

 SAR exploration at a single position of the psoralen ring led to improved selectivity to the chymotrypsin-like (β5i) subunit of the immunoproteasome.
SAR exploration at a single position of the psoralen ring led to improved selectivity to the chymotrypsin-like (β5i) subunit of the immunoproteasome.
Abstract
The immunoproteasome is a multicatalytic protease that is predominantly expressed in cells of hematopoietic origin. Its elevated expression has been associated with autoimmune diseases, various types of cancer, and inflammatory diseases. The development of immunoproteasome-selective inhibitors with non-peptidic scaffolds remains a challenging task. Here, we describe a focused series of psoralen-based inhibitors of the β5i subunit of the immunoproteasome with different substituents placed at position 4′. The most promising compound was further evaluated through changes at position 3 of the psoralen ring. Despite a small decrease in the inhibitory potency in comparison with the parent compound, we were able to improve the selectivity against other subunits of both the immunoproteasome and the constitutive proteasome. The most potent compounds discriminated between both proteasome types in cell lysates and also showed a decrease in cytokine secretion in peripheral blood mononuclear cells.
Introduction
The heart of the ubiquitin–proteasome system, the 26S proteasome, is responsible for the regulation of various cell processes and the maintenance of protein homeostasis in cells. It is the most important non-lysosomal proteolytic complex degrading misfolded or damaged proteins.1 The 26S proteasome consists of a 20S core and 19S regulatory sections, with the 20S core comprising three active subunits with distinct catalytic activities, i.e. β1 (caspase-like), β2 (trypsin-like), and β5 (chymotrypsin-like).2,3 Two major types of 20S cores exist in vertebrates: the constitutive proteasome (cCP), expressed in all cell types, and the immunoproteasome (iCP), expressed mostly in hematopoietic cells. The latter can also be found in other cell types upon induction by the tumour necrosis factor-α and interferon-γ during acute immune and inflammatory responses.4,5 Of note, in the iCP, the catalytically active subunits β are replaced by their β1i, β2i, and β5i counterparts.
Extensive research in the proteasome field in the last two decades resulted in the development of non-selective proteasome inhibitors, such as bortezomib and carfilzomib, used in the treatment of multiple myeloma and mantle-cell lymphoma.6 However, the clinical use of such conventional, non-selective proteasome inhibitors is often limited due to the side effects caused by the non-selective inhibition of protein degradation.7 While the cCP is expressed in all eukaryotic cells, the iCP expression is mostly upregulated during the course of disease processes,8–10 leading to the assumption that its selective inhibition is expected to cause fewer side effects. The latest studies showed that co-inhibition of two subunits of the iCP is required to show beneficial effects in autoimmune diseases, i.e. either by combining β1i- and β5i-selective inhibitors in a synergistic approach11 or with a single molecule that is able to simultaneously inhibit two catalytically active subunits of the iCP.12 The excellent work by the groups of Overkleeft,13,14 Groll,15 Lin,16,17 and Johnson12,18 yielded selective inhibitors of all subunits of the iCP (Fig. 1) and these represent both very valuable molecular tools in the studies of iCP biology and lead compounds towards new drugs (Fig. 1). It is noteworthy that KZR-616, a β1i- and β5i-targeting compound, is currently being evaluated in clinical studies for the treatment of autoimmune and inflammatory diseases.12,19
Fig. 1. Examples of peptidic subunit-selective inhibitors of iCP; original labelling of compounds by the authors is used here. For more details, the corresponding references are given with each inhibitor.

From a structural perspective, the majority of these inhibitors of the iCP have a peptidic backbone, and are thus prone to poor metabolic stability and low bioavailability leading to lack of oral exposure.20,21 With an attempt to circumvent these limitations, several lines of research by various groups are devoted to the development of non-peptidic inhibitors of the iCP (Fig. 2).22–26 Despite this, the number of such inhibitors is scarce and, interestingly, only inhibitors of the β5i subunit were discovered so far.
Fig. 2. A representative set of non-peptidic selective inhibitors of the iCP; original labelling of compounds by the authors is used here for the bottom three compounds. For more details, the corresponding references are given with each inhibitor.

Previously, we reported a series of psoralens as selective inhibitors of the β5i subunit.22 These compounds were discovered through a structure-based virtual screening of the ZINC drug-like compound library. A library of psoralen-based compounds was subsequently synthesised for the initial structure–activity relationship (SAR) studies. The best non-covalent inhibitor was transformed into a covalent inhibitor through the addition of an electrophilic warhead on position 3 of the psoralen ring. The resulting compounds exhibited selectivity to β5i compared to the β2i and β1i subunits, as well as compared to all cCP subunits. The most selective covalent inhibitor, a psoralen with an oxathiazolone as the electrophilic warhead (psoralen-based inhibitor, Fig. 2) and a phenyl group at position 4′, exhibited more than 80-fold selectivity to β5i compared to the other iCP and cCP subunits.22 Of note, oxathiazolone as a covalent warhead had been used previously in inhibitors of Mycobacterium tuberculosis proteasome27 and oxathiazolone-based compounds were also described as iCP-selective by the same group.25
Herein, we present a focused SAR study of psoralen-based β5i inhibitors with oxathiazolone at position 3 (Fig. 3). A series of compounds with various substituents at position 4′ of the psoralen ring is described and characterised to evaluate the effect of these modifications on the activity, selectivity, and biochemical characteristics of psoralen-based compounds. The best performing 4′-substituted inhibitor was further modified at position 3 to evaluate the influence of different electrophilic groups at this position.
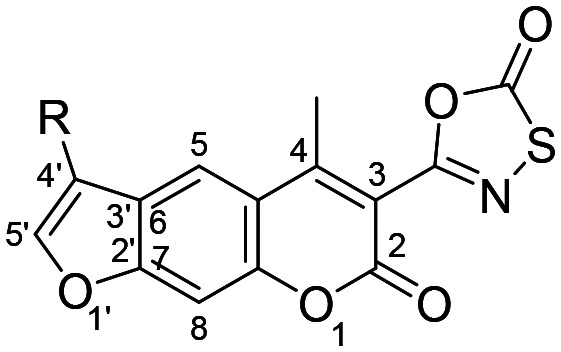
Fig. 3. Schematic representation of the work disclosed in this study. The numbering system for the psoralen ring is shown for clarity.

