

Arising from Niknafs et al. Nature Communications 10.1038/s41467-019-13100-w (2019)
Niknafs et al. describe evolutionary trajectories in pancreatic cancer using mouse models with engineered KrasG12D and Trp53R172H mutations (KPC model). As an additional aspect, the study reports frequent homozygous deletions at the Nlrp1 locus, which are interpreted as a somatic driver event in pancreatic cancer. We observed that the origin of this Nlrp1 alteration is strain-specific germline variation, having profound impact on the interpretation of its biological relevance. Beyond this specific locus, we show that strain-specific germline variation is a general confounder of genome analyses in mouse models of cancer.
In line with Niknafs et al.1, we also observed frequent changes at the Nlrp1 locus in our own cohorts of KPC mice. However, Nlrp1 changes were invariably associated with a series of unusual characteristics. First, the deletion encompasses the exact same genomic region on chromosome 11 in all affected cancers (Fig. 1a, d). These identical breakpoints in independent cancers do not reflect the typical “stepped” pattern of somatic losses at tumor suppressor loci (Fig. 1a shows such a pattern of overlaid copy number profiles). Second, the exact same deletion can also be found in other cancer entities induced in Trp53 mutant mice, as revealed in our own studies (pancreatic cancer, osteosarcoma, lung adenocarcinoma, cutaneous squamous cell carcinoma) as well as through re-analysis of publicly available datasets (lymphomas, hepatocellular carcinomas2–4). Somatic acquisition of absolutely identical homozygous deletions in different cancers, models, entities, and laboratories is rather unlikely. Third, we observed Nlrp1 locus alterations only in mouse models with engineered mutant or floxed Trp53 alleles (Trp53ENG). More specifically, Nlrp1 locus alterations were only detected in heterozygous Trp53ENG tumors, but never in mice, which were crossed to Trp53ENG homozygosity (n = 0/27, own cohort).
