

Abstract
Older patients with obesity/type II DM frequently present with advanced non-alcoholic steatohepatitis (NASH). Whether this is due to specific molecular pathways that accelerate fibrosis during aging, is unknown. Activation of the Src homology 2 domain containing collagen-related (Shc) proteins and redox stress have been recognized in aging, however their link to NASH has not been explored. Shc expression increased in livers of older patients with NASH, as assessed by RTqPCR or western blots. Fibrosis, Shc expression, markers of senescence and NADPH oxidases (NOXs) were studied in young/old mice on fast food diet (FFD). To inhibit Shc in old mice LV-shShc vs. control-LV were used during FFD. For hepatocyte-specific effects, fl/fl Shc mice on FFD were injected with AAV8-TBG-Cre vs. control. Fibrosis was accelerated in older mice on FFD, and Shc inhibition by LV in older mice, or hepatocyte-specific deletion resulted in significantly improved inflammation, reduction in senescence markers in older mice, lipid peroxidation and fibrosis. To study NOX2 activation, the interaction of p47phox (NOX2 regulatory subunit) and p52Shc was evaluated by proximity ligation, and co-IPs. Palmitate induced p52Shc binding to p47phox activating the NOX2 complex, more so at older age. Kinetics of binding were assessed in SH2 or PTB deletion mutants by biolayer interferometry, revealing the role of SH2 and the PTB domains. Lastly, an in silico model of p52Shc/p47phox interaction using RosettaDock was generated.
Conclusion:
Accelerated fibrosis in the aged is modulated by p52Shc/NOX2. We show a novel pathway for direct activation of the phagocytic NOX2 in hepatocytes by p52Shc binding and activating the p47phox subunit that results in redox stress, and accelerated fibrosis in the aged.
Keywords: liver fibrosis, NADPH oxidase, Src homology 2 domain containing collagen-related (Shc) proteins
Non-alcoholic steatohepatitis (NASH) and associated cirrhosis are the leading causes of liver-related mortality in the US and worldwide (1). The incidence of NASH rises strikingly with age, contributing significantly to elder mortality (2). Older patients are often excluded from liver transplantation because of comorbidities and the age limit for transplantation; therefore, a significant increase in NASH-related mortality is expected since NASH has no approved medical treatment. Most of the preclinical data in NASH/fibrosis research is derived from experiments employing young mice. In clinical trials however, the target population is usually middle-aged or elderly. To address this contradiction, we focused on the elucidating age-related pathways that contribute to accelerated fibrosis progression. Oxidative stress is a key to NASH progression and anecdotally has been linked to aging pathways in different organs (3). In the liver, the source of oxidative radicals during aging as well as the signaling pathways linking to a more profibrogenic phenotype have not been well defined.
Src homology 2 domain containing collagen-related (Shc) proteins regulate multiple aging-related pathways, serving as adaptors to receptor tyrosine kinase signals (4), controlling metabolic pathways including insulin signals (5), and modulating oxidative pathways (6). They derive from the ShcA locus: p46 and p52Shc are generated from different start codons within the same transcript, whereas p66Shc is a product of alternative splicing. The domain structure of Shc consists of 2 major domains: the phosphotyrosine-binding domain (PTB), and the Src Homology 2 domain (SH2) connected via a CH1 domain/fragment. The PTB domain is responsible for binding to phosphorylated receptor tyrosine kinases, facilitating the membrane translocation of Shc. SH2 is a binding domain to other proteins and is available for binding in only open conformation (7). Downregulation of Shc mitigates aging-related dysregulation of energy metabolism (8). Several studies with the p66ShcKO mice have been published, however, in reality, these are hypomorphs with a significant decrease in p46 and p52Shc expression (9). Because of the low p66Shc expression in the liver, the other isoforms should be taken into consideration in the context of liver injury and fibrosis during aging.
In this study, we focused on the role of p52Shc and discovered the critical link between p52Shc and the activation of the phagocytic NADPH oxidase (NOX2) in non-phagocytic hepatocytes during aging. Aged mice exhibited more pronounced inflammatory, oxidative and fibrogenic injury and an increase in senescence compared to young animals. We show that p46Shc and p52Shc are induced in NASH patients during aging, and in older mice on fast food diet (FDD). Lentiviral (LV)-mediated short hairpin RNA to Shc (LV-ShShc) reduced inflammation, markers of senescence and fibrosis in old mice. In hepatocytes, the phagocytic NOX2 enzyme was activated by an interaction with p52Shc via the regulatory p47phox subunit of NOX2. The importance of the p52Shc isoform was also corroborated by the direct binding assays where mutations in the p52Shc (SH2 or PTP binding domains) has affected binding efficiency and kinetics. During aging, p52Shc/NOX2-mediated redox stress is central to hepatocyte injury and disease progression.
Experimental Procedures:
Human liver biopsy samples.
Liver biopsy samples were obtained from the Stanford Pathology Department, the XenoTech Research Biobank (Kansas City), and the UC Davis Cancer Center Biorepository (funded by the NCI). Frozen liver samples (from surgical resection) from 12 individuals of different ages (6 samples with normal histology, obtained from peritumoral tissues, and 6 patients with NASH) were processed for western blots. Formalin fixed paraffin-embedded (FFPE) biopsy samples from 4 additional patients (normal histology), and 3 NASH patients were processed for RT-qPCR. All samples were de-identified and exempted. Patient demographics, histology, NAS score and fibrosis stage are provided in Supplementary Table I.
Animal experiments.
