

Abstract
Hemorrhagic fevers caused by viruses were identified in the late 1950s in South America. These viruses have existed in their hosts, the New World rodents, for millions of years. Their emergence as infectious agents in humans coincided with changes in the environment and farming practices that caused explosions in their host rodent populations. Zoonosis into humans likely occurs because the pathogenic New World arenaviruses use human transferrin receptor 1 to enter cells. The mortality rate after infection with these viruses is high, but the mechanism by which disease is induced is still not clear. Possibilities include direct effects of cellular infection or the induction of high levels of cytokines by infected sentinel cells of the immune system, leading to endothelia and thrombocyte dysfunction and neurological disease. Here we provide a review of the ecology and molecular and cellular biology of New World arenaviruses, as well as a discussion of the current animal models of infection. The development of animal models, coupled with an improved understanding of the infection pathway and host response, should lead to the discovery of new drugs for treating infections.
Keywords: arenavirus, transferrin receptor, animal models
INTRODUCTION
There are a large number of arenaviruses found in different regions worldwide. These include the New World (NW) and Old World (OW) arenaviruses, which largely infect different rodent species endemic to their respective geographic locations. More recently, snake arenaviruses have been identified in cases of inclusion body disease, a progressive and sometimes fatal disease best described for members of the Boidae and Pythonidae families (1). With the discovery of snake arenaviruses, a new taxonomy has been suggested, which places the mammalian and snake viruses into the Mammarenavirus and Reptarenavirus genera, respectively (2). The mammarenaviruses normally produce persistent infections in their rodent hosts, with chronic viremia that is not generally pathogenic, spreading virus through urine, feces, and saliva to other rodents (3). Most of the NW mammarenaviruses have rodent hosts, but Tacaribe virus (found in Trinidad) and Ocozocoautla de Espinosa virus (found in Mexico) may be carried by bats (4–6).
The NW arenaviruses Junín virus, Machupo virus, Sabiá virus, and Guanarito virus, which infect rodents of the Cricetidae family, Sigmodontinae subfamily, found in Argentina, Bolivia, Brazil, and Venezuela, respectively, cause hemorrhagic fever in humans with about 30% mortality (7). The geographic distribution of each arenavirus is assumed to be determined by the habitat range of its reservoir species (8, 9). Humans may become infected through direct contact with infected animals, including bites, or through inhalation of infectious rodent excreta and secreta. The domestic and peridomestic behavior of these rodent species is a major contributing factor facilitating viral transmission from rodent to human (8).
Argentine hemorrhagic fever, a disease endemic to the Pampa region of Argentina, with about five million people at risk, is caused by Junín virus (10). Although an effective live attenuated Junín virus vaccine jointly developed by the Argentinian and US governments, called Candid #1, has decreased the incidence of Argentine hemorrhagic fever from about ~1,000 cases each year, there are still approximately 30–50 sporadic cases of infection with Junín virus as well as the other known and novel clade B arenaviruses for which there are no vaccines (11, 12). Indeed, in 2007–2008, there were more than 200 reported cases of Bolivian hemorrhagic fever, caused by Machupo virus infection, in several outbreaks in Bolivia (12), and in recent years there have been more than 40 cases annually of Guanarito virus infection in Venezuela (e.g., see 13). In 2004, a second fatal hemorrhagic fever arenavirus, Chaparé virus, was discovered in Bolivia (14), and it has been suggested that a novel NW arenavirus was responsible for a hemorrhagic fever outbreak in Chiapas, Mexico, in the late 1960s (4).
Because they can be readily transmitted by aerosols, hemorrhagic fever arenaviruses are potential bioterrorism agents and are included in the list of agents in the Material Threat Determinations and Population Threat Assessments issued by the US Department of Homeland Security (15). Thus, research with the human pathogenic NW arenaviruses must be conducted under biosafety level 4 (BSL-4)/animal biosafety level 4 (ABSL-4) conditions. Recovered patient serum has been successfully used to treat Junín virus infection, bringing mortality down from approximately 30% to 1% (11); whether it would be effective in the treatment of other NW arenaviruses is not known. About 10% of infected individuals treated with convalescent serum develop long-term neurological symptoms of unknown etiology (16). Ribavirin is currently the only antiviral drug in use for therapeutic or postinfection prophylactic treatment of arenavirus infection, although it has mixed efficacy and significant side effects (17). Both ribavirin and convalescent serum must be administered within the first 7 to 10 days after infection to be effective. As such, there is a great need for a better understanding of NW arenavirus infection and the development of new antiarenaviral therapeutics.
TAXONOMY AND DISTRIBUTION
The mammarenaviruses are classified into two groups according to their antigenic properties. The Tacaribe (NW) serocomplex includes viruses indigenous to the Americas, and the Lassa-lymphocytic choriomeningitis serocomplex (OW) includes viruses indigenous to Africa, such as Lassa fever virus and the ubiquitous lymphocytic choriomeningitis virus (LCMV). The Tacaribe serocomplex is the most genetically diverse group of the Mammarenavirus genus, composed of ~18 species divided into four lineages: clades A, B, C, and D (Figure 1a) (2). The NW lineage A includes five South American viruses: Flexal virus (Brazil), Pichinde virus (Colombia), Paraná virus (Paraguay), Allpahuayo virus (Perú), and Pirital virus (Venezuela). Viruses of this lineage generally do not infect humans. Lineage B includes eight South American viruses: Junín virus (Argentina); Chaparé and Machupo viruses (Bolivia); Sabiá, Cupixi, and Amapari viruses (Brazil); Tacaribe virus (Trinidad); and Guanarito virus (Venezuela) (Figure 1b). This clade includes the NW arenaviruses that cause hemorrhagic fever in humans: Junín, Machupo, Chaparé, Guanarito, and Sabiá viruses. The other members of clade B do not cause human disease, likely due to their poor ability to infect human cells (see below) (5). Lineage C NW arenaviruses, which are also not human pathogens, are the smallest clade within the genus, with only two described members: Latino virus (Bolivia) and Oliveros virus (Argentina) (8, 18). Bear Canyon, Tamiami, and Whitewater Arroyo viruses in the United States were originally termed clade A/B but have more recently been termed clade D (2); the latter virus was implicated in three fatalities in California in the 1990s (19, 20). The individuals in these cases had symptoms similar to those seen with the other NW hemorrhagic fever viruses.
Figure 1.
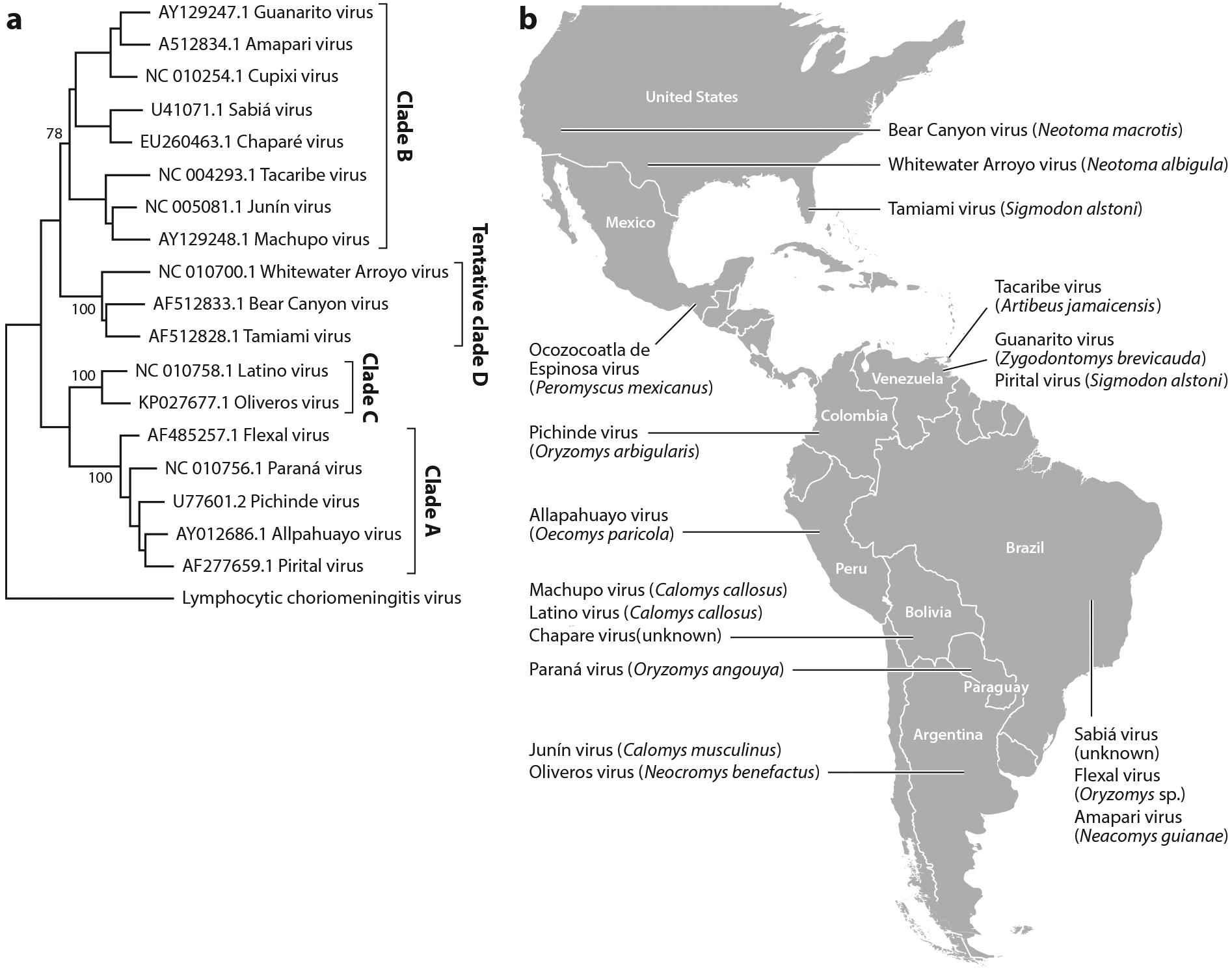
New World arenavirus taxonomy and geographic distribution. (a) Cladogram inferred by using the maximum-likelihood method with the Kimura two-parameter and gamma distribution substitution model. Phylogenetic reconstruction was performed using the full-length glycoprotein precursor (GPC) gene. Bootstrap values are shown for the main nodes. The GenBank accession number and origin of each sequence are denoted, as are the clade names. A strain of lymphocytic choriomeningitis virus was used as outgroup.
