

Abstract
Patient: Male, 25-year-old
Final Diagnosis: Acquired von Willebrand deficiency
Symptoms: Bleeding • chest discomfort
Medication: —
Clinical Procedure: Bleeding • chest discomfort
Specialty: Laboratory Diagnostics • Hematology • General and Internal Medicine • Physiology
Objective:
Rare co-existance of disease or pathology
Background:
Essential thrombocythemia (ET) is a type of myeloproliferative neoplasm (MPN) characterized by sustained thrombocytosis in peripheral blood. Patients typically have gene mutations like JAK2V617F, CALR, and MPLW515L/K. This report describes a young man with ET without any of the above mutations who had paradoxical bleeding due to acquired Von-Willebrand disease.
Case Report:
A young man with a medical history of thrombocytosis on aspirin presented with acute chest pain and was found to have had a myocardial infarction. Emergency cardiac catheterization revealed a thrombotic occlusion of the left anterior descending (LAD) artery and the right posteriolateral system, with an ejection fraction of 25%. He underwent thrombectomy and balloon angioplasty with LAD stenting, and an Impella 2.5 was inserted due to severe left ventricular dysfunction with akinesia. Aspirin and ticagrelor were administered, but the patient later experienced postoperative bleeding from the site of the Impella device. The bleeding was attributed to acquired Von-Willebrand disease secondary to ET. Emergency plateletpheresis was recommended. Further workup demonstrated that he was triple-negative for JAK2, MPL, and CALR gene mutations.
Conclusions:
The paradoxical bleeding resulting from acquired Von-Willebrand disease was likely an entirely separate entity from the hyper-thrombotic state expected from ET. Careful assessment of clinical symptoms and laboratory markers, in addition to a high degree of suspicion, are needed to diagnose acquired Von-Willebrand disease as a complication of ET.
MeSH Keywords: Janus Kinase 2; Thrombocythemia, Essential; von Willebrand Diseases; von Willebrand Factor
Background
Essential thrombocythemia (ET) is a type of myeloproliferative neoplasm (MPN) characterized by sustained thrombocytosis, defined as ≥450,000 platelets/µl blood, as well as an increased number of mature megakaryocytes in bone marrow [1–3]. This chronic clonal stem cell disorder has a prevalence of 0.6 to 2.4 per 100,000 people [1,2,4]. Bone marrow biopsy is usually recommended for diagnosis, after excluding secondary causes of thrombocytosis [5]. The etiology of ET is primarily genetic, with most patients having mutations in the JAK2, CALR or MPL gene, including 50–60% with JAK2 V617F, 15–30% with CALR, and (1–5%) with MPL mutations, with studies showing differences in the prevalence of JAK2 mutations [3,5–13]. Some patients with ET, however, lack mutations in all three genes [14], although symptoms in these “triple-negative” patients are similar to those in patients with mutations.
ET results in high susceptibility to vascular thromboses [2,3]. Paradoxically, an acquired version of von Willebrand’s disease may manifest in patients with elevated platelet count [14], as excess platelet activation depletes clotting factors and high molecular weight multimers of von Willebrand factor (VWF) [15]. This rare phenomenon has been reported associated with myelo and lymphoproliferative disorders, causing unusual bleeding after major and minor surgical procedures [15–18]. ET should be strongly suspected in patients with a hypercoagulable state who are also susceptible to bleeding.
ET is manageable provided it is diagnosed and treated before the development of severe symptoms of a hypercoagulable state or bleeding. Depending on risk stratification, patients are usually managed on an outpatient basis, with the objective of therapy being the prevention of vascular complications [19,20]. Low-dose aspirin, cytoreductive therapy with hydroxyurea, inter-feron-alpha, and anagrelide have been effective in preventing thrombotic episodes on an outpatient basis, whereas platelet-pheresis and VWF-containing concentrates are generally reserved for life-threatening bleeding [1,21–24].
This report describes a young man with triple-negative ET who presented with thrombosis. He later developed severe bleeding due to acquired Von-Willebrand disease and required emergency plateletpheresis [22,24].
