

Abstract
Idiopathic pulmonary fibrosis is a chronic and irreversible respiratory disease with a high incidence worldwide and no specific treatment. Currently, the etiology and pathogenesis of this disease remain largely unknown. In main purpose of this study, bioinformatics analysis was used to uncover key genes and pathways related to idiopathic pulmonary fibrosis (IPF). Gene expression profiles of GSE2052 and GSE35145 were obtained. After combining the 2 chip groups; then, we normalized the data, eliminating batch difference. R software was used to process and to screen differentially expressed genes (DEGs) between the IPF and normal tissues. Then, functional enrichment analysis of these DEGs was carried out, and a protein-protein interaction network (PPI) was also constructed. A total of 276 DEGs (152 up and 134 down-regulated genes) were identified in the IPF lung samples. The PPI network was established with 227 nodes and 763 edges. The top 10 hub genes were CAM1, CDH1, CXCL12, JUN, CTGF, SERPINE1, CXCL1, EDN1, COL1A2, and SPARC. Analyzing the PPI network modules with close interaction, the 3 key modules in the whole PPI network were screened out. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways enriched for the module containing DEGs contained the viral protein interaction with cytokine and the cytokine receptor, the TNF signaling pathway, and the chemokine signaling pathway. The identified key genes and pathways may play an important role in the occurrence and development of IPF, and may be expected to be biomarkers or therapeutic targets for the diagnosis of IPF.
Keywords: bioinformatics, genes, idiopathic pulmonary fibrosis, protein-protein interaction
1. Introduction
Idiopathic pulmonary fibrosis (IPF) is defined as a specific type of chronic, progressive, and fibrous interstitial pneumonia of unknown etiology, which is characterized by fibroblast proliferation and extracellular matrix deposition.[1] Its clinical features are persistent, progressive dyspnea of unknown cause, often accompanied by cough, Velcro rale at the end of the inspiratory breath, which causes diffuse pulmonary fibrosis, and, ultimately, respiratory failure.[2] The incidence of this disease is high, but its onset is insipid, and its clinical manifestations lack specificity. When diagnosed, patients are often in the terminal stage of the disease, and there is still no effective treatment other than lung transplantation.[3] Prognosis is extremely poor, and the median survival is only 3 to 5 years.[4] Currently, many factors are believed to contribute to the pathogenesis of rheumatoid arthritis (RA), including genetics, age, smoking, and environmental exposure; nonetheless, the pathogenesis of IPF has thus far remained unclear.
Persistent inflammatory response leading to lung injury and fibrosis formation is the major mechanism driving IPF.[5] However, recent studies have shown that epithelial injury and dysfunction may be more important factors in the development of pulmonary fibrosis.[6] Currently, the diagnosis of IPF relies on medical history, physical examination, lung function, high-resolution CT, and even lung biopsy.[2] However, high-resolution CT has certain limitations when diagnosing IPF, and lung biopsy specimens are not easily obtainable. Therefore, early diagnosis and treatment intervention is essential for improving prognosis.
High-throughput microarray technology and bioinformatics analyses have been widely applied in the pathogenesis, molecular diagnosis, and prognosis of diseases. Through genome-wide association studies (GWAS), some researchers have found that telomerase reverse transcriptase (TERT) and telomerase RNA component (TERC) gene mutations, caused by telomerase shortening, were related to a higher incidence of IPF.[7,8] However, despite having identified these target genes, we still lack a comprehensive representation of the key genes and pathways implicated in IPF.
In this study, gene chipsets expression profile were used for bioinformatics analysis to uncover the genes that differ between normal tissues and IPF tissues. We performed functional enrichment analysis on differentially expressed genes and constructed PPI networks. It provides a theoretical basis for exploring the molecular mechanism of the generation and development of IPF and provides a new idea for the early diagnosis of IPF.
2. Material and methods
Ethical approval or patient consent was not required because the data for the present research were obtained from a public database, and the data are available without personal identifiers.
