

 Close-shelled carbocations DNA intercalators Pr-ADOTA and Pr-DAOTA are very cytotoxic against the cancer cell lines MDA-MB-231 (breast) and HCT116 (colon).
Close-shelled carbocations DNA intercalators Pr-ADOTA and Pr-DAOTA are very cytotoxic against the cancer cell lines MDA-MB-231 (breast) and HCT116 (colon).
Abstract
TOTA (trioxatriangulenium ion) is a close-shelled carbocation known to intercalate strongly with the DNA double helix (J. Reynisson, G. B. Schuster, S. B. Howerton, L. D. Williams, R. N. Barnett, C. L. Cleveland, U. Landman, N. Harrit, J. B. Chaires, J. Am. Chem. Soc. 2003, 125, 2072). The cytotoxicity of TOTA and its four close structural analogues, ADOTA, Pr-ADOTA, Pr-DAOTA and n-Butyl-TATA were tested against the breast cancer cell line MDA-MB-231 and colon cancer cell line HCT116. The most potent derivatives Pr-ADOTA and Pr-DAOTA had IC50 values of ∼80 nM for MDA-MB-231 but slightly higher for HCT116 in the low hundreds nM range. A 3D model assay of HCT116 spheroids was also used, mimicking a tumour environment, again both Pr-ADOTA and Pr-DAOTA were very active with IC50 values of 38 nM and 21 nM, respectively. Molecular modelling suggest that the planar derivatives intercalate between the base pairs of the DNA double helix. However, only modest DNA double stranded DNA cleavage was observed using the γH2AX assay as compared to camptothecin, a topoisomerase I poison suggesting a different mechanism. Finally, a robust density functional theory (DFT) model was built to predict the pKR+ stability values, i.e., to design derivatives, which predominantly have a non-intercalating buckled form in healthy tissues followed by a nucleophilic attach of water on the central carbon, but a planar form at relatively low pH values rendering them only cytotoxic in the interior of tumours.
Introduction
Trioxatriangulenium ion (TOTA, 4,8,12-trioxa-4,8,12,12c-tetrahydrodibenzo[cd,mn]pyrene, Scheme 1) is a closed-shell planar carbocation with its positive charge delocalised throughout the molecule.1 It has interesting biological properties such as being an excellent DNA intercalator, which was demonstrated with a number of techniques using calf thymus DNA (CT-DNA).2 Titration of TOTA with CT-DNA monitored by UV-vis absorption and fluorescence showed clear evidence of ground state complexation, which was supported by increase in the melting point (Tm) of an 12-mer oligonucleotide from 52.8 to 57.3 °C.2 Viscosity measurements with CT-DNA indicated that intercalation was the preferred binding mode, groove binding does not lead to increase viscosity whereas intercalation does by stiffening and elongating the DNA double helix.2TOTA is an achiral molecule but in the presence of CT-DNA, a chiral macromolecule, an induced signal was observed in a circular dichroic (CD) spectra, the relative weakness suggesting intercalation.2 Finally, the X-ray structure of TOTA intercalated into a hexameric duplex d[CGATCG]2 confirmed this binding mode.2 A binding dialysis analysis using a collection of different oligonucleotides revealed that TOTA has a strong preference for guanine–cytosine rich double stranded DNA segments compared to adenine–thymine as well as strong affinity for triplex DNA and M. lysodeikticus bacterial DNA.2 The binding constant of KB = 4.1 × 104 M–1 was derived from the UV-vis and dialysis experiments.2 Interestingly, photo induced DNA damaged by electron transfer to TOTA from a duplex oligonucleotide was reported supporting the evidence of intercalation since overlap of the π-orbitals of the bases and TOTA is necessary to facilitate charge transfer.3 Also, using electrospray ionisation mass spectrometry with duplex DNA the stoichiometry and fragmentation patterns observed were commensurate with an intercalative binding mode of TOTA.4
Scheme 1. Nucleophilic attack on the central carbon atom of TOTA, providing leuco ether TOTA-OR.

Another interesting aspect of the propertied of TOTA is that it can undergo nucleophilic attack on its central carbon atom from alcohol solvents to form leuco ethers (TOTA-OR) as shown in Scheme 1.5 However, this reaction is slow, in the timescale of hours and attempts to measure equilibria using UV-vis was not successful.5 In order to derive meaningful equilibrium constants 1H-NMR was used (K = [TOTA-OR][D+]/[TOTA+][ROD]) with the methanol equilibrium constant reported as K = 1.38 × 10–6 and for ethanol at K = 1.06 × 10–5.5 Considering that K = kforward/kreverse strongly suggest that TOTA is a very stable cation.5 Additionally, no formation of the corresponding leuco alcohol was seen when the experiment was conducted in water. The formation of the TOTA-OR leuco ethers resulted in a 1 ppm upfield shift of the 1H-NMR absorption peaks caused by a structure alteration to a “buckled” or “umbrella” shape of the molecule.5
In general, potent DNA intercelators, such as ethidium bromide, have a flat polycyclic moiety lodged between the base pairs of the double helix.6 It can therefore be reasoned that the buckled TOTA-OR is less likely, or even incapable, of intercalating into DNA base pair stack. It can also be argued that the formation of the leuco alcohol can be facilitated by increasing the pH of the solution, i.e., the hydroxyl ion is a much better nucleophile than a water molecule. Therefore, it should be possible to design TOTA derivatives, which are more susceptible to a nucleophilic water attack by, e.g., substituting some of the hydrogen atoms with electron withdrawing groups. Until now most of the effort on TOTA analogues has been on making them more stable.7,8 Interestingly, TOTA and its derivatives have been developed and used for various applications including biosensor probes due their excellent photo-physical properties.8E.g., endocytosis has been studied utilising the pH difference in endo- and lysosomes as compared to the general cytosol.9 Considering that the environment within solid tumours is acidic,10,11 differing to blood and other tissues that have a close to neutral pH, a TOTA derivative can therefore be conceived that only adopts the planar DNA intercalating shape in the acidic environment leaving it in the benign buckled leuco alcohol form in healthy tissues. Therefore, in theory, the pH gradient can be used to selectively target solid tumours for DNA damage (see ref. 10 and references therein). This is similar to the approach taken where the hypoxic nature solid tumours is used for bio-reduction of, e.g., the well-established alkylating agent mitomycin C and the experimental drug tirapazamine.12–15 Regrettably, small molecular therapy using classical DNA intercalating therapeutic agents such as mitomycin C and doxorubicin for treating solid tumours is not as effective cure as required in clinical practice.16 Therefore, there is still an acute need for selective and potent therapeutic agents for treating solid tumours.
