 N HCl, 1 N NaOH, and water, and then finally dried with anhydrous sodium sulfate. The product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, gradient elution).
4.1.4. General procedure for the synthesis of intermediate compounds 5a–5d (ref. 37 and 39)
The reaction was performed at 0 ° C, under an Ar2 atmosphere. LiAlH4 (70 mg, 1.87 mmol) and 5 mL anhydrous THF were added to the reaction flask. A THF solution (4 mL) of compounds 4a–4d (0.35 mmol) was added dropwise at 0 °C. The reaction was stirred at room temperature for 2 days and then analyzed using TLC. After the reaction was completed, 70 μL water, 70 μL 15% NaOH(aq), and 210 μL water were added slowly at 0 °C, stirred for 5 min and then filtered using a short Celite plug. The filtrate was extracted with diethyl ether, washed with brine, and dried with anhydrous sodium sulfate. The product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, gradient elution).
4.1.5. General procedure for the synthesis of target compounds 1–7 (ref. 25)
The reaction was performed at –78 ° C, under an Ar2 atmosphere. HMPA (195 μL, 1.12 mmol) was added to 3.5 mL anhydrous THF solution for the last step compounds (5a–5d) (0.37 mmol). Then 0.83 mL KHMDS (0.5 M in toluene solution) was added dropwise. The mixture was stirred at –22 ° C for 30 min. Then, substituted acid chloride (0.57 mmol) was added at –78 ° C, and then stirred at room temperature for 2–3 h. TLC was then performed. After the reaction was completed, 9 mL saturated NaHCO3(aq) was added. The mixture was extracted with ethyl acetate, washed with brine, and dried with anhydrous sodium sulfate. The product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, gradient elution).
4.1.6. General procedure for the synthesis of intermediate compounds 6a–6d (ref. 40)
The reaction was performed at 0 ° C, under an Ar2 atmosphere. 5a–5d (1.79 mmol), NaH (75 mg, 2.51 mmol) and 15 mL anhydrous DMF were added to the reaction flask and stirred. After 30–50 min, tert-butyl bromoacetate (524 μL, 3.58 mmol) was added at room temperature, and then heated to 50 ° C for 12 h and analyzed using TLC. The flask was then cooled down to 0 °C, and the reaction was quenched with 10 mL ethyl acetate. 30 mL water was added to the mixture, which was then extracted with ethyl acetate, washed with water and brine, and then finally dried with anhydrous sodium sulfate. The product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, gradient elution).
4.1.7. General procedure for the synthesis of intermediate compounds 7a–7d (ref. 40)
1 mmol compounds 6a–6d and 15 mL CH2Cl2/CF3COOH (v : v = 1 : 1) mixed solution were stirred at room temperature for 16 h, and then analyzed using TLC. After completion, the solvent was removed under vacuum. An equal volume of diethyl ether was added to the residue and then sonicated to slowly obtain a white solid. It was then filtered, washed with diethyl ether and dried. The product was recrystallized from diethyl ether.
4.1.8. General procedure for the synthesis of target compounds 8–35 (ref. 38)
Compounds 7a–7d (0.99 mmol) were dissolved in 5.5 mL DMF at 0 °C. Then, EDC·HCl (380 mg, 1.98 mmol) and a catalytic amount of DMAP were added sequentially and stirred for 30 min before a nitrogen-containing compound (1.19 mmol) was added. The reaction was stirred overnight at room temperature and analyzed using TLC. After reaction completion, 11 mL water was added. The mixture was extracted with ethyl acetate, washed with water and brine, and dried with anhydrous sodium sulfate. The product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, gradient elution).
4.1.9. General procedure for the synthesis of intermediate compounds 8a–8c (ref. 41)
The reaction was performed at 0 ° C, under an Ar2 atmosphere. 2a–2c (12.13 mmol), NaH (510 mg, 16.98 mmol) and 60 mL anhydrous DMF were added to the reaction flask and stirred. After 30–40 min, N-(2-chloroethyl) morpholine (2.18 g, 14.57 mmol) was added, and then heated to 45 ° C. The reaction was stirred at 45 ° C for 4–5 h and then analyzed using TLC. After completion, the flask was cooled to 0 °C. The reaction was quenched with 20 mL ethyl acetate. After ethyl acetate and DMF were removed under vacuum, water was added to the mixture (the volume of water was about twice the residue), which was then extracted with ethyl acetate, washed with water and brine, and then dried with anhydrous sodium sulfate. The product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, gradient elution).