2.1. Microarray dataset and data pre-processing
The gene expression profiles of IPF were downloaded from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/), which is a gene expression database created and maintained by the national center for biotechnology information (NCBI). The acquisition conditions of the gene chips data were: (1) from the original data of a human gene expression chip; (2) from IPF lung samples and normal lung tissue samples. GSE2052[9–11] and GSE35145[12] datasets were obtained from GEO, based on the GPL1793 and GPL10558 platforms. Firstly, the gene probe IDs from the raw data were converted to gene symbol codes. Secondly, the 2 databases were merged; and, finally, the data of the 2 combined chips were batched and normalized to eliminate batch differences using the SVA package in R software v3.5.2 (https://www.r-project.org).
2.2. Analysis and identification of DEGs
The DEGs amongst IPF and normal samples were analyzed using R software with a limma package from the Bioconductor project. Probe sets that did not have a corresponding gene symbol or a gene with multiple probe sets were removed or averaged. Fold-changes (FCs) in the gene expression values were calculated. |log2 FC| ≥ 1 and adjusted P values <.05 were considered the cut-off criteria for identifying DEGs. Visualization of the DEGs was illustrated by using the heatmap package in R software.
2.3. GO and KEGG enrichment analyses of DEGs
GO analysis is a common and useful tool which can annotate genes and analyze their biological processes; it includes the following 3 aspects: biological process (BP), molecular function (MF), and cellular component (CC). Kyoto Encyclopedia of Genes and Genomes (KEGG) is a utility database resource for understanding advanced functions and biological systems, which stores extensive data concerning genomes, biological pathways, diseases, and chemical substances. The GO enrichment and KEGG analyses of the DEGs were conducted by using the clusterProfiler package in R software. The GOplot package within R software allowed us to visualize the results from the GO and KEGG analyses. A P value <.05 was considered statistically significant.
2.4. PPI network construction and hub gene analyses
The screened DEGs were input in the STRING database (http://string-db.org/) to construct PPI network. Subsequently, the results of PPI network constructed from the STRING database were imported into Cytoscape software for visual analysis. The cytoHubba app, a plugin for Cytoscape, was applied to calculate the hub genes based on the overlapping results obtained by Degree topological analysis methods. Then, Molecular Complex Detection (MCODE) was performed to monitor the PPI network modules with Cytoscape. The selection criteria of the modules were as follows: node score cutoff = 0.2, degree cutoff = 2, maximum depth = 100, and k-core = 2. The functional enhancement analysis of the DEGs in the top module was performed using the clusterProfiler package within R software.
3. Results
3.1. Identification of the DEGs
Two microarray datasets (GSE2052 and GSE35145) were selected for this study. The GSE2052 dataset included 11 normal lung histology samples and 13 IPF lung samples, while GSE35145 included 4 normal specimens and 4 IPF specimens. After we standardized the batch of merged microarray databases, 276 DEGs were be identified by the limma package in R software, including 152 upregulated genes and 124 downregulated genes in IPF samples compared with normal lung samples. The heatmaps of the up- and downregulated genes amongst the DEGs are shown in Figure 1. The volcano plot of the DEGs is presented in Figure 2.
Figure 1.
