

Graphical Abstract

INTRODUCTION.
Botulinum neurotoxins (BoNTs) are the etiological agents of botulism, a potentially fatal disease characterized by flaccid muscle paralysis. BoNT serotype A (BoNT/A), used therapeutically as Botox™, is the most potent known toxin with an estimated intravenous (iv) 50% lethal dose (LD50) of 1–2 ng/kg and an inhalation LD50 of 100–120 ng/kg in humans.1 Due to the toxin’s long half-life in vivo and the slow-turnover of peripheral muscle neurons, intoxicated patients are often hospitalized for weeks to months, requiring mechanical ventilation and enteral or parenteral nutrition.1 Yearly botulism cases in the United States number in the low hundreds, with most cases resulting from infant botulism. However, the potential use of BoNT/A in a bioterrorism attack remains a serious concern, as evidenced by its classification by the Centers for Disease Control and Prevention (CDC) as a Category A threat.2
A prophylactic pentaserotype vaccine for BoNTs A-E was available for at-risk populations under a CDC Investigational New Drug (IND) application; however, reduced potency over time prompted the vaccine’s discontinuation.3 Currently, there are no Federal Drug Administration (FDA)-approved vaccines for BoNT. The current standard of treatment for botulism relies on antibody therapy; an equine-derived antitoxin can be administered post-exposure to patients with non-infant botulism. However, use of BoNT antitoxin is limited by high cost, potential for adverse reactions, and a short window of opportunity. Specifically, the antitoxin is only effective when administered within 12–24 hours post-exposure due to the inability of antibodies to enter the neuronal compartment.4 A small molecule inhibitor would overcome the limitations of antitoxin because small molecules remain effective after neuronal intoxication, prevent further paralysis, and shorten patient rehabilitation time. To that end, the Janda Laboratory has worked for over fifteen years on small molecule BoNT/A inhibitor discovery.
Herein, we describe the efforts made by the Janda Laboratory towards the discovery of a clinically-viable BoNT/A inhibitor. Through high-throughput screening (HTS) methods and rational design using structure activity relationship (SAR) studies, our group has developed some of the most potent reported inhibitors of BoNT/A. This account discusses inhibitors with multiple mechanisms of action (MOA), including active site binders with zinc chelating and/or covalent MOAs, α-exosite and β-exosite binders, and small molecules with unique MOAs.
STRUCTURAL AND MECHANISTIC CONSIDERATIONS.
BoNT/A is a 150 kDa protein composed of a heavy chain (HC, 100 kDa) linked via a disulfide bond to a light chain (LC, 50 kDa). The HC promotes toxin endocytosis by binding ecto-acceptors on the presynaptic membrane.6–7 Once in the endosome, pH-dependent conformational changes to the LC facilitate its passage into the neuronal cytosol through a channel formed by the HC (Figure 1A).8–9 Cytosolic BoNT/A LC cleaves SNAP-25 (synaptosomal-associated protein of 25-kDa), a SNARE (soluble N-ethylmaleimide sensitive factor attachment protein receptor) protein required for vesicle fusion and release of acetylcholine into the postsynaptic cleft.10
Figure 1.
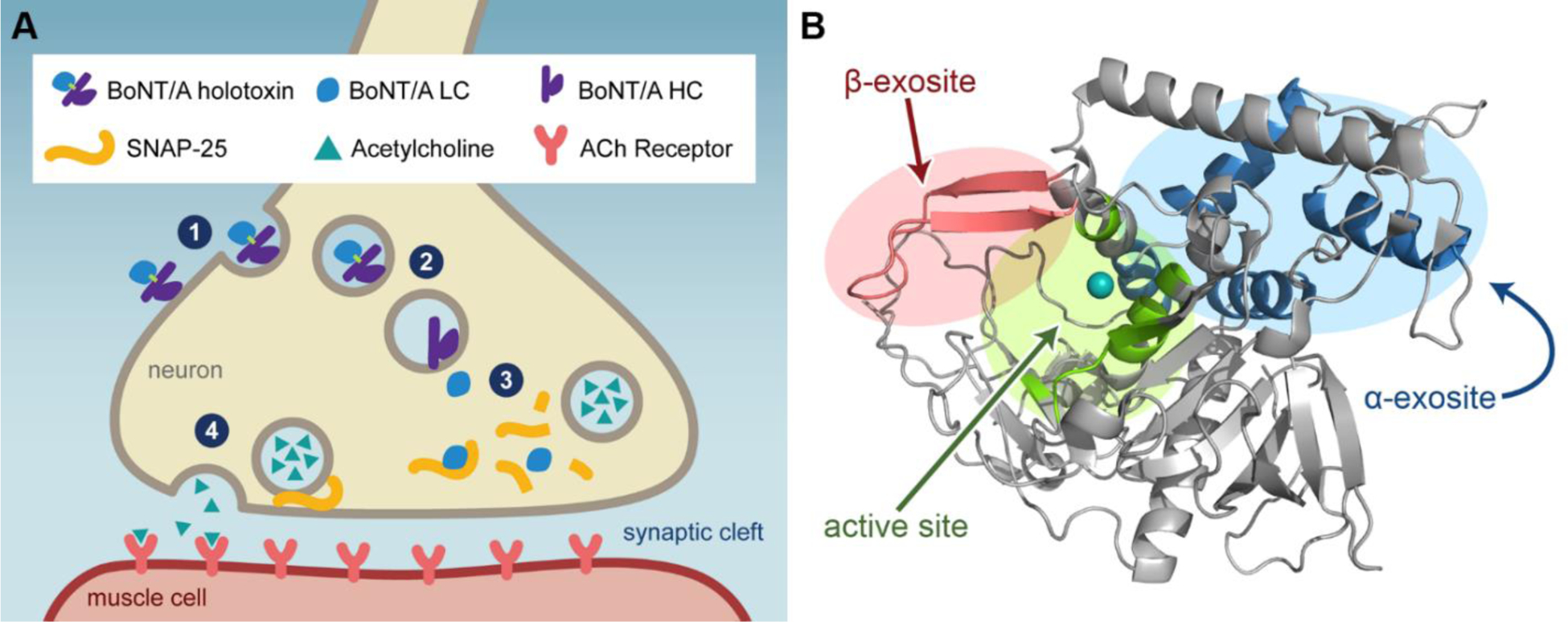
A) Mechanism of BoNT/A action on neurons: (1) BoNT/A holotoxin is recognized at the HC receptor binding domain and internalized. (2) Reduction of the disulfide bond allows the translocation domain of the HC to move BoNT/A LC into the cytosol. (3) Cleavage of SNAP-25 by the LC prevents acetylcholine-carrying vesicle fusion. (4) In a non-intoxicated neuron, SNAP-25 is part of the SNARE complex that allows vesicle docking, fusion, and subsequent release of acetylcholine. B) Structure of BoNT/A LC and the locations of its three major substrate recognition domains. Zinc is shown as a teal sphere in the active site. PDB: 1ZTG.5
The active site of BoNT/A LC contains a Zn2+ atom, which is required for catalytic activity but not the enzyme’s tertiary structure (Figure 1B).11 The residues that line the active site are mostly hydrophobic and form a tight pocket. Interestingly, the enzyme is quite flexible; BoNT/A co-crystal structures demonstrate appreciable changes in the overall shape of the active site dependent on the ligand under investigation.12 In addition to the active site, the other major binding sites are the α-exosite and the β-exosite. The former is a hydrophobic patch at the interface of four α-helices, the latter is comprised of an antiparallel β-sheet that includes the ‘250-loop’ and the ‘370-loop’ of the protein.5, 13 The exosites are located on opposite sides of the enzyme, with the β-exosite located adjacent to the active site and the α-exosite located on the opposing face. Substrate binding to the exosites induces a conformational change in the active site, allowing SNAP-25 to bind with higher affinity.5 It has been shown that excision of a-exosite binding residues from SNAP-25 reduces kcat/Km and point mutations in the β-exosite decrease kcat nearly 50-fold.5, 14
IN VITRO AND IN VIVO ASSAYS FOR SMALL MOLECULE EVALUTION
Throughout the course of our BoNT/A drug discovery program, we have made use of various assays to probe the MOA and efficacy of our inhibitors. The most basic enzyme assay, referred to as the “SNAPtide assay”, is a fluorescence resonance energy transfer (FRET) assay that utilizes truncated BoNT/A LC (1–425) and the FRET substrate SNAPtide to provide a facile and efficient system amenable to HTS.15 SNAPtide is a 13aa (amino acid) modified peptide that contains a FRET pair, commonly FITC/DABCYL, and mimics a portion of BoNT/A’s physiological substrate SNAP-25; BoNT/A LC cleaves the scissile bond between Gln197 and Arg198 of SNAP-25.16 Small molecule inhibitors that bind in the enzyme active site and compete with SNAP-25 can thus be identified with the SNAPtide assay, as well as inhibitors that compromise enzyme integrity allosterically.