Synthesis
The target oxathiazolone-based psoralens were synthesised using a straightforward synthetic route (Scheme 1). The 1-substituted-2-bromo-ethan-1-ones 2–7 were either commercially available or prepared from corresponding ethan-1-ones by bromination with copper(ii) bromide (2, 3, and 4) or bromine (5, 6, and 7). These were then reacted with ethyl 7-hydroxy-4-methyl-2-oxo-2H-chromene-3-carboxylate (1), which was prepared under anhydrous conditions from 1-(2,4-dihydroxyphenyl)ethan-1-one and diethyl malonate using a microwave reactor, to yield alkylated compounds 8–17.
Scheme 1. Synthesis of psoralen-based compounds with an oxathiazolone warhead. Reagents and conditions: (a) diethyl malonate, KOtBu, molecular sieves (3 Å), MW (100 °C, 50 W, 15 min); (b) EtOAc, CuBr2, 85 °C, 24 h (for 2, 3, and 4) or MeOH, Br2, 0 °C to rt, 48 h (for 5, 6, and 7); (c) 1, dioxane, K2CO3, KI, 100 °C, 24 h; (d) propan-2-ol, 1 M NaOH, 100 °C, 5–20 min; (e) dioxane, Boc2O, pyridine, ammonium acetate, rt, 24 h; (f) anhydrous THF, chlorocarbonylsulfenyl chloride, 80 °C, 1.5–2 h.

To obtain the psoralen ring (compounds 18–27), cyclisation under basic conditions was performed. The temperature needed to be maintained at 100 °C and the reaction times were carefully controlled as extensive heating resulted in the formation of undesired side products and low yields. Compounds 18–27 were then treated with Boc2O, pyridine and ammonium acetate to yield 4-methyl-3-carboxamide substituted psoralens 28–37, which were finally reacted with chlorocarbonylsulfenyl chloride under carefully controlled anhydrous conditions. The formation of oxathiazolone-based compounds 38–47 was confirmed with the disappearance of the amide signals in the 1H NMR spectra and the observance of oxathiazolone carbonyl signals in the 170–176 ppm range in the 13C NMR spectra.
The psoralens with the cyclohexyl moiety at position 4′ with variation at position 3 were synthesised through a different, but nevertheless, straightforward synthetic route (Scheme 2). Compound 7 was reacted with ethyl 2-(7-hydroxy-4-methyl-2-oxo-2H-chromen-3-yl)acetate (48) or ethyl 3-(7-hydroxy-4-methyl-2-oxo-2H-chromen-3-yl)propanoate (49) as described previously,22 to yield alkylated compounds 50 and 51. To obtain the psoralen ring (compounds 52 and 53), cyclisation was performed in the same manner as described in Scheme 1. Variation at position 3 was performed through an EDC-mediated coupling with N-hydroxysuccinimide, 2-(methylamino)acetonitrile, or 2-aminoacetonitrile.
Scheme 2. Synthesis of psoralens with the cyclohexyl moiety at position 4′ with variation at position 3. Reagents and conditions: (a) 75% H2SO4, diethyl 2-acetylsuccinate (for 48) or diethyl 2-acetylglutarate (for 49), rt, 24 h; (b) MeOH, Br2, 0 °C to rt, 48 h; (c) 48 or 49, dioxane, K2CO3, KI, 100 °C, 24 h; (d) propan-2-ol, 1 M NaOH, 100 °C, 5–20 min; (e) 52 or 53, THF, N-hydroxysuccinimide, EDC, Et3N, 0 °C to rt, 24 h; (f) 52 or 53, THF, 2-(methylamino)acetonitrile, EDC, Et3N, 0 °C to rt, 24 h; (g) 52 or 53, THF, 2-aminoacetonitrile, EDC, Et3N, 0 °C to rt, 24 h.

Biochemical evaluation
The synthesized compounds with various substituents at position 4′ and oxathiazolone at position 3 (38–47) were first evaluated for their inhibition of β5i activity using succinyl-Leu-Leu-Val-Tyr-7-amino-4-methylcoumarin (Suc-LLVY-AMC) as a fluorogenic substrate (Table 1).
Table 1. Structures and in vitro inhibitory potencies of compounds 38–47 against the β5i subunit of human iCP.
| ||
| Compound | R | IC50 for β5i (nM) |
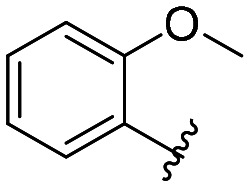
| 38 |
|
227 ± 32 |
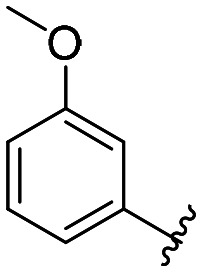
| 39 |
|
226 ± 21 |
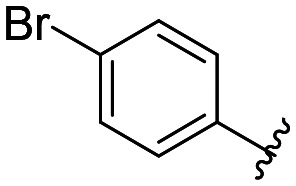
| 40 |
|
351 ± 4 |
| 41 |
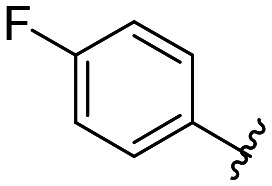
|
200 ± 13 |
| 42 |
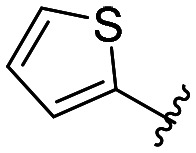
|
141 ± 6 |
| 43 |
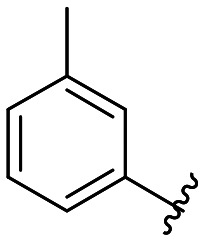
|
365 ± 5 |
| 44 |
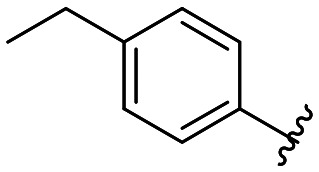
|
174 ± 5 |
| 45 |
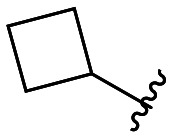
|
433 ± 13 |
| 46 |
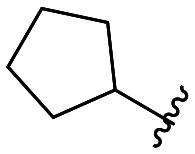
|
264 ± 3 |
| 47 |
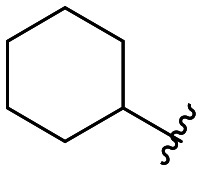
|
106 ± 4 |
All compounds showed sub-micromolar inhibitory activity, with compounds 42, 44, and 47 showing the strongest β5i inhibition with IC50 values of 141 nM, 174 nM, and 106 nM, respectively (Table 1). The position of the methoxy group on the 4′-phenyl ring did not influence the activity as exemplified with compounds 38 and 39, which showed the same inhibition of β5i. When methyl was placed at the meta position (compound 43) instead of the methoxy group (compound 39), a decrease in the inhibitory activity was observed (Table 1). Interestingly, the compound with bromine at the para position (40, IC50 (β5i) = 351 nM) was less active than the corresponding para fluoro (41, IC50 (β5i) = 200 nM) and para ethyl (44, IC50 (β5i) = 174 nM) derivatives. In a series of cycloalkyl derivatives 45–47, a clear trend of improved β5i inhibition with increasing size of substituents at position 4′ of the psoralen ring was observed (Table 1). Additionally, the time-dependence of β5i inhibition by compounds 42, 44, and 47 was determined by IC50 shift assay (Table S3†); i.e. a clear increase in the IC50 values was observed when the compounds were not pre-incubated with the enzyme prior to the addition of the substrate. Moreover, the irreversibility of oxathiazolones 42, 44, and 47 was determined by the rapid dilution assay (Table S4†). In Fig. 4, representative time course curves obtained in the rapid dilution assays for compound 47 and DPLG-3, one of the most selective and reversible β5i inhibitors known,17 are shown. We should state here that DPLG-3 is not commercially available, so we resynthesised it in our lab (see the ESI,† Scheme S1).