4.1.10. General procedure for the synthesis of intermediate compounds 9a–9c (ref. 41)
Compounds 8a–8c (1 mmol) were dissolved in 3 mL ethanol, and then 6 N NaOH (0.27 mL) was added. The reaction was stirred for 3 h at room temperature. Afterwards ethanol was removed under vacuum. Then 5 mL water and 5 mL ethyl acetate were added to the mixture. The pH was adjusted to 5–6 with 1 N HCl at 0 °C. The large amount of white solid that was produced was filtered and dried.
4.1.11. General procedure for the synthesis of target compounds 36–56 (ref. 38)
Compounds 9a–9c (1 mmol) were dissolved in 5 mL DMF at 0 °C. Then, EDC·HCl (269 mg, 1.4 mmol) and a catalytic amount of DMAP were added sequentially. It was then stirred for 30 min and a nitrogen-containing compound (1.2 mmol) was added at 0 °C. The reaction was stirred overnight at room temperature and then analyzed using TLC. After reaction completion, 10 mL water was added. The mixture was extracted with ethyl acetate, washed with water and brine, and then dried with anhydrous sodium sulfate. The product was purified by flash chromatography on silica gel (EtOAc/petroleum ether, gradient elution).
4.2. Assessment of in vitro CB1 and CB2 receptor functional activities
A cAMP Hunter™ assay enzyme fragment complementation chemiluminescence detection kit was used to determine the functional activity of CB1 or CB2 receptor-expressing cell lines.42 In brief, CHO cells expressing human CB2 or CB1 (Euroscreen) donated by Professor Hui Ji (China Pharmaceutical University, Nanjing, China) were plated at a density of 5000 cells per well in 384-well plates and incubated overnight at 37 °C. The medium was then removed and the cells were treated with test compounds diluted in stimulation buffer containing 1 mM IBMX, 0.25% BSA and 10 lM forskolin. The cells were incubated for 30 min at 37 °C and then lysed and cAMP concentrations were measured using a DiscoveRx XS+ cAMP kit, following the manufacturer's instructions. The maximal amount of cAMP produced by forskolin compared to the level of cAMP inhibited by 200 nM CP-55940 was defined as 100%. Using a four-parameter logistic model, the EC50 value for each test compound was determined as the concentration at which 50% of the forskolin-stimulated cAMP produced was inhibited. The reproducibility of the cAMP assays was assessed using the control compound CP-55940 which was replicated twice on every assay plate to rule out plate artefacts. Efficacy was expressed as a percentage relative to the efficacy of CP-55940. Each compound was tested in triplicate for each dilution. The results are the mean values of the measurements where the individual values for the compounds did not differ by more than a factor of three from the mean.
4.3. In vivo evaluation of inflammatory hyperalgesia and pro-inflammatory cytokine secretion
4.3.1. Animal care
Male Sprague-Dawley rats (180–200 g) were purchased from Jiangning Qinglongshan Animal Cultivation Farm (Nanjing, China). The rats were bred under standard laboratory conditions and housed in a 12 h light/dark schedule with free access to food and water. All animal protocols (No: 2017000037, 03/06/2017) were approved by the China Pharmaceutical University in accordance with the guidelines of the National Institutes of Health throughout the study.
4.3.2. Inflammatory hyperalgesia
The complete Freund's adjuvant (CFA) model was established to investigate the efficacy of compound 2 on hyperalgesia with inflammation in rats. Hind paw withdrawal thresholds (PWTs) to a noxious mechanical stimulus were measured using an analgesiometer (model 7200; Ugo Basile, Varese, Italy). The back of the rat's left foot was put under the head of an electronic tenderness apparatus, which then began to apply pressure. The experimenter stopped the pressure when the animals neighed or struggled in pain, and recorded the value of PWT simultaneously. The cut-off was set at 250 g and the endpoint was taken as complete paw withdrawal. Baseline measurements were determined, and the rats were anesthetized with isofluorane and received an intraplantar (i.pl.) injection of 50% CFA (50 μL diluted in 0.9% saline) to the left hind paw. After baseline measurements, 50% CFA (50 μL diluted in 0.9% saline) was injected into the left hind paw via the intraplantar (i.pl.) route. Twenty-four hours following CFA injection, pre-drug PWT was determined. Immediately afterwards, the rats received a single dose of 10, 2.5, 1, 0.625, 0.156, 0.1, 0.039, 0.01 or 0.0025 mg kg–1 of compound 2, 10 mg kg–1 GW405833 (positive control) or 2 mL kg–1 vehicle via the intra-peritoneal (i.p.) route. Post-drug PWT values were measured at different time points between 1 and 24 h post drug administration. Percent reversal of hyperalgesia for each rat was calculated using (postdose threshold – predose threshold)/(baseline threshold – predose threshold) × 100%.