Fig. 1. Strain-specific haplotype variation at the Nlrp1 locus in 129S and C57BL/6J mice.
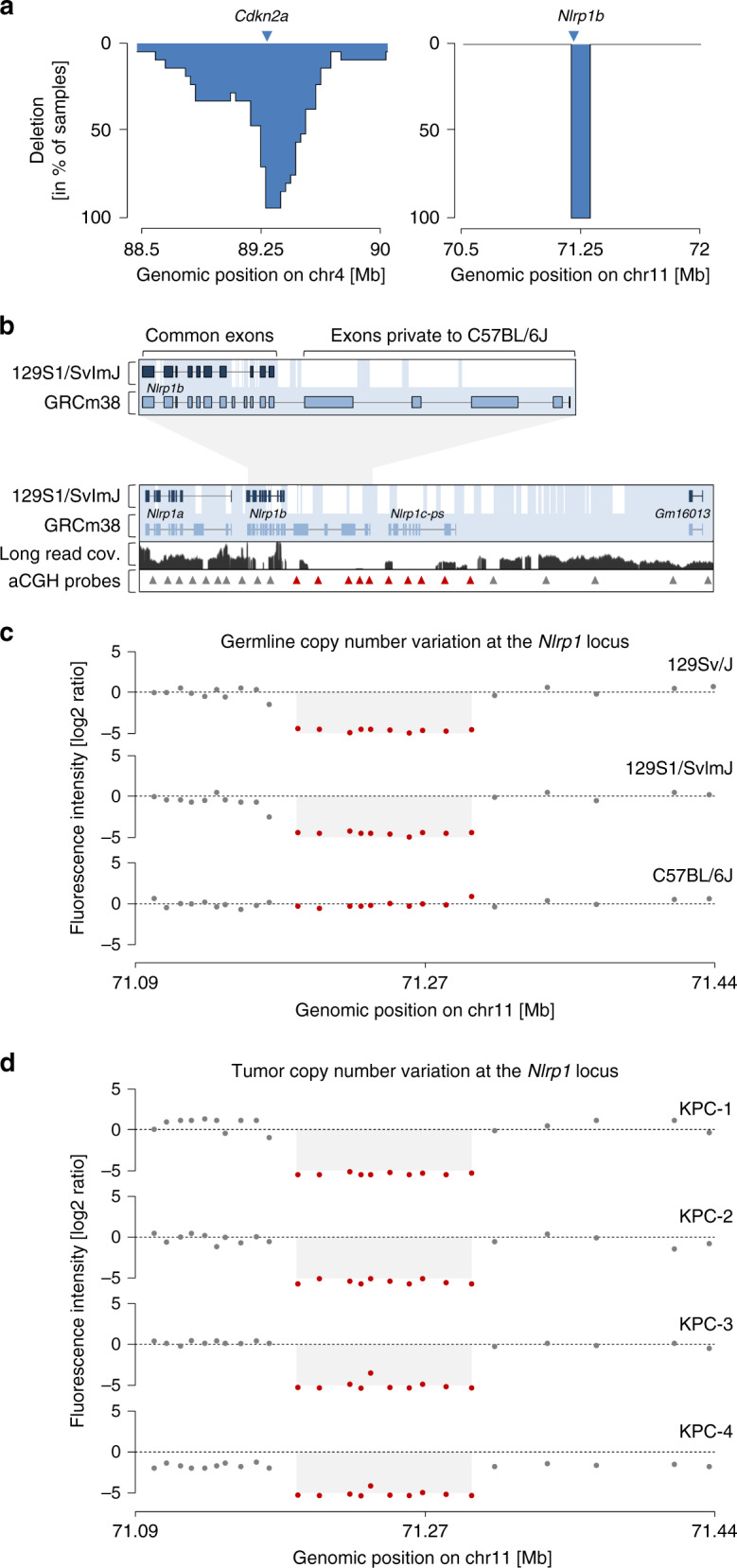
a Overlay of homozygous somatic deletions at the “classic” tumor suppressor locus Cdkn2a is shown (n = 21 KC mice; for each tumor the homozygously deleted region is shown; data from ref. 13). For comparison, overlay of copy number alterations in primary pancreatic cancer cell cultures with Nlrp1 locus deletions, as detected by aCGH (exemplary KPC mice are shown, n = 4; see also details of individual tumors in Fig. 1d). Y axis, frequency of genomic regions homozygously deleted in the cohort. b Strain-specific haplotype diversity at the mouse Nlrp1 locus on chr11. Genomic alignment of the Nlrp1129S1/SvImJ locus to Nlrp1C57BL/6J (GRCm38 mouse reference genome). Sequence homology of C57BL/6J and 129S1/SvImJ is highlighted in light blue. Genomic regions without homology in 129S1/SvImJ are depicted in white (data adapted from ref. 5). Upper panel: zoom-in of Nlrp1b (exon/intron lengths not proportional to genomic distances). Lower panel, middle row: read coverage of the Nlrp1C57BL/6J locus in a KPC mouse with Trp53ENG, as detected by nanopore long-read sequencing. Lower panel, bottom row: genomic position of oligonucleotide probes of the Agilent SurePrint G3 Mouse CGH 240 K array. Red arrowheads, aCGH probes located within the Nlrp1 locus alteration described by Niknafs et al.1 (compare to Fig. 1c, d). c Germline CNV profiles at the Nlrp1 locus in three inbred mouse strains. DNA from indicated strains was hybridized against DNA from C57BL/6J. Red dots, aCGH probes within the Nlrp1 locus alteration (data from ref. 6). d Recurrent Nlrp1 locus alterations in primary pancreatic cancer cell cultures with identical genomic boundaries (exemplary KPC mice are shown). The Agilent SurePrint G3 Mouse CGH 240 K array was used similar to Niknafs et al.1 and Fig. 1c. Red dots, aCGH probes within the Nlrp1 locus alteration.