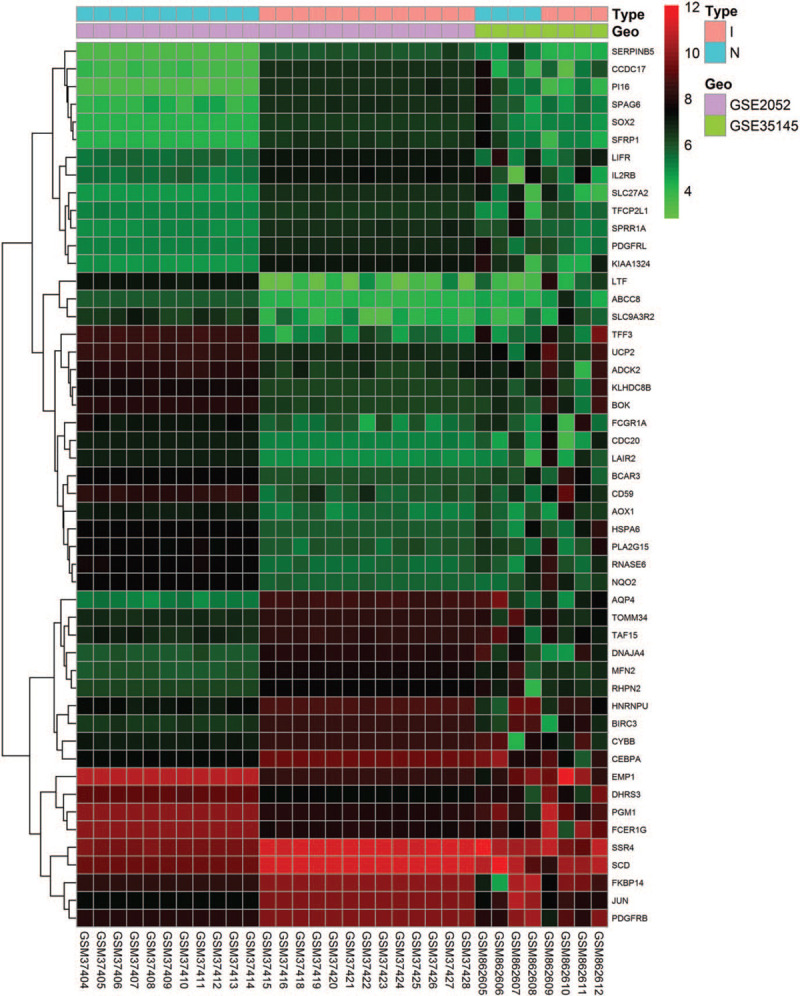
Heatmap of the top 50 DEGs of GSE2052 and GSE35145. The gene expression data are presented in a matrix format. Green color represents a lower expression and red color represents a higher expression. Black color indicates no differential expression.
Figure 2.
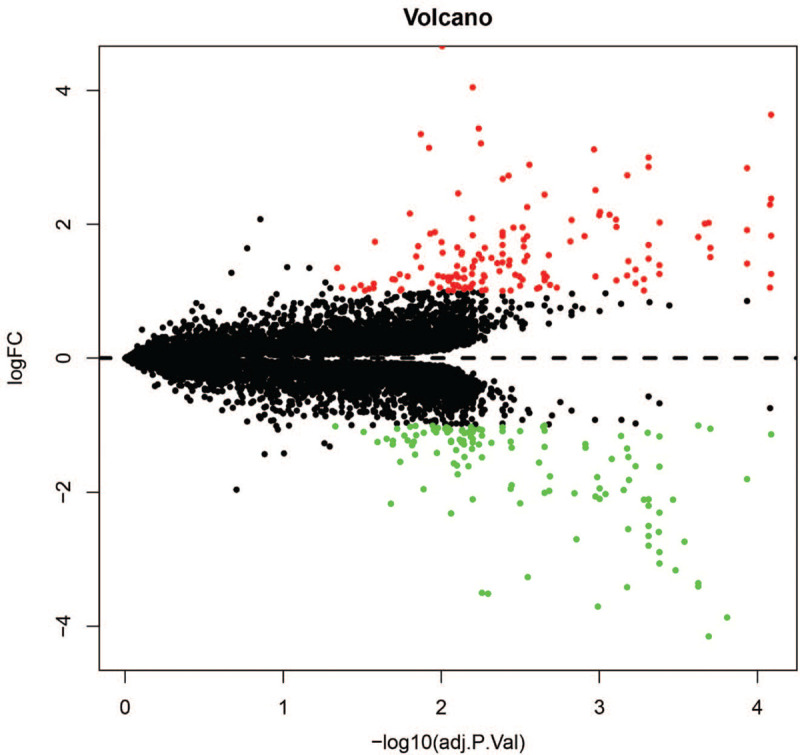
Volcano plot of DEGs. The black dots represent undifferentially expressed genes; the red dots represent up-regulated genes; green is the down-regulated gene.