In order to test the idea whether TOTA can act as a lead compound in an anticancer drug discovery project two main questions needed to be answered. First, is TOTA cytotoxic to cancer cells resulting in DNA damage and, second, does the leuco buckled form intercalate into the DNA helix? To this end, we synthesised TOTA and its known derivatives shown in Fig. 1 and Scheme 1, conducted viability experiments on two cancer cell lines and finally molecular modelling of the derivatives to the DNA double helix to elucidate their modes of action.
Fig. 1. Structure of TOTA and its analogues, ADOTA, Pr-ADOTA and Pr-DAOTA.

Results
Synthesis
TOTA was made using the methodology by Sabacky et al.17 In order to generate a preliminary structural activity relationship (SAR) four well-known analogues of TOTA were synthesised; ADOTA (azadioxatrianguelnium), Pr-ADOTA (propyl-azadioxatrianguelnium), Pr-DAOTA (propyl-diazaoxatriangulenium, Fig. 1) and n-butyl-TATA (n-butyl-triazatriangulenium, Scheme 2) using established methodology.18 Furthermore, the leuco alcohol and ether derivatives TOTA-OH, TOTA-OMe, and TOTA-OEt were prepared for comparison to the planar carbocations. TOTA-OH was obtained through the reaction of tris(2,6-dimethoxyphenyl)methylium tetrafluoroboronate 1 with pyridine·HCl followed by treatment with sodium hydroxide, TOTA-OH was subsequently reacted with methanol and ethanol, providing TOTA-OMe (88%) and TOTA-OEt (64%), respectively. The reaction pathways are shown in Scheme 2.
Scheme 2. Synthesis of TOTA-OH, TOTA-OMe, TOTA-OEt and n-butyl-TATA.

Cell proliferation
Two cancer cell lines HCT116 (colon) and MDA-MB-231 (breast) were used to test the potency of TOTA and its derivatives using the thymidine uptake assay.19 The results are shown in Fig. 2. Unfortunately, ADOTA only showed marginal effect on the cancer cells most likely due to its insolubility (see Fig. S1 in the ESI†). It can be argued that the pKa of ADOTA's labile proton is similar to of pyridine (pKa – 5.2),20 both systems being highly conjugated. Using the density functional theory (DFT) the proton affinities of pyridine (–222.5 kcal mol–1) and ADOTA (–245.1 kcal mol–1) were derived and assuming a linear correlation the pKa value of 5.7 was determined.21 Therefore, at physiological pH (∼7.4) ADOTA is predominantly neutral and poorly soluble in water-based solutions.
Fig. 2. Relative anti-proliferative activity of TOTA and its derivatives against the breast MDA-MB-231 and colon HCT116 cancer cell lines. The mean percentages (%) at 10 μM as compared to untreated cells at 100% growth, i.e., the lower percentage numbers represent greater growth inhibition.

The y-axis in Fig. 2 depicts the thymidine uptake of the cells, the average percentages (%) compared to untreated cells, i.e. 100% growth is the control of untreated cells; thus, the lower the percentage number the greater the inhibition. DMSO is used as a negative control and does not affect the cell viability (see Fig. S1 in the ESI†). Healthy cells use thymidine for their proliferation and its consumption is therefore an excellent measure to gauge the vitality of the cells. As can be seen in Fig. 2, TOTA and its derivatives have a pronounced impact on the viability of the cell lines with less than 1% thymidine incorporation taking place as compared to untreated cells. The only exception to this was ADOTA (not depicted in the graph), which was found to only inhibit ∼12–25% of cell growth. Notable discrepancies are the results for TOTA-OEt and n-butyl-TATA for MDA-MB-231 with large error bars, which could be caused by relative insolubility of the compounds. Very similar results are obtained for TOTA, Pr-ADOTA and Pr-DAOTA reduction in viability as well as for n-butyl-TATA but only for the HCT116 cell line. The leuco derivatives are most likely reverted back to TOTA due its high carbenium stability pKR+ value of 9.1 (ref. 22) during the experiment explaining their high potency.
The quantitative effects on the cancer cells for TOTA, Pr-ADOTA and Pr-DAOTA were determined and their IC50 values (concentration leading to 50% inhibition of activity) were derived. The results are given in Table 1.
Table 1. IC50 values of TOTA, Pr-ADOTA and Pr-DAOTA against breast (MDA-MB-231) and colon (HCT116) cancer cell lines (SD – standard deviation).
| Compound | MDA-MB-231 | SD | HCT116 | SD |
| IC50 (nM) | IC50 (nM) | |||
| Camptothecin | 28.4 | 0.8 | 8.5 | 0.9 |
| TOTA | 448.5 | 46.5 | 707.5 | 64.5 |
| Pr-ADOTA | 87.5 | 37.6 | 256.5 | 3.5 |
| Pr-DAOTA | 77.5 | 27.0 | 106.5 | 5.5 |
From the data in Table 1, it is clear that Pr-DAOTA is overall the most potent of the derivatives with IC50 values around 100 nM for both cell lines closely followed by Pr-ADOTA, which has a similar value for MDA-MB-231 but is less active for HCT116. The least potent of the compounds is the parent TOTA with IC50 values in the mid to high hundreds nano-molar region, which in itself is quite cytotoxic. Camptothecin was used as a control, it is a standard topoisomerase 1 poison and its structural derivative, topotecan and irinotecan, are in clinical use.23 As can be seen in Table 1 camptothecin has better potency than the carbenium ions, in particular for the HCT116 cell line. Nevertheless, the Pr-ADOTA and Pr-DAOTA are very potent, they have not yet been optimised for potency and can therefore be regarded as hits, which need to be developed further.
To examine the bioactivity of the most potent compounds further, we tested the efficacy of the carbenium ions using a spheroid three-dimensional cluster of HCT116 colon cancer cells – a model that is used to mimic the conditions in solid tumours.24 Spheroids simulate physiological barriers to drug delivery in vivo thus serving as an improved assay format for testing efficacy.25
Spheroids develop a low intracellular pH of ∼6.3 and a reduced pH of ∼6.9 in restricted extracellular spaces at their core.26 The diameters of the spheroids lie in the range of 300–500 mm with a hypoxic core of quiescent cells thought to be responsible for resistance to chemo- and radiotherapies. The results for Pr-DAOTA and Pr-ADOTA are shown in Fig. 3.
Fig. 3. IC50 values for HCT116 cell line cultured as monolayer or spheroid are represented on the y-axis. The highest drug concentration is depicted where 50% growth inhibition was not reached.