Case Report
A 25-year-old Caucasian man with a medical history of anxiety presented to the emergency department with acute chest discomfort. Initial examination showed a body temperature of 97.4ºF, a pulse of 98 beats/min, a respiratory rate of 20 breaths/min, a blood pressure of 192/139 mm Hg, and 100% oxygen saturation on room air. A physical examination showed significant diaphoresis and distress. A cardiovascular examination yielded normal results, with regular S1 and S2 and no additional sounds, murmurs, rubs, or gallops. Laboratory tests showed a white blood cell count of 19.8×103/µl (reference 4–9×103/µl), a hemoglobin concentration of of 15.5 g/dl (reference 12–16 g/dl), a platelet count of 1472×103/µl (reference 140–350×103/µl), a PTT of 32.8 sec (reference 25–37 sec), and an INR of 1.1 (reference 0.8–1.1), with other laboratory tests, including blood chemistry and a coagulation panel, yielding results within normal limits. Chest X-rays were negative for any acute pathology. Troponin tended to increase, from 0.41 μg/l to 72 μg/l. An electrocardiogram (EKG) showed significant ST elevation in the anterior precordial leads, consistent with anterior ST-elevated myocardial infarction (STEMI) (Figure 1).
Figure 1.
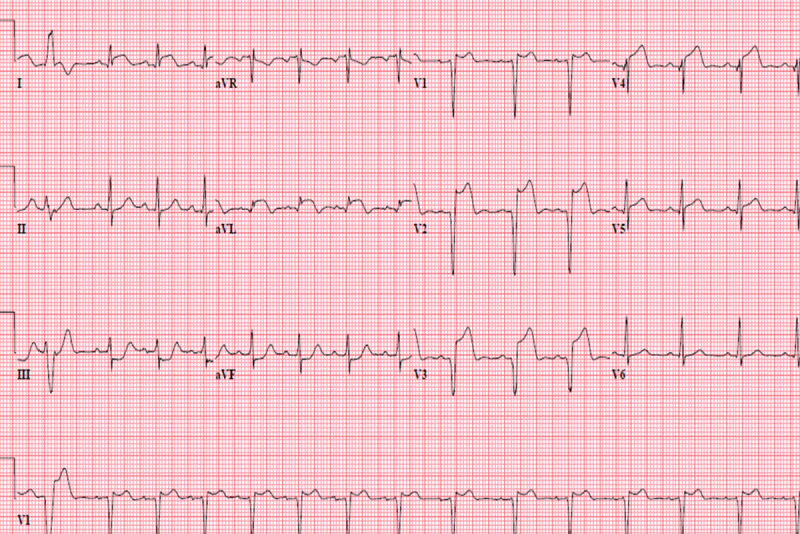
EKG on admission, demonstrating ST segment elevation in V2, V3 and V4, consistent with an anterior myocar-dial infarction in the left anterior descending artery.
Emergency cardiac catheterization revealed thrombotic occlusion of the proximal and apical left anterior descending (LAD) artery, requiring thrombectomy, balloon angioplasty, and drug-eluting stent placement. Moderate disease was observed in his mid-right coronary artery and diagonal branch (Figures 2, 3).
Figure 2.
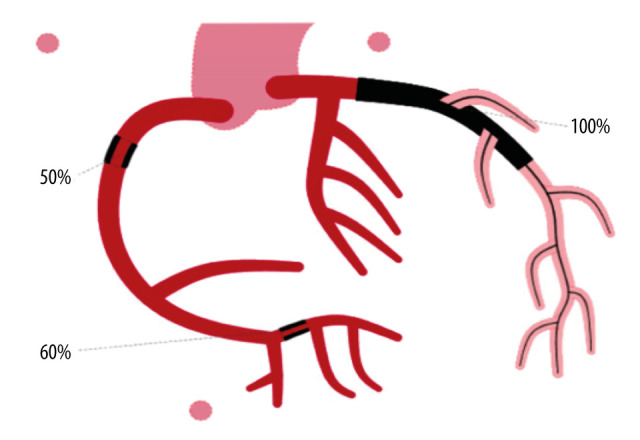
Diagram representing the patient’s condition before cardiac catheterization, showing 100% occlusion of the left anterior descending artery (black).
Figure 3.
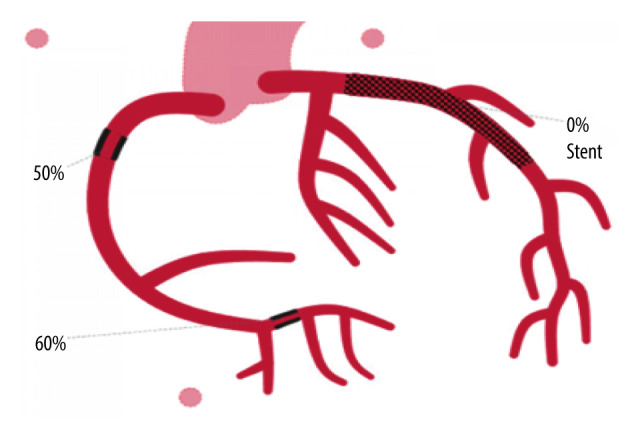
Diagram representing the patient’s condition after cardiac catheterization, showing revascularization of the left anterior descending artery, indicated as a mesh denoting a stent with 0% occlusion.