All animal experiments were conducted according to the experimental procedures approved by the Institutional Animal Care and Use Committee at Stanford University, Palo Alto VA, and at UC Davis. Mice were in standard cages with 12:12 hour light/dark cycles and ad libitum access to water and food. Young (6-week), middle-aged (26-week) and old (48-week) male mice with C57BL/6 background were obtained from Charles River (Wilmington, MA) via the NIA consortium (https://www.nia.nih.gov). Shc hypomorph (“Shc knockdown, or ShcKD”) and wild type (WT) littermates were originally from the Pelicci lab (10), and fl/fl Shc mice were from Dr. Haj (UC Davis). Mice were placed on control chow or FFD ( #1810060, 17.4% protein, 20% fat, and 49.9% carbohydrate, AIN-76A, Richmond, IN), supplemented with high fructose corn syrup in the drinking water at a final concentration of 42 g/L, for 18 weeks. Older mice were injected with short hairpin (Sh) Shc or Sh-scrambled lentiviral vectors (1.0×109 PFU) via the tail vein at the 9th week of feeding.
In a different cohort, male fl/fl Shc mice or littermate controls were fed chow or FFD, for 18 weeks. To generate a hepatocyte specific Shc knockdown, mice were injected with either AAV8-TBG-Cre or control GFP vectors (Vector BioLabs) at 5.0×10e11GC at week 9 of FFD. At the end of experiments, mice were fasted overnight, serum and liver samples were collected for further analysis, and liver/body weight ratios assessed.
Statistical analyses.
All data represent at least three independent experiments using the standard error of the mean (SEM). ANOVA and two-tailed student’s t-test were used to analyze the significance of the data. A value of p< 0.05 was considered significant. Correlation coefficient between Shc expression and patients’ age was analyzed by Pearson r correlation.
The other methods for primary cell isolation and culture, immunohistochemistry, SA-β galactosidase staining, immunofluorescence, confocal microscopy, lucigenin assay, malondialdehyde, triglyceride content assay, real time qPCR, western blotting immunoprecipitation, proximity ligation assay, biolayer interferometry, and in silico modeling are described in detail in the Supplementary Materials and Methods. Primers used for RT-qPCR, antibodies, and SAS interface of p52Shc to the p47phox are listed in Supplementary Tables 2–4.
Results
Older age increases oxidative injury, inflammation, markers of senescence and fibrosis in mice on fast food diet.
To assess age-dependent sensitivity to diet-induced NASH, we fed young (6w at the beginning of feeding), and middle-aged (26w) mice FFD for 18 weeks. This diet closely resembles calorie intake and composition that of human diet linked to NASH and results in weight gain, insulin resistance, and steatohepatitis with fibrosis (11). NAS scores in young mice were 1–4, fibrosis stage 0–1, and in middle-aged mice 5–7, and 1–3, respectively. TNF-α expression (p<0.05) increased more significantly in older vs. young mice on FFD (Fig. 1A), and older mice exhibited an increased number of F4/80 positive macrophages (Fig. 1C). Serum ELISA showed an increase inTNF-α in middle-aged mice (Fig. S1A). There was extensive fibrosis with an increase in procollagen-Iα1, TGF-β, and a more enhanced increase in hydroxy-proline (Fig. 1B), and picrosirius red signal (Fig. 1C) in older mice. An expansion of CK19+ ductular cells was seen in older mice on FFD whereas it was negligible in young mice (Fig. 1C). As senescence has been observed in aging livers in the context of steatosis and alcohol use (12, 13), we examined senescence phenotypes in our models. Middle-aged mice had an increase in p21Cip1 nuclear signal (Fig. 1C), mRNA expression for p21Cip1 and p16INK4a (Fig. 1D), and senescence associated heterochromatin foci (SAHF, Fig 1D). SAHF contain silent hetero-chromatin domains consisting of a di-or tri-methylated lysine 9 of histone H3 (H3K9Me2/3), and histone H2A variant (macroH2A). We observed increased marks in the older cells, and larger nuclei that are described during senescence (14). Senescent cells are also known to excrete various chemokines, proinflammatory cytokines, or proteases acquiring senescence-associated secretory phenotype (SASP). We saw significant increase in IL-1β (p<0.01), and increasing trends in IL-1α, IL-6, and Gro-β (Fig. 1D). Of note, SASP markers showed more significance in old mice (48w) on FFD (Fig. S2D). SA-β-galactosidase showed positive signal in older mice on FFD (Fig. S1B). Middle-aged mice also exhibited more prominent oxidative injury (Fig. 1E). To analyze whether this was linked to NADPH oxidases (NOXs), key sources of redox radicals in NASH, we studied NOX2 (phagocytic NOX), and the non-phagocytic isoforms NOX1 and 4 in the liver. Only NOX2 was induced in older mice whereas NOX1 and 4 were not (NOX4 was induced in NASH both in young/old mice, but the difference was not statistically significant). Steatosis increased in middle-aged mice (Fig. S1C) and more so, in old mice after FFD (Fig. S2F).
Figure 1. Older mice display enhanced necroinflammation, ductular reaction, fibrosis and markers of senescence on fast food diet (FFD).

Young (6-week, at the beginning of FFD), and middle-aged mice (26-week) were fed FFD for 18 weeks. Older mice had more significantly increased mRNA expression for TNF-α (A). Procollagen-Iα1 (col1α1), TGF-β, hydroxy-proline (B), picrosirius red positive area (C), as assessed by NIH ImageJ analysis, have significantly increased compared to young mice on the same diet, and they exhibited expansion of CK19+ ductular cells (counted from 8 random fields of 3–4 mice in each group C). Middle-aged mice had an increased number of F4/80 positive macrophages (bar=100um, quantified by ImageJ from 5 random fields of 3–6 mice in each group, C). Increase in markers of senescence (p21Cip1 positive nuclei, arrows, C) was seen in older mice, and senescence associated histone marks mH2A1 and H3K9Me2/3 had more nuclear signal (D). Markers of SASP IL-1α, IL-6, IL-1β and Gro-β showed an increase in older mice (D). More significant ROS production (E, lucigenin chemiluminescence assay), and induction of the NADPH oxidase (NOX2) was seen on FFD in older mice whereas the non-phagocytic NOX1 and 4 isoforms showed no significant induction compared to young mice (Mean±SEM, N=4–5, *p<0.01, **p<0.01).