3.2. GO and KEGG pathway enrichment of DEGs
The enriched GO terms were divided into BP, CC, and MF ontologies. Within the BP functional group, upregulated DEGs were mainly enriched in the extracellular matrix organization, extracellular structure organization and response to tumor necrosis factor; whereas downregulated DEGs were enriched in the neutrophil activation, neutrophil degranulation, and neutrophil activation involved in the immune response. Upregulated DEGs enriched in CC function were significantly associated with the extracellular matrix, endoplasmic reticulum lumen, and basolateral plasma membrane, whereas downregulated DEGs were enriched in the cytoplasmic vesicle lumen, vesicle lumen, and secretory granule lumen. For the MF functional group, upregulated DEGs were enriched in heparin-binding, sulfur compound binding, and cell adhesion molecule binding, while downregulated DEGs were enriched in the glycosaminoglycan binding, sulfur compound binding, and heparin-binding. In addition, the results of the KEGG pathway analysis showed that upregulated DEGs were mainly enriched in protein digestion and absorption, NF-kappa B signaling pathway, and rheumatoid arthritis. The downregulated DEGs were mainly enriched in aldosterone-regulated sodium reabsorption, complement and coagulation cascades, and amino sugar and nucleotide sugar metabolism. The above functional enrichment results are presented in Tables 1 and 2 respectively.
Table 1.
Top 10 GO terms and pathways enrichment analysis of upregulated DEGs (P < .05).
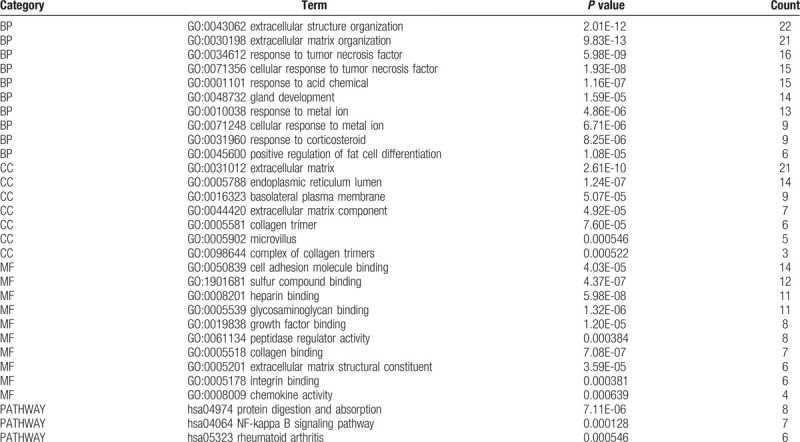
Table 2.
Top 10 GO terms and pathways enrichment analysis of downregulated DEGs (P < .05).

3.3. PPI network and hub genes
PPI network of DEGs was drawn and beautified by STRING database and Cytoscape, as shown in Figure 3A. Using the cytoHubba app in Cytoscape, the top 10 hub genes (VCAM1, CDH1, CXCL12, JUN, CTGF, SERPINE1, CXCL1, EDN1, COL1A2, and SPARC) were identified in in Table 3 and Figure 3 B. Three network modules that satisfied the number of nodes >4 were then selected from the PPI network using MCODE (Fig. 4A–C).
Figure 3.
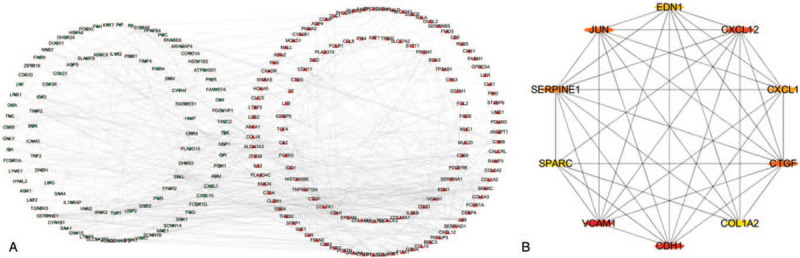
The PPI network and hub genes. (A) PPI networks of all differentially expressed genes were shown in circles, with red representing up-regulated differentially expressed genes and green representing down-regulated genes. (B) The 10 hub genes are represented by circles. The ellipses with different color-differences represent different degrees, and the darker the color, the more important it is, and the interaction evidence degree between proteins is presented as the gray scale of the lines.