From the results shown in Fig. 3 it is clear that both Pr-DAOTA and Pr-ADOTA are more active in the 3D model than in the monolayer format. Pr-DAOTA has IC50 of 21 nM in the spheroid as opposed to 111 nM in 2D and for Pr-ADOTA the effect is even more pronounced (IC50 3D 38 nM and 2D 321 nM). The parent TOTA had IC50 in excess of 1 μM (see Fig. S2 in the ESI†). This potency difference is easy to understand since Pr-DAOTA has additional two n-propyl groups and is the most active closely followed Pr-ADOTA with one n-propyl as compared with TOTA. The alkyl moieties increase the lipophilicity of Pr-DAOTA (log 4.0, see Table 4) and Pr-ADOTA (log P 3.3) compared to TOTA's (log P 2.7) and therefore aid their penetration into the spheroids. Since the pKR+ values of the carbenium ions are much higher than the physiological pH of the cells, it is very unlikely that the reason for the differences in efficacy are due to planar and umbrella forms.
Table 4. The bond dissociation energies (BDEs) of the bonds formed upon a nucleophilic attack on the carbocations in vacuum and in water in parenthesis. All values are in kcal mol–1.
| Analogue | OH– | H2O | MeOH | EtOH | pKR+ a |
| TOTA | –140.7 (–25.7) | 248.1 (285.2) | 246.3 (283.3) | 246.5 (283.3) | 9.1 |
| ADOTA | –132.6 (–17.9) | 256.2 (292.9) | x | x | y |
| Pr-ADOTA | –129.2 (–16.8) | 259.6 (294.1) | x | x | 14.5 |
| Pr-DAOTA | –117.9 (–8.0) | 270.9 (302.9) | x | x | 19.4 |
| n-Butyl-TATA | –109.0 (–2.0) | 279.7 (308.9) | x | x | 23.7 |
aRef: Laursen and Sørensen.22
DNA damage assay
TOTA is an excellent DNA intercalator and can induce DNA oligomer strand cleavage upon radiation.2,3 Furthermore, M2-DAOTA, containing two morpholinoethyl moieties, intercalates into the G-quadruplex structure of DNA forming π–π stacks with guanine residues observed by NMR.27 Also, M2- and Pr-ADOTA intercalate with high affinity of KB = 7.9 × 105 M–1 and KB = 8.1 × 106 M–1 respectively into CT-DNA.28 Thus, a plausible mechanism of action for the cations is DNA intercalation causing DNA damage. To test this hypothesis γ-phosphorylation of the histone protein H2AX was measured, which is a well-established method to quantify DNA double-strand breaks.29
The results are shown in Table 2 for TOTA, Pr-ADOTA and Pr-DAOTA in the MDA-MB-231 human breast cancer cell line. Camptothecin, a topoisomerase I poison was used as a reference compound and its administration resulted in 73.6% double strand break. The carbocations are much less effective even at a concentration ten times higher than camptothecin. Nevertheless, up to 57.5% strand breaks are seen for TOTA, with it being the most potent followed by Pr-ADOTA and with considerably less effective Pr-DAOTA. Considering the very low concentrations needed to impair the cancer cell viability (see Table 1 and Fig. 3) it is clear that DNA damage only plays a modest role in the efficacy of the cations.
Table 2. Induction of DNA double strand break in the MDA-MB-231 breast cancer cell line.
| Compound | Concentration (μM) | Average γH2AX (% of cells) | SD |
| Control | 6.1 | 0.7 | |
| Camptothecin | 1 | 73.6 | 0.6 |
| TOTA | 10 | 43.6 | 0.6 |
| Pr-ADOTA | 10 | 27.8 | 3.0 |
| Pr-DAOTA | 10 | 2.4 | 2.3 |
| TOTA | 5 | 57.5 | 1.3 |
| Pr-ADOTA | 5 | 30.6 | 0.9 |
| Pr-DAOTA | 5 | 1.3 | 0.2 |
| TOTA | 1 | 8.8 | 0.7 |
| Pr-ADOTA | 1 | 12.9 | 1.9 |
| Pr-DAOTA | 1 | 13.6 | 1.1 |
From competition binding studies by dialysis it is clear that TOTA had high affinity towards guanine rich double stranded DNA as well as triplexes and quadruplexes.2 Also, Pr-DAOTA has high affinity towards quadruplexes.28 Indeed, telomere strands have abundance of guanine, making them a potential target for TOTA. The telomere regions protect the genome from oxidative damage.30 The DNA triplex scaffold is associated with many biological processes such as gene expression, DNA damage and repair, RNA processing, folding and chromatin organisation and impair DNA polymerization and influence DNA recombination process (see ref. 31 and references therein). Finally, quadruplexes are associated with gene expression, DNA replication and telomere biology.32 Intercalation into any of these DNA scaffolds can therefore perturb the biological process associated with them and therefore on the viability of the cancer cells explaining the excellent efficacy of the carbocations. Obviously, these are only hypotheses on the mechanism of action and further work is required for their determination, which is outside the scope of this work.
Comparison of flow cytometry profiles following drug treatment for 16 h revealed that TOTA treated cells do not induce S-phase DNA damage, which is specific to camptothecin as shown in Fig. S3 in the ESI.† Interestingly, the dot plots presented in Fig. S3† indicate cell cycle arrest in the G1 and G2 phases and also it is possible that Pr-DAOTA is binding DNA very tightly and therefore halts the DNA damage response in the cells.
Molecular modelling
The crystal structure of TOTA intercalated into a DNA double strand is known with an excellent 1.55 Å resolution.2 Two TOTA molecules are sandwiched between two G–C base pairs in the [d(CGATCG)]2 × TOTA2 hexamer, interacting with the nucleotides via π–π stacking. The GOLD algorithm was used to dock TOTA, and its derivatives, between the base pairs using the four scoring functions available, i.e., GoldScore (GS),33 ChemScore (CS),34,35 piecewise linear potential (ChemPLP)36 and Astex Statistical Potential (ASP).37 To check the prediction power of the scoring functions TOTA was re-docked into its binding site and the root mean square deviation (RMSD) between the poses was derived. An excellent average RMSD value of 0.67 Å was obtained (GS = 0.66 Å, CS = 0.71 Å, ChemPLP = 0.34 Å, ASP = 0.98 Å) supporting the validity of the docking algorithm.
TOTA and eleven of its planar and buckled structures were docked into the intercalating site of the DNA crystal structure and their scores derived. The scores give arbitrary numbers with higher values indicating stronger affinity and the results are given in Table 3.