We also studied liver samples of old mice (48w at the beginning of feeding), after a shorter course of FFD. We opted for a shorter course (8w) as these mice may not survive the entire course of 18w feeding. In these mice NAS scores were 5–7, fibrosis stage 2–3. We confirmed a more enhanced induction of proinflammatory and fibrogenic transcripts in old vs. young mice (Fig. S2A, B). Of note, young mice had no significant increase in profibrogenic transcripts at this early stage whereas there was a significant induction after short feeding in older mice. NOX2 was upregulated in old compared to young mice on FFD (Fig. S2C). There was significant induction of markers of senescence (even compared to middle-aged mice in Fig. 1D), with an upregulation of p16INK4A and P21Cip1 (Fig. S2D) and SASP markers (IL-1α, IL-1β, IL-6, Groβ). SAHF marks were increased, as well (Fig. S2E). Steatosis showed an increase in old mice on FFD (Fig. S2F).
Shc proteins are more induced in older patients and in mice with NASH
Shc proteins are adaptors regulating metabolic signals and Shc dysregulation has been linked to the aging process in several tissues. To study the different Shc isoforms in the liver, we analyzed p66Shc as it has been featured in publications relating to oxidative stress. By analyzing human and mouse samples by western blots, we found that p66Shc was expressed at a low level at baseline (Fig. 2A) and was not induced in NASH (Fig. 2C). In contrast, p46 and 52Shc were the major liver isoforms in both humans and mice. p46 and 52Shc were induced in an age-dependent manner in older patients with NASH (Fig. 2A). Shc expression examined in 10 normal and 9 NASH patient samples by RT-qPCR showed an age-dependent increase, especially in NASH (Fig. 2B). Shc immunostaining depicts an increased signal in NASH patient samples compared to age-matched control (Fig. 2C). γH2aX marker of increased DNA damage in senescence, and SAHF marks mH2A and H3K9Me2/3 had an enhanced nuclear signal in older NASH patients (Fig. 2C). In middle-aged mice on FFD, there was Shc induction at protein and mRNA level (Fig. 2D). Shc was periportal in young mice with NASH whereas in older mice the signal was more extensive, panlobular (Fig. 2E). To better localize Shc, immunofluorescence and confocal microscopy were done using albumin (hepatocytes), F4/80 (Kupffer cells), and CK19 (cholangiocytes) antibodies. Shc was expressed in hepatocytes, and bile ductular cells to a lesser extent. With this method we could not detect a signal in Kupffer cells (Fig. 2F).
Figure 2. p46 and p52Shc are the main liver isoforms that are induced in older NASH patients and in mice on NASH diet.
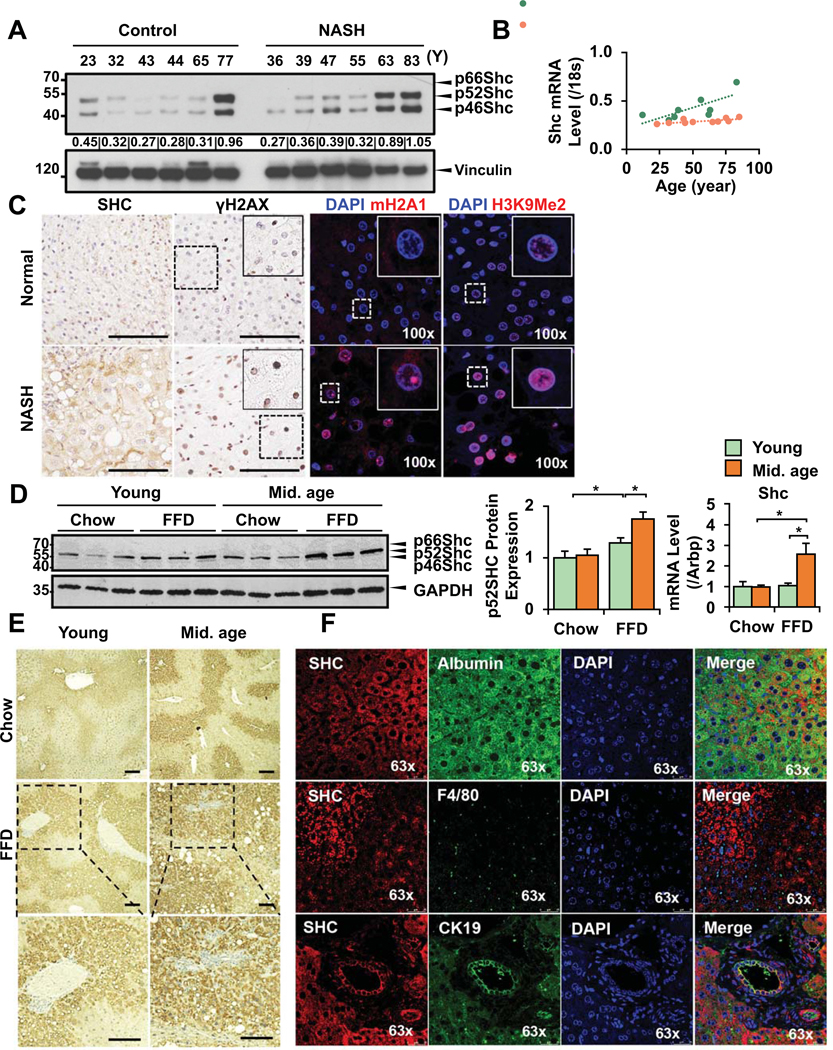
Normal human liver samples (resection samples from peritumoral areas) and samples from NASH patients (NAS score 5–7, fibrosis stage 0–4, see Table I for details) were obtained. By western blot analyses, p46 and p52Shc were expressed at a higher level during aging, and more so in the livers of NASH patients whereas the p66Shc isoform was not induced (A, densitometry data for each patient is included). To have more quantitative assessment, RT-qPCR data from 10 normal and 9 NASH patients were analyzed for Shc and linear regression analysis shows more age-dependent induction in NASH patients (R values as shown, B). Representative Shc immunostaining in control and NASH patients depicts increased signal in NASH (bar=100um, C). There is more intense γH2aX nuclear signal (DNA damage), mH2A1 and H3Me2K9 (SAHF marks) in livers of old NASH patients compared to age-matched controls (C). Young (6-week) and middle-aged