Table 3.
Top ten hub genes with higher degree of connectivity.
Figure 4.
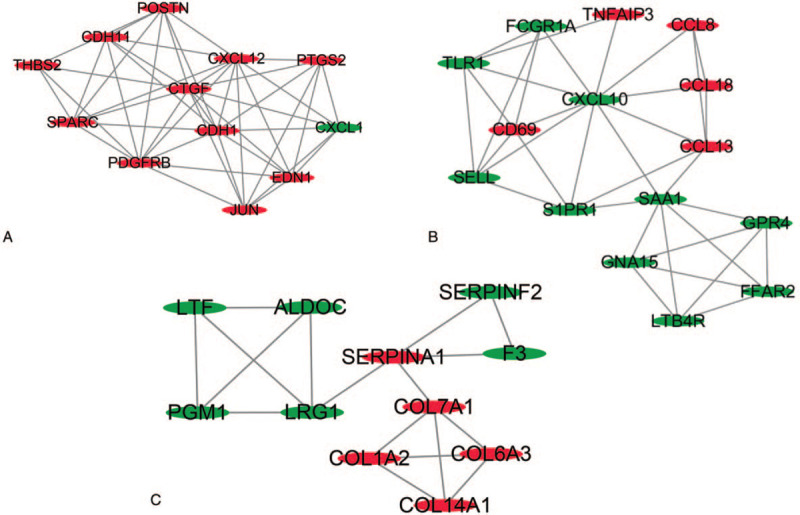
Three key modules of PPI network (A–C). Node size is negatively related to P value; edge color and width are positively related to combined score.
The functional annotation of the module containing the DEGs was carried out. The GO functions enriched for the module containing the DEGs were determined. Regarding the BPs, the DEGs in the PPI networks module were mainly enhanced for leukocyte migration, positive regulation of cell migration, and cell chemotaxis. As for the MF functional group, DEGs were enriched in the protein-coupled receptors binding, receptor-ligand activity, and sulfur compounds binding. Moreover, for the CC functional group, DEGs were enhanced in the extracellular matrix, the cytoplasmic vesicle lumen, and the vesicle lumen. KEGG pathways enriched for the module containing DEGs contained the viral protein interaction with cytokine and the cytokine receptor, TNF signaling pathways, and chemokine signaling pathways. The results are presented in Table 4.
Table 4.
Top 5 GO terms and pathways enrichment analysis of DEGs in the modules (P < .05).

4. Discussion
IPF is a relatively common clinical pattern of interstitial lung disease (ILD) with a high fatality rate. This is a typical age-related disease with onset age of over 60 years old that predominantly affects male patients. With the development of society and the prolonged lifespan of the general population, the incidence of IPF has tended to increase. The average annual incidence of IPF in Europe and North America is 3 parts per 100,000 people and 9 parts per 100,000 people, respectively.[13,14] However, in East Asia and South America, the prevalence of IPF is lower.[15] The pathological process of patients with IPF after initial diagnosis is complex, and the prognosis is poor overall[16]; the average survival time of patients with IPF is only about 3.5 years.[17]
By using bioinformatics, this study analyzed 2 IPF gene chips and normal lung tissue and identified the DEGs between the 2 expression profiles. The results of the microarray analysis revealed the expression of 276 altered DEGs significantly (150 upregulated genes and 236 down-regulated genes). The associations between these genes were revealed by constructing a PPI network. The top 10 genes with the highest degrees wereidentified, including VCAM1, CDH1, CXCL12, JUN, CTGF, SERPINE1, CXCL1, EDN1, COL1A2, and SPARC. Furthermore, 6 modules were selected according to their respective MCODE computed node scores (>4), and their functions were determined by GO and KEGG pathway analyses.
VCAM1 encodes vascular cell adhesion molecule 1 and is a member of the immunoglobulin superfamily.[18] It is an important cell adhesion molecule, which plays an important role in regulating the inflammatory response and immunity.[19] Previous studies have shown that VCAM1 can be used as an indicator to predict the death rate of IPF patients and that the level of VCAM1 in peripheral blood of patients with IPF is positively correlated with the mortality rate of IPF.[20] Similarly, some scholars have confirmed that VCAM1 is a transforming growth factor-β1, which is involved in the proliferation of the fibroblasts related to IPF.[21] These findings suggest that upregulation of the VCAM1 may be used as a biomarker in IPF.