Fig. 4. Recovery of iCP activity in the rapid dilution assay. To a 100-fold concentration of iCP used in the kinetic assay, a 10-fold IC50 concentration of the selected inhibitor (either 45 nM DPLG-3 or 1060 nM compound 47) was added. The reaction mix was immediately (0 min), or after pre-incubation (30 min), diluted 100-fold with the substrate. For the reversible inhibitor DPLG-3 (A), almost a full recovery of iCP activity was observed, while in the case of compound 47 (B) only a 22% recovery was observed, indicating the irreversible mode of inhibition.

To assess the selectivity of these psoralen-based inhibitors 38–47, the compounds were evaluated against all remaining subunits of the iCP and the cCP (Table S1,† data presented as residual activities in the presence of 10 μM of each compound). All the compounds showed preferential inhibition of β5i over the β2i and β1i subunits of the human iCP, as well as over all three subunits of the human cCP. Due to poor solubility of the compounds in higher concentrations that did not allow precise IC50 determinations for inhibition of other subunits, the degree of selectivity for compounds 38–47 can only be assumed; e.g. for compounds 42, 44, and 47 (which were the most potent β5i inhibitors in the series), the selectivity can be estimated to be more than 100-fold. This represents an improvement in comparison with a previously described oxathiazolone-based psoralen (cpd. 42, Fig. 2, 80-fold selectivity, ref. 22), which was, however, more active with an IC50 value for β5i inhibition of 13 nM.
The most promising inhibitor, compound 47, was further modified at position 3 to evaluate the effect of different electrophilic groups at this position on the inhibitory properties of psoralens (compounds 54–59, Table 2). The nitrile-based compounds 56–59 showed no inhibition of β5i, whereas only one succinimidyl-ester exhibited inhibitory properties, albeit to a moderate extent (55, IC50 (β5i) = 1830 nM). These results show that oxathiazolone-based compounds are more preferable to achieve β5i inhibition.
Table 2. Structures and in vitro inhibitory potencies of compounds 54–59 against the β5i subunit of human iCP.
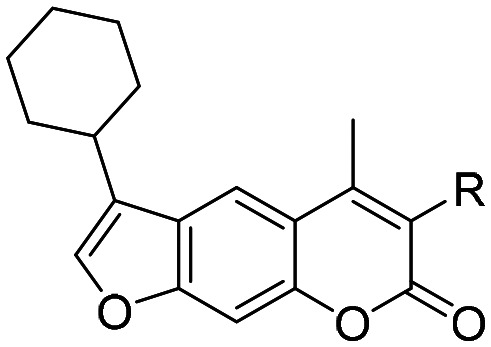
| ||
| Compound | R | RA (%) at 10 μM or IC50 for β5i (nM) |
| 54 |
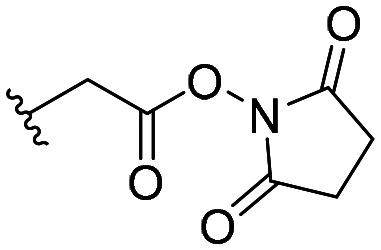
|
62 ± 3 |
| 55 |
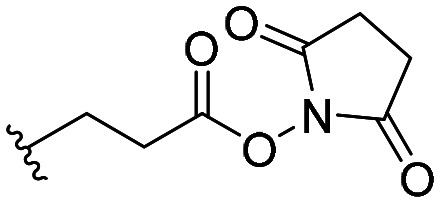
|
IC50 = 1830 ± 380 |
| 56 |
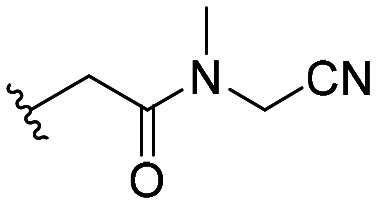
|
101 ± 7 |
| 57 |
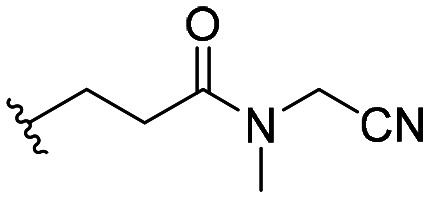
|
111 ± 16 |
| 58 |
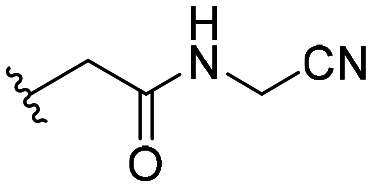
|
99 ± 9 |
| 59 |
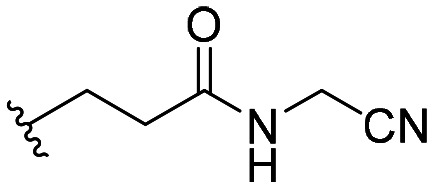
|
102 ± 9 |
Next, we incubated compounds 44 and 47 with cell lysates of HeLa and THP-1 cells to estimate the capability of the compounds to inhibit the iCP in the presence of other cytosolic components. THP-1 cells were used as they are derived from acute monocytic leukemia and express high levels of iCP, whereas HeLa cells predominantly contain cCP.28 Both compounds showed better inhibition of β5 activity in THP-1 cell lysates than in HeLa cell lysates (Table S2†); although, similarly, as with previously described psoralens,22 approximately 10-fold reductions in potency (44: IC50 [THP-1 lysates] = 1.53 μM, 47: IC50 [THP-1 lysates] = 1.01 μM) were observed when compared with those of in vitro assays with purified human iCP (Table 1). Unfortunately, we were not able to solve issues with poor cellular permeability of psoralen-based compounds as worse inhibition of β5 activity was observed for both derivatives in intact cells (Table S2,† only RA values are given due to issues with solubility at higher concentrations of 44 and 47).