(b) Map of the Americas showing the distribution of the New World arenaviruses. Virus names and most frequent host reservoirs are indicated in the boxes.
The parameters used to define an arenavirus species within a clade are (a) association with a specific host species or group of species; (b) presence in a defined geographical area; (c) status as etiological agent (or not) of disease in humans; (d) significant differences in antigenic cross-reactivity, including lack of cross-neutralization activity; (e) significant amino acid sequence differences from other species in the genus [i.e., at least 12% divergence in the nucleoprotein (NP) amino acid sequence] (2). These classifications are sometimes fluid. For example, the genetic distances observed between Pampa virus and its closest relative, Oliveros virus, both found in Argentina, suggested that Pampa virus should be considered as a genotype of Oliveros virus instead of as a new viral entity. This was corroborated by its geographic distribution and mammalian host (Bolomys spp.), which are identical to those of Oliveros virus (21).
ECOLOGY AND EPIDEMIOLOGY
Most of the natural hosts for clade B NW arenaviruses are rodents of the subfamily Sigmodontinae (22), with the exception of Sabiá virus, for which no wild reservoir is known, and Tacaribe and Ocozocoautla de Espinosa viruses, which are thought to be found in bats (4–6). In contrast, OW arenaviruses are hosted by murid rodents of the subfamily Murinae (OW mice and rats).
The animal hosts for the highly pathogenic Junín, Machupo, and Guanarito viruses are Calomys musculinus (drylands vesper mouse), Calomys callosus (large vesper mouse), and Zygodontomys brevicauda (short-tailed cane mouse), respectively (5). C. musculinus is a wild rodent widely distributed throughout Argentina. It is an opportunistic species, characterized by a high reproductive rate and the ability to colonize a wide variety of habitats. These properties of the rodent host have allowed Junín virus to progressively expand its endemic area, which was originally centered in the city of Junín in the late 1950s and grew to cover nearly 150,000 km2 in four Argentinean provinces in the 2000s (22). Modifications of the environment, due to either human activities (modern farming practices) or natural ecological changes (flooding, storms), have been implicated in the emergence of human disease caused by arenaviruses, resulting in increases in the population and changes in the behavior of the reservoir host (8, 22).
Bolivian hemorrhagic fever was first described in 1959, affecting isolated human communities in eastern Bolivia largely through residential rodent infestation and also as a result of socioeconomic upheaval and changes in farming practices (23). The etiologic agent, Machupo virus, was isolated in 1963 from a patient who died from the disease (12). Although Machupo virus emerges only sporadically, the mortality rate associated with infection is high (20%), rendering necessary the development of vaccines and therapies for the disease (12, 24). For decades, Bolivian hemorrhagic fever cases were found only in northeastern Bolivia, and Machupo virus was therefore thought to be the only pathogenic arenavirus found in this country. However, a fatal hemorrhagic fever case occurred in 2003 near Cochabamba Department that was associated with a unique, newly discovered arenavirus, denoted as Chaparé virus (14).
Venezuelan hemorrhagic fever was recognized as a clinical entity in 1989 during an outbreak in the municipality of Guanarito (25). During the period 1989–2006, 618 Venezuelan hemorrhagic fever cases occurred, with a mortality rate of 23%. Up to 99% of the affected individuals lived or worked in rural areas in Guanarito when they became ill, so the etiological agent of the disease was named Guanarito virus (26). For the other lineage B human pathogen, Sabiá virus, only one naturally occurring fatal case has been reported (27). The sporadic appearance of disease caused by established and novel arenaviruses underscores the seriousness of potential outbreaks in South America.
The other lineage B viruses, including Tacaribe virus, and the lineage A Flexal virus have only been associated with febrile and nonfatal laboratory-acquired infections in humans (28). Exposure to the lineage A Pichinde virus has resulted in numerous seroconversions, primarily in laboratory workers, without any noticeable clinical symptoms (29). Tacaribe virus provides an excellent model system to identify the determinants of pathogenesis of the lethal lineage B viruses, without the need for BSL-4 containment facilities. The placement of Tacaribe virus within phylogenetic lineage B is based on the viral glycoprotein and nucleoprotein genes, which encode proteins involved in host cell invasion (8). Thus, the relatedness of the viruses within clade B is not predictive of their ability to cause human disease, although their receptor usage is (see below) (30).
The rodent species that serve as the primary hosts for both the NW and OW arenaviruses are persistently infected by a single viral species and are capable of both vertical and horizontal transmission (8, 9). Humans usually become infected through contact with infected rodents (cuts or bites) or via inhalation of infectious rodent excreta or secreta (21, 31). People with regular direct or indirect contact with wildlife, agricultural workers, and laboratory workers are particularly susceptible (31, 32). There are also rare incidences of human-to-human infection by close contact, nosocomial infection, or possibly organ transplantation, as has been seen with LCMV (33, 34). Other means of infection may include ingestion of infected rodents. For example, white-throated woodrats are consumed by humans in the Potosí-Zacatecan Mexican Plateau, and Mexican woodrats (Neotoma mexicana), Mexican deer mice (Peromyscus mexicanus), and other cricetid rodents are consumed by the Tzeltal Indians in Chiapas, posing a higher risk for these populations to become infected with arenaviruses carried by these rodent vectors (35).
For many decades it was thought that arenavirus-rodent interactions were an example of virus-host codivergence, where one rodent species diverged from another so much that the viruses carried in each host were no longer able to infect the different host species equally well. However, a recent study in NW arenaviruses suggested that host switching is mainly responsible for the host-virus evolution; this emphasizes the potential for arenaviruses to become panzootic (36). This ability to host switch is likely due to a relatively error-prone viral polymerase or to recombination that occurs during coinfection of a single species with multiple different arenaviruses (36, 37). Recombination events require infection of a single cell by two or more different viruses. Although it was originally believed that arenaviruses prevented infection by more than one virus by a mechanism termed superinfection interference, recent work has indicated that cells already infected with one Junín virus do not block infection by a second virus (38). Such coinfection/recombination has recently been suggested to have occurred with the reptarenaviruses (39).
ARENAVIRUS RECEPTORS
Arenaviruses are enveloped RNA viruses whose entry is mediated by the viral glycoprotein (GP), which is generated by proteolytic processing of the precursor GPC by the cellular protease SK1/S1P, which cleaves the precursor into the envelope protein subunits GP1 (the receptor-binding domain) and GP2 (the transmembrane fusion protein), and by the host signal peptidase, which processes membrane proteins to generate the stable signal peptide (SSP), a third subunit required for virus-cell fusion (Figure 2a) (40, 41). Entry of these viruses requires GP1 binding to a cellular surface receptor (Figure 2b). The clade B pathogenic NW arenaviruses use transferrin receptor 1 (TfR1) from humans and from their host species, but not from mice, for cell entry (42). TfR1 is a highly conserved cell surface receptor that is the major means by which cells take up iron in the form of iron-bound transferrin (43). In contrast, three other viruses in this clade (Tacaribe, Amapari, and Cupixi viruses) are nonpathogenic to humans, at least in part because they cannot bind to human TfR1 (44). They do, however, use TfR1 molecules from their host species. The crystal structure of the Machupo virus GP1 bound to human TfR1 revealed that clade B arenaviruses contact human TfR1 on a surface with substantial sequence variability among viruses, which might reflect their independent evolution and adaptation to their host species’ receptors (45).
Figure 2.