These seeming inconsistencies prompted us to examine the locus in detail. Humans have only one gene at this locus, NLRP1. In the mouse reference genome (based on strain C57BL/6J) the Nlrp1 locus comprises three related genes: Nlrp1a, Nlrp1b, and Nlrp1c-ps. Importantly, Trp53 and the Nlrp1 locus are separated by only 1.5 Mb on chromosome 11, causing tight genetic linkage between both loci. The genetically engineered Trp53 allele was generated on a 129S-related background (Trp53ENG-129S). We examined the Nlrp1 locus in 129S genomes (Nlrp1129S) and found that parts of the C57BL/6J sequence have no genomic alignment in the 129S reference assembly5 (Fig. 1b). We also analyzed array comparative genomic hybridization (aCGH) data from a study examining germline copy number variation (CNV) between different mouse strains6. We found that genomes of 129S-related mouse strains contain homozygous deletions of the Nlrp1 locus that were identical to Nlrp1 locus deletions in KPC tumors (Fig. 1c, d) and all other cancer entities mentioned above. Using nanopore long-read sequencing (Fig. 1b), we confirmed the presence of the strain-specific Nlrp1129S locus variant in the engineered Trp53R172H mouse line7 used by us (and by Niknafs et al.1). Thus, the origin of the Nlrp1 locus deletion is not somatic acquisition followed by selection during tumor evolution, but a pre-existing strain-specific germline variant.
After identifying that the Trp53ENG-129S allele is genetically linked to the Nlrp1129S locus (Trp53ENG-129S;Nlrp1129S), we interrogated the status of the second allele in the germline. This consideration is important, because we kept the mice on a mixed 129S;C57BL/6J background (similar to Niknafs et al.1, who used the same Trp53ENG-129S allele on a mixed genetic background). Our analysis revealed two important findings, which explain the genesis of Nlrp1 locus deletions in cancer: First, Nlrp1 locus deletions in cancer were only observed in mice, whose second haplotype is of C57BL/6J origin (Trp53WT;Nlrp1C57BL/6J, Fig. 2a). Second, this C57BL/6J haplotype is lost in the tumor through copy-neutral loss of heterozygosity (CN-LOH). Mechanistically, this reflects selective pressure to lose wild-type Trp53 during tumor progression, which almost invariably occurs in the KPC model8.
Fig. 2. Genetic linkage of strain-specific variants to cancer genes undergoing LOH confound cancer genome analyses.

a, b Trp53;Nlrp1 haplotype reconstruction by WES-based SNP analysis in germline and tumors of KPC mice (a: KPC-5, b: KPC-6). Genomic regions with heterozygous SNPs contain two distinct alleles: one C57BL/6J- and one 129S-specific haplotype (SNP frequencies: ~0.5; light blue background in SNP-plots). Conversely, regions with SNP frequencies of ~1.0 are pure 129S. Regions without values perfectly match the C57BL/6J reference genome (homozygous C57BL/6J). a In the germline, heterozygous SNPs confirm presence of two haplotypes: (1) Trp53ENG-129S;Nlrp1129S (engineered Trp53R172H allele; strain-specific Nlrp1129S variant) and (2) Trp53WT;Nlrp1C57BL/6J. In the tumor, the Trp53WT;Nlrp1C57BL/6J haplotype is lost through CN-LOH (reflecting selective pressure to lose Trp53WT). Homozygosity of the Trp53ENG-129S;Nlrp1129S haplotype in the tumor manifests as a Nlrp1 locus deletion when compared to germline (right; CNV plot based on WES with Nlrp1 locus zoom-in). b In the germline, the Trp53;Nlrp1 haplotype is S129-derived on both copies of chr11. The Nlrp1129S variant is already homozygous in the germline. CNV analyses relying on tumor/germline comparisons thus fail to detect the Nlrp1 alteration in the tumor (right; CNV plot based on WES with Nlrp1 locus zoom-in). c Germline SNP analysis of mouse KPC-7 backcrossed to C57BL/6J for fourteen generations. High SNP densities persist in genomic proximity of engineered alleles. d Germline CNV profiles at the Skint locus in three inbred mouse strains as compared to C57BL/6J. Red dots, aCGH probes within Skint locus alteration (data from ref. 6). e Upper panel: Skint locus CNV profile in primary pancreatic cancer cell culture (compare to germline Skint alterations in Fig. 2d). Lower panels: WES-based Cdkn2a;Skint haplotype reconstruction as in Fig. 2a, b. In the germline, heterozygous SNPs confirm presence of two haplotypes: (1) Cdkn2a;SkintC57BL/6J and (2) Cdkn2a;Skint129S. During tumor evolution, the Cdkn2a locus is first somatically deleted (Cdkn2aΔ) on the chromosome carrying the Skint129S variant (Cdkn2aΔ;Skint129S) followed by CN-LOH of the Cdkn2aΔ;Skint129S allele, reflecting selective pressure to lose Cdkn2aWT. Homozygosity of Skint129S in the tumor manifests as a Skint locus deletion when compared to germline (right; CNV plot based on WES with Skint locus zoom-in).