mice (26-week) were fed FFD for 18 weeks. Shc was studied by western blots, and RTqPCR. p46/p52Shc were induced in older mice on FFD (WB, densitometry), and total Shc by RTqPCR (D). Immunohistochemistry of total Shc (E) shows increased signal in the liver lobules in older mice on FFD (enlarged areas shown in the lower panel, bar=100um). Immunofluorescence analysis and confocal microscopy in young and older mice shows Shc (red) signals relative to hepatocytes (albumin, green), Kupffer cells (green), and cholangiocytes (green), (F). While Shc colocalized with albumin, and CK19, no signal was seen in Kupffer cells by immunoflorescence (Mean±SEM, N=4–5, *p<0.05).
Inhibition of Shc in old mice or Shc hypomorphs (ShcKD) protects against liver injury and fibrosis in NASH.
To study whether Shc inhibition in older mice improves NASH, 48w old mice were fed chow or FFD, and injected with either 1011pfu LV-shShc (or LV-shScr as control), at week 9 of 18. NAS scores in LV-ShScr mice on FFD were 5–7 and in LV-ShShc mice 3–5, fibrosis stage 2–3 and 0–2, respectively. Western blot shows a decrease in Shc in LV-shShc-injected mice (Fig. S3A). ALT levels (Fig. 3A), mRNA expression of MCP1, TNF-α (Fig. 3B), number of F4/80 positive macrophages (Fig. 3C) were significantly reduced in LV-ShShc-injected mice. Procollagen-Iα1, α-SMA, and TGF-β (Fig. 3B) showed significant decrease. Picrosirius red (Fig. 3C) and hydroxy-proline (Fig. 3B) demonstrate that LV-shShc injected mice had less fibrosis. As we observed an increase in senescent cells in middle-aged mice, we studied senescence in this older model. SAHF marks mH2A1 and H3K9Me2/3 increased in control-LV-injected mice on FFD with improvement in LV-ShShc (Fig. 3D). Expression of p16INK4a and P21Cip1 and SASP markers (IL-1α, IL-1β, IL-6, Groβ, Fig. 3E), and γH2aX marks (Fig. S3B) showed reduction in LV-ShShc injected mice. There was also an improvement in serum TNF-α (Fig. S3C), redox radicals, NOX2 expression, TG content (Fig. 3F) and steatosis (Fig. S3D) in LV-ShShc-injected mice.
Figure 3. Shc inhibition reduces inflammation, fibrosis, and senescence markers in FFD-fed old mice.

48w old mice were fed FFD and injected by LV-ShScr or LV-ShShc (1011pfu) at week 9 of the 18w diet. Inhibition of Shc reduced ALT (A), decreased mRNA expression for TNF-α, MCP1 (B), and decreased the number of F4/80 macrophages (C, bar=100um). Procol1α1, α-SMA, TGF-β, OH-proline levels were reduced (B), and picrosirius red (PSR) images (C) revealed less fibrosis in shShc-injected mice. Senescence marker p21Cip1 nuclear signal (arrows, C) as well as mRNA levels increased in LV-ShScr-injected mice and improved after Shc inhibition. SAHF marks mH2A1 and H3K9Me2/3 (red) displayed more nuclear signal in control-vector-injected mice with lower staining after Shc inhibition (blue: DAPI, D). SASP markers IL-1α, IL-6, IL-1β and Gro-β showed a similar trend with an improvement in Shc-inhibited mice (E). Lower oxidative radicals, NOX2 expression and triglyceride levels were seen in ShShc-transduced mice (F) (Mean±SEM, N=4, *p<0.05, **p<0.01).
Total ShcKO mice are embryonically lethal. Past studies investigating the influence of Shc on energy metabolism focused on the p66Shc isoform and used “p66Shc KO” mice. While these lack p66Shc, they also have decreased levels of p52Shc and p46Shc in the liver (Fig S4A), skeletal muscle and heart (6). We placed the Shc hypomorphs (henceforth called ShcKD) and wt age-matched littermates on FFD and found that TNF-α, IL-1β, MCP1, fibrogenic transcripts procollagen-Iα1, TGF-β, and NOX2 were reduced, and TG content has improved (Fig. S4B).