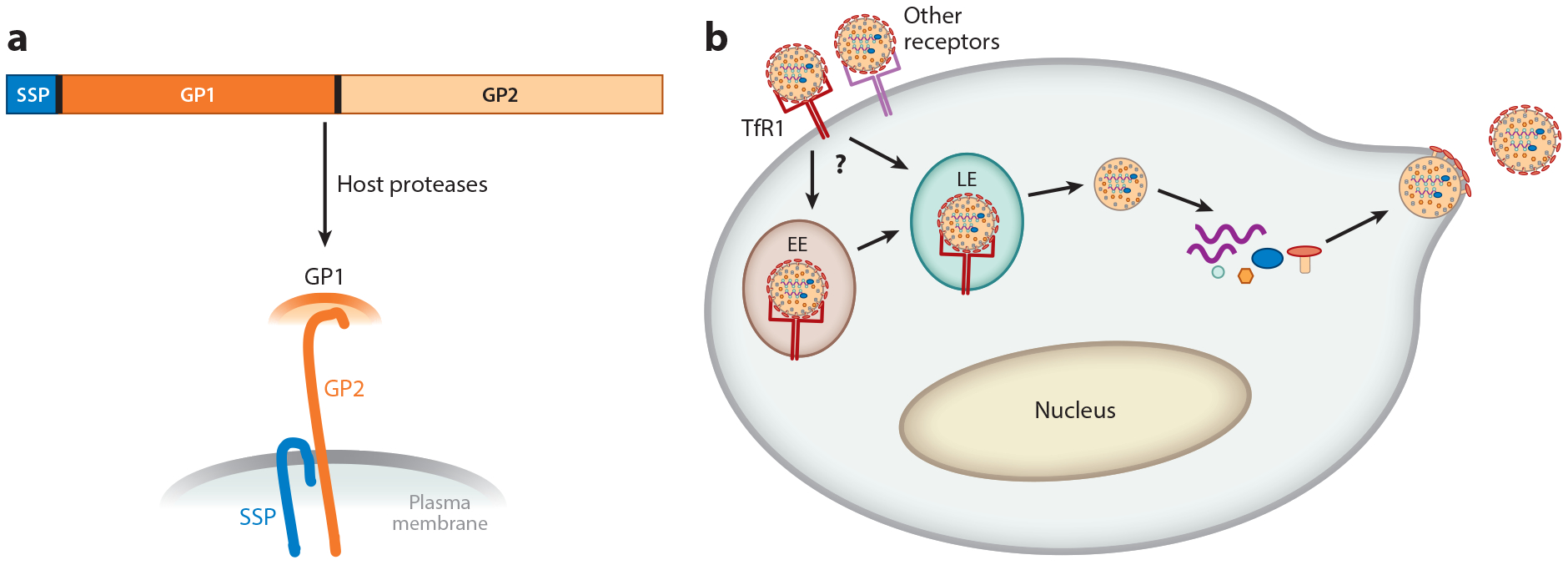
Cellular infection pathway and structure of the arenavirus glycoprotein (GP). (a) The arenavirus glycoprotein precursor (GPC) is translated as a polyprotein, which is synthesized in the endoplasmic reticulum, modified in the Golgi, and then cleaved by the SK1/S1P cellular protease and host signal peptidase to generate the three subunits—SSP, GP1, and GP2—found in the virion membrane.
(b) The pathogenic New World arenaviruses enter cells after binding to transferrin receptor 1 (TfR1) and perhaps other cell surface molecules. After endocytosis to either the early (EE) or late (LE) acidic endosome, which may be cell type dependent, the viral and host membranes fuse and the capsid is released into the cytoplasm. Transcription of the viral RNA follows, to generate the viral genome as well as mRNAs that encode the viral proteins. After synthesis of the proteins, the viral genomic RNAs are packaged into capsids, and the new viruses bud from the plasma membrane, which already contains the viral GP components.
Mutational analysis has shown that the TfR1 apical domain is the principal site of interaction with NW arenavirus GP1, and a short segment of four residues (208–212) in this domain is a critical determinant of virus host specificity (46). Substitutions in the counterpart region of mouse TfR1, based on the human TfR1 sequence, convert this molecule into an efficient receptor for Junín virus (45). Conversely, naturally occurring mutations in the domain of TfR1 that interacts with Machupo virus GP1 are able to prevent viral binding while retaining the receptor functionality (47); these variations might be able to confer protection in nonsusceptible rodents, and similar variants could potentially also protect humans from Machupo virus infection (27, 47). Intriguingly, Amapari and Tacaribe viruses can infect and replicate in human cells, but they do so independently of human TfR1, although they efficiently use the TfR1 of their respective hosts (5, 48). However, a point mutation was enough to convert human TfR1 into an efficient receptor for Tacaribe virus, and four mutations within nucleotides found in the host species Neacomys spinosus TfR1 sequence converted human TfR1 into an efficient receptor for Amapari virus (5). Likewise, human TfR1 variants with only modest changes allow efficient infection by tentative clade D viruses, implying that a small number of mutations in the GP may be sufficient for these viruses to gain use of human TfR1. These data show that TfR1 has an important role in the replication of nonpathogenic NW arenaviruses and suggest that subtle changes in their GPs might adapt them to use human TfR1. Moreover, lack of host species specificity among the clade D viruses may promote cross-species transmission and recombination, which can in turn increase the likelihood of producing viruses that use human TfR1. The North American arenaviruses may infect humans more frequently than previously understood given that only modest changes in receptor recognition are needed to change species tropism (49). This highlights the potential of these viruses to emerge as human pathogens.
The clade B viruses that infect humans (Junín, Machupo, Guanarito, Sabiá, and Chaparé viruses) incidentally acquired the capacity to bind human TfR1 during coevolution with the receptor in their natural rodent hosts, enabling the zoonosis (30, 45). Thus, the ability of an arenavirus to use human TfR1 is predictive of its ability to cause hemorrhagic fevers in humans (30). The close relationship between human TfR1 use and severe human disease suggests that TfR1 regulation plays a pivotal role in disease severity. TfR1 has several properties favorable to arenavirus replication and the development of hemorrhagic fevers: It is rapidly endocytosed into acidic compartments, is expressed on endothelial cells, and is upregulated on rapidly dividing cells including activated lymphocytes (5). The early response to viral infection thus could drive TfR1 expression on activated target cells, which in turn would accelerate viral replication; this feedback mechanism could distinguish clade B hemorrhagic fever arenaviruses from the nonpathogenic variants that have the ability to infect cells using other receptors that may not be upregulated in response to infection (30). Because TfR1 expression is highly regulated by host iron levels, it has been suggested that treatment of infected individuals with iron might ameliorate disease (42, 43, 50).
NW arenaviruses also enter cells via TfR1-independent means (48, 51) and likely use receptors other than TfR1 to infect sentinel cells of the immune system, their probable initial in vivo targets (see below). This must also be the case for the nonpathogenic clade B viruses Tacaribe virus, Amapari virus, and Flexal virus, which can infect human cells or humans independently of TfR1 (28, 48). Indeed, the live attenuated Junín vaccine, Candid #1, was generated in part through extensive passage in neonatal mouse brain, where it was reported to be initially pathogenic (52, 53). C-type lectin receptors such as DC-SIGN/CD209 and L-SIGN/CD209L have been implicated in Junín virus infection, and other cell surface molecules, such as members of the TIMs and Axl family, which bind phosphatidylserine, have been suggested to increase the efficacy of NW arenavirus infection by interaction with the viral GP1 (54, 55). Although human TfR1 is the primary cell entry receptor for these viruses, NW arenaviruses can infect murine cell lines, macrophage primary cultures from rat and C. musculinus, and laboratory mouse models through a TfR1-independent mechanism (48, 51, 56, 57). We as well as others have also shown that Junín and Machupo pseudoviruses can infect Mus musculus in vitro and in vivo, although they are unable to bind to mouse TfR1 (48, 51). We recently suggested that voltage-gated calcium channels serve as additional entry receptors (58), and our unpublished work suggests that they are the major means by which Junín virus enters mouse cells. Some of these other receptors, as well as TfR1, may serve to increase binding of NW arenaviruses to the cell surface, thereby increasing the local concentration of virions and the efficacy of infection.
ARENAVIRUS REPLICATION
Whereas the GP1 subunit primarily determines host and likely tissue tropism by binding to the cell surface receptors, the GP2 subunit mediates membrane fusion, along with the SSP subunit, after virus particles are internalized into acidified endosomes. Subsequent to GP interaction with receptors on the cell surface, trafficking to a pH 5 late endosomal compartment is required for virus-cell membrane fusion and entry of the capsid into cells (Figure 2b) (59–61). Internalization may occur through clathrin-mediated endocytosis, although this has only been shown in immortalized epithelial cells and fibroblasts (62). Although it has been suggested that NW arenaviruses traffic through an early endocytic compartment to reach the late endosome, we as well as others have suggested that the virus bypasses the early compartment, at least in certain cell types (62–64); one possibility is that NW arenavirus entry through intracellular compartments differs depending on the cell type. Both the TfR1-dependent pathway and the TfR1-independent pathway still require entry through a low-pH compartment (56, 64). It has recently been shown that the OW arenavirus Lassa fever virus, in addition to using the cell surface receptor α-dystroglycan to bind to cells and trigger endocytosis, also uses LAMP1 in the acidic compartment to trigger virus entry into the cytoplasm; however, no such molecule has been identified for NW arenavirus infection (65).
Arenavirus replication is confined to the cytoplasm. All arenaviruses contain two ambisense RNA molecules: The large (L) RNA (~7.3 kb) encodes the RNA-dependent viral RNA polymerase, termed L, in the sense orientation as well as the Z matrix protein in the antisense orientation, and the small (S) RNA (~3.5 kb) encodes GPC in the antisense orientation and NP in the sense orientation (Figure 3) (66). Subsequent to entry into the cytoplasm, the L protein begins transcription and replication of the viral antigenome. NP and L are the first to be transcribed and translated; translation of GPC and Z requires synthesis of the viral antigenome and subsequent transcription. Transcription and translation are regulated by 5′ and 3′ untranslated regions, as well as an intergenic region that separates the two genes on the L and S RNAs; the secondary structure formed by the intergenic region is also believed to provide translation termination signals (Figure 3).