Kinetic limitations inherent to the SNAPtide assay prompted our laboratory to develop an LCMS-based assay, the “66mer assay”.17 The 66aa substrate consists of residues 141–206 of the native SNAP-25 sequence (1–206, full-length) and incorporates both the α- and β-exosite binding regions. This robust 66mer assay enables full kinetic characterization of BoNT/A LC inhibitors from multiple classes, including small molecules that disrupt essential protein-protein interactions between the enzyme and substrate distal to the active site (e.g. α-exosite and β-exosite inhibitors). Furthermore, synergistic effects among inhibitors with complementary MOAs can be evaluated.
Cell-based assays provide a model system that requires all steps of BoNT/A intoxication, initiating with cell surface receptor binding and culminating in intracellular SNAP-25 cleavage (Figure 1A). Early efforts with immortal cell lines, such as Neuro-2a, provided valuable information. However, lack of cell sensitivity to BoNT necessitated the simultaneous addition of toxin and compound. Recent advancements in stem cell research led to the development of human-induced pluripotent stem cells (hiPSCs) that can be differentiated into mature human neurons, which retain high sensitivity to BoNT/A.18 Accordingly, our collaborative efforts with the University of Wisconsin resulted in the development of a post-intoxication cell model using hiPSC-derived neurons. In this study, small molecules are evaluated on their ability to rescue intoxicated neurons by Western blot analysis of SNAP-25 cleavage. An alternative cell-based approach takes advantage of a neuron-specific cell line that expresses intracellular full-length SNAP-25 substrate containing N- and C-terminal fluorescent proteins. In contrast to the endpoint cell assays described vide supra, SNAP-25 cleavage can be continuously monitored in real-time BoCell assay.19
To probe the ability of inhibitors to disrupt the translocation event in the intoxication pathway (Figure 1; Step 2), small molecule modulation of holotoxin channels were investigated in a single-molecule assay designed with Professor Montal’s laboratory at the University of California, San Diego.20 In this assay, meticulous mechanistic studies at the single-molecule level are conducted with excised membrane patches from Neuro-2A cells and translocation of the LC through the HC channel is monitored in real-time through electrophysiology recordings.
The complexity of neuromuscular transmission necessitates an assay that evaluates the restoration of muscle contractions post-intoxication. The mouse phrenic hemidiaphragm assay is an established ex vivo model system that is highly sensitive to BoNT/A, and is often considered a replacement assay for in vivo studies.21 Muscle contractions stimulated by the phrenic nerve are measured in hemidiaphragm preparations excised from mice. BoNT/A poisoning causes a dose-dependent decrease in muscle twitch amplitude and paralytic half-time (HPT). Administration of effective therapeutics, including K+ channel blockers and BoNT/A LC inhibitors, can delay the onset of paralysis.
Finally, the mouse bioassay, which evaluates lethality as a result of intoxication, is the gold standard in vivo method to assess BoNT/A activity. In this assay, mice are challenged with ~5–10 LD50 of BoNT/A administered intraperitoneally (ip), after which small molecule is administered. The antidotal potency of treatment is measured as an extension of time to death with respect to toxin control. It is worth noting that mouse bioassay results with pharmacotherapeutic antagonists should not be directly compared to those of immunotherapeutic agents (e.g. antibodies). While antibodies against BoNT/A have been reported to neutralize hundreds to thousands LD50, antibody-toxin complexes are often preformed prior to administration and the site and mechanism of action are distinct.
Thus far, our efforts have uncovered numerous inhibitors of BoNT/A LC with low nanomolar inhibition constants that demonstrate effective protection against BoNT/A-mediated SNAP-25 cleavage in cells. However, only modest extension of time to death has been observed in mouse lethality assays. Despite the aforementioned assay advancements, the disconnect between in vitro results and in vivo studies that plagues researchers has yet to be rectified. Therefore, there remains an urgent unmet need for secondary screening assays that are more predictive of in vivo efficacy.
ACTIVE SITE INHIBITORS.
In 2006, we synthesized BoNT/A inhibitors inspired by Gln and Arg, the amino acids that flank the proteolytic cleavage site in SNAP-25, and Cys (Figure 2A).22 In the first series, Gln and Arg were synthetically modified to incorporate zinc-chelating hydroxamate functionalities.23 While the best inhibitor of this series exhibited only a modest Ki of 60 μM (1), this study marked the first use of hydroxamic acids as metal binding inhibitors of BoNT/A LC. Later that year, evaluation of a series of carboxylate and amine-protected amino acids identified Cys as a BoNT/A LC inhibitory fragment.24 Further elaboration of Cys to H-Cys-OMe, Fmoc-Cys(trt)-OH, and Fmoc-D-Cys(trt)-OH improved the BoNT/A inhibitory potency from IC50 = 144 ± 10 μM to 87 ± 9 μM, 40 ± 4 μM and 15 ±3 μM, respectively (SNAPtide). Most importantly, Fmoc-D-Cys(trt)-OH almost completely inhibited BoNT/A LC-catalyzed SNAP-25 cleavage in Neuro-2a cells at 30 μM, marking the first demonstration of BoNT/A LC inhibition in a cell-based assay.
Figure 2.

Development of zinc-binding, active site inhibitors of BoNT/A LC from initial hits, with representative compounds. A) arginine-hydroxamate inhibitors; B) cinnamic hydroxamate inhibitors; C) adamantane hydroxamate inhibitors; D) quinolinol inhibitors. a Values obtained from the SNAPtide assay. b Values obtained from the 66mer assay. c Values obtained from the hiPSC-derived cellular assay. d Values obtained from the ex vivo hemidiaphragm assay.