An Impella 2.5 was inserted because his ejection fraction was severely decreased, at 25%, accompanied by akinesis of the inferior apex and the mid to distal anterior wall. After the procedure, the patient was continued on the antiplatelet agent Tirofiban (Aggrastat), in addition to a loading dose of dual antiplatelet therapy (DAPT) with aspirin and ticagrelor. He started bleeding from the Impella insertion site, and the bleeding could not be controlled with FemoStop or manual pressure application. Despite placement of a 50-pound bag, blood constantly oozed from the site, but there was no active bleeding at any other site. He was suspected of having acquired Von-Willebrand’s syndrome (AVWS) secondary to his thrombocytosis, in addition to acute platelet dysfunction secondary to treatment with aspirin and ticagrelor. We elected not to treat his AVWS with 1-desamino-8-d-arginine-vasopressin (DDAVP) or the antihemophilic factor/Von Willebrand factor complex Humate P due to the risk that they would increase thrombogenicity. Rather, emergency plateletpheresis was deemed necessary.
In addition to undergoing a workup for ET, this patient had been diagnosed with thrombocythemia 1 month before presentation and had been prescribed oral aspirin 81 mg/day. A peripheral blood smear showed prominent thrombocytosis with occasional giant forms. A bone marrow aspirate and biopsy showed significant numbers of atypical megakaryocytes (data not shown). Genetic testing showed that he was negative for JAK2, MPL, and CALR gene mutations. Further testing during hospitalization revealed low VWF ristocetin cofactor activity, consistent with AVWS. His platelet count was 1393×103/µl two weeks after discharge, decreasing significantly to 652×103/µl three months after discharge. The patient continued treatment with aspirin and ticagrelor.
Discussion
Identifying patients with ET who are at high risk of developing AVWS can facilitate appropriate treatment and improve outcomes [15]. AVWS presents similar to the hereditary form of VWS, except that the acquired form is diagnosed at an older age as these patients tend not to have a family history or prior bleeding episodes.
Our patient had triple-negative ET, which usually presents at a younger age and is associated with fewer thrombotic events and favorable overall survival than patients with the genetic form of ET [2]. The incidence of vascular complications has been reported higher among patients positive than negative for JAK2 mutations, for reasons that remain unclear, indicating the need for routine genetic testing to anticipate the risk of complications and treat high-risk individuals aggressively. Vascular complications, both thrombosis and bleeding, are major causes of death among patients with ET [20]. Major risk factors for thrombosis include prior thrombotic episodes and age >60 years. Cardiovascular risk factors, including diabetes, hypertension, and hyperlipidemia, should also be evaluated in ET patients, although the effect of cardiovascular risk factors on the risk of thrombosis in low to intermediate-risk groups remains uncertain. Leukocytosis and mutational status may be additional risk factors [22,25], although further validation is needed.
Younger age, elevated hemoglobin concentration and/or platelet counts, and the presence of a JAK2 mutation have been associated with a higher risk for AVWS, although the evidence remains uncertain [16,26–28]. Current guidelines recommend routine testing of VWF ristocetin cofactor activity in patients with platelet counts over 1000×109/L, although a significant proportion of patients who developed AVWS had lower platelet counts [15,16]. These findings indicate a need for retrospective analysis to modify guidelines and affect management with antiplatelet agents. Low platelet count may protect against the development of AVWS [16]. We recommend adequate control of ET with stable low platelet count and testing for AVWS when there is evidence of bleeding, irrespective of platelet count [15]. Guidelines indicate that the functional activity of VWF be tested with the VWF ristocetin cofactor assay and that factor VIII and VWF levels be measured [16,15,29]. Interestingly antiplatelet agents do not interfere with VWF ristocetin cofactor assays or Factor VIII levels.
Patients with JAK2 mutations are at greater risk of developing AVWS than patients with CALR mutations despite the former having lower platelet counts [16]. Retrospective studies have found concomitant Von Willebrand disease in as many as 20% of patients with ET, indicating the need to consider AVWS when managing patients with ET [15,30].
Aspirin is recommended for all patients with ET if there is no contraindication, although a platelet count >1000×109/L is considered a relative contraindication [21]. A randomized study of high-risk patients showed that treatment with hydroxyurea significantly reduced the rate of thrombotic events when compared with a control group [21].