CDH1, also known as cadherins 1, is a calcium-dependent cell adhesion protein.[22]CDH1 is an important molecule that maintains the phenotype of epithelial cells; its deficiency promotes epithelial-mesenchymal transition (EMT) and decreases intercellular adhesion.[23] One study has shown that EMT is one of the important factorsin the pathogenesis of IPF.[24] Therefore, it may be hypothesized that CDH1 may play a significant role in IPF.
CXCL12 is a small-molecule cytokine that belongs to the CXC chemokine family. CXCL12 is the main ligand of CXCR4 and can recruit fibroblasts and participate in the fibrosis process once it ligates to the receptor.[25] Phillips et al. found that, in a mouse model with pulmonary fibrosis and human pulmonary fibrosis lesions, the CXCL12/CXCR4 biological axis plays a key role in transforming fiber cells into myofibroblasts.[26]CXCL1 is also a member of the CXC chemokine family, but there are no relevant studies of this gene in lung fibrosis.
JUN is a subunit of the activator protein-1 (AP-1) and is also known as Jun AP-1 transcription factor subunit.[27] AP-1 is a homologous or heterodimer composed of JUN, Fos, ATF, and MAF protein families; nonetheless, JUN remains the predominant part.[28] One study has reported that AP-1 is considered an important requirement for TGF-β to lead to excessive deposition of collagen in lung tissues and changes in pulmonary fibrosis.[29] Recently, it has been demonstrated that CXCL12 activates Rac/ERK and JNK signaling pathways through CXCR4 and then initiates c-Jun phosphorylation, recruits c-Jun and c-Fos to the CTGF promoter, and, finally, induces the expression of CTGF in human lung fibroblasts.[30] Given these findings, the JUN gene is closely related to CXCL12 and GTGF in the formation of pulmonary fibrosis.
CTGF, also known as calponin 2 (CCN2), is a growth factor that promotes fibrocyte cell division and collagen deposition.[31] In vitro experiments have confirmed that expression of CTGF in rat and human lung fibroblasts stimulates mitosis, adhesion, apoptosis, production of the extracellular matrix, and migration of various cell-types.[32,33] CCN2 is considered an indicator to diagnose IPF and is used to monitor the progression of IPF.[34]
SPARC is a matricellular molecule that can regulatethe interaction between cells and the extracellular matrix topromote cell adhesion and induce cell migration.[35] Early studies have found that SPARC was mainly located in the cytoplasm and active fibroblasts within the fibroblastic foci in IPF lung tissues.[36] A recent publication objectively described the role of SPARC in pulmonary fibrosis, suggesting that SPARC may contribute to the formation of pulmonary fibrosis, but this finding requires further verification.[37] Therefore, the SPARC gene may be involved in forming IPF.
In the module containing DEGs, GO enhancement showed that these genes were mainly linked to leukocyte migration, positive regulation of cell migration, cell chemotaxis, protein-coupled receptor binding, receptor-ligand activity, sulfur compound binding, the extracellular matrix, the cytoplasmic vesicle lumen, and the vesicle lumen; KEGG pathway showed that these genes were mainly linked to the viral protein interaction with cytokine the cytokine receptor, the TNF signaling pathway, and the chemokine signaling pathway, suggesting that they pose an important role in the pathogenesis of IPF. However, there are certain limitations to the present study. Firstly, the current sample size of these datasets was small. Therefore, further studies using high-throughput sequencing experiments with larger clinical samples would be valuable. Secondly, some key genes and pathways were not found to be associated with IPF in previous studies.
5. Conclusions
This study makes a comprehensive analysis of IPF related genes and pathways, which is conducive to the future research on the pathogenesis of IPF. For future research, the target genes and pathways that we have identified here should be confirmed through in vitro studies and functional studies to determine the molecular mechanism in the pathogenesis of IPF. Overall, our results provided new insights into the potential targets for IPF diagnosis and treatment.