Despite poor permeability, we did test the ability of compounds 44 and 47 (both compounds at 10 μM with 24-hour incubation) to inhibit cytokine secretion. Here, we used DPLG-3 as a positive control. In lipopolysaccharide (LPS) stimulated peripheral blood mononuclear cells, we determined that all compounds, to a small extent, affected the secretion of IL-6 and IL-10 (Fig. 5), while other assayed cytokines (IL-1β, IL-8, IL-12p70, and TNFα) were not affected (Table S5†). The results obtained for DPLG-3 and compounds 44 and 47 are not only in line with permeability issues for compounds 44 and 47, but also corroborate previously determined data12 that besides β5i, another iCP subunit must be inhibited to achieve notable cellular effects in terms of decreasing the pro-inflammatory cytokine production.
Fig. 5. The effect of compounds 44, 47, and DPLG-3 on the secretion of cytokines. The results present values relative to LPS treated cells. The cells were pre-treated for 1 h with DPLG (50 nM), compound 44 (10 μM), or 47 (10 μM). Afterwards, LPS was added and the concentrations of cytokines were determined in the supernatants after additional 24 h of treatment. The results are expressed as means of duplicates ± SEM of two independent experiments, * is p < 0.05.

Conclusions
Discovery of non-peptidic and selective inhibitors of the iCP is an extremely challenging task, so such incremental steps, as shown in this study, are needed to optimize a given compound class. We showed that small changes in the structures can lead to further improvements in their selectivity for the inhibition of the β5i subunit of iCP. In addition, we calculated parameters to evaluate pharmacokinetics and drug-likeness with the SwissADME Web tool29 (ESI,† section 9); these indicate slightly better overall characteristics and predicted water solubility for structures with saturated rings attached to psoralen. However, problems with poor cellular permeability remain unsolved; therefore, more pronounced changes in the psoralen structure are needed in the future to tackle this problem. Of note, our results confirmed the recently disclosed data,12 in which simultaneous inhibition of two iCP subunits is necessary to achieve a significant anti-inflammatory effect. Therefore, two separate issues, i.e. permeability and simultaneous inhibition of two iCP subunits, will represent our main focus during the design of next generation non-peptidic iCP inhibitors.
Experimental
Synthetic procedures and analyses for a representative set of compounds are reported in the main text. General chemistry methods, all biochemistry experimental data, spectroscopic analyses, and representative NMR, HRMS, and HPLC spectra are available in the ESI.†
Procedure for the synthesis of ethyl 7-hydroxy-4-methyl-2-oxo-2H-chromene-3-carboxylate 1
To a mixture of 1-(2,4-dihydroxyphenyl)ethan-1-one (10 mmol, 1 equiv.), diethyl malonate (2.3 equiv.) and KOtBu (0.22 equiv.), activated molecular sieves (pore diameter of 3 Å) were added. The mixture was reacted in a microwave reactor at 100 °C and 50 W for 15 min. Compound 1 was purified by column chromatography (n-hexane/ethyl acetate = 4 : 1, gradient to 1 : 1).
Off-white solid; yield 33%; 1H NMR (400 MHz, acetone-d6) δ 1.34 (t, J = 7.1 Hz, 3H, CH2CH[combining low line]3), 2.43 (s, 3H, Ar–CH[combining low line]3), 4.35 (q, J = 7.1 Hz, 2H, CH[combining low line]2CH3), 6.76 (d, J = 2.4 Hz, 1H, Ar–H[combining low line]), 6.91 (dd, J = 8.8 Hz, 2.4 Hz, 1H, Ar–H[combining low line]), 7.73 (d, J = 8.8 Hz, 1H, Ar–H[combining low line]), 9.68 (br s, 1H, OH[combining low line]). HRMS (m/z) (ESI): calcd for C13H12O5 [M + H]+: 249.0763, found: 249.0765.
General procedure for the synthesis of 1-substituted-2-bromo-ethan-1-ones 2–4
The corresponding ethan-1-ones (starting amount ranging from 6.7 to 7.9 mmol, 1 equiv.) were dissolved in EtOAc (50 mL). Copper(ii) bromide (1.7 equiv.) was added slowly and the reaction mixture was stirred at 85 °C for 24 h. The solvent was evaporated under reduced pressure and the solid residue was washed with EtOAc and dried under reduced pressure to obtain a crude product that was used in the next step without further purification.
2-Bromo-1-(4-ethylphenyl)ethan-1-one 4
Brown oil; yield 100% (crude, unpurified product); 1H NMR (400 MHz, DMSO-d6) δ 1.21 (t, J = 7.0 Hz, 3H, CH[combining low line]3), 2.70 (q, J = 7.0 Hz, 2H, CH[combining low line]2CH3), 4.91 (s, 2H, CH[combining low line]2), 7.40 (d, J = 7.7 Hz, 2H, Ar–H[combining low line]), 7.94 (d, J = 7.7 Hz, 2H, Ar–H[combining low line]). HRMS (m/z) (ESI): calcd for C10H11BrO [M + H]+: 227.0072, found: 227.0069.