4.3.3. Measurement of IL-1β, IL-6 and TNF-α cytokine levels
After 1 h post drug administration, rats were euthanized and paw skin samples were harvested for pro-inflammatory cytokine measurements. The samples were homogenized in ice-cold buffer with a protease inhibitor followed by centrifugation. The supernatants from paw skin homogenates were used to determine the concentration of IL-1β, IL-6 and TNF-α using enzyme-linked immunosorbent assay (ELISA) kits (Biolegend, United States) based on the manufacturer's instructions.
4.4. Molecular docking studies
The crystal structure of the human cannabinoid receptor CB2 (PDB ID: 5ZTY) was retrieved from the Protein Data Bank server (; www.pdb.org).
The protein preparation module of MOE is used to hydrogenate and protonize the protein, and to correct errors, so as to optimize the treatment before docking and remove the water molecules in the protein. A virtual cavity was built, all small molecular ligands were prepared, and MOE's Dock module to induced docking was used.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The project was supported by The Natural Science Foundation of the Jiangsu Higher Education Institutions of China (No. 17KJB350002, 17KJD520002), Science Foundation of Huaihai Institute of Technology (No. Z2015012, Z2015015), Jiangsu Key Laboratory of Marine Pharmaceutical Compound Screening (No. HY2015B03, HY201703) Haiyan Plan of Jiangsu Province Lianyungang City in 2017 and the Fifth progress of “521” Project Funding of Lianyungang City in 2017.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c9md00173e
References
- Pertwee R. G. Addiction. 1999;94:317–320. doi: 10.1080/09652149933801. [DOI] [PubMed] [Google Scholar]
- Matsuda L. A., Lolait S. J., Brownstein M. J., Young A. C., Bonner T. I. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- Munro S., Thomas K. L., Abu-Shaar M. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- Ryberg E., Larsson N., Sjögren S. Br. J. Pharmacol. 2007;152:1092–1101. doi: 10.1038/sj.bjp.0707460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann A. G., Suplita R. L., Bolton N. M. Nature. 2005;435:1108. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- Shao Z., Yin J. Nature. 2016;540:602–606. doi: 10.1038/nature20613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Hua T., Vemuri K. Cell. 2019;176:1–9. [Google Scholar]
- Howlett A. C. Prostaglandins Other Lipid Mediators. 2002;68:619–631. doi: 10.1016/s0090-6980(02)00060-6. [DOI] [PubMed] [Google Scholar]
- Rajesh M., Mukhopadhyay P. Br. J. Pharmacol. 2008;153:347–357. doi: 10.1038/sj.bjp.0707569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picone R. P., Kendall D. A. Mol. Endocrinol. 2015;29:801–813. doi: 10.1210/me.2015-1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scutt A., Williamson E. M. Calcif. Tissue Int. 2007;80:50–59. doi: 10.1007/s00223-006-0171-7. [DOI] [PubMed] [Google Scholar]
- Arevalo-Martin A., Vela J. M. J. Neurosci. 2003;23:2511–2516. doi: 10.1523/JNEUROSCI.23-07-02511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D., Pryce G. Nature. 2000;404:84–87. doi: 10.1038/35003583. [DOI] [PubMed] [Google Scholar]
- Kenneth D., Bromberg A. M. Science. 2008;320:903–909. doi: 10.1126/science.1152662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratzke C., Streng T. J. Urol. 2011;185:731–736. doi: 10.1016/j.juro.2010.09.080. [DOI] [PubMed] [Google Scholar]
- Belvisi M. G., Patel H. J. Br. J. Pharmacol. 2008;155:547–557. doi: 10.1038/bjp.2008.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquini S., Mugnaini C. J. Med. Chem. 2012;55:5391–5402. doi: 10.1021/jm3003334. [DOI] [PubMed] [Google Scholar]
- Soethoudt M., Grether U., Fingerle J. Nat. Commun. 2017;8:13958. doi: 10.1038/ncomms13958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales P., Hernandez-Folgado L., Goya P. Expert Opin. Ther. Pat. 2016;26:843–856. doi: 10.1080/13543776.2016.1193157. [DOI] [PubMed] [Google Scholar]
- Chadha N., Silakari O. Eur. J. Med. Chem. 2017;134:159–184. doi: 10.1016/j.ejmech.2017.04.003. [DOI] [PubMed] [Google Scholar]
- Pagé D., Yang H., Brown W., Walpole C., Fleurent M., Fyfe M. Bioorg. Med. Chem. Lett. 2007;17:6183–6187. doi: 10.1016/j.bmcl.2007.09.019. [DOI] [PubMed] [Google Scholar]
- Diaz P., Xu J., Astruc-Diaz F., Pan H.-M., Brown D. L., Naguib M. J. Med. Chem. 2008;51:4932–4947. doi: 10.1021/jm8002203. [DOI] [PubMed] [Google Scholar]
- D'Ambra T. E., Estep K. G., Bell M. R., Eissenstat M. A., Josef K. A. J. Med. Chem. 1992;35:124–135. doi: 10.1021/jm00079a016. [DOI] [PubMed] [Google Scholar]
- Ross R. A., Brockie H. C., Stevenson L. A., Murphy V. L., Templeton F., Makriyannis A., Pertwee R. G. Br. J. Pharmacol. 1999;126:665–672. doi: 10.1038/sj.bjp.0702351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallant M., Gareau Y., Guay D., Labelle M. and Prasit P., Indole derivatives with affinity for the cannabinoid receptor, US5532237, 1996.