To conclude 2006, we published 3 (Figure 2B), which remains one of the most potent BoNT/A inhibitors to date.25 In this study, a diverse library of 150 were synthesized and screened by SNAPtide assay.25 With an IC50 value of 0.41 ± 0.03 μM, 3 became the first non-peptidic BoNT/A LC inhibitor with sub-micromolar potency. Importantly, 3 partially restored in vitro muscle twitches in BoNT/A-poisoned mouse nerve-muscle preparations at 5 μM and protected 16% of mice from lethal BoNT/A challenge at 1 mM.26–27 Co-crystallization of BoNT/A LC with 3 confirmed hydroxamate coordination of zinc and depicted the 2,4-dichlorocinnamic pharmacophore entirely filling the shallow, hydrophobic P1 pocket.12 SAR investigation revealed that the 4-Cl substituent is essential to activity.25 Further conclusions about SAR, however, were elusive, potentially due to the flexibility of the enzyme active site.12, 25, 28 A series of second-generation inhibitors was synthesized with emphasis on varying the ortho substituent, which interacts with Arg363.12 Interestingly, while the most potent analogues incorporated ortho electron-withdrawing substitutions (−Br, −NO2, −CF3), a methyl substituted derivative performed equipotently, demonstrating no obvious trend between inhibitory potency and electronegativity.29 In an attempt to increase the stability of the enzyme-inhibitor complex by reducing conformational flexibility and engaging Tyr366 through π-stacking, derivatives with fused ring systems were investigated (4, Figure 2B). These modifications, however, reduced inhibitory activity >40-fold.29 Finally, the X-ray co-crystal structure of 1 suggested that displacement of a catalytically-important water molecule from the active site could improve Ki.30–31 We hypothesized that appending a hydroxyl group to the backbone of 3 would provide the desired hydrogen bonding interactions. Indeed, it was found that chiral inhibitor (R)-5 exhibited an improved Ki of 0.16 ± 0.02 μM.32
During development of the 66mer assay described vide supra, we elucidated adamantane as a bioisosteric substitute for the phenyl ring of 3.33 A series of inhibitors based on the adamantane pharmacophore was accomplished with the intent of probing the hydrophobic pocket. Initial lead 6 displayed a Ki of 0.46 μM and a co-crystal structure of 6 and BoNT/A LC was obtained. As observed in the co-crystal structure of 3 and BoNT/A LC, 6 induced rotation of Phe369 and Asp370, altering the active site environment from polar to hydrophobic.12, 34 Leveraging this induced conformational change, efforts to increase the hydrophobicity of 6 by incorporating substituents able to π-stack with Phe163, Phe194, and Phe369 were investigated. Ultimately, substitution of phenyl substituents at the 3-position of adamantane resulted in significant increases to potency, with the best inhibitor (7) exhibiting an in vitro Ki of 27 nM. Unfortunately, this adamantane series did not inhibit BoNT/A LC in cells.34 Theorizing that the discrepancy between observed enzymatic and cellular inhibition resulted from poor cell permeability, the polar hydroxamate in 6 was masked as a carbamate prodrug (8). Gratifyingly, this molecule inhibited BoNT/A LC in hiPSC-derived neurons with an EC50 of 20 μM.35
Because of the assumed limitations of hydroxamates as clinical therapeutics, particularly regarding off-target effects and metabolic toxicities, we sought to explore new chemical space by leveraging an alternative metal binding group. Quinolinol-containing small molecules had been previously reported to inhibit BoNT/A LC, with the best-in-class exhibiting sub-micromolar IC50 values (SNAPtide).36–38 Thus, we synthesized and screened a quinolinol library with 5-position modifications, heretofore unexplored, ultimately accomplishing 9 with an IC50 of 3.66 ± 0.08 μM (SNAPtide).39 Molecular modeling guided the synthesis of a second generation series elaborated at the amine with the intention of engaging nearby Glu262. Inhibitors 10-12 (Figure 2) exhibited the most potent BoNT/A inhibition with IC50 values of 3.36 ± 0.09 μM, 1.55 ± 0.05 μM, and 3.26 ± 0.01 μM, respectively. Quinolinol leads 9–12 were subsequently evaluated for BoNT/A inhibitory activity in the fluorescent BoCell assay. While 11 did not significantly reduce SNAP-25 cleavage, leads 9, 10 and 12 demonstrated decreased SNAP-25 cleavage at 25 μM. Quinolinols 9–12 were also evaluated in an ex vivo hemidiaphragm assay for BoNT/A activity. Interestingly, all inhibitors exhibited extended time to paralysis. The discrepancy in the inhibitory activity of 11 between the cellular and ex vivo assays could result from a secondary MOA; certain quinolines have been reported to impair endosomal translocation of BoNT/A LC, an inhibitory strategy described in greater detail below.21, 40
Inhibitors with non-metal binding mechanisms have also been investigated. Early efforts in the Janda Laboratory investigated betulin, a pentacyclic triterpene alcohol natural product, and 39 betulin derivatives for BoNT/A inhibition. While multiple betulins exhibited low to sub-micromolar Ki’s for BoNT/A and a mechanism of competitive inhibition in vitro, this series failed to protect rat cerebellar neurons from BoNT/A intoxication, likely resulting from poor pharmacokinetic properties.41
The single class of non-metal binding, non-covalent active site inhibitors stems from an unconventional source. A SNAPtide screen of a triazole-based “click” library serendipitously revealed that group 11 and 12 metals, particularly Cu(II) and Hg(II), inhibit BoNT/A LC. Further investigation into the mechanism of Cu(II)-mediated BoNT/A LC inhibition, in which Hg(II) was abandoned due to its associated toxicity, demonstrated that Cu(II) cations exhibit noncompetitive LC inhibition with a Ki = 1 μM.42 X-ray co-crystallography and point mutations of C165, E164, K166, C134, M106, and M344 identified Cu(II)-sulfur chelation with Cys165 as the key inhibitory binding interaction. Delivery of Cu(II) to hiPSCs-derived neurons using the ligand-copper complexes Cu(PTB)2 (13), Cu(GTSM) (14), Cu(MEDTC)2 (15) inhibited SNAP-25 cleavage at low micromolar concentrations, and impressively, 14 and 15 delayed BoNT/A-induced fatality in mice two-fold (Figure 3).
Figure 3.

Development of other active site binding inhibitors of BoNT/A LC from initial hits, with representative compounds. A) Betulin; B) Copper-ligand complexes. aValues obtained from the SNAPtide assay. bValues obtained from the 66mer assay. cValues obtained from the hiPSC-derived neuronal assay. dTime to death was measured relative to treatment with vehicle. Mice were administered ip with 5 LD50’s of BoNT/A LC, then treated one hour post-toxin with a copper dose of 1 mg/kg.
Most recently, the Janda Laboratory renewed interest in covalent inhibition of BoNT/A LC as a strategy to address extraordinarily long toxin half-life. To date, we have reported two covalent BoNT/A LC inhibitor classes: benzylidene cyclopentenediones and benzoquinones. The benzylidene cyclopentenediones were rationally designed to bind the P1 binding pocket, with 2,4-dichlorobenzyl decorated electrophiles based on the structure of 3. The cyclopentenedione warhead reacts with a nucleophilic amino acid, hypothesized to be Cys165 in the context of BoNT/A LC. The best-in-class cyclopentenedione 16 was found to have a kinact/Ki of 520 ± 50 M−1 s−1 (Figure 4). After treatment with 16, exhaustive dialysis of BoNT/A LC to remove the inhibitor failed to rescue enzymatic activity, indicating irreversible covalent inhibition. Unfortunately, the compound’s inhibitory effect was significantly compromised in cells, and poor pharmacological profiling barred its further development.43
Figure 4.