General procedure for the synthesis of 1-substituted-2-bromo-ethan-1-ones 5–7
The corresponding ethan-1-ones (starting amount ranging from 2.4 to 10.2 mmol, 1 equiv.) were dissolved in MeOH (volume ranging from 2 to 8 mL). The solution was cooled to 0 °C and bromine (1 equiv.) was added dropwise. The mixture was stirred for 3 h at a temperature below 15 °C and then H2O (2 mL) was added. The mixture was stirred for additional 24–48 h at rt. H2O (25 mL) was subsequently added and the aqueous layer was extracted with EtOAc (3 × 25 mL). The combined organic phases were washed with an aqueous solution of Na2CO3 (10%, 50 mL), dried with anhydrous Na2SO4, filtered, and evaporated under reduced pressure to obtain crude products that were used in the next step without further purification.
2-Bromo-1-cyclohexylethan-1-one 7
Orange oil; yield 90% (crude, unpurified product); 1H NMR (400 MHz, DMSO-d6) δ 1.15–1.48 (m, 5H, cyclohexyl-CH[combining low line]2), 1.62–1.70 (m, 1H, cyclohexyl-CH[combining low line]), 1.75–1.82 (m, 2H, cyclohexyl-CH[combining low line]2), 1.84–1.92 (m, 2H, cyclohexyl-CH[combining low line]2), 2.71 (sym m, 1H, cyclohexyl-CH[combining low line]), 3.97 (s, 2H, COCH[combining low line]2Br). HRMS (m/z) (ESI): calcd for C8H14OBr [M + H]+: 206.0922, found: 206.0918.
General procedure for the synthesis of compounds 8–17, 50 and 51
To a solution of 7-hydroxycoumarin 1, 48 or 49 (starting amount ranging from 0.93 to 2.0 mmol, 1 equiv.) in dioxane (50 mL), K2CO3 (4 equiv.) and KI (0.1 equiv.) were added. After 5 min, the corresponding α-haloketone (1.5 equiv.) was added and the reaction mixture was stirred at 100 °C for 24 h. The solvent was then removed under reduced pressure, followed by the addition of H2O (100 mL) to the residue. The aqueous phase was extracted with EtOAc (3 × 100 mL), and the combined organic layers were washed with brine (200 mL), dried with anhydrous Na2SO4, filtered, and evaporated under reduced pressure. Compounds 8–17, 50 and 51 were subsequently purified by crystallization from EtOH.
Ethyl 7-(2-(4-ethylphenyl)-2-oxoethoxy)-4-methyl-2-oxo-2H-chromene-3-carboxylate 14
Yellow solid; yield 74%; 1H NMR (400 MHz, DMSO-d6) δ 1.22 (t, J = 7.5 Hz, 3H, Ar–CH2CH[combining low line]3), 1.30 (t, J = 7.1 Hz, 3H, COOCH2CH[combining low line]3), 2.44 (s, 3H, Ar–CH[combining low line]3), 2.72 (q, J = 7.5 Hz, 2H, Ar–CH[combining low line]2CH3), 4.33 (q, J = 7.1 Hz, 2H, COOCH[combining low line]2CH3), 5.77 (s, 2H, CH[combining low line]2), 7.09–7.15 (m, 2H, Ar–H[combining low line]), 7.44 (d, J = 8.0 Hz, 2H, Ar–H[combining low line]), 7.83 (d, J = 9.0 Hz, 1H, Ar–H[combining low line]), 7.97 (d, J = 8.0 Hz, 2H, Ar–H[combining low line]). HRMS (m/z) (ESI): calcd for C23H22O6 [M + H]+: 395.1495, found: 395.1491.
Ethyl 7-(2-cyclohexyl-2-oxoethoxy)-4-methyl-2-oxo-2H-chromene-3-carboxylate 17
Orange solid; yield 78%; 1H NMR (400 MHz, DMSO-d6) δ 1.15–1.34 (m, 8H, CH2CH[combining low line]3 and cyclohexyl-CH[combining low line]2), 1.62–1.88 (m, 5H, cyclohexyl-CH[combining low line]2), 2.43 (s, 3H, Ar–CH[combining low line]3), 2.53–2.60 (m, 1H, cyclohexyl-CH[combining low line]), 4.33 (d, J = 7.1 Hz, 2H, CH[combining low line]2CH3), 5.16 (s, 2H, CH[combining low line]2), 6.96–7.03 (m, 2H, Ar–H[combining low line]), 7.81 (d, J = 8.8 Hz, 1H, Ar–H[combining low line]). HRMS (m/z) (ESI): calcd for C21H24O6 [M + H]+: 373.1651, found: 373.1651.
Spectroscopic analyses of compounds 50 and 51 were previously reported.22
General procedure for the synthesis of 4-methyl-3-carboxyalkyl substituted psoralens 18–27, 52 and 53
To a suspension of the desired O-alkylated compound 8–17, 48 or 49 (starting amount ranging from 0.56 to 1.45 mmol, 1 equiv.) in propan-2-ol (volume ranging from 20–30 mL), an aqueous solution of NaOH (1 M, 10 equiv.) was added. The reaction mixture was stirred at 100 °C for 5–20 min. After the reaction was complete (monitored by TLC), propan-2-ol was evaporated under reduced pressure. The aqueous residue was acidified with 5 M HCl to pH 1, and the precipitate that formed was filtered off. In cases where there was no precipitate, the aqueous layer was extracted with EtOAc (2 × 50 mL), and the combined organic layers were washed with brine (100 mL), dried with anhydrous Na2SO4, filtered, and evaporated under reduced pressure to obtain a crude product that was used in the next step without further purification.
3-(4-Ethylphenyl)-5-methyl-7-oxo-7H-furo[3,2-g]chromen-6-carboxylic acid 24
Brown oil; yield 100% (crude, unpurified product); 1H NMR (400 MHz, DMSO-d6) δ 1.20 (t, J = 7.5 Hz, 3H, CH2CH[combining low line]3), 2.45 (s, 3H, Ar–CH[combining low line]3), 2.68 (q, J = 7.5 Hz, 2H, CH[combining low line]2CH3), 7.31 (s, 1H, Ar–H[combining low line]), 7.40–7.47 (m, 4H, Ar–H[combining low line]), 8.26 (s, 1H, Ar–H[combining low line]), 8.49 (s, 1H, Ar–H[combining low line]). HRMS (m/z) (ESI): calcd for C21H16O5 [M + H]+: 349.1076, found: 349.1081.