Multiple variants have been observed in patients with triple-negative disease, but the clinical correlations of these variants with symptoms and the development of vascular complications are unclear, indicating the need for further exploration of the genetic component of this disease [31]. Next generation sequencing has shown mutations in several genes, including the TET2, SH2B3, and ASXL1 genes, in patients with triple-negative disease, although these results require validation in large cohorts [32].
Conclusions
Adequate management of ET can help prevent the development of AVWS. Patients with ET and high platelet counts who experience bleeding complications should be suspected of having AVWS, as prompt treatment with plateletpheresis is life-saving. Patients with ET should undergo routine testing with the VWF ristocetin cofactor assay. The findings in our patient indicate that clinicians should have a high degree of suspicion of AVWS if patients with ET present with bleeding in addition to, or instead of, clotting.
Footnotes
Department and Institution where work was done
Department of Internal Medicine, UPMC Pinnacle, Harrisburg, PA, U.S.A.; and Department of Hematology/Oncology, UPMC Hillman Cancer Center, Camp Hill, PA, U.S.A.
Conflict of interest
None.
References:
- 1.Beer PA, Erber WN, Campbell PJ, Green AR. How I treat essential thrombocythemia. Blood. 2011;117:1472–82. doi: 10.1182/blood-2010-08-270033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akcan T, Strati P, Yan M, Idowu M. A rare case of triple-negative essential thrombocythemia in a young postsplenectomy patient: A diagnostic challenge. Case Rep Hematol. 2018;2018:9079462. doi: 10.1155/2018/9079462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 4.Hehlmann R, Jahn M, Baumann B, Köpcke W. Essential thrombocythemia. Clinical characteristics and course of 61 cases. Cancer. 1988;61:2487–96. doi: 10.1002/1097-0142(19880615)61:12<2487::aid-cncr2820611217>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 5.Yokus O, Gedik H. Jak-2 mutation frequency in patients with thrombocytosis. Caspian J Intern Med. 2018;9:189–93. doi: 10.22088/cjim.9.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cetin G, Özkan T, Turgut S, et al. Evaluation of clinical and laboratory findings with JAK2 V617F mutation as an independent variable in essential thrombocytosis. Mol Biol Rep. 2014;41:6737–42. doi: 10.1007/s11033-014-3559-x. [DOI] [PubMed] [Google Scholar]
- 7.Cervantes F. Management of essential thrombocythemia. Hematology Am Soc Hematol Educ Program. 2011;2011:215–21. doi: 10.1182/asheducation-2011.1.215. [DOI] [PubMed] [Google Scholar]
- 8.Karkucak M, Yakut T, Ozkocaman V, et al. Evaluation of the JAK2-V617F gene mutation in Turkish patients with essential thrombocythemia and polycythemia vera. Mol Biol Rep. 2012;39:8663–67. doi: 10.1007/s11033-012-1721-x. [DOI] [PubMed] [Google Scholar]
- 9.National Organisation of Rare Disorders (NORD) Tefferi A. Essential thrombocythemia. Updated 2018 https://rarediseases.org/rare-diseases/essential-thrombocythemia/
- 10.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–90. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 11.Baxter EJ, Scott LM, Campbell PJ, et al. Cancer Genome Project Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–61. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 12.Langabeer S, Ni Ainle F, Conneally E, Lawler M. Incidence and significance of the JAK2 V617F mutation in patients with chronic myeloproliferative disorders. Ir J Med Sci. 2007;176:105–9. doi: 10.1007/s11845-007-0026-x. [DOI] [PubMed] [Google Scholar]
- 13.Basquiera AL, Soria NW, Ryser R, et al. Clinical significance of V617F mutation of the JAK2 gene in patients with chronic myeloproliferative disorders. Hematology. 2009;14:323–30. doi: 10.1179/102453309X12473408860226. [DOI] [PubMed] [Google Scholar]
- 14.Van Genderen PJJ, Budde U, Michiels JJ, et al. The reduction of large von Willebrand factor multimers in plasma in essential thrombocythaemia is related to the platelet count. Br J Haematol. 1996;93:962–65. doi: 10.1046/j.1365-2141.1996.d01-1729.x. [DOI] [PubMed] [Google Scholar]