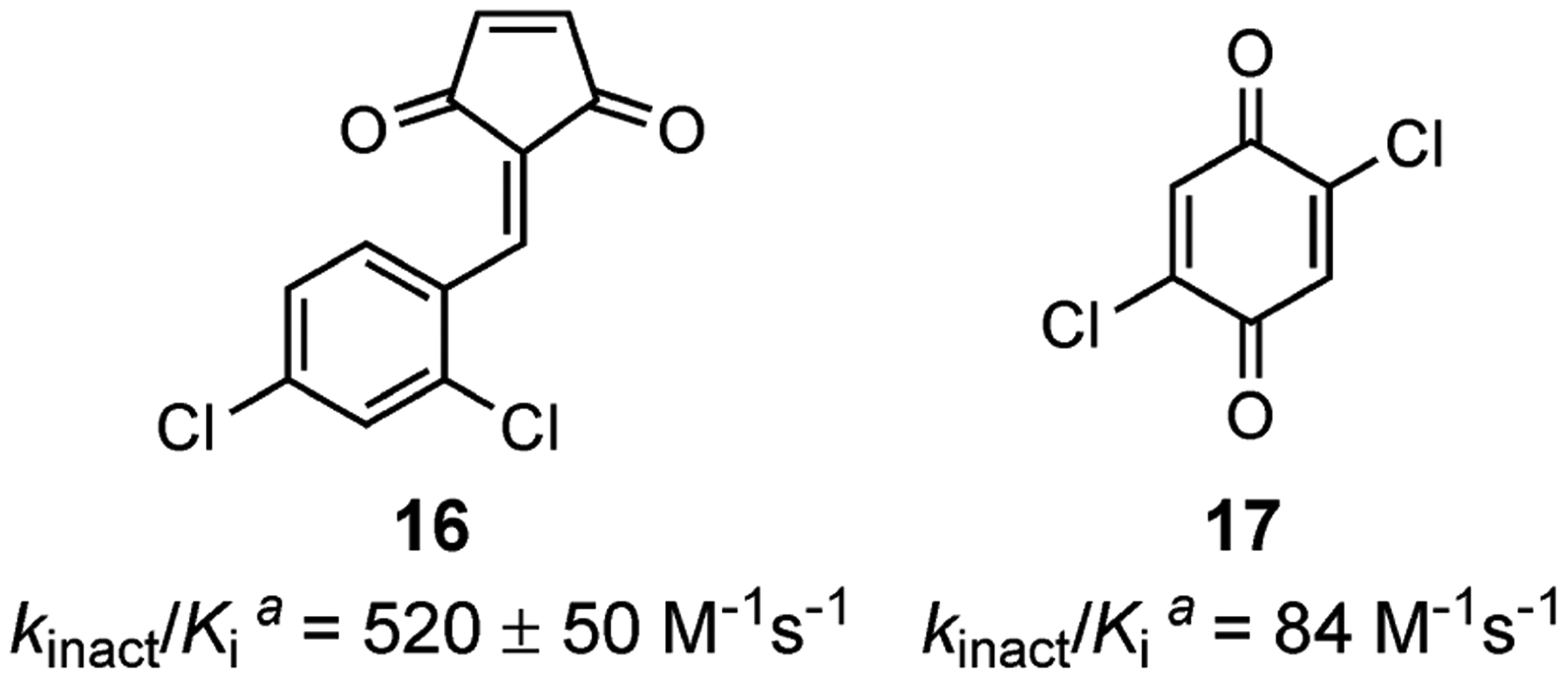
Covalent inhibitors developed in the Janda group. aValues obtained from the 66mer assay.
Our search for therapeutically-relevant covalent inhibitors continued with a screen of electrophilic fragments. The modest inhibitory activity of N-ethylmaleimide prompted us to investigate benzoquinone as a covalent inhibitor of BoNT/A LC. While benzoquinone did exhibit irreversible inhibition of BoNT/A LC with a kinact/Ki = 12.4 M−1 s−1, further attempts to optimize inhibitory potency in cell-based BoNT/A assays have not been successful (Figure 4, 17).44
EXOSITE INHIBITORS.
Because active site inhibition of BoNT/A is presently limited to few MOAs, namely metal-binding, metal-based and irreversible covalent strategies, BoNT/A exosite inhibitors may offer an advantageous approach to therapeutic discovery. Exosite inhibitors, which target two specific folds found only within the BoNT family, may be more selective and cause fewer off-target effects compared to active site inhibitors which may be promiscuous with other Zn2+ metalloproteases. The first report of a BoNT/A LC exosite inhibitor was the discovery of Echinacea derived D-chicoric acid (18).45 Kinetic analysis revealed that D-chicoric acid is a partial inhibitor of BoNT/A LC and exerts its effect synergistically and non-exclusively with 3, implying non-active site binding. To elucidate the D-chicoric binding pocket, BoNT/A LC α-exosite probe 24 (BAP-24, Ac-ARENEMDENLEQVSGIIGNLRHMAib-NH2) was synthesized. This probe consists of the helical SNAP-25 sequence (24aa) that binds to the α-exosite.46 Competition studies between 18 and BAP-24 revealed mutually exclusive binding, indicating that 18 is an α-exosite inhibitor. Alkylation of 18 to give the i-Pr ester analogue 19 yielded an inhibitor with a Ki = 1.8 ± 0.3 μM and complete inhibition.47 Further SAR investigation on the chicoric acid scaffold revealed that the caffeic acid moieties are necessary for inhibitory activity, indicating favorable hydrogen binding interactions with the helices of the a-exosite. Additionally, substitution of the tartaric acid linker with hydrophobic esters, of which 20 is representative, improved inhibitory activity (Figure 5).48
Figure 5.

Development of BoNT/A LC exosite inhibitors. A) α-exosite inhibitors; B) β-exosite inhibitors. aValues obtained from the 66mer assay. bPartial inhibition (~60%).
In investigating compounds structurally similar to chicoric acid, Dyngo-4a™ (21) was found to inhibit BoNT/A LC in vitro47 Dyngo-4a™ is a dynamin inhibitor with anti-BoNT/A activity previously attributed to inhibition of BoNT/A LC endocytosis.49 However, we found Dyngo-4a™ also exhibits a Ki = 0.32 ± 0.05 μM for BoNT/A enzymatic activity. SAR investigation into the hydroxyphenyl substructure of Dyngo-4a™ determined the trihydroxy architecture is required for inhibitory activity; removing any of the phenyl ring substitutions reduces Ki by at least 100-fold. Despite conflicting results in cell-based assays, Dyngo-4a™ rescued 25% of mice from lethal BoNT/A challenge at a 1 mg dose. As Dyngo-4a™ was administered 2.5–3 hours post BoNT/A intoxication, it is hypothesized that the observed effects of Dyngo-4a™ were enzymatic and not dynamin-based.47 Although evidence that Dyngo-4a™ binds the α-exosite is not yet available, the structural similarities between Dyngo-4a™ and chicoric acid formulate this hypothesis.