Figure 3.
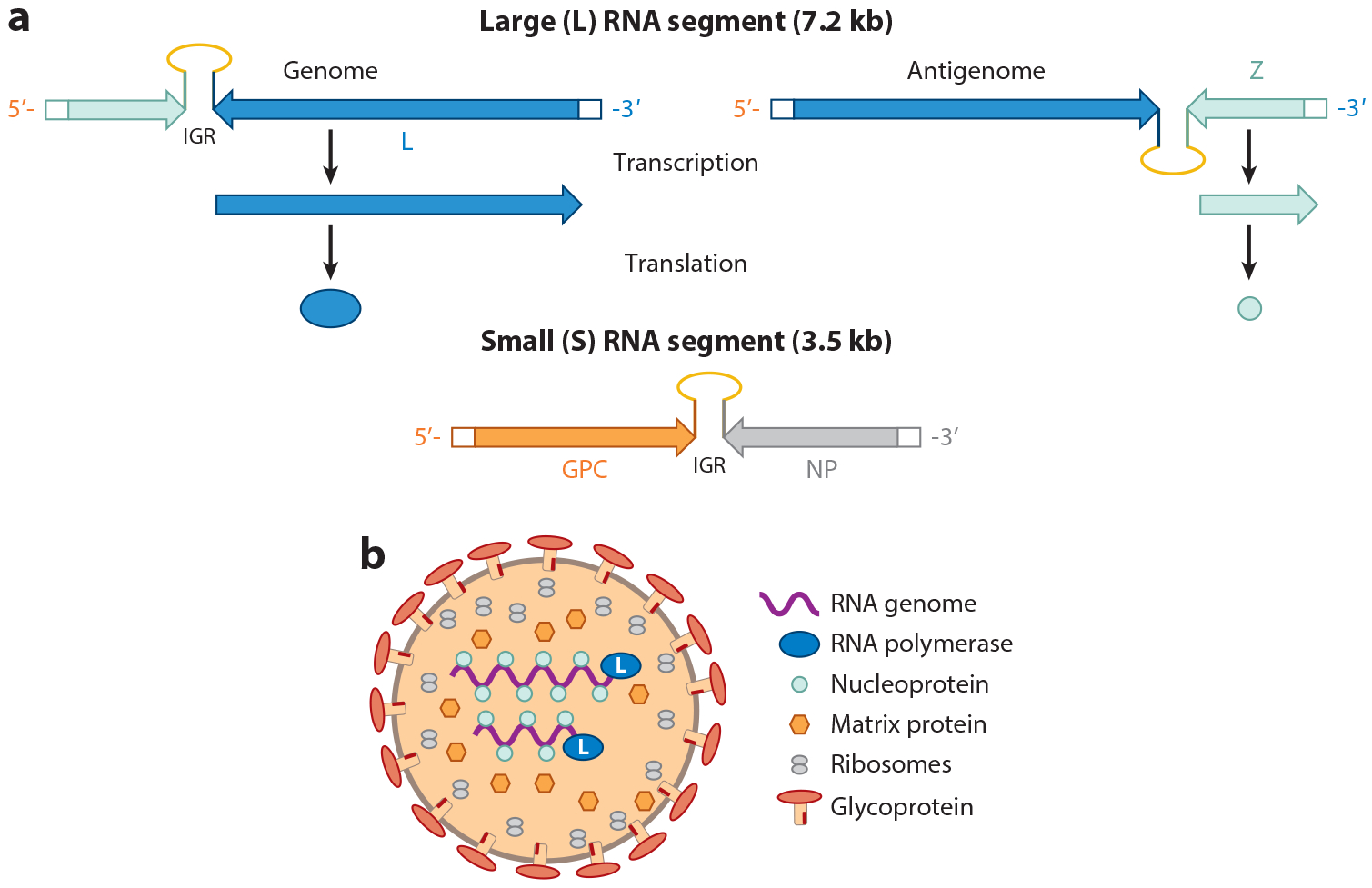
Arenavirus genome and virion structure. (a) Genome organization of the two RNA subunits packaged into arenavirus particles. The large (L) RNA encodes the matrix (Z) and polymerase (L), and the small (S) RNA encodes the glycoprotein precursor (GPC) and nucleoprotein (NP). Both RNAs are ambisense. The scheme for generating the mRNAs for the L and Z RNAs is shown; a similar process occurs for the GPC and NP RNAs. The intergenic region (IGR) between the RNAs controls both RNA transcription and translation.
(b) Diagram of the assembled viral particle.
Both NP and L are required for viral RNA synthesis and transcription. As discussed above, the L polymerase is likely error-prone, generating a relatively high mutation frequency with the potential to produce viruses with novel host tropism. Although this has not been examined for the NW arenaviruses, the mutation rate for the OW arenavirus LCMV has been estimated to be on the order of 1–5 × 10−4 (67). In addition to synthesizing the viral RNAs, L has endonuclease activity, which is likely required for cap-snatching of the 5′–7-methylguanosine cap structure needed for translation from host cellular mRNAs (68–70). NP and Z are also believed to be important for mediating viral RNA synthesis and translation (71, 72). The cap-binding activity of the NW arenavirus NP appears to allow these viruses to translate their RNAs independently of the cellular eIF4E protein (73).
Once sufficient amounts of NP and Z are synthesized, capsid assembly occurs in the cytoplasm (Figures 2b and 3b). Although the process is not well studied for the NW arenaviruses, it is assumed that, similar to what occurs with the OW arenavirus LCMV, interaction between NP and viral RNAs allows their packaging within the capsid, while Z and NP binding causes virion core assembly (66). The viruses then bud from the plasma membrane, which has already incorporated the viral GP1 and GP2 glycoprotein subunits in association with the SSP glycoprotein subunit (Figure 2b) (74). The budding process is facilitated by interactions between the Z protein and cellular ESCRT proteins (75). The name arenaviruses derives from the sandy (arena in Greek) appearance of the capsid interior in electron micrographs (66) (Figure 3b). The particles that lend this sandy appearance are packaged host ribosomes; the role, if any, of these ribosomes in virus replication is not known.
NEW WORLD ARENAVIRUS PATHOGENESIS IN HUMANS AND ANIMAL MODELS
Like many viral hemorrhagic fevers, the early clinical signs of NW arenavirus infections include flu-like symptoms such as fever, malaise, headache, conjunctivitis, nausea, vomiting, and diarrhea, so NW arenavirus disease is difficult to diagnose in the absence of a known outbreak (76). These symptoms last approximately 7 to 10 days, at which point ~75% of patients show signs of improvement; the remaining 25–30% progress and develop bleeding and neurological disorders in addition to the fever. Patient survival is characterized by a robust neutralizing antibody response (11). Outright hemorrhaging from multiple mucosal surfaces is rare in NW arenavirus infection. More common are petechiae in oral mucosa and the chest, arms, and axillary region, as well as gingival bleeding. Untreated, severe disease is often accompanied by ataxia, mental confusion, increased irritability, and tremors and can progress to seizures and coma. The causes of these symptoms are not well understood. Endothelial cell and platelet dysfunction caused by direct viral infection have been documented in vitro, but whether this occurs in vivo is not known (77–79). Indeed, there is little or no evidence of overt endothelial damage in infected humans. In patients treated with convalescent serum, the mortality rate from Junín virus infection can be reduced to 1%; about 10% of patients receiving this treatment develop long-term neurological syndromes of unknown cause (11, 80).
While NW arenaviruses infect many different cell types in vitro and ex vivo, the exact targets during human infection are not clear. This lack of in vitro specificity may be linked to the high levels of TfR1 expression on cells in culture; TfR1 is highly upregulated in actively dividing cells because it is the major means by which cells take up iron (43). In contrast, most cells in vivo express low or no TfR1 unless activated or dividing. This includes lymphocytes and sentinel cells of the immune system as well as mucosal epithelia. There is evidence from both human and animal studies that macrophages and dendritic cells are major primary targets of NW arenavirus infection after airborne infection, likely those cells resident in the lung (81–86). Interestingly, this is reminiscent of another virus, the milkborne mouse mammary tumor virus, which also uses TfR1 for entry, and whose initial in vivo targets are dendritic cells located in the intestinal mucosa (87, 88). Infection of macrophages and dendritic cells may lead to aberrant cytokine production and bystander effects on endothelial and other cells affected by NW arenavirus infection (78). Indeed, it has been reported that humans infected with Junín virus exhibit high levels of type I interferon in serum, and primary human and mouse dendritic cells produce interferons and other cytokines upon infection with Junín or Machupo virus (51, 89–91).
There are a number of animal models that recapitulate some but not all of the pathologies associated with NW arenavirus infection in humans. Although OW mice (Mus musculus/Mus domesticus) are not efficiently infected with the clade B NW arenaviruses, both wild-type and mutant Mus species have been used extensively to examine pathology and immune response. Indeed, as mentioned above, the initial step in developing the Candid #1 vaccine was 44 passages of the pathogenic Romero strain of Junín virus in newborn mouse brain, until the initial associated pathologies were lost (53, 92). This resulted in only twelve amino acid differences between the Candid #1 vaccine and pathogenic strains of Junín virus—six in GP and six in L—as well as changes in the nucleotide sequence of both the coding and the 5′ and 3′ untranslated regions, but not the intergenic regions (52). Surprisingly, passaging of Junín virus in mice did not lead to a mouse-adapted virus. In fact, the amino acid changes in the Candid #1 GP appear to have made this strain more dependent on human TfR1 than the wild-type virus from which it was derived; however, these changes were not sufficient to completely attenuate the virus in a guinea pig or mouse model (93–95). Whether the amino acid changes in the L gene encoding the viral polymerase or alterations in RNA secondary structure due to nucleotide changes are also needed for full attenuation is not clear (52, 53). When the Candid #1 L gene was substituted for the pathogenic Romero L gene it did not attenuate viral replication kinetics in vitro, but whether this substitution would affect in vivo infection is not known (96).