Table 3. The results of the four scoring functions for the planar and buckled TOTA analogues. GoldScore (GS), ChemScore (CS), piecewise linear potential (ChemPLP) and Astex Statistical Potential (ASP) were used.
| Compound | GS | CS | ChemPLP | ASP |
| TOTA | 70.7 | 16.2 | 77.7 | 42.9 |
| TOTA-OH | 61.4 | 14.3 | 59.0 | 31.8 |
| TOTA-OMe | 61.4 | 11.8 | 54.2 | 29.5 |
| TOTA-OEt | 61.5 | 12.7 | 53.5 | 28.4 |
| ADOTA | 72.7 | 17.3 | 79.6 | 47.0 |
| ADOTA-OH | 65.0 | 16.6 | 65.5 | 36.3 |
| Pr-ADOTA | 79.0 | 14.5 | 91.5 | 43.5 |
| Pr-ADOTA-OH | 70.0 | 14.7 | 64.1 | 31.7 |
| Pr-DAOTA | 82.0 | 15.2 | 97.0 | 42.1 |
| Pr-DAOTA-OH | 74.4 | 14.2 | 72.8 | 28.6 |
| n-Butyl-TATA | 101.7 | 18.7 | 119.2 | 40.6 |
| n-Butyl-TATA-OH | 88.9 | 16.5 | 100.8 | 34.8 |
Upon docking to DNA, some analogues fitted into the intercalator site with similar poses as TOTA, whilst others had different binding modes but with good predicted affinity. As expected, the planar carbocations had superior scores as compared to their umbrella shaped counterparts for all of the derivatives except for Pr-ADOTA's CS prediction. The superior scores for the planar form was true in particular for TOTA-OH, -OMe and -OEt as compared to TOTA.
Both GS and ChemPLP predict better binding for all the planar analogues (ADOTA, Pr-ADOTA, Pr-DAOTA and n-butyl-TATA) than TOTA, and all the hydroxy-umbrella analogues (ADOTA-OH, Pr-ADOTA-OH, Pr-DAOTA-OH, n-butyl-TATA-OH) than TOTA-OH. All the scoring functions predicted better binding with substitution of oxygen with nitrogen atoms, as exemplified by TOTA and TOTA-OHvs.ADOTA and ADOTA-OH, respectively. While GS and ChemPLP predicted better binding of analogues with a propyl substituent on the nitrogen, over hydrogen, CS and ASP both predicted that the compounds with propyl substituents on the nitrogen atoms binds less tightly to the intercalator site. In general, GS and ChemPLP predicted compounds with more nitrogen atoms bind more effectively than with fewer, whilst this trend was not observed for the CS and ASP algorithms. The predicted binding mode of Pr-DAOTA, as compared to TOTA, is shown in Fig. 4.
Fig. 4. (A) The docked configuration of Pr-DAOTA in the binding site of DNA using the ChemPLP scoring function. Pr-DAOTA shown in the binding pocket overlain with TOTA in green. (B) π–π stacking depicted as purple lines between the ligand and the base pairs.

Thermochemistry
In order to understand mechanism of the nucleophilic attack on the central carbon of the cations DFT calculations were performed and the thermochemical profiles of the reactions derived. As can be seen in Scheme 3 upon the nucleophilic attack a new carbon–oxygen bond is formed. The bond dissociation energies (BDEs) are reliably calculated using the DFT method when correlated with their experimental counterparts.38,39
Scheme 3. The nucleophilic attach by water on the carbocations and a new carbon–oxygen bond is formed.

The BDEs for all of the bond formations were calculated for both hydroxyl ion and water (see Table 4). Not only were the water additions derived but also the attacks by methanol and ethanol on TOTA.
For the most potent nucleophile, the hydroxyl anion, a very exothermic reaction is observed with the in vacuum values well in excess of 100 kcal mol–1 as expected since two oppositely charged entities are brought together forming a new bond (see Table 4). The large exothermicity is diminished in water due to the penalty paid for eliminating two soluble charged molecules resulting in a neutral organic compound with a much smaller solvation energy. The carbocations are stabilised as nitrogen atoms are substituted for oxygen as well as introduction of propyl groups, which contribute electron density onto the ring scaffold, thus lower exothermic values are seen. When the less potent nucleophiles, water and the alcohols, are considered very high endothermic values are seen. However, in the water phase, the huge solvation energy of the proton (263.9 kcal mol–1)40 has to be considered, which makes these reactions possible albeit very slow, which is in agreement with experimental observations.
When the BDEs from Table 4 are correlated with the experimental carbenium stability pKR+ values straight lines are obtained for all scenarios as shown in Fig. 5 for the hydroxyl anion in vacuum.
Fig. 5. The correlation of the BDEs of the carbocations and the hydroxyl anion against the experimentally determined carbenium stability (pKR+).

For the BDE derived values, hydroxyl and water, an excellent correlation was obtained with the Pearson's correlation coefficient >0.999. This trend line can therefore be used to predict the pKR+ value of TOTA derivatives, in general, making it a superb design tool. E.g., the pKR+ value of ADOTA can be confidently be predicted to be 14.3.
The pKR+ values for TOTA, TOTA-OMe and TOTA-OEt were measured. Very similar results were obtained for all three, with an average value of 9.0, almost identical to the reported literature value of TOTA (see Table 4). The reason for the leuco derivatives giving the same value as its parent is due to their instability so the values pertain to TOTA.
Chemical space
As we are investigating the possibility of the TOTA derivatives as possible therapeutic agents, it was pertinent to assess their molecular properties against the benchmarks of lead-like, drug-like and known drug space (KDS) chemical spaces (for the definition of lead-like, drug-like and KDS regions see ref. 41 and in Table S1 in the ESI†).
The calculated molecular descriptors; molecular weight (MW), number of hydrogen bond donors (HD donor), number of hydrogen bond acceptors (HA acceptor), lipophilicity (log P), polar surface area (PSA) and number of rotatable bonds for TOTA and its derivatives in both planar and buckled forms are given in Table 5.
Table 5. Calculated molecular descriptors for the studied compounds.
| Compound | MW g mol–1 | HD donor | HA acceptor | log P | PSA Å2 | Rot. bonds |
| TOTA | 285.3 | 0 | 3 | 2.7 | 39.4 | 0 |
| ADOTA | 284.3 | 1 | 2 | 2.7 | 42.1 | 0 |
| Pr-ADOTA | 326.4 | 0 | 2 | 3.3 | 31.2 | 2 |
| Pr-DAOTA | 367.5 | 0 | 1 | 4.0 | 23.0 | 4 |
| n-Butyl-TATA | 450.6 | 0 | 0 | 5.1 | 14.8 | 9 |
| TOTA-OH | 302.3 | 1 | 4 | 2.5 | 54.1 | 0 |
| ADOTA-OH | 301.3 | 2 | 4 | 2.5 | 58.4 | 0 |
| Pr-ADOTA-OH | 343.4 | 1 | 4 | 3.2 | 48.7 | 2 |
| Pr-DAOTA-OH | 384.5 | 1 | 4 | 3.8 | 43.3 | 4 |
| n-Butyl-TATA-OH | 467.6 | 1 | 0 | 5.0 | 37.9 | 9 |
| TOTA-OMe | 316.3 | 0 | 4 | 2.8 | 43.1 | 1 |
| TOTA-OEt | 330.3 | 0 | 4 | 3.0 | 43.1 | 2 |
From Table 5 it can be stated that the compounds are medium sized with molecular weights within drug-like chemical space (MW < 500 g mol–1) or even in lead-like chemical space (MW < 300 g mol–1). The log P values range from 2.5 to 5.1 with only n-butyl-TATA peeking into KDS, i.e., above 5. Interestingly, all the compounds have PSA values within lead-like chemical space (≤60 Å2). The rest of the molecular descriptors are within drug-like chemical space making the compounds reasonably compatible with biological systems.