Because detection of CNVs is based on the comparison of tumor to germline, these findings explain: (1) why a single somatic event (loss of Trp53WT;Nlrp1C57BL/6J through CN-LOH) manifests as a focal homozygous deletion (Fig. 2a), (2) why the Nlrp1129S variant is not detected (despite being present) in tumors from mice crossed to Trp53ENG homozygosity (Trp53ENG-129S;Nlrp1129S already homozygous in the germline) or other more rare scenarios (Fig. 2b) and (3) why the coordinates of the Nlrp1129S variant are identical in tumors across models and entities (see Fig. 1a, d).
Is the Nlrp1129S variant biologically relevant and thus selected for during tumor evolution? Several lines of evidence suggest that interpretation of the Nlrp1129S variant as a cancer driver requires further functional validation: First, the pre-existing Nlrp1129S variant does not arise through somatic mutation (it is already present in the germline of 129S-related mouse strains). LOH at the locus is explained by the tight genetic linkage of Nlrp1129S to Trp53ENG-129S. Second, in our own cohorts of several hundred KrasG12D-driven mouse pancreatic cancers, there are no Nlrp1 locus deletions without associated LOH of mutant Trp53ENG. Third, large transposon-based pancreatic cancer gene discovery screens performed on a mixed 129S;C57BL/6J genetic background did not find common insertions in the wild-type, hemizygous Nlrp1C57BL/6J allele9–11. Fourth, in human pancreatic cancer, isolated, deep NLRP1 deletions that spare TP53, have been only reported in 1/109 cases in one study (PDA_078 in UTSW cohort12; cbioportal.org). In fact, re-analysis of raw-sequencing data with manual inspection of the NLRP1 locus in PDA_078 did not confirm the presence of an isolated NLRP1 deletion in our hands.
Beyond the Nlrp1 locus, our findings highlight an important—and so far underappreciated—confounder of mouse cancer genome analysis. The basis of this confounder is the widespread use of inbred mice with extensive inter-strain haplotype variation5. Typical experimental cohorts are derived from few generations of crosses involving few different inbred strains, with inheritance of large strain-specific haplotype blocks. Thus, when genetically linked to a cancer gene undergoing LOH, any strain-specific deletion/insertion variant will appear as a somatically acquired CNV in related tumors. Importantly, this CNV will be hugely recurrent in the cohort. Backcrossing of mice/alleles to a single genetic background can substantially reduce the amount of strain-specific germline haplotypes. However, this confounder can persist in direct genomic proximity to engineered alleles (which are bred/genotyped for) even after extensive backcrossing (Fig. 2c).