Mice with genetic modifications, particularly in innate immune response pathway genes, are more susceptible to Junín virus infection. For example, we showed that an early step in infection was interaction of either the pathogenic Junín virus or Candid #1 GP with the pathogen-associated molecular pattern receptor Toll-like receptor 2 (TLR2), leading to the production of cytokines (97). As a result, knockout mice lacking this receptor sustained a more long-term infection with Candid #1 than did wild-type mice, due at least in part to diminished CD8 T cell responses. However, both wild-type and TLR2 knockout mice mounted strong humoral immune antibody responses that rapidly cleared virus. Interestingly, direct intracranial inoculation of Candid #1 into wild-type mice generated a TLR2-dependent cytokine response that did not diminish infection levels, suggesting that the virus could replicate in the brain unimpeded by the host innate immune response (97). In this model, both astrocytes and microglia were infected. Whether outright infection of the brain occurs in human infection, leading to the neurological syndromes, is not known.
Mice lacking type I and type III interferon receptors have also been shown to be highly susceptible to infection with pathogenic Junín virus and to rapidly succumb to infection, although these mice did not recapitulate the human disease—e.g., no hemorrhagic lesions were found in any tissue (98). Pathogenesis in humans, including endothelial dysfunction, may rely on high cytokine levels induced by the virus, and mice lacking interferon receptors would therefore lack this aspect of the infection. It is not known what receptor NW arenaviruses use in mice, although, as mentioned above, the L-type voltage-gated calcium channels can function as entry receptors in mouse cells (58). Because NW arenaviruses use alternate receptors in mice, the in vivo cell type tropism may differ from that in humans; this could also contribute to the altered pathogenic phenotype.
While the currently studied mouse models with immunological dysfunction may be useful to test antiviral therapeutics, because of the differences in disease outcome and the lack of a full immune response, they most likely are not useful for understanding the effects of in vivo infection. Guinea pigs, on the other hand, have been infected with a number of NW arenaviruses, including Junín virus and Machupo virus, and their disease more faithfully mimics that seen in humans, although infection is 100% fatal, likely due to the lack of a humoral antibody response to infection (11, 99–101). Other animal models, including hamsters, succumb to infection with NW arenaviruses that do not cause disease in humans, including the lineage A Pirital virus, an arenavirus isolated in western Venezuela during epidemiologic studies of Venezuelan hemorrhagic fever (102).
Nonhuman primates are susceptible to infection by NW arenaviruses, and prior to its use in humans, the Candid #1 vaccine was shown to protect rhesus macaques against lethal challenge with virulent Junín virus (103). Similarly, ribavirin was also shown to be effective in monkeys (104, 105). Although primates are clearly useful for testing therapeutics and vaccine strategies, the difficulty of working with ABSL-4 agents in large animals precludes their usefulness as an experimental model to study disease progression.
VIRAL PROTEINS THAT COUNTERACT THE HOST RESPONSE
A number of viral proteins have been implicated in suppressing the host immune response to NW arenavirus infection. Interferon-β treatment of Junín virus–infected primary mouse fibroblasts inhibited virus replication, suggesting that the virus must avoid the host innate immune response in order to establish persistence (91, 106, 107). Both the NP and Z proteins of NW arenaviruses have been shown to block type I interferon production, both in tissue culture and in animal models (108, 109). The NP proteins of a number of NW arenaviruses, including Junín, Machupo, White Water Arroyo, Tacaribe, and Latino viruses, have been shown to inhibit induction of interferon transcription by suppressing nuclear translocation of the transcription factor interferon regulatory factor 3 (IRF3) (110).
Although this has not been formally demonstrated for the pathogenic NW arenaviruses, the C-terminal segments of some arenaviruses’ NPs have been shown to have exonuclease activity that counteracts the host response, likely by preventing recognition of viral RNA by the host retinoic acid-inducible gene I (RIG-I)-like proteins (111–113). RIG-I-like proteins recognize foreign RNAs with unusual secondary structure or lacking 5′ caps (114, 115). However, the short dsRNAs with 5′ ppp-G overhangs found at the ends of arenavirus RNAs, as well as the capped viral mRNAs, are not efficiently recognized by RIG-I-like proteins (116, 117). The Junín virus Z protein is also thought to inhibit RIG-I-like receptor-dependent interferon production (108); activation of the RIG-I-like pathway likely occurs late in infection when high levels of viral RNA are present (51, 97). However, given the high levels of interferon and cytokine induced by virus in cultured cells, in animal models, and in humans, as discussed above, it is not entirely clear whether the virus uses these antihost mechanisms in in vivo infection.
NEW WORLD ARENAVIRUS TREATMENT
Although the Candid #1 vaccine has been highly effective in reducing the incidence of Argentine hemorrhagic fever, there are few treatments available for unvaccinated individuals or for those infected with the other hemorrhagic fever viruses found in South America (4–6). Antibody therapy is promising, given the success of Junín virus convalescent therapy in treating early viral infection. Indeed, a number of new monoclonal antibodies have been developed that show promise for treatment of Junín virus-infected individuals (118). However, it has never been formally tested whether convalescent serum would work to treat hemorrhagic fevers caused by the other NW arenaviruses, and as of yet, no similar monoclonals to the other viruses have been developed. Moreover, there is the potential for new NW arenavirus zoonoses, as discussed above. Whether it will be possible to develop pan-NW arenavirus antibodies to be used in treatment is not known.
Thus, new antiarenaviral therapies are still needed. Several promising leads have been developed over recent years, resulting from both small interfering RNA (siRNA) and drug high-throughput screens. Several groups have focused on inhibiting viral entry, and inhibitors to this step in infection have been developed, although only a few have been tested in mouse models as well as tissue culture cells (58, 119–122). Some of these inhibitors, such as gabapentin, which targets the voltage-gated calcium channel implicated as an entry receptor, are highly specific for NW arenaviruses, whereas others likely target a more general entry step because they are effective on multiple virus families (58, 122).
Ribavirin is a nucleoside analog that inhibits the viral L polymerase of all arenaviruses, with significant side effects in some individuals (17, 123). Several novel inhibitors of the polymerase, including favipiravir and A3, a pyrimidine biosynthesis inhibitor, have shown promise in guinea pig and tissue culture models, respectively (124, 125). A compound that inhibits virus budding has also been identified by in silico screening (126). Like some of the inhibitors of entry, these polymerase and exit molecules are likely to function as pan-virus inhibitors.
SUMMARY AND FUTURE DIRECTIONS
Although much has been learned in recent years about the biology of the NW pathogenic arenaviruses, they still remain of serious concern to human health because of the lack of effective vaccines and the relative dearth of antiviral pharmaceuticals. Additionally, there are a large number of these viruses residing in rodent hosts endemic to different regions of the Americas, with the potential to zoonose into humans. The development of tissue culture cell and animal models has provided much information about the biology of these viruses, but there is still much to be learned about how they cause disease in humans. A better understanding of NW arenavirus-host interactions and pathogenesis is needed for the development of therapeutics that can be used to treat infection.
FUTURE ISSUES.
Are novel mammarenaviruses generated only by error-prone viral polymerases, or can they also be generated by recombination after coinfection of a single host with two viruses?
What receptors allow the viruses to enter cells of nonhost species, and are additional subcellular or endosomal proteins involved in entry?
How can mouse models be generated that faithfully recapitulate human disease? Would expression of human TfR1 in a mouse render it susceptible to NW arenavirus-induced disease?
Which cells are infected in vivo, and how does this influence pathogenicity?
Is pathogenesis directly caused by viral infection, or does the host immune response also play a role?
ACKNOWLEDGMENTS
We thank the members of our lab for helpful discussions and Christian Cuevas for some of the figures. Work in our lab has been supported by National Institutes of Health grants U54 AI 057168 and R21 AI 112696.