Discussion
The results presented here establish that TOTA and its analogues are very cytotoxic against cancer cells and therefore of interest for further development as potential therapeutic agents. As for the mode of action DNA intercalation is the primary hypothesis as the molecular modelling results predict that the carbocations have an excellent fit between the G–C base pairs in line with experimental finding for TOTA and Pr-DAOTA.2,27,28 Unfortunately, the is no experimental data available for the interaction mode to DNA of ADOTA, Pr-ADOTA and n-butyl-TATA. Only relatively modest double stranded DNA strand breaks are observed using the γH2AX assay suggesting a different mechanism of action than for camptothecin, i.e., topoisomerase 1 poison. Both TOTA and Pr-DAOTA have high affinity towards the elaborate DNA scaffolds of triplexes and quadruplexes,2,27,28 which are linked to many biological processes and when perturbed can result in the reduced viability of the cancer cells. It is clear that further work is needed to elucidate the precise mechanism of action for the carbenium ions.
The carbenium ions tested are not expected to form the buckled umbrella form in the experiments reported here, due to their high pKR+ values, but a model based on DFT calculations was developed to predict suitable pKR+ values with electron donating substituents. According the literature, intracellular pH in solid tumours is <7.0 and ∼7.5 in normal tissue, albeit with a large range with values reported as low at 5.8 for tumours.10 More recently, the pH on the cell surface of highly metastatic solid tumours was given in the range of 6.1 to 6.4 pH and 6.7 to 6.8 in non-metastatic tumours.42 Thus, designing the hypothetical carbenium ion to form its planar state with a relatively narrow pH range could prove challenging. Another consideration for the approach proposed is that not only the interior of hypoxic tumours are relatively acidic, but potentially other parts of the human physiology, which could be affected by the carbocation intercalators. Nevertheless, the TOTA derivatives have been proven to be very cytotoxic and an excellent prediction model built to design suitable derivatives only adopting planar conformations in weakly acidic media.
An interesting idea emerged regarding the close structural relative of the triangulenes, helicenes are also highly stable carbocations with helical conformations.8 They have two heteroatom bridges connecting the three phenyl rings resulting in a chiral compounds. It can be postulated that by introducing substituents at the ortho positions that react forming a planar carbocation at lower pH values can also be used as a potential strategy to make the triangulenes only cytotoxic in tumour tissue.
Conclusion
In this work it is established that TOTA, and in particular its Pr-ADOTA and Pr-DAOTA analogues are very potent cytotoxic agents against the cancer cell lines MDA-MB-231 (triple negative breast) and HCT116 (colon). When tested in spheroids of the colon cell line even more potency was achieved, IC50 values of 21 nM for Pr-DAOTA and 38 nM for Pr-ADOTA were achieved. The docking of the triangulenes between the base pairs of double stranded DNA suggest that the mode of action is intercalation. As compared to camptothecin only modest DNA double stranded cleavage was observed using the γH2AX assay excluding topoisomerase I poisoning as the main mechanism of action. It is known that TOTA and Pr-DAOTA have high affinity for DNA triplexes and quadruplexes,2,27,28 which can result in the disruption of the biological processes of the cancer cells and resulting in the potent cytotoxicity of the triangulenes. The BDEs of the covalent bond between the buckled umbrella shaped form and a water molecule formed upon a nucleophilic attach by the latter on the carbenium ion has a very good correlation with the pKR+ experimentally derived stability values, making the calculations of the BDEs an excellent prediction tool to design new derivatives of TOTA that are only planar in mildly acidic environment but buckled at neutral physiological pH values. Such derivatives have the potential to be only cytotoxic in tumours, which have an acidic interior, healthy tissues at neutral, or slightly alkaline, conditions would not be affected.
Experimental details
Synthesis of compounds
Tris(2,6-dimethoxyphenyl)methylium tetrafluoroboronate 1
To a solution of dimethoxybenzene (2.4 mL, 18.11 mmol) in dry ether (12 mL) under an atmosphere of nitrogen was added nBuLi (2 M in cyclohexane, 9 mL, 18 mmol) and the reaction was stirred r.t. for 3 h. Diethylcarbonate (0.65 mL, 5.4 mmol) was then added to the reaction mixture, which was then stirred for 3 days at 45 °C. The reaction was then cooled to room temperature and water (15 mL) added. The layers were separated and the aqueous layer further extracted with ether (2 × 15 mL). The combined organic extracts were dried (MgSO4) and filtered. To the filtrate was added HBF4 (50% aq. sol., 1.3 mL). The mixture was stirred for 10 min and the resulting precipitate was removed by filtration and dried to give the title product (1.02 g, 33%) as a dark green solid. Mp 185–187 °C; IR: νmax(film)/cm–1; 2949, 1589, 1471, 1416, 1252, 1099, 1050, 1031, 1011 and 745. δH (400 MHz, CDCl3): 7.59 (3H, t, J = 8.0 Hz, Ar–H), 6.53 (6H, d, J = 8.0 Hz, Ar–H), 3.59 (18H, s, OCH3). δC (100.5 MHz, CDCl3): 181.6 (C-1′), 162.7 (C-2, C-6), 142.5 (C-4), 125.4 (C-1), 105.0 (C-3, C-5), 56.9 (OCH3); m/z 423 [MH]+ (100), 151 (90); HRMS (ESI+) found [MH+] 423.1806; C25H27O6 requires 423.1802.
TOTA-OH
A mixture of pyridine·HCl (4.07 g, 35.4 mmol) and tris(2,6-dimethoxyphenyl)methylium tetrafluoroboronate 1 (0.75 g, 1.77 mmol) was heated at 205 °C for 1.5 h. The resulting mixture was poured onto water and the resulting solution was filtered to remove the resultant precipitate. The filtrate was then basified (1 M NaOH) and the resulting precipitate collected and dried to give the TOTA-OH (80 mg, 27%) as an off-white solid. Mp: 200 °C (decomp.) (lit.: 200–229 °C (decomp.)17); IR: νmax(film)/cm–1; 3474, 3399, 1616, 1458, 1265, 1016 and 741; δH (400 MHz, CDCl3): 7.38 (3H, t, J = 8.0 Hz, Ar–H), 7.06 (6H, d, J = 8.0 Hz, Ar–H), 2.27 (1H, s, OH); δC (100.5 MHz, CDCl3): 152.3 (C-3a), 129.8 (C-2), 111.4 (C-1, C-3), 110.6 (C-3a1), 50.6 (C-3a2). m/z 325 [MNa]+ (20), 219 (100); HRMS (ESI+) found [MNa+] 325.0459; C19H10NaO4 requires 325.0471.