3-Cyclohexyl-5-methyl-7-oxo-7H-furo[3,2-g]chromen-6-carboxylic acid 27
Orange solid; yield 100% (crude, unpurified product); 1H NMR (400 MHz, DMSO-d6) δ 1.26–1.31 (m, 4H, cyclohexyl-CH[combining low line]2), 1.42–1.49 (m, 4H, cyclohexyl-CH[combining low line]2), 2.05–2.07 (m, 2H, cyclohexyl-CH[combining low line]2), 2.43 (s, 3H, Ar–CH[combining low line]3), 2.82–2.87 (m, 1H, cyclohexyl-CH[combining low line]), 7.57 (s, 1H, Ar–H[combining low line]), 7.82 (d, J = 0.8 Hz, 1H, Ar–H[combining low line]), 7.93 (s, 1H, Ar–H[combining low line]). HRMS (m/z) (ESI): calcd for C19H18O5 [M + H]+: 327.1232, found: 327.1235.
Spectroscopic analyses of compounds 52 and 53 were previously reported.22
General procedure for the synthesis of 4-methyl-3-carboxamide substituted psoralens 28–37
To a suspension of the desired 4-methyl-3-carboxyalkyl substituted psoralen 18–27 (starting amount ranging from 0.29 to 2.10 mmol, 1 equiv.) in dioxane (volume ranging from 4–20 mL), Boc2O (2 equiv.), pyridine (4 equiv.) and ammonium acetate (6 equiv.) were added. The reaction mixture was stirred at rt for 24 h. The solvent was evaporated under reduced pressure and pyridine was removed by co-evaporation with toluene. CH2Cl2 (50 mL) was added and the organic phase was washed with water (2 × 50 mL). The organic layer was washed with brine (100 mL), dried with anhydrous Na2SO4, filtered, and evaporated under reduced pressure. The compounds were purified by column chromatography (EtOAc/n-hexane = 2 : 1).
3-(4-Ethylphenyl)-5-methyl-7-oxo-7H-furo[3,2-g]chromen-6-carboxamide 34
Orange solid; yield 6%; 1H NMR (400 MHz, CDCl3) δ 1.32 (t, J = 7.6 Hz, 3H, CH2CH[combining low line]3), 2.75 (q, J = 7.6 Hz, 2H, CH[combining low line]2CH3), 2.90 (s, 3H, Ar–CH[combining low line]3), 7.36–7.41 (m, 2H, CONH[combining low line]2), 7.53 (s, 1H, Ar–H[combining low line]), 7.54–7.57 (m, 2H, Ar–H[combining low line]), 7.72–7.76 (m, 2H, Ar–H[combining low line]), 7.84 (s, 1H, Ar–H[combining low line]), 8.20 (s, 1H, Ar–H[combining low line]). HRMS (m/z) (ESI): calcd for C21H17NO4 [M + H]+: 348.1236, found: 348.1241.
3-Cyclohexyl-5-methyl-7-oxo-7H-furo[3,2-g]chromen-6-carboxamide 37
White solid; yield 27%; 1H NMR (400 MHz, DMSO-d6) δ 1.42–1.52 (m, 4H, cyclohexyl-CH[combining low line]2), 1.73–1.83 (m, 4H, cyclohexyl-CH[combining low line]2), 2.05–2.09 (m, 2H, cyclohexyl-CH[combining low line]2), 2.54 (s, 3H, Ar–CH[combining low line]3), 2.83–2.91 (m, 1H, cyclohexyl-CH[combining low line]), 7.67–7.69 (m, 1H, CONH[combining low line]2), 7.71 (s, 1H, Ar–H[combining low line]), 7.87–7.89 (m, 2H, Ar–H[combining low line] and CONH[combining low line]2), 8.11 (s, 1H, Ar–H[combining low line]). HRMS (m/z) (ESI): calcd for C19H19NO4 [M + H]+: 326.1392, found: 326.1395.
General procedure for the synthesis of oxathiazolone-based compounds 38–47
To a suspension of the desired 4-methyl-3-carboxamide substituted psoralen 28–37 (starting amount ranging from 0.09 to 0.47 mmol, 1 equiv.) in anhydrous THF (volume ranging from 3 to 5 mL), chlorocarbonylsulfenyl chloride (4 equiv.) was added under argon at rt. The resulting solution was stirred at 80 °C for 1.5–2 h. After the reaction was complete (monitored by TLC), the solvent was removed under reduced pressure. Compounds 38–47 were subsequently purified by crystallization from a mixture of EtOAc/n-hexane and column chromatography (EtOAc/n-hexane = 1 : 3).
5-(3-(4-Ethylphenyl)-5-methyl-7-oxo-7H-furo[3,2-g]chromen-6-yl)-1,3,4-oxathiazol-2-one 44
Yellow solid; yield 16%; mp 67.5–68.8 °C; 1H NMR (400 MHz, CDCl3) δ 1.32 (t, J = 7.6 Hz, CH2CH[combining low line]3), 2.65 (s, 3H, Ar–CH[combining low line]3), 2.75 (q, J = 7.6 Hz, 2H, CH[combining low line]2CH3), 7.36–7.41 (m, 2H, Ar–H[combining low line]), 7.53–7.56 (m, 2H, Ar–H[combining low line]), 7.57 (s, 1H, Ar–H[combining low line]), 7.85 (s, 1H, Ar–H[combining low line]), 8.12 (s, 1H, Ar–H[combining low line]). 13C NMR (100 MHz, CDCl3) δ 172.93, 158.17, 156.21, 153.57, 151.41, 149.65, 144.69, 143.35, 128.90 (2C), 127.75, 127.64 (2C), 125.16, 122.39, 117.64, 115.50, 112.71, 100.55, 28.72, 17.16, 15.60. HRMS (m/z) (ESI): calcd for C22H16NO5S [M + H]+: 406.07437, found: 406.07355. Purity by HPLC: 100%.