The general relevance of the considerations raised in our commentary is evident at many strain-specific loci linked to LOH of a cancer driver. The driver can be an engineered allele (like Trp53ENG), but also a somatically acquired cancer gene alteration. For example, in mouse pancreatic cancer cohorts we observed Skint locus deletions with equivalent characteristics to Nlrp1 alterations: (1) identical genomic deletion coordinates in unrelated tumors, (2) corresponding strain-specific locus variation in the germline, and (3) genetic linkage to a cancer gene (Cdkn2a) undergoing LOH in the tumor. As for Nlrp1, the Skint locus deletion is present in the germline of 129S-related mouse strains but not in C57BL/6J (Fig. 2d, e).
In conclusion, our observations highlight the importance of considering sequence diversity of inbred mouse strains when analyzing cancer genomes in typical experimental settings/cohorts. With sequencing costs dropping, genomic analyses in mouse models of human cancer are increasing at a rapid pace. So far, strain-specific germline variants have obtained little attention in mouse cancer genome sequencing studies. Their potential interpretation as somatically acquired cancer drivers is a common problem, reinforcing the need to raise awareness of this confounder.
Methods
Datasets and data analyses
Data and conclusions of this commentary are based on the systematic genetic analysis of our own cohort of over 1000 mouse cancers derived from a variety of distinct mouse models covering different cancer entities. This large cohort of mouse cancers comprise primary cell cultures as well as tissues and was characterized by a series of methods, including array comparative genomic hybridization (aCGH), whole-exome sequencing (WES), long-read sequencing and/or quantitative insertion-site sequencing (QiSeq). Animal experiments, primary mouse pancreatic cancer culture preparation, and maintenance, gDNA isolation were performed as described in detail before13,14. Genome-wide identification of transposon integration sites in transposon-based mouse models was assessed by using QiSeq and custom bioinformatic analyses as described previously11,15. Animal studies were ethically approved by the Institutional Animal Care and Use Committees (IACUC) of Technische Universität München, Regierung von Oberbayern and the UK Home Office.
Whole-exome sequencing
Raw WES data of mouse pancreatic cancers from our cohorts and from Niknafs et al.1 were analyzed by using a workflow adapted to the analysis of mouse cancer sequencing data which we described elsewhere in detail14 (source code: https://github.com/roland-rad-lab/MoCaSeq). In brief, reads were trimmed using Trimmomatic 0.38. BWA-MEM 0.7.17 was used to align reads to the mouse reference genome GRCm38.p6 (with alternate contigs). Picard 2.20.0 and GATK 4.1.0.0 were used for postprocessing (CleanSam, MarkDuplicates, BaseRecalibrator). For LOH analyses from WES data, germline SNP calling was performed with Mutect2 which removes the vast majority of sequencing artifacts. The high number of pseudogenes and segmental duplications in the mouse genome (as compared to the human genome) increases the chance of read mis-mapping. To avoid ambiguous SNP positions resulting from mis-mapping, only reads with a mapping quality of 60 were included in LOH analyses14. For CNV detection in mouse and human pancreatic cancers, we used CopywriteR 2.6.1.216 which is based on the analysis of “off-target” reads. “Off-targets” (such as intronic reads), which represent ~20% of all reads in typical WES data sets (due to incomplete removal during standard library preparation), are not affected by variation in capture efficiencies. CopywriteR outperforms algorithms based on the analysis of “on-target” reads (exonic-read based algorithms) for CNV calling from human and mouse WES data14,16.
Array comparative genomic hybridization
aCGH data from Niknafs et al.1, Maser et al.2, Foijer et al.3, Foijer et al.4, Cutler et al.6, Mueller et al.13 and our own KPC cohort was analyzed using Agilent Genomic Workbench software version 7.0.4.0. Importantly, the identical aCGH array (Agilent, SurePrint G3 Mouse CGH 240 K) was used in all studies, allowing for the direct comparison of strain-specific germline variation at Nlrp1 and Skint loci across all mouse cancer cohorts/genomes.