Hepatocyte-specific Shc deleted mice show improvement in NASH and fibrosis
Since hypomorph or LV-injected mice have reduction of Shc in all liver cells, we first focused on stellate cells (HSC) and monocytes/macrophages as potential sources for Shc-mediated proinflammatory and fibrogenic events. Shc was not induced in culture-activated primary, HSC; and the induction of profibrogenic transcripts was not different in young vs. old cells (Fig. S5A). HSC from ShcKD mice showed no significant difference in procollagen-Iα1 or α-SMA (Fig. S5B). In a murine macrophage cell line Shc was required for macrophage differentiation thus it was conceivable that Shc in macrophages could age-dependently modulate phagocytic activity. However, oxidative burst upon efferocytosis was not affected in macrophages isolated from older mouse livers (Fig. S5C). While cholangiocytes expressed Shc, no NOX2 signal was visible in these cells by immunostaining (Fig. S5D).
Therefore, it was plausible that hepatocyte Shc had a major role in driving proinflammatory and fibrogenic responses. Hepatocyte-specific Shc deletion by AAV8-TBG-Cre in FFD-fed Shc fl/fl mice reduced NAS scores (from 5–6 in AAV8-GFP-injected mice to 3 in AAV8-Cre mice), fibrosis (from 2–3 to 0–1), mRNA expression of proinflammatory transcripts (Fig. 4A), F4/80 positive macrophage numbers (Fig. 4D), fibrogenic transcripts, hydroxy-proline (Fig. 4B, C), and redox radicals and NOX2 (Fig. 4C). Picrosirius red, 4-hydroxynoneal assay revealed that AAV8-TBG-Cre injected mice displayed reduced fibrosis, lipid peroxidation (Fig. 4D), and steatosis (Fig. S6A). Senescence markers showed no significant increase in nuclear p21Cip1 (Fig. S6B), but early changes in SAHF marks (Fig. S6C) were detected. No significant change in SASP was seen (data not shown) however, these mice were much younger than those injected with LV. Sirt1 is a histone-deacetylase involved in the transcriptional control of metabolism and its low activity has been associated with senescent phenotypes in several organs (15, 16). Supporting aging-mediated changes in Sirt1, we found that it was downregulated in older mice on FFD and that Shc downregulation in the LV or AAV8-injected model reversed this (Fig. S7A). Sirt1 is known to be a master regulator of lipid metabolism (17). In our models age-mediated increase in steatosis was seen that improved in Shc-inhibited mice (Fig. S3D). In hepatocytes isolated from wt and ShcKD mice palmitate-induced Shc expression improved in ShcKD. LXRα and important nuclear receptor targeted by Sirt1 showed similar trend suggesting Shc effects on lipid metabolism (Fig. S7B). Liver/body weight ratios have not changed significantly between genotypes (Fig. S8).
Figure 4: Shc deletion from hepatocytes by an AAV8-TBG-Cre approach in FFD-fed Shc fl/fl mice reduced inflammation, fibrosis, and lipid peroxidation.

fl/fl Shc mice (12w-old) were placed on chow diet or FFD. At week 9 of the 18w FFD, a group of fl/fl mice were injected with either AAV8-GFP (control) or AAV8-TBG-Cre via the tail vein. Proinflammatory transcripts (A, TNF-α, IL-1β, MCP1), and F4/80 positive macrophage numbers (D) show significant improvement in AAV8-TBG-Cre-injected mice. Fibrogenic transcripts Col1α1, α-SMA, and TGF-β (B) were induced by FFD in control AAV8-injected mice, and were significantly reduced after AAV8-TBG-Cre injection. ROS production, NOX2 expression and OH-proline contents were reduced after Shc deletion (C). Picrosirius red and 4-hydroxynoneal images (D, bar=100um) demonstrate that AAV8-Cre-injected mice displayed less fibrosis and lipid peroxidation (Mean±SEM, N=4–6, *p<0.05, **p<0.01).
Palmitate-induced binding of p52Shc to the regulatory p47phox subunit of NOX2 is enhanced in old hepatocytes
Older mice presented with more prominent oxidative injury, and NOX2 induction. Hence, we asked the question: what is the role and mechanism of NOX2 activation in non-phagocytic hepatocytes? NOX2 is classically activated by phagocytosis of pathogens or apoptotic bodies (efferocytosis) in monocytes/macrophages or neutrophils. We previously showed that stellate cells become phagocytic and profibrogenic upon efferocytosis (18). However, in non-phagocytic epithelial cells such as hepatocytes the presence and mode of activation of NOX2 posed an interesting question. p52Shc is an important adaptor protein that upon phosphorylation the Y317 or Y239/240 residues translocates to the membrane. Thus, it was conceivable that p52Shc may interact with the NOX2 complex, activate the enzyme and induce oxidative burst. P47phox is a key regulatory subunit of NOX2 that upon serine phosphorylation associates to p67phox and other cytosolic factors activating the NOX2 complex and superoxide production (19). We performed proximity ligation (PLA) experiments on primary hepatocytes (1 day after isolation) from young (6w) or old (20m) mice to study the binding of p52Shc to p47phox upon palmitate challenge. There was more intense signal in older hepatocytes after palmitate exposure (Fig. 5A, B). Of note, isolated hepatocytes from old mice showed higher level of p52Shc expression similar to liver tissues (Fig. 5C). To address whether the NOX2 complex was functionally more active in a Shc-dependent way in older cells, first, we isolated membrane fractions from BSA or palmitate-treated wt. or ShcKD hepatocytes, and performed lucigenin chemiluminescence assay (Fig. 5D). Significant reduction of ROS production was seen following palmitate exposure in ShcKD hepatocytes. To analyze the p52Shc-dependence of our findings, we generated mutant SH2 domain-deleted p52Shc. Primary hepatocytes were transfected with ΔSH2p52Shc also had significantly reduced ROS production (Fig. 5D). Immunoprecipitation (IP) of NOX2 (gp91phox) from membrane fractions of BSA vs. palmitate-exposed hepatocytes and immunoblots of p-serine p47phox that denotes the activated state of the enzyme complex revealed that the enzyme activity was reduced in ShcKD hepatocytes (Fig. 5E). We performed an IP of NOX2 (gp91phox) from membrane fractions of AAV8-GFP or AAV8-TBG-Cre injected Shc fl/fl livers and found that serine phosphorylation of the complex was much reduced after AAV8-TBG-Cre injection (Fig. 5F).