Acknowledgments
The authors wish to thank the library of Jiangxi University of Traditional Chinese Medicine.
Author contributions
Conceptualization: Zhongbo Xu, Xin Feng.
Data curation: Lisha Mo, Mingru Huang.
Formal analysis: Lisha Mo.
Methodology: Lin Li, Xin Feng, Lisha Mo.
Resources: Mingru Huang.
Software: Mingru Huang, Xin Feng, Lisha Mo.
Supervision: Lin Li.
Validation: Lin Li.
Visualization: Xin Feng.
Writing – original draft: Zhongbo Xu, Xin Feng, Lisha Mo.
Writing – review & editing: Zhongbo Xu.
Footnotes
Abbreviations: BP = biological process, CC = cellular component, DEG = differently expressed gene, GEO = gene expression omnibus, GO = gene ontology, IPF = idiopathic pulmonary fibrosis, KEGG = Kyoto Encyclopedia of Genes and Genomes, MCODE = molecular complex detection, MF = molecular function, PPI = protein-protein interaction, STRING = search tool for the retrieval of interacting genes.
How to cite this article: Xu Z, Mo L, Feng X, Huang M, Li L. Using bioinformatics approach identifies key genes and pathways in idiopathic pulmonary fibrosis. Medicine. 2020;99:36(e22099).
ZX and LM contributed equally to this article.
The authors have no funding and conflicts of interest to disclose.
The datasets generated during and/or analyzed during the current study are publicly available.
References
- [1].Lasky JA, Ortiz LA, Tonthat B, et al. Connective tissue growth factor mRNA expression is upregulated in bleomycin-induced lung fibrosis. Am J Physiol 1998;275:L365–71.. [DOI] [PubMed] [Google Scholar]
- [2].Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nature Cell Biol 2002;4:E131–6.. [DOI] [PubMed] [Google Scholar]
- [3].Ye N, Ding Y, Wild C, et al. Small molecule inhibitors targeting activator protein 1 (AP-1). J Med Chem 2014;57:6930–48.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Phillips RJ, Burdick MD, Hong K, et al. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest 2004;114:438–46.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Richards TJ, Kaminski N, Baribaud F, et al. Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2012;185:67–76.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wang XM, Zhang Y, Kim HP, et al. Caveolin-1: a critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J Exp Med 2006;203:2895–906.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2018;198:e44–68.. [DOI] [PubMed] [Google Scholar]
- [8].Meigs TE, Fedor-Chaiken M, Kaplan DD, et al. Galpha12 and Galpha13 negatively regulate the adhesive functions of cadherin. J Biol Chem 2002;277:24594–600.. [DOI] [PubMed] [Google Scholar]
- [9].Mushiroda T, Wattanapokayakit S, Takahashi A, et al. A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J Med Genet 2008;45:654–6.. [DOI] [PubMed] [Google Scholar]
- [10].Salton F, Volpe MC, Confalonieri M. Epithelial- mesenchymal transition in the pathogenesis of (Kaunas) 2019;55:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Brekken RA, Sage EH. SPARC, a matricellular protein: at the crossroads of cell-matrix communication. Matrix Biol 2001;19:816–27.. [DOI] [PubMed] [Google Scholar]
- [12].Chen HY, Lin CH, Chen BC. ADAM17/EGFR-dependent ERK activation mediates thrombin-induced CTGF expression in human lung fibroblasts. Exp Cell Res 2018;370:39–45.. [DOI] [PubMed] [Google Scholar]
- [13].Pardo A, Gibson K, Cisneros J, et al. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med 2005;2:e251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Englert JM, Hanford LE, Kaminski N, et al. A role for the receptor for advanced glycation end products in idiopathic pulmonary fibrosis. Am J Pathol 2008;172:583–91.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Alder JK, Chen JJ, Lancaster L, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci USA 2008;105:13051–6.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gjonbrataj J, Choi WI, Bahn YE, et al. Incidence of idiopathic pulmonary fibrosis in Korea based on the 2011 ATS/ERS/JRS/ALAT statement. Int J Tuberc Lung Dis 2015;19:742–6.. [DOI] [PubMed] [Google Scholar]