TOTA-OMe
TOTA-OH (60 mg, 0.19 mmol) was added to methanol (15 mL) and heated at reflux until the solid was completely dissolved. The mixture was then allowed to cool and the resulting precipitate was removed by filtration and dried to give the TOTA-OMe (55 mg, 88%) as an off white solid. Mp 185–187 °C; IR: νmax(film)/cm–1; 2939, 1616, 1457, 1263, 1013, 901, 776 and 728; δH (400 MHz, CDCl3): 7.39–7.41 (3H, m, Ar–H), 7.05 (6H, d, J = 8.0 Hz, Ar–H), 2.88 (3H, s, OCH3); δC (100.5 MHz, CDCl3): 152.9 (C-3a), 130.0 (C-2), 111.1 (C-1, C-3), 108.1 (C-3a1), 55.6 (C-3a2), 49.1 (OCH3); m/z 339 [MNa]+ (10), 285 (100), 227 (10); HRMS (ESI+) found [MNa+] 339.0620; C20H12NaO4 requires 339.0628.
TOTA-OEt
TOTA-OH (33 mg, 0.11 mmol) was added to ethanol (20 mL) and heated at reflux until the solid was completely dissolved. The mixture was then allowed to cool and the resulting precipitate was removed by filtration and dried to give the TOTA-OEt (23 mg, 64%) as a white solid. Mp: 195 °C (decomp.); IR: νmax(film)/cm–1; 2989, 2976, 1616, 1486, 1456, 1259, 1067, 1054, 1013, 942, 934, 775 and 739; δH (400 MHz, CDCl3): 7.37 (3H, t, J = 8.0 Hz, Ar–H), 7.04 (6H, d, J = 8.0 Hz, Ar–H), 3.06–3.12 (2H, m, OCH2CH3), 0.88 (3H, t, J = 8.0 Hz, OCH2CH3); δC (100.5 MHz, CDCl3): 153 (C-3a), 129.7 (C-2), 111.1 (C-1, C-3), 108.9 (C-3a1), 56.9 (OCH2CH3), 55.2 (C-3a2), 15.2 (OCH2CH3); m/z 353 [MNa]+ (40), 285 (100); HRMS (ESI+) found [MNa+] 353.0793; C21H14NaO4 requires 353.0784.
n-Butyl-TATA-tetrafluoroboronate
Benzoic acid (1.26 g, 10.4 mmol) and n-butylamine (1.4 mL, 14.1 mmol) were added to a solution of tris(2,6-dimethoxyphenyl)methylium tetrafluoroboronate 1 (0.20 g, 0.47 mmol) in NMP (3.6 mL). The reaction mixture was heated at reflux for 24 h and after cooling to room temperature, the reaction mixture was poured onto ice to give a precipitate which was collected through filtration and dried to give the n-Butyl-TATA (85 mg, 40%) as a red solid. Mp: >230 °C; IR: νmax(film)/cm–1; 2954, 2930, 2870, 1609, 1533, 1334, 1248, 1177, 1047, 1034, 823 and 756; δH (400 MHz, CDCl3): 8.06 (3H, t, J = 8.0 Hz, Ar–H), 7.35 (6H, d, J = 8.0 Hz, Ar–H), 4.33 (6H, t, J = 12.0 Hz, NCH2), 1.75–1.77 (6H, m, NCH2CH2), 1.56–1.61 (6H, m, NCH2CH2CH2CH3), 1.02 (9H, t, J = 12.0 Hz, NCH2CH2CH2CH3); δC (100.5 MHz, CDCl3): 140 (C-3a), 139.72 (C-3a2), 137.68 (C-2), 109.9 (C-3a1), 105.1 (C-1, C-3); m/z 451 [MH]+ (100); HRMS (ESI+) Found [MH+] 450.2904; C31H36N3 requires 450.2901.18
Cell proliferation assay
As described previously,43,44 cell proliferation was measured by the degree of incorporation of 3H-thymidine into DNA of S-phase cells. Briefly, 3000 cells per well were seeded in 96 well plates that were tissue culture-treated for monolayer culture and incubated for 3 days. Alternatively, 4000 cells per well were seeded in 96 well plates (Corning Costar Ultra-Low attachment). The spheroid formation was initiated by centrifugation of the plates at 1000 × g for 10 min with swinging buckets. After 3 days, cells were exposed to the ligands for another four days. 3H-Thymidine (0.04 μCi per well for monolayer culture or 0.08 μCi per well for spheroid culture) was added (5 hours for monolayer culture or 16 hours for spheroid culture) prior to harvest. The human breast cancer cell line MDA-MB-231 and the colon cancer cell line HCT116 were purchased from the American Type Culture Collection (ATCC).
Flow cytometric analysis
As described in detail previously,45 cells (106 cells per well) were grown in 6 well plates and incubated with inhibitors for the indicated time. Cells were harvested, washed and re-suspended in 1 mL of blocking buffer (1% FCS/PBS), and incubated with antibody to γH2AX (phosphorylated Ser139) (Millipore, USA) in blocking buffer (1 : 500 dilution) at room temperature for 2 h. Cells were washed, incubated with goat anti-mouse Alex 488 Fab fragment secondary antibody (Invitrogen, New Zealand) (1 : 400 in blocking buffer for 1 h, at room temperature; dark), washed and resuspended in 1 ml of blocking buffer containing RNase (1 μg mL–1) and propidium iodide (PI) (10 μg mL–1) for 30 min at room temperature. Cells were analysed in a Becton–Dickinson LSRII and profiles were analysed with ModFit LT 3 software.