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Stenglein MD, Sanders C, Kistler AL, Ruby JG, Franco JY, et al. 2012. Identification, characterization, and in vitro culture of highly divergent arenaviruses from boa constrictors and annulated tree boas: candidate etiological agents for snake inclusion body disease. mBio 3:e00180–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Radoshitzky SR, Bao Y, Buchmeier MJ, Charrel RN, Clawson AN, et al. 2015. Past, present, and future of arenavirus taxonomy. Arch. Virol 160:1851–74 [DOI] [PubMed] [Google Scholar]
- 3.Peralta LA, Laguens RP, Cossio PM, Sabattini MS, Maiztegui JI, Arana RM. 1979. Presence of viral particles in the salivary gland of Calomys musculinus infected with Junin virus by a natural route. Intervirology 11:111–16 [DOI] [PubMed] [Google Scholar]
- 4.Cajimat MN, Milazzo ML, Bradley RD, Fulhorst CF. 2012. Ocozocoautla de Espinosa virus and hemorrhagic fever, Mexico. Emerg. Infect. Dis 18:401–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abraham J, Kwong JA, Albarino CG, Lu JG, Radoshitzky SR, et al. 2009. Host-species transferrin receptor 1 orthologs are cellular receptors for nonpathogenic new world clade B arenaviruses. PLOS Pathog. 5:e1000358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai Y, Yu S, Mazur S, Dong L, Janosko K, et al. 2013. Nonhuman transferrin receptor 1 is an efficient cell entry receptor for Ocozocoautla de Espinosa virus. J. Virol 87:13930–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gomez RM, Jaquenod de Giusti C, Sanchez Vallduvi MM, Frik J, Ferrer MF, Schattner M. 2011. Junín virus. A XXI century update. Microbes Infect. 13:303–11 [DOI] [PubMed] [Google Scholar]
- 8.Charrel RN, de Lamballerie X. 2010. Zoonotic aspects of arenavirus infections. Vet. Microbiol 140:213–20 [DOI] [PubMed] [Google Scholar]
- 9.Salazar-Bravo J, Ruedas LA, Yates TL. 2002. Mammalian reservoirs of arenaviruses. Curr. Top. Microbiol. Immunol 262:25–63 [DOI] [PubMed] [Google Scholar]
- 10.Mills JN, Ellis BA, Childs JE, McKee KT Jr., Maiztegui JI, et al. 1994. Prevalence of infection with Junin virus in rodent populations in the epidemic area of Argentine hemorrhagic fever. Am. J. Trop. Med. Hygiene 51:554–62 [PubMed] [Google Scholar]
- 11.Enria DA, Briggiler AM, Sanchez Z. 2008. Treatment of Argentine hemorrhagic fever. Antivir. Res 78:132–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aguilar PV, Camargo W, Vargas J, Guevara C, Roca Y, et al. 2009. Reemergence of Bolivian hemorrhagic fever, 2007–2008. Emerg. Infect. Dis 15:1526–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.ProMED-mail. 2012. Venezuelan hemorrhagic fever—Venezuela: (PO). Arch. No. 20120313.1069429, March 3, ProMED-mail, Int. Soc. Infect. Dis., Brookline, MA: http://www.promedmail.org [Google Scholar]
- 14.Delgado S, Erickson BR, Agudo R, Blair PJ, Vallejo E, et al. 2008. Chapare virus, a newly discovered arenavirus isolated from a fatal hemorrhagic fever case in Bolivia. PLOS Pathog. 4:e1000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.US Dep. Health Hum. Serv. (HHS). 2007. HHS public health emergency medical countermeasure enterprise implementation plan for chemical, biological, radiological, and nuclear threats. US Dep. HHS, Off. Public Health Emerg. Med. Countermeas., Off. Assist. Secr. Prep. Response, Washington, DC: https://www.medicalcountermeasures.gov/barda/documents/phemce_implplan_041607final.pdf [Google Scholar]
- 16.Maiztegui JI. 1975. Clinical and epidemiological patterns of Argentine haemorrhagic fever. Bull. World Health Organ 52:567–75 [PMC free article] [PubMed] [Google Scholar]
- 17.Charrel RN, Coutard B, Baronti C, Canard B, Nougairede A, et al. 2011. Arenaviruses and hantaviruses: from epidemiology and genomics to antivirals. Antivir. Res 90:102–14 [DOI] [PubMed] [Google Scholar]
- 18.Fulhorst CF, Cajimat MN, Milazzo ML, Paredes H, de Manzione NM, et al. 2008. Genetic diversity between and within the arenavirus species indigenous to western Venezuela. Virology 378:205–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Enserink M 2000. New arenavirus blamed for recent deaths in California. Science 289:842–43 [DOI] [PubMed] [Google Scholar]
- 20.Irwin NR, Bayerlova M, Missa O, Martinkova N. 2012. Complex patterns of host switching in New World arenaviruses. Mol. Ecol 21:4137–50 [DOI] [PubMed] [Google Scholar]
- 21.Charrel RN, de Lamballerie X, Emonet S. 2008. Phylogeny of the genus Arenavirus. Curr. Opin. Microbiol 11:362–68 [DOI] [PubMed] [Google Scholar]
- 22.Chiappero MB, Gardenal CN. 2003. Restricted gene flow in Calomys musculinus (Rodentia, Muridae), the natural reservoir of Junin virus. J. Hered 94:490–95 [DOI] [PubMed] [Google Scholar]
- 23.Strauch TY. 1998. The history of Machupo virus in Bolivia: arenavirus hemorrhagic fever. Sloping Halls Rev. 1998:97–109 [Google Scholar]
- 24.Charrel RN, de Lamballerie X. 2003. Arenaviruses other than Lassa virus. Antivir. Res 57:89–100 [DOI] [PubMed] [Google Scholar]
- 25.Salas R, de Manzione N, Tesh RB, Rico-Hesse R, Shope RE, et al. 1991. Venezuelan haemorrhagic fever. Lancet 338:1033–36 [DOI] [PubMed] [Google Scholar]
- 26.Fulhorst CF, Ksiazek TG, Peters CJ, Tesh RB. 1999. Experimental infection of the cane mouse Zygodontomys brevicauda (family Muridae) with Guanarito virus (Arenaviridae), the etiologic agent of Venezuelan hemorrhagic fever. J. Infect. Dis 180:966–69 [DOI] [PubMed] [Google Scholar]
- 27.McLay L, Liang Y, Ly H. 2014. Comparative analysis of disease pathogenesis and molecular mechanisms of New World and Old World arenavirus infections. J. Gen. Virol 95:1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bowen MD, Peters CJ, Nichol ST. 1996. The phylogeny of New World (Tacaribe complex) arenaviruses. Virology 219:285–90 [DOI] [PubMed] [Google Scholar]
- 29.Buchmeier M, Adam E, Rawls WE. 1974. Serological evidence of infection by Pichinde virus among laboratory workers. Infect. Immun 9:821–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choe H, Jemielity S, Abraham J, Radoshitzky SR, Farzan M. 2011. Transferrin receptor 1 in the zoonosis and pathogenesis of New World hemorrhagic fever arenaviruses. Curr. Opin. Microbiol 14:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Childs JE, Mills JN, Glass GE. 1995. Rodent-borne hemorrhagic fever viruses: a special risk for mammalogists? J. Mammal 76:664–80 [Google Scholar]
- 32.Kilgore PE, Peters CJ, Mills JN, Rollin PE, Armstrong L, et al. 1995. Prospects for the control of Bolivian hemorrhagic fever. Emerg. Infect. Dis 1:97–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palacios G, Druce J, Du L, Tran T, Birch C, et al. 2008. A new arenavirus in a cluster of fatal transplant-associated diseases. N. Engl. J. Med 358:991–98 [DOI] [PubMed] [Google Scholar]
- 34.Schafer IJ, Miller R, Stroher U, Knust B, Nichol ST, et al. 2014. Notes from the field: a cluster of lymphocytic choriomeningitis virus infections transmitted through organ transplantation—Iowa, 2013. Morb. Mortal. Wkly. Rep 63:249. [PMC free article] [PubMed] [Google Scholar]
- 35.Inizan CC, Cajimat MN, Milazzo ML, Barragan-Gomez A, Bradley RD, Fulhorst CF. 2010. Genetic evidence for a Tacaribe serocomplex virus, Mexico. Emerg. Infect. Dis 16:1007–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zapata JC, Salvato MS. 2013. Arenavirus variations due to host-specific adaptation. Viruses 5:241–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sevilla N, de la Torre JC. 2006. Arenavirus diversity and evolution: quasispecies in vivo. Curr. Top. Microbiol. Immunol 299:315–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gaudin R, Kirchhausen T. 2015. Superinfection exclusion is absent during acute Junin virus infection of Vero and A549 cells. Sci. Rep 5:15990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stenglein MD, Jacobson ER, Chang LW, Sanders C, Hawkins MG, et al. 2015. Widespread recombination, reassortment, and transmission of unbalanced compound viral genotypes in natural arenavirus infections. PLOS Pathog. 11:e1004900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nunberg JH, York J. 2012. The curious case of arenavirus entry, and its inhibition. Viruses 4:83–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rojek JM, Lee AM, Nguyen N, Spiropoulou CF, Kunz S. 2008. Site 1 protease is required for proteolytic processing of the glycoproteins of the South American hemorrhagic fever viruses Junin, Machupo, and Guanarito. J. Virol 82:6045–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Radoshitzky SR, Abraham J, Spiropoulou CF, Kuhn JH, Nguyen D, et al. 2007. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 446:92–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ponka P, Lok CN. 1999. The transferrin receptor: role in health and disease. Int. J. Biochem. Cell Biol 31:1111–37 [DOI] [PubMed] [Google Scholar]