Figure 5. The NOX2 complex regulatory subunit p47phox binds to p52Shc.

Primary hepatocytes from young (6w) or old (20m) mice 1 day after isolation were treated with palmitate (Palm) (200uM) or BSA, and Proximity Ligation Assay (PLA) was done (A). More intense binding of p52Shc with p47phox is seen in old cells (red signal; green: hepatocyte autofluorescence, blue: DAPI; BSA: bovine serum albumin, N=3, bar=20um). Calculation of PLA+ signals was done in 5 cells/experiment, N=3, using NIH ImageJ program (B). WB shows that older hepatocytes retain higher expression level of p52Shc (C). Palmitate increases ROS production in WT hepatocytes but not in ShcKD cells (D). We generated a binding mutant of p52Shc (SH2 domain-deleted) and primary hepatocytes were transfected with either ΔSH2p52Shc or full length p52Shc, then exposed to palmitate. Lucigenin assay demonstrates significantly reduced ROS in ΔSH2p52Shc-transfected cells (D). To study NOX2 activity, membrane preps from primary BSA or palmitate-exposed hepatocytes was performed. After immunoprecipitation (IP) of NOX2 (gp91phox) immunoblots were done to detect P-serine p47phox which denotes the activated state of the enzyme. Decreased P-serine p47phox was seen in ShcKD cells after palmitate (E). IP of gp91phox was performed on membrane preps from chow, AAV-GFP and AAV8-TBG-Cre-injected Shc fl/fl mice on FFD. IB with p-serine was performed showing a decreased signal in AAV8-Cre injected mice (F) (Mean±SEM, N=4–6, *p<0.05, **p<0.01).
p52Shc binds to p47phox via its PTB and SH2-domains
To analyze the areas in p52Shc that are requisite for binding to p47phox, we generated mutants of p52Shc lacking either the PTB or SH2 binding domains (Fig. 6A). Using Biolayer Interferometry (BLI), we determined that p52Shc can directly interact with p47phox, evaluated binding kinetics, and measured the affinity of p52Shc to p47phox. There was strong binding of the full-length p52Shc to p47phox with an affinity of 200 nM. p52ShcΔPTB exhibited about 10 times weaker affinity in the micromolar range. Deletion of the SH2 domain (p52ShcΔSH2) abrogated the interaction with p47phox, and the affinity was very weak, EC50> 50 uM (Fig. 6B, C). Thus, the SH2 domain is a key for the interaction with p47phox whereas the presence of PTB domain is required for successful phosphorylation of Shc and its full biological activity. Treating Shc with alkaline phosphatase prior to the reaction resulted in much decreased affinity. The non-phosphorylated full size p52Shc interacted with p47phox with an affinity of only 780 nM (Fig. 6D).
Figure 6. Biolayer interferometry demonstrates binding of p47phox to p52Shc.

p52Shc mutants for the phosphotyrosine-binding domain (PTB) and SH2 domain were generated (A). Bio-Layer Interferometry (BLI) was performed to study the interaction between p52Shc and p47phox. Proteins were expressed in HEK T293 cells. Terminally biotinylated p52Shc protein, p52Shc lacking the PTB domain (p52ShcΔPTB), p52Shc lacking SH2 domain (p52ShcΔSH2), GFP and not biotinylated p47phox::His were produced. The contact of the p52Shc-loaded biosensors with p47phox resulted in classical association and dissociation kinetics, whereas the p52Shc deletion mutants produced much smaller responses (B). The maximal p47phox-Shc interaction is depicted, measured in a range of concentrations (C). The kinetics of association was p52Shc>p52ShcΔPTB>p52ShcΔSH2, and in the case of p52ShcΔSH2, essentially no binding was seen. The Kd of the p52Shc to p47phox interaction had high affinity, 3.7E−7M. Inhibiting phosphorylation of p52Shc by alkaline phosphatase (AP) resulted in the loss p47phox binding (D).
To model the docking of p47phox to p52Shc, RosettaDock was used that identifies low energy conformations of protein interactions. The results in Supplementary Table 4 represent the Solvent Accessible Surface Area (SAS) interface of p52Shc to the p47phox (20). Dock1 interface predicts 1045 Å2 while the Dock2 interface predicts 650 Å2 buried surface area. The model predicted to share SAS similarity to 32 structural protein-protein SAS interfaces (21). p52Shc for both docked poses buried SH2 domain interface to p47phox. Dock1 interface buried the SH2 domain of p52Shc to the PX domain of p47phox, while the Dock2 interface buried SH2 to the region close to the SH3 domain of p47phox. For the first dock pose, there were several buried residues: G460 of the SH2 domain in p52Shc can make contact to F14, A87 of p47phox. On the other hand, hydrophilic residues (S417, E462, R433, and G460) of SH2 in p52Shc could make contact to E88, D82, G83, E137, and R90 of p47phox. For the second dock pose, residues T406, Q409, G408 of SH2 in p52Shc could make contact to R306, H309, Q313 of p47phox (Fig. 7).
Figure 7. Docking model of p47phox and p52Shc.