A single BoNT/A β-exosite inhibitor has been reported; lomofungin (22), an antibiotic produced by Streptomyces lomodensis, inhibits BoNT/A LC.50–51 Initially identified in a HTS of the Johns Hopkins Clinical Compound Library, lomofungin was found to have a Ki of 6.7 ± 0.7 μM. Lomofungin displays kinetic activity consistent with non-competitive inhibition. Competition studies with BAP-24 produced data that fits a non-mutually exclusive model, implying that lomofungin does not bind the α-exosite.46 Lomofungin, which incorporates a quinolinol substructure capable of chelating zinc, also exhibits no zinc sequestration capability. In an attempt to further explore the chemical space for β-exosite binding, a picolinic acid pharmacophore was used as a mimetic of the lomofungin scaffold.52 Cross-coupling indolyl fragments to bromopicolinates via the Suzuki-Miyaura reaction allowed for efficient synthesis of derivatives. Gratifyingly, we determined through competition studies that these compounds do bind mutually exclusively with lomofungin. The best inhibitor found, CBIP (23), exhibited an IC50 of 2.7 ± 0.2 μM, about 8-fold more potent than lomofungin.
INHIBITORS WITH UNIQUE MECHANISMS OF ACTION.
Inhibitors of BoNT LC do not present the only opportunity for anti-BoNT pharmacotherapy; inhibitors of BoNT holotoxin and of host processes essential to BoNT intoxication present viable avenues for pharmacological intervention (Figure 6). The triterpenoid toosendanin (24), a traditional Chinese medicine derived from the bark of M. toosendan trees, had been reported to protect primates from BoNT-induced fatality post BoNT/A/B/E exposure.53 Using a library of semi-synthetic toosendanin analogues, we identified essential features of the limonoid’s molecular architecture and elucidated toosendanin’s MOA. Three functionalities unique to limonoid steroids, i.e. the hemiacetal bridge between C4 and C10 of the AB ring system, the C17 heterocyclic furan, and the epoxide at C14 and C15, were evaluated for their role in anti-BoNT activity. In comparison to toosendanin, which extends time to death four-fold in mice at 7.5 mg/kg doses, only modification to the C17 furan ring was tolerated. A single molecule assay of BoNT/A and BoNT/E translocation reported that toosendanin arrests LC translocation with sub-nanomolar potency by stabilizing occluded conformations of the HC channel.20 Importantly, this BoNT inhibition strategy was effective against immunologically distinct BoNT/A and BoNT/E proteins, alluding to its potential utility as a generally-applicable, prophylactic anti-botulinum agent. Further medicinal chemistry efforts to identify substructures of toosendanin responsible for anti-BoNT activity, which in turn accomplished the first syntheses of toosendanin’s AB and CD rings, demonstrated that the complete cyclopentanoperhydrophenanthrene core is required for inhibition.54–55
Figure 6.

Anti-BoNT small molecules with non-LC MOAs. A) Toosendanin inhibits LC translocation by stabilizing the closed conformation of the HC channel; B) Aminopyridines inhibit Kv to induce muscle contractions; C) 4-Acetoxybenzyl-^-hydroxysuccinimide irreversibly inhibits TrxR, preventing cleavage of the LC from the HC.
BoNT intoxication can also be thwarted by molecules that block voltage-gated K+ channels (Kv). Recall that BoNTs induce muscle paralysis by cleaving SNARE proteins, which are responsible for mediating acetylcholine release at neuromuscular junctions.56 Degradation of SNARE proteins can be overcome by enhancing Ca2+ influx via Kv inhibition. 4-Aminopyridine (4-AP, 25) and 3,4-diaminopyridine (3,4-DAP, 26) can induce muscle contractions in BoNT/A poisoned neurons by generating action potentials (Figure 6). Their therapeutic utility, however, is limited by aminopyridine-associated toxicities, specifically seizures resulting from the ability of 4-AP and 3,4-DAP to cross the blood brain barrier (BBB). The Janda Laboratory has invested considerable efforts to limit BBB penetration of these aminopyridines while maintaining anti-BoNT activity through the following strategies: synthesis of less BBB penetrant analogues, prodrug design, and safe delivery platforms. Despite previous reports that suggested medicinal chemistry modifications to 3,4-DAP were unable to eliminate central nervous system (CNS) toxicity, we employed crystallographic and modeling data to rationally design 3,4,5-triaminopyridine (27) and 3,4-diamino-1-(prop-2-ynyl)pyridinium. These were ultimately determined to block Kv en route to reversing BoNT/A-induced muscle paralysis in phrenic nerve hemidiaphragm preparations. Moreover, 27 relieved BoNT/A poisoning in a mouse lethality assay. This pharmacological activity coupled with an absence of detectable brain exposure hypothetically results from reduced lipophilicity and an increased propensity to hydrogen bond, resulting from the addition of a third amine. In addition to reporting 3,4-DAP analogues with improved safety profiles, we investigated carbamate, amide, and amino acid prodrugs of 3,4-DAP.57–58 Prodrugs of 3,4-DAP would allow localized enhancement of acetylcholine release at intoxicated neuromuscular junctions without BBB penetration. Despite our efforts, optimization of 3,4-DAP prodrugs is still required for successful in vivo translation. In a complementary approach, we reported a non-allergenic and non-toxic, pH-dependent lycopodium clavatum exine microcapsule oral delivery system for controlled drug release that successfully slowed 3,4-DAP absorption, reduced seizure activity, and enabled delivery of therapeutic doses to prolong mouse survival after lethal BoNT/A challenge.59
Our final class of identified small molecules that protect SNAP-25 from BoNT cleavage include inhibitors of thioredoxin reductase (TrxR), a host enzyme required for BoNT/A protease translocation. This inhibitor class was serendipitously identified as a sub-scaffold of a “hit” from a cell-based screen for BoNT/A inhibition.60 The active pharmacophore, 4-acetoxybenzyl N-hydroxysuccinimide (28), exhibited single-digit micromolar inhibition of BoNT/A in cells; however, showed no inhibitory activity against recombinant BoNT/A LC (SNAPtide/66mer). Because the compound was inactive against BoNT/A LC in vitro, we surmised that the inhibitors either perturbed LC modification by host cytosolic enzymes or interfered with LC translocation after endocytosis, analogous to toosendanin’s MOA. We observed the succinimide series exhibited reduced protection of SNAP-25 cleavage over time, and accordingly, hypothesized that inhibition stems from translocation. Using mass spectrometry-based proteomics for target identification, we identified two putative targets for succinimide inhibition: TrxR 1 isoform 2 and/or protein disulfide isomerase ERp61 precursor. It had been reported that BoNT/A hijacks TrxR to cleave the disulfide bond between HC and LC during translocation, and that inhibition of TrxR leads to SNAP-25 protection in neurons. Ultimately, we demonstrated that this class inhibits TrxR through a covalent mechanism where acetyl and quinone methide metabolites label the TrxR nucleophilic residues Cys and Sec. This mechanism was confirmed through competition experiments with the pH-sensitive affinity reagent BIAM (N-(biotinoyl-N’-(iodoacetyl)ethylenediamine) and LCMS analysis of adducts formed by the reaction of succinimide metabolites and the TrxR active site sequence GCSecG. The kinetic parameters of the two lead inhibitors were characterized as Ki = 4.2 ± 0.3 μM and 51 ± 8 μM and kinact = (0.78 ± 0.09) × 10−3 and (3.2 ± 0.05) × 10−3 s−1.