- 44.Shao J, Liang Y, Ly H. 2015. Human hemorrhagic fever causing arenaviruses: molecular mechanisms contributing to virus virulence and disease pathogenesis. Pathogens 4:283–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abraham J, Corbett KD, Farzan M, Choe H, Harrison SC. 2010. Structural basis for receptor recognition by New World hemorrhagic fever arenaviruses. Nat. Struct. Mol. Biol 17:438–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Radoshitzky SR, Kuhn JH, Spiropoulou CF, Albarino CG, Nguyen DP, et al. 2008. Receptor determinants of zoonotic transmission of New World hemorrhagic fever arenaviruses. PNAS 105:2664–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Demogines A, Abraham J, Choe H, Farzan M, Sawyer SL. 2013. Dual host-virus arms races shape an essential housekeeping protein. PLOS Biol. 11:e1001571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flanagan ML, Oldenburg J, Reignier T, Holt N, Hamilton GA, et al. 2008. New World clade B arenaviruses can use transferrin receptor 1 (TfR1)-dependent and -independent entry pathways, and glycoproteins from human pathogenic strains are associated with the use of TfR1. J. Virol 82:938–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zong M, Fofana I, Choe H. 2014. Human and host species transferrin receptor 1 use by North American arenaviruses. J. Virol 88:9418–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Casey JL, Koeller DM, Ramin VC, Klausner RD, Harford JB. 1989. Iron regulation of transferrin receptor mRNA levels requires iron-responsive elements and a rapid turnover determinant in the 3′ untranslated region of the mRNA. EMBO J. 8:3693–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cuevas CD, Lavanya M, Wang E, Ross SR. 2011. Junín virus infects mouse cells and induces innate immune responses. J. Virol 85:11058–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goni SE, Iserte JA, Ambrosio AM, Romanowski V, Ghiringhelli PD, Lozano ME. 2006. Genomic features of attenuated Junín virus vaccine strain candidate. Virus Genes 32:37–41 [DOI] [PubMed] [Google Scholar]
- 53.Goni SE, Iserte JA, Stephan BI, Borio CS, Ghiringhelli PD, Lozano ME. 2010. Molecular analysis of the virulence attenuation process in Junín virus vaccine genealogy. Virus Genes 40:320–28 [DOI] [PubMed] [Google Scholar]
- 54.Jemielity S, Wang JJ, Chan YK, Ahmed AA, Li W, et al. 2013. TIM-family proteins promote infection of multiple enveloped viruses through virion-associated phosphatidylserine. PLOS Pathog. 9:e1003232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martinez MG, Bialecki MA, Belouzard S, Cordo SM, Candurra NA, Whittaker GR. 2013. Utilization of human DC-SIGN and L-SIGN for entry and infection of host cells by the New World arenavirus, Junín virus. Biochem. Biophys. Res. Commun 441:612–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martin VK, Droniou-Bonzom ME, Reignier T, Oldenburg JE, Cox AU, Cannon PM. 2010. Investigation of clade B New World arenavirus tropism by using chimeric GP1 proteins. J. Virol 84:1176–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grant A, Seregin A, Huang C, Kolokoltsova O, Brasier A, et al. 2012. Junín virus pathogenesis and virus replication. Viruses 4:2317–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lavanya M, Cuevas CD, Thomas M, Cherry S, Ross SR. 2013. siRNA screen for genes that affect Junín virus entry uncovers voltage-gated calcium channels as a therapeutic target. Sci. Transl. Med 5:204ra131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oldenburg J, Reignier T, Flanagan ML, Hamilton GA, Cannon PM. 2007. Differences in tropism and pH dependence for glycoproteins from the clade B1 arenaviruses: implications for receptor usage and pathogenicity. Virology 364:132–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Castilla V, Palermo LM, Coto CE. 2001. Involvement of vacuolar proton ATPase in Junin virus multiplication. Arch. Virol 146:251–63 [DOI] [PubMed] [Google Scholar]
- 61.York J, Nunberg JH. 2006. Role of the stable signal peptide of Junín arenavirus envelope glycoprotein in pH-dependent membrane fusion. J. Virol 80:7775–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rojek JM, Sanchez AB, Nguyen NT, de la Torre JC, Kunz S. 2008. Different mechanisms of cell entry by human-pathogenic Old World and New World arenaviruses. J. Virol 82:7677–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martinez MG, Cordo SM, Candurra NA. 2007. Characterization of Junín arenavirus cell entry. J. Gen. Virol 88:1776–84 [DOI] [PubMed] [Google Scholar]
- 64.Roldan JS, Martinez MG, Forlenza MB, Whittaker GR, Candurra NA. 2016. Human transferrin receptor triggers an alternative Tacaribe virus internalization pathway. Arch. Virol 161:353–63 [DOI] [PubMed] [Google Scholar]
- 65.Jae LT, Raaben M, Herbert AS, Kuehne AI, Wirchnianski AS, et al. 2014. Lassa virus entry requires a trigger-induced receptor switch. Science 344:1506–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Buchmeier MJ, de la Torre JC, Peters CJ. 2007. Arenaviridae: the viruses and their replication In Fields Virology, ed. Knipe DL, Howley PM, pp. 1791–828. Philadelphia: Lippincott Williams & Wilkins [Google Scholar]
- 67.Grande-Perez A, Martin V, Moreno H, de la Torre JC. 2016. Arenavirus quasispecies and their biological implications. Curr. Top. Microbiol. Immunol 392:231–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Raju R, Raju L, Hacker D, Garcin D, Compans R, Kolakofsky D. 1990. Nontemplated bases at the 5′ ends of Tacaribe virus mRNAs. Virology 174:53–59 [DOI] [PubMed] [Google Scholar]
- 69.Kranzusch PJ, Schenk AD, Rahmeh AA, Radoshitzky SR, Bavari S, et al. 2010. Assembly of a functional Machupo virus polymerase complex. PNAS 107:20069–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morin B, Coutard B, Lelke M, Ferron F, Kerber R, et al. 2010. The N-terminal domain of the arenavirus L protein is an RNA endonuclease essential in mRNA transcription. PLOS Pathog. 6:e1001038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cornu TI, de la Torre JC. 2001. RING finger Z protein of lymphocytic choriomeningitis virus (LCMV) inhibits transcription and RNA replication of an LCMV S-segment minigenome. J. Virol 75:9415–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.D’Antuono A, Loureiro ME, Foscaldi S, Marino-Buslje C, Lopez N. 2014. Differential contributions of Tacaribe arenavirus nucleoprotein N-terminal and C-terminal residues to nucleocapsid functional activity. J. Virol 88:6492–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Linero F, Welnowska E, Carrasco L, Scolaro L. 2013. Participation of eIF4F complex in Junin virus infection: Blockage of eIF4E does not impair virus replication. Cell. Microbiol 15:1766–82 [DOI] [PubMed] [Google Scholar]
- 74.Groseth A, Wolff S, Strecker T, Hoenen T, Becker S. 2010. Efficient budding of the Tacaribe virus matrix protein Z requires the nucleoprotein. J. Virol 84:3603–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wolff S, Ebihara H, Groseth A. 2013. Arenavirus budding: a common pathway with mechanistic differences. Viruses 5:528–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kerber R, Reindl S, Romanowski V, Gomez RM, Ogbaini-Emovon E, et al. 2015. Research efforts to control highly pathogenic arenaviruses: a summary of the progress and gaps. J. Clin. Virol 64:120–27 [DOI] [PubMed] [Google Scholar]
- 77.Lander HM, Grant AM, Albrecht T, Hill T, Peters CJ. 2014. Endothelial cell permeability and adherens junction disruption induced by Junín virus infection. Am. J. Trop. Med. Hygiene 90:993–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pozner RG, Ure AE, Jaquenod de Giusti C, D’Atri LP, Italiano JE, et al. 2010. Junín virus infection of human hematopoietic progenitors impairs in vitro proplatelet formation and platelet release via a bystander effect involving type I IFN signaling. PLOS Pathog. 6:e1000847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gomez RM, Pozner RG, Lazzari MA, D’Atri LP, Negrotto S, et al. 2003. Endothelial cell function alteration after Junin virus infection. Thromb. Haemost 90:326–33 [DOI] [PubMed] [Google Scholar]
- 80.Moraz ML, Kunz S. 2011. Pathogenesis of arenavirus hemorrhagic fevers. Exp. Rev. Anti-Infect. Ther 9:49–59 [DOI] [PubMed] [Google Scholar]
- 81.Gonzalez PH, Cossio PM, Arana R, Maiztegui JI, Laguens RP. 1980. Lymphatic tissue in Argentine hemorrhagic fever. Pathologic features. Arch. Pathol. Lab. Med 104:250–54 [PubMed] [Google Scholar]
- 82.Gonzalez PH, Lampuri JS, Coto CE, Laguens RP. 1982. In vitro infection of murine macrophages with Junin virus. Infect. Immun 35:356–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Laguens M, Chambo JG, Laguens RP. 1983. In vivo replication of pathogenic and attenuated strains of Junin virus in different cell populations of lymphatic tissue. Infect. Haemost 41:1279–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ambrosio AM, Enria DA, Maiztegui JI. 1986. Junin virus isolation from lympho-mononuclear cells of patients with Argentine hemorrhagic fever. Intervirology 25:97–102 [DOI] [PubMed] [Google Scholar]
- 85.Ambrosio M, Vallejos A, Saavedra C, Maiztegui JI. 1990. Junin virus replication in peripheral blood mononuclear cells of patients with Argentine haemorrhagic fever. Acta Virol. 34:58–63 [PubMed] [Google Scholar]
- 86.Maiztegui JI, Laguens RP, Cossio PM, Casanova MB, de la Vega MT, et al. 1975. Ultrastructural and immunohistochemical studies in five cases of Argentine hemorrhagic fever. J. Infect. Dis 132:35–53 [DOI] [PubMed] [Google Scholar]