- Frost J. M., Dart M. J., Tietje K. R., Garrison T. R., Grayson G. K., Daza A. V. J. Med. Chem. 2008;51:1904–1912. doi: 10.1021/jm7011613. [DOI] [PubMed] [Google Scholar]
- Wrobleski S. T., Chen P., Hynes J. J., Lin S., Norris D. J., Pandit C. R. J. Med. Chem. 2003;46:2110–2116. doi: 10.1021/jm020329q. [DOI] [PubMed] [Google Scholar]
- Pasquini S., Mugnaini C., Ligresti A. J. Med. Chem. 2012;55:5391–5402. doi: 10.1021/jm3003334. [DOI] [PubMed] [Google Scholar]
- Zindell R., Riether D., Bosanac T., Berry A., Gemkow M. J., Ebneth A. Bioorg. Med. Chem. Lett. 2009;19:1604–1609. doi: 10.1016/j.bmcl.2009.02.033. [DOI] [PubMed] [Google Scholar]
- Ermann M., Riether D. Bioorg. Med. Chem. Lett. 2008;18:1725–1729. doi: 10.1016/j.bmcl.2008.01.042. [DOI] [PubMed] [Google Scholar]
- Ohta H., Ishizaka T., Tatsuzuki M. Bioorg. Med. Chem. Lett. 2007;17:6299–6304. doi: 10.1016/j.bmcl.2007.09.004. [DOI] [PubMed] [Google Scholar]
- Valenzano K. J., Tafesse L., Lee G. Neuropharmacology. 2005;48:658–672. doi: 10.1016/j.neuropharm.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Rabelo T. K., Guimarães A. G., Oliveira M. A. Int. Immunopharmacol. 2016;39:97–105. doi: 10.1016/j.intimp.2016.07.016. [DOI] [PubMed] [Google Scholar]
- Singh A. K., Vinayak M. Phytother. Res. 2016;30:1164–1171. doi: 10.1002/ptr.5624. [DOI] [PubMed] [Google Scholar]
- Da S. M. D., Bobinski F., Sato K. L. Mol. Neurobiol. 2015;51:19–31. doi: 10.1007/s12035-014-8790-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menciu C., Duflos M., Fouchard F. J. Med. Chem. 1999;42:638–648. doi: 10.1021/jm981079+. [DOI] [PubMed] [Google Scholar]
- Elliott S. J. Am. Chem. Soc. 1955;77:4319–4324. [Google Scholar]
- Collins J. L., Shearer B. G., Oplinger J. A. J. Med. Chem. 1998;41:2858–2871. doi: 10.1021/jm980072p. [DOI] [PubMed] [Google Scholar]
- Kalgutkar A. S., Crews B. C., Saleh S. Bioorg. Med. Chem. 2005;13:6810–6822. doi: 10.1016/j.bmc.2005.07.073. [DOI] [PubMed] [Google Scholar]
- Steven H., Barbara P., Salvatore A., Marcello D. F. J. Med. Chem. 2005;48:1314–1317. [Google Scholar]
- Rulin Z., Bei W., Hong W., John Jr. H. ARKIVOC. 2010;vi:89–95. [Google Scholar]
- Huang Z., Wang H., Wang J. Sci. Rep. 2016;6:29757. doi: 10.1038/srep29757. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.