The most probable complex that represents the SH2 domain of p52Shc binding with p47phox is selected. The p47phox is in a space-filling model, and p52Shc is in ribbon model, where PX domain is colored in violet, SH2 is green, PTB and SH3/C-terminus is in grey. Zoom-in views of dock 2 and dock 3 represent the close contact residues from p47phox and SH2 of p52Shc which may interact at the protein-protein interface.
Discussion
NASH often presents at advanced stages in older patients, many of whom are excluded from liver transplantation. Most studies in NASH/fibrosis research however, use young mice as animal models. To overcome this contradiction, we carried out this study on age-related NASH progression and the involvement of Shc proteins. We found that both p52Shc and NOX2/ROS production were key to fibrosis in aging and NASH, furthermore we uncovered a novel mechanism of non-phagocytic activation of NOX2 and ROS production in hepatocytes via p52Shc. Aging mice exhibited more inflammation, steatosis and fibrosis in Shc-dependent manner, and they also had more extensive ductular reaction, and displayed a senescent phenotype. NOXs catalyze the reduction of molecular oxygen to superoxide using NADPH as an electron donor. NOXs are important sources of ROS in NASH (18). In the liver hepatocytes, hepatic stellate cells (HSC) and macrophages express the phagocytic NOX2 (22). While the role of NOX2 in macrophages and HSC has been established, its role in non-phagocytic hepatocytes has not been studied. We identified that the aging protein p52Shc binds to and activates the NOX2 enzyme complex via the p47phox subunit. Although binding occurs in younger cells, it is more intense and further augmented by the increased expression of both Shc and NOX2 in old hepatocytes. As p47phox is also a regulatory unit for NOX1, it is possible that the Shc-mediated regulatory effect extends to this NOX further exacerbating the production of ROS.
We discovered the direct interaction of p47phox to p52Shc through several methods. BLI revealed the key role of the SH2 domain whereas deleting the PTB domain reduced binding suggesting that phosphorylation of Shc was important to conformational changes and interaction/NOX2 enzyme activation to occur. PLA experiments showed more intense binding signal in old hepatocytes and immunoprecipitation revealed serine phosphorylation of p47phox. Based on docking analyses, the SH2 domain can favorably dock within the PX domain of p47phox. Further studies delineating the exact residues would be required, as these data may form a basis for future targeting studies.
P52Shc also modulates insulin signaling (6) thus it is tempting to speculate that it is a central adaptor linking redox burst to dysregulated insulin responses through redoxosomal compartmentalization. Here we focused on p52Shc as the isoform that had a potential of regulating NOX2 activity through its adaptor function. Previously, the p66Shc isoform was linked to aging-mediated redox stress in multiple organs however we show that human or mouse livers express low levels of p66Shc and it is not induced in patients or mice with NASH. We saw that p46Shc was also induced in aging which raises the question regarding its role in NASH. In fact, p46Shc dysregulated mitochondrial β-oxidation by inhibiting 3-ketoacylCoA thiolase (ACAA2) (23) thereby increasing the FFA load in hepatocytes. More work would be required to decipher the respective roles of p46 and p52Shc isoforms in NASH. To study the role of Shc in hepatocytes, we generated an AAV8-TBG-Cre deleted model. Of note, here middle-aged mice were studied as we did not have access to old floxed mice. However, these studies already showed significant protection after Shc deletion, and in conjunction with the LV-deleted and ShcKD models in old mice the in vivo data point to Shc as key factor in NASH progression during aging.
Aging and oxidative damage can promote senescence characterized by cell cycle arrest, DNA damage response, development of SAHF, and acquiring SASP. In our middle-aged mice on NASH diet, we saw an increase in some of the senescence markers whereas old mice had more significant induction of SASP and SAHF. Interestingly, reducing Shc in old mice on FFD reversed changes in SAHF, p21Cip1, p16INKa, DNA damage marker γH2AX and SASP. Repression of Shc has been previously associated to delaying replicative senescence in fibroblasts (24). In our case, because the effect on steatosis it was tempting to speculate that the Shc/NOX2 axis is potentially involved in Sirt1 dysregulation. To support this notion we found that Sirt1 was reduced during aging and this was reversed by inhibition of Shc in old mice. LXRα followed a similar Shc-dependent pattern thus it is likely that Shc plays a role in dysregulation of lipid metabolism during aging, via NOX2/ROS, or an independent pathway.
In conclusion, we describe a non-phagocytic, p52Shc-dependent pathway of NOX2 activation in hepatocytes that is more enhanced during aging. Targeting the interaction between Shc/p47phox or lowering Shc would be a rationale therapeutic options in aging and NASH.
Supplementary Material
Figure 8. Working model of p52Shc and p47phox interaction and subsequent NADPH oxidase 2 activation in NASH hepatocytes.

Binding of p52Shc via its SH2 domain to the NOX2 subunit p47phox and its subsequent serine phosphorylation induces the assembly of NOX2 enzyme complex, resulting superoxide production.
Acknowledgements.
We thank Dr. Fawaz Haj (UC Davis, Dept. of Nutrition) for the floxed Shc mice, and Dr. Yanan Wang for the Image J analyses. The NIA consortium (https://www.nia.nih.gov/research/dab/aged-rodent-colonies) facilitated obtaining the aged mice.
Financial Support:
This research was supported by funding from the NIDDK R01 DK083283 (N.J.T), NIA 1RO1AG060726 (G.C and N.J.T), I01 BX002418 (N.J.T), and NIA PO1 AG025532 (J.R. and G.C.).