- 87.Burzyn D, Rassa JC, Kim D, Nepomnaschy I, Ross SR, Piazzon I. 2004. Toll-like receptor 4-dependent activation of dendritic cells by a retrovirus. J. Virol 78:576–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Courreges MC, Burzyn D, Nepomnaschy I, Piazzon I, Ross SR. 2007. Critical role of dendritic cells in mouse mammary tumor virus in vivo infection. J. Virol 81:3769–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Levis SC, Saavedra MC, Ceccoli C, Falcoff E, Feuillade MR, et al. 1984. Endogenous interferon in Argentine hemorrhagic fever. J. Infect. Dis 149:428–33 [DOI] [PubMed] [Google Scholar]
- 90.Levis SC, Saavedra MC, Ceccoli C, Feuillade MR, Enria DA, et al. 1985. Correlation between endogenous interferon and the clinical evolution of patients with Argentine hemorrhagic fever. J. Interferon Res 5:383–89 [DOI] [PubMed] [Google Scholar]
- 91.Huang C, Kolokoltsova OA, Yun NE, Seregin AV, Ronca S, et al. 2015. Highly pathogenic New World and Old World human arenaviruses induce distinct interferon responses in human cells. J. Virol 89:7079–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Enria DA, Barrera Oro JG. 2002. Junin virus vaccines. Curr. Top. Microbiol. Immunol 263:239–61 [DOI] [PubMed] [Google Scholar]
- 93.Droniou-Bonzom ME, Reignier T, Oldenburg JE, Cox AU, Exline CM, et al. 2011. Substitutions in the glycoprotein (GP) of the Candid#1 vaccine strain of Junin virus increase dependence on human transferrin receptor 1 for entry and destabilize the metastable conformation of GP. J. Virol 85:13457–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Seregin AV, Yun NE, Miller M, Aronson J, Smith JK, et al. 2015. The glycoprotein precursor gene of Junin virus determines the virulence of the Romero strain and the attenuation of the Candid #1 strain in a representative animal model of Argentine hemorrhagic fever. J. Virol 89:5949–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Koma T, Huang C, Aronson JF, Walker AG, Miller M, et al. 2016. The ectodomain of glycoprotein from the Candid#1 vaccine strain of Junin virus rendered Machupo virus partially attenuated in mice lacking IFN-αβ/γ receptor. PLOS Negl. Trop. Dis 10:e0004969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Emonet SF, Seregin AV, Yun NE, Poussard AL, Walker AG, et al. 2011. Rescue from cloned cDNAs and in vivo characterization of recombinant pathogenic Romero and live-attenuated Candid #1 strains of Junin virus, the causative agent of Argentine hemorrhagic fever disease. J. Virol 85:1473–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cuevas CD, Ross SR. 2014. Toll-like receptor 2-mediated innate immune responses against Junín virus in mice lead to antiviral adaptive immune responses during systemic infection and do not affect viral replication in the brain. J. Virol 88:7703–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kolokoltsova OA, Yun NE, Poussard AL, Smith JK, Smith JN, et al. 2010. Mice lacking alpha/beta and gamma interferon receptors are susceptible to Junin virus infection. J. Virol 84:13063–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yun NE, Linde NS, Dziuba N, Zacks MA, Smith JN, et al. 2008. Pathogenesis of XJ and Romero strains of Junin virus in two strains of guinea pigs. Am. J. Trop. Med. Hygiene 79:275–82 [PMC free article] [PubMed] [Google Scholar]
- 100.Laguens RM, Avila MM, Samoilovich SR, Weissenbacher MC, Laguens RP. 1983. Pathogenicity of an attenuated strain (XJCl3) of Junin virus. Morphological and virological studies in experimentally infected guinea pigs. Intervirology 20:195–201 [DOI] [PubMed] [Google Scholar]
- 101.Kenyon RH, Green DE, Eddy GA, Peters CJ. 1986. Treatment of Junin virus-infected guinea pigs with immune serum: development of late neurological disease. J. Med. Virol 20:207–18 [DOI] [PubMed] [Google Scholar]
- 102.Xiao SY, Zhang H, Yang Y, Tesh RB. 2001. Pirital virus (Arenaviridae) infection in the Syrian golden hamster, Mesocricetus auratus: a new animal model for arenaviral hemorrhagic fever. Am. J. Trop. Med. Hygiene 64:111–18 [DOI] [PubMed] [Google Scholar]
- 103.McKee KT Jr., Oro JG, Kuehne AI, Spisso JA, Mahlandt BG. 1992. Candid No. 1 Argentine hemorrhagic fever vaccine protects against lethal Junin virus challenge in rhesus macaques. Intervirology 34:154–63 [DOI] [PubMed] [Google Scholar]
- 104.Weissenbacher MC, Calello MA, Merani MS, McCormick JB, Rodriguez M. 1986. Therapeutic effect of the antiviral agent ribavirin in Junín virus infection of primates. J. Med. Virol 20:261–67 [DOI] [PubMed] [Google Scholar]
- 105.Weissenbacher MC, Avila MM, Calello MA, Merani MS, McCormick JB, Rodriguez M. 1986. Effect of ribavirin and immune serum on Junin virus-infected primates. Med. Microbiol. Immunol 175:183–86 [DOI] [PubMed] [Google Scholar]
- 106.Huang C, Kolokoltsova OA, Yun NE, Seregin AV, Poussard AL, et al. 2012. Junín virus infection activates the type I interferon pathway in a RIG-I-dependent manner. PLOS Negl. Trop. Dis 6:e1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Huang C, Walker AG, Grant AM, Kolokoltsova OA, Yun NE, et al. 2014. Potent inhibition of Junín virus infection by interferon in murine cells. PLOS Negl. Trop. Dis 8:e2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xing J, Ly H, Liang Y. 2015. The Z proteins of pathogenic but not nonpathogenic arenaviruses inhibit RIG-I-like receptor-dependent interferon production. J. Virol 89:2944–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fan L, Briese T, Lipkin WI. 2010. Z proteins of New World arenaviruses bind RIG-I and interfere with type I interferon induction. J. Virol 84:1785–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rodrigo WW, Ortiz-Riano E, Pythoud C, Kunz S, de la Torre JC, Martinez-Sobrido L. 2012. Arenavirus nucleoproteins prevent activation of nuclear factor kappa B. J. Virol 86:8185–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hastie KM, Kimberlin CR, Zandonatti MA, MacRae IJ, Saphire EO. 2011. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3′ to 5′ exonuclease activity essential for immune suppression. PNAS 108:2396–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Reynard S, Russier M, Fizet A, Carnec X, Baize S. 2014. Exonuclease domain of the Lassa virus nucleoprotein is critical to avoid RIG-I signaling and to inhibit the innate immune response. J. Virol 88:13923–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Huang Q, Shao J, Lan S, Zhou Y, Xing J, et al. 2015. In vitro and in vivo characterizations of Pichinde viral nucleoprotein exoribonuclease functions. J. Virol 89:6595–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Goubau D, Deddouche S, Reis e Sousa C. 2013. Cytosolic sensing of viruses. Immunity 38:855–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Loo Y, Gale M. 2011. Immune signaling by RIG-I-like receptors. Immunity 34:680–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Marq JB, Hausmann S, Veillard N, Kolakofsky D, Garcin D. 2011. Short double-stranded RNAs with an overhanging 5′ ppp-nucleotide, as found in arenavirus genomes, act as RIG-I decoys. J. Biol. Chem 286:6108–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Marq JB, Kolakofsky D, Garcin D. 2010. Unpaired 5′ ppp-nucleotides, as found in arenavirus double-stranded RNA panhandles, are not recognized by RIG-I. J. Biol. Chem 285:18208–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zeitlin L, Geisbert JB, Deer DJ, Fenton KA, Bohorov O, et al. 2016. Monoclonal antibody therapy for Junin virus infection. PNAS 113:4458–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lee AM, Rojek JM, Spiropoulou CF, Gundersen AT, Jin W, et al. 2008. Unique small molecule entry inhibitors of hemorrhagic fever arenaviruses. J. Biol. Chem 283:18734–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Larson RA, Dai D, Hosack VT, Tan Y, Bolken TC, et al. 2008. Identification of a broad-spectrum arenavirus entry inhibitor. J. Virol 82:10768–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Iwasaki M, Ngo N, de la Torre JC. 2013. Sodium hydrogen exchangers contribute to arenavirus cell entry. J. Virol 88:643–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chou YY, Cuevas C, Carocci M, Stubbs SH, Ma M, et al. 2016. Identification and characterization of a novel broad-spectrum virus entry inhibitor. J. Virol 90:4494–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.McCormick JB, King IJ, Webb PA, Scribner CL, Craven RB, et al. 1986. Lassa fever. Effective therapy with ribavirin. N. Engl. J. Med 314:20–26 [DOI] [PubMed] [Google Scholar]
- 124.Furuta Y, Gowen BB, Takahashi K, Shiraki K, Smee DF, Barnard DL. 2013. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antivir. Res 100:446–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gowen BB, Juelich TL, Sefing EJ, Brasel T, Smith JK, et al. 2013. Favipiravir (T-705) inhibits Junín virus infection and reduces mortality in a guinea pig model of Argentine hemorrhagic fever. PLOS Negl. Trop. Dis 7:e2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lu J, Han Z, Liu Y, Liu W, Lee MS, et al. 2014. A host-oriented inhibitor of Junin Argentine hemorrhagic fever virus egress. J. Virol 88:4736–43 [DOI] [PMC free article] [PubMed] [Google Scholar]