5-(3-Cyclohexyl-5-methyl-7-oxo-7H-furo[3,2-g]chromen-6-yl)-1,3,4-oxathiazol-2-one 47
Off-white solid; yield 45%; mp 124.8–125.8 °C; 1H NMR (400 MHz, CDCl3) δ 1.28–1.39 (m, 1H, cyclohexyl-CH[combining low line]2), 1.47–1.55 (m, 4H, cyclohexyl-CH[combining low line]2), 1.83–1.94 (m, 3H, cyclohexyl-CH[combining low line]2), 2.13–2.15 (m, 2H, cyclohexyl-CH[combining low line]2), 2.70 (s, 3H, Ar–CH[combining low line]3), 2.76–2.86 (m, 1H, cyclohexyl-CH[combining low line]), 7.49 (s, 1H, Ar–H[combining low line]), 7.50 (d, J = 0.8 Hz, 1H, Ar–H[combining low line]), 7.91 (s, 1H, Ar–H[combining low line]). 13C NMR (100 MHz, CDCl3) δ 173.00, 158.08, 156.29, 153.72, 151.23, 142.57, 126.54, 126.03, 116.97, 114.80, 112.38, 111.47, 100.31, 34.07, 33.09 (2C), 26.46 (2C), 26.18, 17.21. HRMS (m/z) (ESI): calcd for C20H17NO5S [M + H]+: 384.0906, found: 384.0899. Purity by HPLC: 96%.
General procedure for the synthesis of coumarin derivatives 48 and 49
To a solution of resorcinol (45.41 mmol, 1 equiv.) in H2SO4 (75%, 50 mL), diethyl 2-acetylsuccinate or diethyl 2-acetylglutarate (1.1 equiv.) was added. The reaction mixture was stirred at room temperature for 24 h. After the reaction was complete, the solution was poured onto crushed ice. The solid that formed was filtered off, washed with water and dried. Spectroscopic analyses of compounds 48 and 49 were previously reported.22
General procedure for the synthesis of psoralens with the cyclohexyl moiety at position 4′ with variation at position 3 (compounds 54–59)
The starting compound 52 or 53 (1 equiv.) was transferred to a dry flask. A suitable nucleophilic reagent (N-hydroxysuccinimide (1 equiv.), 2-(methylamino)acetonitrile (1.25 equiv.) or 2-aminoacetonitrile (2 equiv.)) was added. THF (5 mL) was added under an argon atmosphere at 0 °C. After stirring for 5 min at 0 °C, EDC (1.25 equiv.) and Et3N (2.5 equiv.) were added. The reaction mixture was stirred at room temperature for 24 h. The solvent was evaporated and HCl (0.01 M, 20 mL) was added. The reaction mixture was extracted with EtOAc (3 × 30 mL). The combined organic layers were dried with anhydrous Na2SO4, filtered, and evaporated under reduced pressure. The compounds were purified by column chromatography (EtOAc or EtOAc/n-hexane = 2 : 1).
2,5-Dioxopyrrolidin-1-yl 3-(3-cyclohexyl-5-methyl-7-oxo-7H-furo[3,2-g]chromen-6-yl)propanoate 55
White solid; yield 32%; mp 150.1–151.3 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.25–1.30 (m, 1H, cyclohexyl-H[combining low line]), 1.36–1.50 (m, 4H, cyclohexyl-H[combining low line]), 1.70–1.85 (m, 3H, cyclohexyl-H[combining low line]), 2.02–2.09 (m, 2H, cyclohexyl-H[combining low line]), 2.58 (s, 3H, Ar–CH[combining low line]3), 2.80 (br s, 4H, COCH[combining low line]2CH[combining low line]2CO), 2.84–2.90 (m, 1H, cyclohexyl-H[combining low line]), 2.95 (sym m, 4H, CH[combining low line]2), 7.65 (s, 1H, Ar–H[combining low line]), 7.85 (s, 1H, Ar–H[combining low line]), 8.07 (s, 1H, Ar–H[combining low line]); 13C NMR (100 MHz, DMSO-d6) δ 170.12, 168.22, 160.52, 155.44, 149.65, 149.10, 142.37, 126.20, 124.44, 120.49, 116.40, 116.12, 98.96, 33.02, 32.50, 28.68, 25.95, 25.69, 25.37, 22.58, 15.36. HRMS (m/z) (ESI): calcd for C25H26NO7 [M + H]+: 452.1723, found: 452.1716. Elemental analysis: found: C, 66.20; H, 5.50; N, 3.41; calcd for C25H25NO7: C, 66.51; H, 5.58; N, 3.10%.
Conflicts of interest
The authors declare no competing interests.
Supplementary Material
Acknowledgments
We sincerely thank Assoc. Prof. Dr. Urban Švajger (Center for Development of Advanced Therapy Medicinal Products, Blood Transfusion Center of Slovenia) for the assistance in performing the cytokine secretion assay. The authors acknowledge the support from the Slovenian Research Agency: research core funding No. P1-0208, and grant number N1-0068 (B) to S.G.: identification of non-peptidic inhibitors of the immunoproteasome using fragment based drug discovery methods.