CRITICAL PERSPECTIVE AND FUTURE WORK.
The Janda Laboratory has invested nearly two decades into the research of small molecule therapeutics for the reversal of BoNT intoxication and achieved the discovery of multiple inhibitory strategies: metal-binding, metal-dependent, and covalent active site binders, α and β-exosite binders, translocation inhibitors and Kv channel blockers. Despite these efforts, no anti-BoNT small molecules have advanced to clinical trials and nearly all reported classes are handicapped. For example, active site metal binding pharmacophores are prone to off-target toxicities as highly conserved Zn2+ binding motifs are present in essential human metalloproteinases. Analogously, highly reactive, electrophilic small molecules exhibit promiscuous reactivity with many in vivo nucleophiles, and metals ions can coordinate various biologically-relevant thiols. Unless localized delivery of these inhibitor classes can be accomplished, such limitations may preference future investigation of exosite inhibitors. The accomplishment of X-ray co-crystallographic data on α and β-exosite binding inhibitors has, however, proven elusive to date; an X-ray co-crystal structure will ultimately be required to drive the evolution of these inhibitors. Finally, the non-LC interacting inhibitor classes including toosendanins, aminopyridines, and succinimides are not without limitations. In particular, the CNS toxicity of the leading aminopyridine 3,4-DAP and the promiscuous reactivity of 4-acetoxybenzyl N-hydroxysuccinimide and its metabolites diminish therapeutic potential.
The next decade in BoNT inhibitor discovery must build on the foundation described herein to overcome the inherent challenges of these inhibitor classes and arrive at a clinically-relevant BoNT antagonist. Two of the greatest challenges for the future of BoNT inhibitor discovery are the disconnect in inhibitor efficacy between cell-based and animal models, and the reconciliation of toxin half-life with in vivo pharmaceutical lifetimes. Continued research efforts are required to deliver new in vitro assays that more accurately predict in vivo performance. To address BoNT/A’s extraordinarily long half-life, we are especially interested in the development of next-generation covalent inhibitors with tunable reactivity. Though drug discovery for BoNT faces on-going challenges, formidable progress has been achieved, paving the way for the next decade of BoNT inhibitor research.
Funding and Acknowledgments
Research was supported by the National Institutes of Health grants R01 AI119564 (to KDJ), R21 AI137709 (to KDJ), and F32 DA044692 (to MEO), the Natural Sciences and Engineering Research Council of Canada (NSERC) grant PGSD3-502274 (to LL), and the Skaggs Institute for Chemical Biology (to LL). This is manuscript #29841 from the Scripps Research Institute.
Biography
Lucy Lin received her BSc in Pharmaceutical Chemistry from the University of Toronto in 2016, where she researched nanoparticle drug formulations with Prof. Xiao Yu Wu, and polymer-based drug delivery with Prof. Ping Lee. She is currently pursuing a PhD in Medicinal Chemistry in the laboratory of Prof. Kim D. Janda at The Scripps Research Institute (TSRI), where she is developing BoNT/A inhibitors.
Margaret E. Olson received her BA in Biology from Illinois Wesleyan University in 2010, where she worked with Prof. Ram S. Mohan on the development of environmentally-friendly organic methodologies. She went on to pursue graduate studies with Prof. Daniel A. Harki at the University of Minnesota and received her PhD in Medicinal Chemistry in 2015. Her dissertation investigated high-throughput screening and lead validation for the discovery of APOBEC3 DNA cytosine deaminase inhibitors. In 2016, she joined the laboratory of Prof. Kim D. Janda at TSRI for postdoctoral studies. Recently, she began her independent career at Roosevelt University College of Pharmacy as an Assistant Professor of Medicinal Chemistry. Her research interests lie in infectious disease and chemical immunology, especially the synthesis and biological evaluation of novel antibacterial agents.
Lisa M. Eubanks received her BA in Chemistry with a minor in Biology from Huntingdon College. She went on to pursue graduate studies with C. Dale Poulter at the University of Utah investigating enzymes in the isoprenoid biosynthetic pathways. After receiving her PhD in Biological Chemistry, she joined the laboratory of Prof. Kim D. Janda at TSRI as a NIH postdoctoral research fellow. She continues to conduct research at TSRI in the areas of drugs of abuse and infectious disease.
REFERENCES
- 1.Arnon SS; Schechter R; Inglesby TV; Henderson DA; Bartlett JG; Ascher MS; Eitzen E; Fine AD; Hauer J; Layton M; Lillibridge S; Osterholm MT; O’Toole T; Parker G; Perl TM; Russell PK; Swerdlow DL; Tonat K, Botulinum toxin as a biological weapon: medical and public health management. J. Amer. Med. Assoc 2001, 285 (8), 1059–70. [DOI] [PubMed] [Google Scholar]
- 2.(CDC), C. f. D. C. a. P. Botulism Annual Summary, 2016; Center for Disease Control and Prevention (CDC): Atlanta, Georgia, 2017; p 10. [Google Scholar]
- 3.Sundeen G; Barbieri JT, Vaccines against Botulism. Toxins 2017, 9 (9), 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tacket CO; Shandera WX; Mann JM; Hargrett NT; Blake PA, Equine antitoxin use and other factors that predict outcome in type A foodborne botulism. Am. J. Med 1984, 76 (5), 794–798. [DOI] [PubMed] [Google Scholar]
- 5.Breidenbach MA; Brunger AT, Substrate recognition strategy for botulinum neurotoxin serotype A. Nature 2004, 432, 925. [DOI] [PubMed] [Google Scholar]
- 6.Pellizzari R; Rossetto O; Schiavo G; Montecucco C, Tetanus and botulinum neurotoxins: mechanism of action and therapeutic uses. Philos. Trans. Royal Soc. B 1999, 354 (1381), 259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schiavo G; Matteoli M; Montecucco C, Neurotoxins Affecting Neuroexocytosis. Physiol. Rev 2000, 80 (2), 717–766. [DOI] [PubMed] [Google Scholar]
- 8.Lacy DB; Tepp W; Cohen AC; DasGupta BR; Stevens RC, Crystal structure of botulinum neurotoxin type A and implications for toxicity. Nat. Struct. Biol 1998, 5, 898. [DOI] [PubMed] [Google Scholar]
- 9.Koriazova LK; Montal M, Translocation of botulinum neurotoxin light chain protease through the heavy chain channel. Nat. Struct. Biol 2002, 10, 13. [DOI] [PubMed] [Google Scholar]
- 10.Rizo J; Sudhof TC, Snares and munc18 in synaptic vesicle fusion. Nat. Rev. Neurosci 2002, 3, 641. [DOI] [PubMed] [Google Scholar]