Molecular modelling
The compounds were docked to the known crystal structure of DNA co-crystallised with TOTA.2 The Scigress Ultra version FJ 2.6 program46 was used to prepare the crystal structure for docking, i.e., hydrogen atoms were added and the intercalated TOTA was removed. The Scigress software suite was also used to build the inhibitors and the MO-G-PM3 force47 was used to optimise the structures. The centre of the binding pocket was defined as the position of the cation carbon of TOTA (x = 3.095, y = 13.893, z = 46.979). Fifty docking runs were allowed for each ligand with default search efficiency (100%). The GoldScore (GS),33 ChemScore (CS),34,35 piecewise linear potential (ChemPLP)36 and Astex Statistical Potential (ASP)37 scoring functions were implemented to validate the predicted binding modes and relative energies of the ligands using the GOLD v5.2 software suite. The DNA is complexed with TOTA which has interactions with many residues and re-docking reproduced these interactions with all four docking scores with an average RMSD 0.67 Å (GS = 0.66 Å, CS = 0.71 Å, ChemPLP = 0.34 Å, ASP = 0.98 Å). In order to generate the six molecular descriptors the software package Dragon v7.0 was used.48
Density functional theory
The geometry optimisation and energy calculations were performed with Gaussian 09 (ref. 49) software using restricted density functional theory. The B3LYP (ref. 50 and 51) functional hybrid method was employed and the standard 6-3+G(d, p)52,53 diffuse basis set was used for the geometry optimisation and frequency analysis in both vacuum and aqueous phase using integral equation formalism polarizable continuum model (IEFPCM).54 The zero-point vibrational energies (ZPE) were scaled according to Wong (0.9804).55 In all cases, the normal modes revealed no imaginary frequencies indicating that they represent minima on the potential energy surface. The subsequent energy calculations were performed with the larger 6-311+G(2df, p) basis set in vacuum and water (IEFPCM). The BDEs calculated as in references by Yu and Drew.38,39 All the single point energies and ZPEs are given in Table S2 in the ESI.†
Determination of pKR+ values
The absorbance at 330 nm of various solutions of TOTA (1 mg mL–1 in DMSO with various volumes of, 0, 20 μL, 40 μL and 60 μL, of 0.1 M tetramethylammonium hydroxide) was determined by spectrophotometric measurement, 330 nm being where the carbonium ion shows intense absorption and the carbinol species does not absorb. Subsequently, a calibration curve for the molar absorbance for the 330 nm peak was created by a study of TOTA in several concentrations of 0.1 M acetic acid, in which the alcohol is completely ionised. The pKR was evaluated from the relationship in eqn (1), following the procedures outlined in the literature.56,57
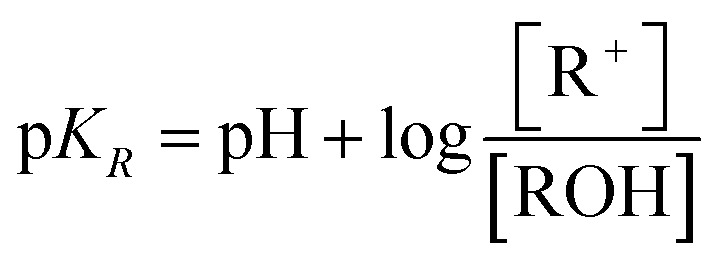 |
1 |
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary Material
Acknowledgments
The authors are very grateful to Professor Bo W. Laursen, Nano-Science Centre and Department of Chemistry, University of Copenhagen, Denmark for providing us with samples of TOTA, ADOTA, Pr-ADOTA and Pr-DAOTA. There are no conflicts of interest to declare.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c9md00305c
References
- Reynisson J., Balakrishnan G., Wilbrandt R., Harrit N. J. Mol. Struct. 2000;520:63–73. [Google Scholar]
- Reynisson J., Schuster G. B., Howerton S. B., Williams L. D., Barnett R. N., Cleveland C. L., Landman U., Harrit N., Chaires J. B. J. Am. Chem. Soc. 2003;125:2072–2083. doi: 10.1021/ja0211196. [DOI] [PubMed] [Google Scholar]
- Pothukuchy A., Ellapan S., Gopidas K. R., Slazar M. Bioorg. Med. Chem. Lett. 2003;13:1491–1494. doi: 10.1016/s0960-894x(03)00121-5. [DOI] [PubMed] [Google Scholar]
- Pothukuchy A., Mazzitelli C. L., Rodriguez M. L., Tuesuwan B., Salazar M., Brodbelt J. S., Kerwin S. M. Biochemistry. 2005;44:2163–2172. doi: 10.1021/bi0485981. [DOI] [PubMed] [Google Scholar]
- Reynisson J., Wilbrandt R., Brinck V., Laursen B. W., Nørgaard K., Harrit N., Brouwer A. M. Photochem. Photobiol. Sci. 2002;1:763–773. doi: 10.1039/b204954f. [DOI] [PubMed] [Google Scholar]
- Martinez R., Chacon-Garcia L. Curr. Med. Chem. 2005;12:127–151. doi: 10.2174/0929867053363414. [DOI] [PubMed] [Google Scholar]
- Laursen B. W., Reynisson J., Mikkelsen K. V., Bechgaard K., Harrit N. Photochem. Photobiol. Sci. 2005;4:568–576. doi: 10.1039/b501584g. [DOI] [PubMed] [Google Scholar]
- Bosson J., Gouin J., Lacour J. Chem. Soc. Rev. 2014;43:2824–2840. doi: 10.1039/c3cs60461f. [DOI] [PubMed] [Google Scholar]
- Wallabregue A., Moreau D., Sherin P., Moneva Lorente P., Jarolímová Z., Bakker E., Vauthey E., Gruenberg J., Lacour J. J. Am. Chem. Soc. 2016;138:1752–1755. doi: 10.1021/jacs.5b09972. [DOI] [PubMed] [Google Scholar]
- Tannock I. F., Rotin D. Cancer Res. 1989;49:4373–4384. [PubMed] [Google Scholar]
- Shirmanova M. V., Druzhkova I. N., Lukina M. M., Matlashov M. E., Belousov V. V., Snopova L. B., Prodanetz N. N., Dudenkova V. V., Lukyanov S. A., Zagaynova E. V. Biochim. Biophys. Acta, Gen. Subj. 2015;1850:1905–1911. doi: 10.1016/j.bbagen.2015.05.001. [DOI] [PubMed] [Google Scholar]
- Verweij J., Pinedo H. Anti-Cancer Drugs. 1990;1:5–14. [PubMed] [Google Scholar]
- Hume P. A., Brimble M. A., Reynisson J. Comput. Theor. Chem. 2013;1005:9–15. [Google Scholar]
- Rischin D., Peters L., Fisher R., Macann A., Denham J., Poulsen M., Jackson M., Kenny L., Penniment M., Corry J., Lamb D., McClure B. J. Clin. Oncol. 2005;23:79–87. doi: 10.1200/JCO.2005.01.072. [DOI] [PubMed] [Google Scholar]
- von Pawel J., von Roemeling R., Gatzemeier U., Boyer M., Elisson L. O., Clark P., Talbot D., Rey A., Butler T. W., Hirsh V., Olver I., Bergman B., Ayoub J., Richardson G., Dunlop D., Arcenas A., Vescio R., Viallet J., Treat J. J. Clin. Oncol. 2000;18:1351–1359. doi: 10.1200/JCO.2000.18.6.1351. [DOI] [PubMed] [Google Scholar]