- [17].Ley B, Collard HR. Risk prediction in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2012;185:6–7.. [DOI] [PubMed] [Google Scholar]
- [18].Pan LH, Yamauchi K, Uzuki M, et al. Type II alveolar epithelial cells and interstitial fibroblasts express connective tissue growth factor in IPF. Eur Respir J 2001;17:1220–7.. [DOI] [PubMed] [Google Scholar]
- [19].Ley B, Collard HR, King TE., Jr Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011;183:431–40.. [DOI] [PubMed] [Google Scholar]
- [20].Eickelberg O. Endless healing: TGF-beta, SMADs, and fibrosis. FEBS Lett 2001;506:11–4.. [DOI] [PubMed] [Google Scholar]
- [21].Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med 2001;134:136–51.. [DOI] [PubMed] [Google Scholar]
- [22].Agassandian M, Tedrow JR, Sembrat J, et al. VCAM-1 is a TGF-beta1 inducible gene upregulated in idiopathic pulmonary fibrosis. Cell Signal 2015;27:2467–73.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lim ST, Miller NL, Chen XL, et al. Nuclear-localized focal adhesion kinase regulates inflammatory VCAM-1 expression. J Cell Biol 2012;197:907–19.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lin CH, Shih CH, Tseng CC, et al. CXCL12 induces connective tissue growth factor expression in human lung fibroblasts through the Rac1/ERK, JNK, and AP-1 pathways. PLoS One 2014;9:e104746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Raghu G, Weycker D, Edelsberg J, et al. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2006;174:810–6.. [DOI] [PubMed] [Google Scholar]
- [26].Andersson-Sjoland A, de Alba CG, Nihlberg K, et al. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int J Biochem Cell Biol 2008;40:2129–40.. [DOI] [PubMed] [Google Scholar]
- [27].Kono M, Nakamura Y, Suda T, et al. Plasma CCN2 (connective tissue growth factor; CTGF) is a potential biomarker in idiopathic pulmonary fibrosis (IPF). Clin Chim Acta 2011;412:2211–5.. [DOI] [PubMed] [Google Scholar]
- [28].Wong SL, Sukkar MB. The SPARC protein: an overview of its role in lung cancer and pulmonary fibrosis and its potential role in chronic airways disease. Br J Pharmacol 2017;174:3–14.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Harari S, Caminati A. IPF: new insight on pathogenesis and treatment. Allergy 2010;65:537–53.. [DOI] [PubMed] [Google Scholar]
- [30].Schmitz B, Vischer P, Brand E, et al. Increased monocyte adhesion by endothelial expression of VCAM-1 missense variation in vitro. Atherosclerosis 2013;230:185–90.. [DOI] [PubMed] [Google Scholar]
- [31].McAnulty RJ, Laurent GJ. Pathogenesis of lung fibrosis and potential new therapeutic strategies. Exp Nephrol 1995;3:96–107.. [PubMed] [Google Scholar]
- [32].Harari S, Caminati A, Madotto F, et al. Epidemiology, survival, incidence and prevalence of idiopathic pulmonary fibrosis in the USA and Canada. Eur Respir J 2017;49:1601504. [DOI] [PubMed] [Google Scholar]
- [33].Ma F, Li W, Liu C, et al. MiR-23a promotes TGF-beta1-induced EMT and tumor metastasis in breast cancer cells by directly targeting CDH1 and activating Wnt/beta-catenin signaling. Oncotarget 2017;8:69538–50.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sanders YY, Ambalavanan N, Halloran B, et al. Altered DNA methylation profile in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2012;186:525–35.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183:788–824.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hutchinson J, Fogarty A, Hubbard R, et al. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 2015;46:795–806.. [DOI] [PubMed] [Google Scholar]
- [37].Kuhn C, Mason RJ. Immunolocalization of SPARC, tenascin, and thrombospondin in pulmonary fibrosis. Am J Pathol 1995;147:1759–69.. [PMC free article] [PubMed] [Google Scholar]