- 11.Li; Singh BR, Role of Zinc Binding in Type A Botulinum Neurotoxin Light Chain’s Toxic Structure. Biochemistry 2000, 39 (34), 10581–10586. [DOI] [PubMed] [Google Scholar]
- 12.Silvaggi NR; Boldt GE; Hixon MS; Kennedy JP; Tzipori S; Janda KD; Allen KN, Structures of Clostridium botulinum Neurotoxin Serotype A Light Chain Complexed with Small-Molecule Inhibitors Highlight Active-Site Flexibility. Chem. Biol 2007, 14 (5), 533–542. [DOI] [PubMed] [Google Scholar]
- 13.Chen S; Barbieri JT, Unique Substrate Recognition by Botulinum Neurotoxins Serotypes A and E. J. Biol. Chem 2006, 281 (16), 10906–10911. [DOI] [PubMed] [Google Scholar]
- 14.Sukonpan C; Oost T; Goodnough M; Tepp W; Johnson EA; Rich DH, Synthesis of substrates and inhibitors of botulinum neurotoxin type A metalloprotease*. J. Pept. Sci 2008, 63 (2), 181193. [DOI] [PubMed] [Google Scholar]
- 15.Boldt GE; Kennedy JP; Hixon MS; McAllister LA; Barbieri JT; Tzipori S; Janda KD, Synthesis, Characterization and Development of a High-Throughput Methodology for the Discovery of Botulinum Neurotoxin A Inhibitors. Journal of Combinatorial Chemistry 2006, 8 (4), 513521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shine NRLA, CA), Crawford,Karen Renee (Saratoga, CA), Eaton, Linda Jo Ann (San Jose, CA) Substrate peptides and assays for detecting and measuring proteolytic activity of serotype A neurotoxin from clostridium botulinum. 2003. [Google Scholar]
- 17.Ćapková K; Hixon MS; McAllister LA; Janda KD, Toward the discovery of potent inhibitors of botulinum neurotoxin A: development of a robust LC MS based assay operational from low to subnanomolar enzyme concentrations. Chemical Communications 2008, (30), 3525–3527. [DOI] [PubMed] [Google Scholar]
- 18.Whitemarsh RCM; Strathman MJ; Chase LG; Stankewicz C; Tepp WH; Johnson EA; Pellett S, Novel Application of Human Neurons Derived from Induced Pluripotent Stem Cells for Highly Sensitive Botulinum Neurotoxin Detection. Toxicological Sciences 2012, 126 (2), 426–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dong M; Tepp WH; Johnson EA; Chapman ER, Using fluorescent sensors to detect botulinum neurotoxin activity in vitro and in living cells. Proceedings of the National Academy of Sciences of the United States of America 2004, 101 (41), 14701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer A; Nakai Y; Eubanks LM; Clancy CM; Tepp WH; Pellett S; Dickerson TJ; Johnson EA; Janda KD; Montal M, Bimodal modulation of the botulinum neurotoxin protein-conducting channel. Proc. Natl. Acad. Sci. U. S. A 2009, 106 (5), 1330–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheridan RE; Deshpande SS; Nicholson JD; Adler M, Structural features of aminoquinolines necessary for antagonist activity against botulinum neurotoxin. Toxicon 1997, 35 (9), 1439–1451. [DOI] [PubMed] [Google Scholar]
- 22.Boldt GE; Kennedy JP; Hixon MS; McAllister LA; Barbieri JT; Tzipori S; Janda KD, Synthesis, Characterization and Development of a High-Throughput Methodology for the Discovery of Botulinum Neurotoxin A Inhibitors. J. Comb. Chem 2006, 8 (4), 513–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rao BG, Recent Developments in the Design of Specific Matrix Metalloproteinase Inhibitors aided by Structural and Computational Studies. Curr. Pharm. Design 2005, 11 (3), 295–322. [DOI] [PubMed] [Google Scholar]
- 24.Boldt GE; Eubanks LM; Janda KD, Identification of a botulinum neurotoxin A protease inhibitor displaying efficacy in a cellular model. Chem. Commun 2006, (29), 3063–3065. [DOI] [PubMed] [Google Scholar]
- 25.Boldt GE; Kennedy JP; Janda KD, Identification of a Potent Botulinum Neurotoxin A Protease Inhibitor Using in Situ Lead Identification Chemistry. Org. Lett 2006, 8 (8), 1729–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eubanks LM; Hixon MS; Jin W; Hong S; Clancy CM; Tepp WH; Baldwin MR; Malizio CJ; Goodnough MC; Barbieri JT; Johnson EA; Boger DL; Dickerson TJ; Janda KD, An in vitro and in vivo disconnect uncovered through high-throughput identification of botulinum neurotoxin A antagonists. Proc. Natl. Acad. Sci. U. S. A 2007, 104 (8), 2602–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thyagarajan B; Potian JG; Garcia CC; Hognason K; Capkova K; Moe ST; Jacobson AR; Janda KD; McArdle JJ, Effects of hydroxamate metalloendoprotease inhibitors on botulinum neurotoxin A poisoned mouse neuromuscular junctions. Neuropharmacology 2010, 58 (8), 1189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith GR; Caglič D; Čapek P; Zhang Y; Godbole S; Reitz AB; Dickerson TJ, Reexamining hydroxamate inhibitors of botulinum neurotoxin serotype A: Extending towards the β-exosite. Bioorg. Med. Chem. Lett 2012, 22 (11), 3754–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Capkova K; Yoneda Y; Dickerson TJ; Janda KD, Synthesis and structure-activity relationships of second-generation hydroxamate botulinum neurotoxin A protease inhibitors. Bioorg. Med. Chem. Lett 2007, 17 (23), 6463–6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu Z; Chen S; Baldwin MR; Boldt GE; Crawford A; Janda KD; Barbieri JT; Kim J-JP, Light Chain of Botulinum Neurotoxin Serotype A: Structural Resolution of a Catalytic Intermediate. Biochemistry 2006, 45 (29), 8903–8911. [DOI] [PubMed] [Google Scholar]
- 31.Silvaggi NR; Wilson D; Tzipori S; Allen KN, Catalytic Features of the Botulinum Neurotoxin A Light Chain Revealed by High Resolution Structure of an Inhibitory Peptide Complex. Biochemistry 2008, 47 (21), 5736–5745. [DOI] [PubMed] [Google Scholar]
- 32.Stowe GN; Šilhár P; Hixon MS; Silvaggi NR; Allen KN; Moe ST; Jacobson AR; Barbieri JT; Janda KD, Chirality Holds the Key for Potent Inhibition of the Botulinum Neurotoxin Serotype A Protease. Org. Lett 2010, 12 (4), 756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Čapková K; Hixon MS; McAllister LA; Janda KD, Toward the discovery of potent inhibitors of botulinum neurotoxin A: development of a robust LC MS based assay operational from low to subnanomolar enzyme concentrations. Chem. Commun 2008, (30), 3525–3527. [DOI] [PubMed] [Google Scholar]