Footnotes
†Electronic supplementary information (ESI) available: General chemistry methods, all biochemistry experimental data, spectroscopic analyses, and representative NMR, HPLC, and HRMS spectra. See DOI: 10.1039/c9md00365g
References
- Collins G. A., Goldberg A. L. Cell. 2017;169:792–806. doi: 10.1016/j.cell.2017.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budenholzer L., Cheng C. L., Li Y., Hochstrasser M. J. Mol. Biol. 2017;429:3500–3524. doi: 10.1016/j.jmb.2017.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt C. S., Hochstrasser M. Proc. Natl. Acad. Sci. U. S. A. 1997;94:7156–7161. doi: 10.1073/pnas.94.14.7156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groettrup M., Khan S., Schwarz K., Schmidtke G. Biochimie. 2001;83:367–372. doi: 10.1016/s0300-9084(01)01251-2. [DOI] [PubMed] [Google Scholar]
- Griffin T. A., Nandi D., Cruz M., Fehling H. J., Van Kaer L., Monaco J. J., Colbert R. A. J. Exp. Med. 1998;187:97–104. doi: 10.1084/jem.187.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sledź P., Baumeister W. Annu. Rev. Pharmacol. Toxicol. 2016;56:191–209. doi: 10.1146/annurev-pharmtox-010814-124727. [DOI] [PubMed] [Google Scholar]
- Micale N., Scarbaci K., Troiano V., Ettari R., Grasso S., Zappalà M. Med. Res. Rev. 2014;34:1001–1069. doi: 10.1002/med.21312. [DOI] [PubMed] [Google Scholar]
- Kuhn D. J., Hunsucker S. A., Chen Q., Voorhees P. M., Orlowski M., Orlowski R. Z. Blood. 2009;113:4667–4676. doi: 10.1182/blood-2008-07-171637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlati F., Lee S. J., Aujay M., Suzuki E., Levitsky K., Lorens J. B., Micklem D. R., Ruurs P., Sylvain C., Lu Y., Shenk K. D., Bennett M. K. Blood. 2009;114:3439–3447. doi: 10.1182/blood-2009-05-223677. [DOI] [PubMed] [Google Scholar]
- Thibaudeau T. A., Smith D. M. Pharmacol. Rev. 2019;71:170–197. doi: 10.1124/pr.117.015370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler M., Lindstrom M. M., LaStant J. J., Bradshaw J. M., Owens T. D., Schmidt C., Maurits E., Tsu C., Overkleeft H. S., Kirk C. J., Langrish C. L., Groettrup M. EMBO Rep. 2018;19:e46512. doi: 10.15252/embr.201846512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson H. W., Lowe E., Anderl J. L., Fan A., Muchamuel T., Bowers S., Moebius D. C., Kirk C., McMinn D. L. J. Med. Chem. 2018;61:11127–11143. doi: 10.1021/acs.jmedchem.8b01201. [DOI] [PubMed] [Google Scholar]
- de Bruin G., Hubert E. M., Xin B.-T., van Rooden E., Al-Ayed K., Kim K.-B., Kisselev A. F., Driessen C., van der Stelt M., van der Marel G. A., Groll M., Overkleeft H. S. J. Med. Chem. 2014;57:6197–6209. doi: 10.1021/jm500716s. [DOI] [PubMed] [Google Scholar]
- Xin B.-T., Huber E. M., de Bruin G., Heinemeyer W., Maurits E., Espinal C., Du Y., Janssens M., Weyburne E. S., Kisselev A. F., Florea B. I., Driessen C., van der Marel G. A., Groll M., Overkleeft H. S. J. Med. Chem. 2019;62:1626–1642. doi: 10.1021/acs.jmedchem.8b01884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubiella C., Baur R., Cui H., Huber E. M., Groll M. Angew. Chem., Int. Ed. 2015;54:15888–15891. doi: 10.1002/anie.201506631. [DOI] [PubMed] [Google Scholar]
- Singh P. K., Fan H., Jiang X., Shi L., Nathan C. F., Lin G. ChemMedChem. 2016;11:2127–2131. doi: 10.1002/cmdc.201600384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karreci E. S., Fan H., Uehara M., Mihali A. B., Singh P. K., Kurdi A. T., Solhjou Z., Riella L. V., Ghobrial I., Laragione T., Routray S., Assaker J. P., Wang R., Sukenick G., Shi L., Barrat F. J., Nathan C. F., Lin G., Azzi J. Proc. Natl. Acad. Sci. U. S. A. 2016;113:E8425–E8432. doi: 10.1073/pnas.1618548114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson H. W., Anderl J. L., Bradley E. K., Bui J., Jones J., Arastu-Kapur S., Kelly L. M., Lowe E., Moebius D. C., Muchamuel T., Kirk C., Wang Z., McMinn D. ACS Med. Chem. Lett. 2017;8:413–417. doi: 10.1021/acsmedchemlett.6b00496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettari R., Previti S., Bitto A., Grasso S., Zappala M. Curr. Med. Chem. 2016;23:217–238. doi: 10.2174/0929867323666160318173706. [DOI] [PubMed] [Google Scholar]
- Kisselev A. F., Groettrup M. Curr. Opin. Chem. Biol. 2014;23:16–22. doi: 10.1016/j.cbpa.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber E. M., Groll M. Angew. Chem., Int. Ed. 2012;51:8708–8720. doi: 10.1002/anie.201201616. [DOI] [PubMed] [Google Scholar]
- Sosič I., Gobec M., Brus B., Knez D., živec M., Konc J., Lešnik S., Ogrizek M., Obreza A., žigon D., JaneŽič D., Mlinarič-Raščan I., Gobec S. Angew. Chem., Int. Ed. 2016;55:5745–5748. doi: 10.1002/anie.201600190. [DOI] [PubMed] [Google Scholar]
- Kasam V., Lee N.-R., Kim K.-B., Zhan C.-G. Bioorg. Med. Chem. Lett. 2014;24:3614–3617. doi: 10.1016/j.bmcl.2014.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H., Baur R., Le Chapelain C., Dubiella C., Heinemeyer W., Huber E. M., Groll M. ChemBioChem. 2017;18:523–526. doi: 10.1002/cbic.201700021. [DOI] [PubMed] [Google Scholar]
- Fan H., Angelo N. G., Warren J. D., Nathan C. F., Lin G. ACS Med. Chem. Lett. 2014;5:405–410. doi: 10.1021/ml400531d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosc E., Nastri J., Lefort V., Valli M., Contiguiba F., Pioli R., Furlan M., da Silva Bolzani V., El Amri C., Reboud-Ravaux M. Biochem. Biophys. Res. Commun. 2018;496:961–966. doi: 10.1016/j.bbrc.2018.01.100. [DOI] [PubMed] [Google Scholar]
- Lin G., Li D., de Carvalho L., Deng H., Tao H., Vogt G., Wu K., Schneider J., Chidawanyika T., Warren J., Li H., Nathan C. Nature. 2009;461(7264):621–626. doi: 10.1038/nature08357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewerth D., Jansen G., Riethoff L. F. V., van Meerloo J., Kale A. J., Moore B. S., Assaraf Y. G., Anderl J. L., Zweegman S., Kaspers G. J. L., Cloos J. Mol. Pharmacol. 2014;86:12–19. doi: 10.1124/mol.114.092114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A., Michielin O., Zoete V. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.