- Maeda H., Khatami M. Clin. Transl. Med. 2018;7:11. doi: 10.1186/s40169-018-0185-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabacky M. J., Johnson C. S., Smith R. G., Gutowsky H. S., Martin J. C. J. Am. Chem. Soc. 1967;89:2054–2058. [Google Scholar]
- Lemke S., Ulrich S., Claußen F., Bloedorn A., Jung U., Herges R., Magnussen O. M. Surf. Sci. 2015;632:71–76. [Google Scholar]
- Goncharov D. A., Goncharova E. A., Krymskaya V. P., Panettieri R. A., Eszterhas A., Lim P. Nat. Protoc. 2007;1:2905–2908. doi: 10.1038/nprot.2006.433. [DOI] [PubMed] [Google Scholar]
- Dean J. A., Lange's Handbook of Chemistry, McGraw Hill Book Company, New York, NY, 1979. [Google Scholar]
- Ayine-Tora D., Reynisson J. Aust. J. Chem. 2018;71:580–586. [Google Scholar]
- Laursen B. W., Sørensen T. J. J. Org. Chem. 2009;74:3183–3185. doi: 10.1021/jo9002486. [DOI] [PubMed] [Google Scholar]
- Pommier Y. Nat. Rev. Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- Han X., Wei B., Fang J., Zhang S., Zhang F., Zhang H., Lan T., Lu H., Wei H. PLoS One. 2013;8:e73341. doi: 10.1371/journal.pone.0073341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta G., Hsiao A. Y., Ingram M., Luker G. D., Takayama S. J. Controlled Release. 2012;164:192–204. doi: 10.1016/j.jconrel.2012.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swietach P., Patiar S., Supuran C. T., Harris A. L., Vaughan-Jones R. D. J. Biol. Chem. 2009;284:20299–20310. doi: 10.1074/jbc.M109.006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotar A., Wang B., Shivalingam A., Gonzalez-Garcia J., Vilar R., Plavec J. Angew. Chem., Int. Ed. 2016;55:12508–12511. doi: 10.1002/anie.201606877. [DOI] [PubMed] [Google Scholar]
- Shivalingam A., Vyšniauskas A., Albrecht T., White A. J. P., Kuimova M. K., Vilar R. Chem. – Eur. J. 2016;22:4129–4139. doi: 10.1002/chem.201504099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi J., Tauchi H., Sakamoto S., Nakamura A., Ken-ichi Morishima K., Matsuura S., Kobayashi T., Tamai K., Tanimoto K., Komatsu K. Curr. Biol. 2002;12:1846–1851. doi: 10.1016/s0960-9822(02)01259-9. [DOI] [PubMed] [Google Scholar]
- Oikawa S., Tada-Oikawa S., Kawanishi S. Biochemistry. 2001;40:4763–4768. doi: 10.1021/bi002721g. [DOI] [PubMed] [Google Scholar]
- Goldsmith G., Rathinavelan T., Yathindra N. PLoS One. 2016;11:e0152102. doi: 10.1371/journal.pone.0152102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes D., Lipps H. J. Nucleic Acids Res. 2015;43:8627–8637. doi: 10.1093/nar/gkv862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G., Willet P., Glen R. C., Leach A. R., Taylor R. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Eldridge M. D., Murray C., Auton T. R., Paolini G. V., Mee P. M. J. Comput.-Aided Mol. Des. 1997;11:425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- Verdonk M. L., Cole J. C., Hartshorn M. J., Murray C. W., Taylor R. D. Proteins. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- Korb O., Stützle T., Exner T. E. J. Chem. Inf. Model. 2009;49:84–96. doi: 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]
- Mooij W. T. M., Verdonk M. L. Proteins. 2005;61:272–287. doi: 10.1002/prot.20588. [DOI] [PubMed] [Google Scholar]
- Yu B., Reynisson J. Eur. J. Med. Chem. 2011;46:5833–5837. doi: 10.1016/j.ejmech.2011.09.044. [DOI] [PubMed] [Google Scholar]
- Drew K. L. M., Reynisson J. Eur. J. Med. Chem. 2012;56:48–55. doi: 10.1016/j.ejmech.2012.08.017. [DOI] [PubMed] [Google Scholar]
- Tissandier M. D., Cowen K. A., Feng W. Y., Gundlach E., Cohen M. H., Earhart A. D., Coe J. V., Tuttle T. R. J. Phys. Chem. A. 1998;102:7787–7794. [Google Scholar]
- Zhu F., Logan G., Reynisson J. Mol. Inf. 2012;31:847–855. doi: 10.1002/minf.201200103. [DOI] [PubMed] [Google Scholar]
- Anderson M., Moshnikova A., Engelman D. M., Reshetnyak Y. K., Andreev O. A. Proc. Natl. Acad. Sci. U. S. A. 2016;113:8177–8181. doi: 10.1073/pnas.1608247113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung E., Kim J. E., Rewcastle G. W., Finlay G. J., Baguley B. C. Cancer Biol. Ther. 2011;11:938–946. doi: 10.4161/cbt.11.11.15527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung E. Y., Askarian-Amiri M. E., Sarkar D., Ferraro-Peyret C., Joseph W. R., Finlay G. J., Baguley B. C. Front. Oncol. 2017;7:184. doi: 10.3389/fonc.2017.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung E., Rewcastle G. W., Joseph W. R., Rosengren R. J., Larsen L., Baguley B. C. Invest. New Drugs. 2012;30:2103. doi: 10.1007/s10637-011-9768-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scigress, version FJ 2.6 (EU 3.1.7), Fujitsu Limited, Tokyo, Japan, 2008–2016.
- Stewart J. J. Mol. Model. 2004;10:155–164. doi: 10.1007/s00894-004-0183-z. [DOI] [PubMed] [Google Scholar]
- Todeschini R., Lasagni M., Marengo E. J. Chemom. 1994;8:263–272. [Google Scholar]
- Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery Jr J. A., Peralta J. E., Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin K. N., Staroverov V. N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam J. M., Klene M., Knox J. E., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J. J., Dapprich S., Daniels A. D., Farkas O., Foresman J. B., Ortiz J. V., Cioslowski J. and Fox D. J., Gaussian 09, revision D. 01, Gaussian, Inc., Wallingford, CT, 2010.
- Becke A. D. Phys. Rev. A: At., Mol., Opt. Phys. 1988;38:3098. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- Becke A. D. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- Frisch M. J., Pople J. A., Binkley J. S. J. Chem. Phys. 1984;80:3265–3269. [Google Scholar]
- Hariharan P. C., Pople J. A. Theor. Chim. Acta. 1973;28:213–222. [Google Scholar]
- Klamt A., Mennucci B., Tomasi J., Barone V., Curutchet C., Orozco M., Luque F. J. Acc. Chem. Res. 2009;42:489–492. doi: 10.1021/ar800187p. [DOI] [PubMed] [Google Scholar]
- Wong M. W. Chem. Phys. Lett. 1996;256:391–399. [Google Scholar]
- Martin J. C., Smith R. G. J. Am. Chem. Soc. 1964;86:2252–2256. [Google Scholar]
- Deno N. C., Jaruzelski J. J., Schriesheim A. J. Org. Chem. 1954;19:155–167. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.