- 34.Šilhár P; Silvaggi NR; Pellett S; Capkova K; Johnson EA; Allen KN; Janda KD, Evaluation of adamantane hydroxamates as botulinum neurotoxin inhibitors: Synthesis, crystallography, modeling, kinetic and cellular based studies. Bioorg. Med. Chem 2013, 21 (5), 1344–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Šilhár P; Eubanks LM; Seki H; Pellett S; Javor S; Tepp WH; Johnson EA; Janda KD, Targeting Botulinum A Cellular Toxicity: A Prodrug Approach. J. Med. Chem 2013, 56 (20), 7870–7879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caglič D; Krutein MC; Bompiani KM; Barlow DJ; Benoni G; Pelletier JC; Reitz AB; Lairson LL; Houseknecht KL; Smith GR; Dickerson TJ, Identification of Clinically Viable Quinolinol Inhibitors of Botulinum Neurotoxin A Light Chain. Journal of Medicinal Chemistry 2014, 57 (3), 669–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roxas-Duncan V; Enyedy I; Montgomery VA; Eccard VS; Carrington MA; Lai H; Gul N; Yang DCH; Smith LA, Identification and Biochemical Characterization of Small-Molecule Inhibitors of Clostridium Botulinum Neurotoxin Serotype A. Antimicrob. Agents Chemother 2009, 53 (8), 3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lai H; Feng M; Roxas-Duncan V; Dakshanamurthy S; Smith LA; Yang DCH, Quinolinol and peptide inhibitors of zinc protease in botulinum neurotoxin A: Effects of zinc ion and peptides on inhibition. Archives of Biochemistry and Biophysics 2009, 491 (1), 75–84. [DOI] [PubMed] [Google Scholar]
- 39.Bremer PT; Adler M; Phung CH; Singh AK; Janda KD, Newly Designed Quinolinol Inhibitors Mitigate the Effects of Botulinum Neurotoxin A in Enzymatic, Cell-Based, and ex Vivo Assays. Journal of Medicinal Chemistry 2017, 60 (1), 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deshpande SS; Sheridan RE; Adler M, Efficacy of certain quinolines as pharmacological antagonists in botulinum neurotoxin poisoning. Toxicon 1997, 35 (3), 433–445. [DOI] [PubMed] [Google Scholar]
- 41.Silhar P; Alakurtti S; Capkova K; Xiaochuan F; Shoemaker CB; Yli-Kauhaluoma J; Janda KD, Synthesis and evaluation of library of betulin derivatives against the botulinum neurotoxin A protease. Bioorg. Med. Chem. Lett 2011, 21 (8), 2229–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bremer PT; Pellett S; Carolan JP; Tepp WH; Eubanks LM; Allen KN; Johnson EA; Janda KD, Metal Ions Effectively Ablate the Action of Botulinum Neurotoxin A. Journal of the American Chemical Society 2017, 139 (21), 7264–7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Capkova K; Hixon MS; Pellett S; Barbieri JT; Johnson EA; Janda KD, Benzylidene cyclopentenediones: First irreversible inhibitors against botulinum neurotoxin A’s zinc endopeptidase. Bioorg. Med. Chem. Lett 2010, 20 (1), 206–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bremer PT; Hixon MS; Janda KD, Benzoquinones as inhibitors of botulinum neurotoxin serotype A. Bioorg. Med. Chem 2014, 22 (15), 3971–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silhar P; Capkova K; Salzameda NT; Barbieri JT; Hixon MS; Janda KD, Botulinum Neurotoxin A Protease: Discovery of Natural Product Exosite Inhibitors. J. Am. Chem. Soc 2010, 132 (9), 2868–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xue S; Javor S; Hixon MS; Janda KD, Probing BoNT/A Protease Exosites: Implications for Inhibitor Design and Light Chain Longevity. Biochemistry 2014, 53 (43), 6820–6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seki H; Xue S; Hixon MS; Pellett S; Reme M; Johnson EA; Janda KD, Toward the discovery of dual inhibitors for botulinum neurotoxin A: concomitant targeting of endocytosis and light chain protease activity. Chem. Commun 2015, 51 (28), 6226–6229. [DOI] [PubMed] [Google Scholar]
- 48.Xue S; Seki H; Remes M; Silhar P; Janda K, Examination of a-exosite inhibitors against Botulinum neurotoxin A protease through structure-activity relationship studies of chicoric acid. Bioorganic & Medicinal Chemistry Letters 2017, 27 (22), 4956–4959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harper CB; Martin S; Nguyen TH; Daniels SJ; Lavidis NA; Popoff MR; Hadzic G; Mariana A; Chau N; McCluskey A; Robinson PJ; Meunier FA, Dynamin Inhibition Blocks Botulinum Neurotoxin Type A Endocytosis in Neurons and Delays Botulism. J. Biol. Chem 2011, 286 (41), 35966–35976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fraser RSS; Creanor J; Mitchison JM, Rapid and Selective Inhibition of the Synthesis of High Molecular Weight RNA in Yeast by Lomofungin. Nature 1973, 244, 222. [DOI] [PubMed] [Google Scholar]
- 51.Eubanks LM; Silhar P; Salzameda NT; Zakhari JS; Xiaochuan F; Barbieri JT; Shoemaker CB; Hixon MS; Janda KD, Identification of a Natural Product Antagonist against the Botulinum Neurotoxin Light Chain Protease. ACS Med. Chem. Lett 2010, 1 (6), 268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bremer PT; Xue S; Janda KD, Picolinic acids as P-exosite inhibitors of botulinum neurotoxin A light chain. Chem. Commun 2016, 52 (84), 12521–12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zou J; Miao WY; Ding FH; Meng JY; Ye HJ; Jia GR; He XY; Sun GZ; Li PZ, The effect of toosendanin on monkey botulism. J. Tradit. Chin. Med 1985, 5 (1), 29–30. [PubMed] [Google Scholar]
- 54.Nakai Y; Pellett S; Tepp WH; Johnson EA; Janda KD, Toosendanin: synthesis of the AB-ring and investigations of its anti-botulinum properties (Part II). Bioorg. Med. Chem 2010, 18 (3), 1280–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakai Y; Tepp WH; Dickerson TJ; Johnson EA; Janda KD, Function-oriented synthesis applied to the anti-botulinum natural product toosendanin. Bioorg. Med. Chem 2009, 17 (3), 1152–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mayorov AV; Willis B; Di Mola A; Adler D; Borgia J; Jackson O; Wang J; Luo Y; Tang L; Knapp RJ; Natarajan C; Goodnough MC; Zilberberg N; Simpson LL; Janda KD, Symptomatic relief of botulinum neurotoxin/a intoxication with aminopyridines: a new twist on an old molecule. ACS Chem. Biol 2010, 5 (12), 1183–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zakhari JS; Kinoyama I; Hixon MS; Di Mola A; Globisch D; Janda KD, Formulating a new basis for the treatment against botulinum neurotoxin intoxication: 3,4-Diaminopyridine prodrug design and characterization. Bioorg. Med. Chem 2011, 19 (21), 6203–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harris TL; Lowery CA; Hixon MS; Janda KD, A platform stratifying a sequestering agent and a pharmacological antagonist as a means to negate botulinum neurotoxicity. ACS Chem. Neurosci 2014, 5 (8), 632–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harris TL; Wenthur CJ; Diego-Taboada A; Mackenzie G; Corbitt TS; Janda KD, Lycopodium clavatum exine microcapsules enable safe oral delivery of 3,4-diaminopyridine for treatment of botulinum neurotoxin A intoxication. Chem. Commun 2016, 52 (22), 4187–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Seki H; Xue S; Pellett S; Silhar P; Johnson EA; Janda KD, Cellular Protection of SNAP-25 against Botulinum Neurotoxin/A: Inhibition of Thioredoxin Reductase through a Suicide Substrate Mechanism. J. Am. Chem. Soc 2016, 138 (17), 5568–5575. [DOI] [PMC free article] [PubMed] [Google Scholar]