List of Abbreviations:
- NASH
non-alcoholic steatohepatitis
- Shc
Src homology 2 domain containing collagen-related
- FFD
fast food diet
- NOXs
NADPH oxidases
- LV
lentivirus
- AAV8
adeno-associated virus
- PLA
proximity ligation assay
- IP
immunoprecipitation
- PTB
phosphotyrosine-binding domain
- SH2
Src Homology 2 domain
- TNF-α
tumor necrosis factor alpha
- IL-1β
Interleukin 1 beta
- TGF-β
transforming growth factor beta
- SASP
senescence-associated secretory phenotype
- SAHF
senescence associated heterochromatin foci
- H3K9Me2/3
di-or tri-methylated lysine 9 of histone H3
- macroH2A
histone H2A variant
- γH2aX
gamma histone 2 AX
References
- 1.Angulo P, Machado MV, Diehl AM. Fibrosis in nonalcoholic Fatty liver disease: mechanisms and clinical implications. Semin Liver Dis 2015;35:132–145. [DOI] [PubMed] [Google Scholar]
- 2.Bertolotti M, Lonardo A, Mussi C, Baldelli E, Pellegrini E, Ballestri S, Romagnoli D, et al. Nonalcoholic fatty liver disease and aging: epidemiology to management. World J Gastroenterol 2014;20:14185–14204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shaw PX, Werstuck G, Chen Y. Oxidative stress and aging diseases. Oxid Med Cell Longev 2014;2014:569146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tiganis T, Bennett AM, Ravichandran KS, Tonks NK. Epidermal growth factor receptor and the adaptor protein p52Shc are specific substrates of T-cell protein tyrosine phosphatase. Mol Cell Biol 1998;18:1622–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hagopian K, Kim K, Lopez-Dominguez JA, Tomilov AA, Cortopassi GA, Ramsey JJ. Mice with low levels of Shc proteins display reduced glycolytic and increased gluconeogenic activities in liver. Biochem Biophys Rep 2016;7:273–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tomilov AA, Ramsey JJ, Hagopian K, Giorgio M, Kim KM, Lam A, Migliaccio E, et al. The Shc locus regulates insulin signaling and adiposity in mammals. Aging Cell 2011;10:55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou MM, Ravichandran KS, Olejniczak EF, Petros AM, Meadows RP, Sattler M, Harlan JE, et al. Structure and ligand recognition of the phosphotyrosine binding domain of Shc. Nature 1995;378:584–592. [DOI] [PubMed] [Google Scholar]
- 8.Ranieri SC, Fusco S, Panieri E, Labate V, Mele M, Tesori V, Ferrara AM, et al. Mammalian life-span determinant p66shcA mediates obesity-induced insulin resistance. Proc Natl Acad Sci U S A 2010;107:13420–13425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomilov A, Bettaieb A, Kim K, Sahdeo S, Tomilova N, Lam A, Hagopian K, et al. Shc depletion stimulates brown fat activity in vivo and in vitro. Aging Cell 2014;13:1049–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, et al. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature 1999;402:309–313. [DOI] [PubMed] [Google Scholar]
- 11.Charlton M, Krishnan A, Viker K, Sanderson S, Cazanave S, McConico A, Masuoko H, et al. Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol 2011;301:G825–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huda N, Liu G, Hong H, Yan S, Khambu B, Yin XM. Hepatic senescence, the good and the bad. World J Gastroenterol 2019;25:5069–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wan Y, McDaniel K, Wu N, Ramos-Lorenzo S, Glaser T, Venter J, Francis H, et al. Regulation of Cellular Senescence by miR-34a in Alcoholic Liver Injury. Am J Pathol 2017;187:2788–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aravinthan A, Scarpini C, Tachtatzis P, Verma S, Penrhyn-Lowe S, Harvey R, Davies SE, et al. Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease. J Hepatol 2013;58:549–556. [DOI] [PubMed] [Google Scholar]
- 15.Fusco S, Maulucci G, Pani G. Sirt1: def-eating senescence? Cell Cycle 2012;11:4135–4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pibiri M Liver regeneration in aged mice: new insights. Aging (Albany NY) 2018;10:1801–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yin H, Hu M, Liang X, Ajmo JM, Li X, Bataller R, Odena G, et al. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology 2014;146:801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bettaieb A, Jiang JX, Sasaki Y, Chao TI, Kiss Z, Chen X, Tian J, et al. Hepatocyte Nicotinamide Adenine Dinucleotide Phosphate Reduced Oxidase 4 Regulates Stress Signaling, Fibrosis, and Insulin Sensitivity During Development of Steatohepatitis in Mice. Gastroenterology 2015;149:468–480 e410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Babior BM. NADPH oxidase. Curr Opin Immunol 2004;16:42–47. [DOI] [PubMed] [Google Scholar]
- 20.Shrake A, Rupley JA. Environment and exposure to solvent of protein atoms. Lysozyme and insulin. J Mol Biol 1973;79:351–371. [DOI] [PubMed] [Google Scholar]
- 21.Jones S, Thornton JM. Principles of protein-protein interactions. Proc Natl Acad Sci U S A 1996;93:13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reinehr R, Becker S, Eberle A, Grether-Beck S, Haussinger D. Involvement of NADPH oxidase isoforms and Src family kinases in CD95-dependent hepatocyte apoptosis. J Biol Chem 2005;280:27179–27194. [DOI] [PubMed] [Google Scholar]
- 23.Tomilov A, Tomilova N, Shan Y, Hagopian K, Bettaieb A, Kim K, Pelicci PG, et al. p46Shc Inhibits Thiolase and Lipid Oxidation in Mitochondria. J Biol Chem 2016;291:12575–12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu F, Pang L, Cai X, Liu X, Yuan S, Fan X, Jiang B, et al. let-7-repressesed Shc translation delays replicative senescence. Aging Cell 2014;13:185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.