- 15.Mital A, Prejzner W, Bieniaszewska M, Hellmann A. Prevalence of acquired von Willebrand syndrome during essential thrombocythemia: A retrospective analysis of 170 consecutive patients. Pol Arch Med Wewn. 2015;125:914–20. doi: 10.20452/pamw.3211. [DOI] [PubMed] [Google Scholar]
- 16.Rottenstreich A, Kleinstern G, Krichevsky S, et al. Factors related to the development of acquired von Willebrand syndrome in patients with essential thrombocythemia and polycythemia vera. Eur J Intern Med. 2017;41:49–54. doi: 10.1016/j.ejim.2016.11.011. [DOI] [PubMed] [Google Scholar]
- 17.Federici AB, Rand JH, Bucciarelli P, et al. Subcommittee on von Willebrand Factor: Acquired von Willebrand syndrome: Data from an international registry. Thromb Haemost. 2000;84:345–49. [PubMed] [Google Scholar]
- 18.Franchini M, Lippi G. Acquired von Willebrand syndrome: An update. Am J Hematol. 2007;82:368–75. doi: 10.1002/ajh.20830. [DOI] [PubMed] [Google Scholar]
- 19.Tefferi A, Vannucchi AM, Barbui T. Essential thrombocythemia treatment algorithm 2018. Blood Cancer J. 2018;8:2. doi: 10.1038/s41408-017-0041-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cazzola M, Kralovics R. From Janus kinase 2 to calreticulin: The clinically relevant genomic landscape of myeloproliferative neoplasms. Blood. 2014;123:3714–19. doi: 10.1182/blood-2014-03-530865. [DOI] [PubMed] [Google Scholar]
- 21.Harrison CN, Bareford D, Butt N, et al. British Committee for Standards in Haematology: Guideline for investigation and management of adults and children presenting with a thrombocytosis. Br J Haematol. 2010;149:352–75. doi: 10.1111/j.1365-2141.2010.08122.x. [DOI] [PubMed] [Google Scholar]
- 22.Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood. 2014;124:2507–13. doi: 10.1182/blood-2014-05-579136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolanskyj AP, Schwager SM, McClure RF, et al. Essential thrombocythemia beyond the first decade: Life expectancy, long-term complication rates, and prognostic factors. Mayo Clin Proc. 2006;81:159–66. doi: 10.4065/81.2.159. [DOI] [PubMed] [Google Scholar]
- 24.Ueki T, Takeshige K, Sumi M, et al. [Successful management of intraperitoneal bleeding with platelet apheresis and von Willebrand factor supplementation in a patient with essential thrombocythemia and acquired von Willebrand syndrome] Rinsho Ketsueki. 2017;58:922–26. doi: 10.11406/rinketsu.58.922. [in Japanese] [DOI] [PubMed] [Google Scholar]
- 25.Carobbio A, Finazzi G, Guerini V, et al. Leukocytosis is a risk factor for thrombosis in essential thrombocythemia: interaction with treatment, standard risk factors, and Jak2 mutation status. Blood. 2007;109:2310–13. doi: 10.1182/blood-2006-09-046342. [DOI] [PubMed] [Google Scholar]
- 26.Budde U, Schaefer G, Mueller N, et al. Acquired von Willebrand’s disease in the myeloproliferative syndrome. Blood. 1984;64:981–85. [PubMed] [Google Scholar]
- 27.Budde U, Dent JA, Berkowitz SD, et al. Subunit composition of plasma von Willebrand factor in patients with the myeloproliferative syndrome. Blood. 1986;68:1213–17. [PubMed] [Google Scholar]
- 28.Fabris F, Casonato A, Grazia del Ben M, et al. Abnormalities of von Willebrand factor in myeloproliferative disease: A relationship with bleeding diathesis. Br J Haematol. 1986;63:75–83. doi: 10.1111/j.1365-2141.1986.tb07497.x. [DOI] [PubMed] [Google Scholar]
- 29.van Genderen PJJ, Prins FJ, Lucas IS, et al. Decreased half-life time of plasma von Willebrand factor collagen binding activity in essential thrombocythaemia: Normalization after cytoreduction of the increased platelet count. Br J Haematol. 1997;99:832–36. doi: 10.1046/j.1365-2141.1997.4823285.x. [DOI] [PubMed] [Google Scholar]
- 30.Van Genderen PJJ, Leenknegt H, Michiels JJ, Budde U. Acquired von Willebrand disease in myeloproliferative disorders. Leuk Lymphoma. 1996;22(Suppl. 1):79–82. doi: 10.3109/10428199609074364. [DOI] [PubMed] [Google Scholar]
- 31.Zaidi U, Shahid S, Fatima N, et al. Genomic profile of a patient with triple negative essential thrombocythemia, unresponsive to therapy: A case report and literature review. J Adv Res. 2017;8:375–78. doi: 10.1016/j.jare.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ju M, Fu R, Li H, et al. Mutation profiling by targeted sequencing of “triple-negative” essential thrombocythaemia patients. Br J Haematol. 2018;181:857–60. doi: 10.1111/bjh.14723. [DOI] [PubMed] [Google Scholar]