Long-read nanopore sequencing
Long-read sequencing libraries were prepared using the Oxford Nanopore LSK109 kit (ONT, Oxford, UK). A total of 400 ng of library was loaded on a promethION flowcell and run for 72 h on a promethION beta sequencer (ONT, Oxford, UK). Base-calling was performed on the promethION compute unit’s GPU using guppy 3.2.8 basecaller. The resulting FASTQ file was mapped to the reference genome GRCm38.p6 using Minimap2 (option map-ont). Read coverage was extracted using bam-read count (minimum mapping quality 60, minimum base quality 5).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
D.S. is supported by the European Research Council (Consolidator Grant 648521) and the Deutsche Forschungsgemeinschaft (SA1374/4-2; SFB 1321). R.R. is supported by the European Research Council (Consolidator Grants PACA-MET and MSCA-ITN-ETN PRECODE), the Deutsche Forschungsgemeinschaft (DFG RA1629/2-1; SFB1243; SFB1321; SFB1335), the German Cancer Consortium Joint Funding Program, and the Deutsche Krebshilfe (70112480).
Author contributions
S.M. and S.L. analyzed the data. S.M. and K.C. curated the data and performed experiments. S.L. performed bioinformatic analyses. S.K. and H.B. performed nanopore sequencing. G.S., L.R. and D.S. provided biological resources and critical input during protocol development. R.R. supervised the project. S.M., S.L. and R.R. wrote the manuscript.
Data availability
NGS and aCGH data from Niknafs et al.1 is available from the National Center for Biotechnology Information Sequence Read Archive (NCBI SRA) using study accession PRJNA546566 and from the Gene Expression Omnibus (GEO) database using the accession GSE132235. WES and aCGH data of our KC cohort, described by Mueller et al.13 is available from the European Nucleotide Archive using study accession PRJEB23787 and from the GEO database using accession GSE107458. Nanopore sequencing, WES and aCGH data of our KPC cohort is available from the European Nucleotide Archive using study accession PRJEB39427, PRJEB39429 and from the GEO database using accession GSE154537, respectively. aCGH data from Maser et al.2, Foijer et al.3, Foijer et al.4, and Cutler et al.6 is available from the GEO database using study accession GSE7615, GSE57334, GSE63686, and GSE9186, respectively. Human pancreatic cancer WES data of Witkiewicz et al.12 is available from NCBI SRA using study accession PRJNA278883.
Code availability
The source code for WES analyses pipelines is available at https://github.com/roland-rad-lab/MoCaSeq.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Nature Communications thanks Anton Berns and Amanda Toland for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Sebastian Mueller, Sebastian Lange.
Supplementary information
Supplementary information is available for this paper at 10.1038/s41467-020-18095-3.
References
- 1.Niknafs N, et al. Characterization of genetic subclonal evolution in pancreatic cancer mouse models. Nat. Commun. 2019;10:5435. doi: 10.1038/s41467-019-13100-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maser RS, et al. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature. 2007;447:966–971. doi: 10.1038/nature05886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Foijer F, et al. Chromosome instability induced by Mps1 and p53 mutation generates aggressive lymphomas exhibiting aneuploidy-induced stress. Proc. Natl Acad. Sci. USA. 2014;111:13427–13432. doi: 10.1073/pnas.1400892111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foijer, F. et al. Deletion of the MAD2L1 spindle assembly checkpoint gene is tolerated in mouse models of acute T-cell lymphoma and hepatocellular carcinoma. Elife10.7554/eLife.20873 (2017). [DOI] [PMC free article] [PubMed]
- 5.Lilue J, et al. Sixteen diverse laboratory mouse reference genomes define strain-specific haplotypes and novel functional loci. Nat. Genet. 2018;50:1574–1583. doi: 10.1038/s41588-018-0223-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cutler G, Marshall LA, Chin N, Baribault H, Kassner PD. Significant gene content variation characterizes the genomes of inbred mouse strains. Genome Res. 2007;17:1743–1754. doi: 10.1101/gr.6754607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olive KP, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–860. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 8.Hingorani SR, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 9.Perez-Mancera PA, et al. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature. 2012;486:266–270. doi: 10.1038/nature11114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mann KM, et al. Sleeping Beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proc. Natl Acad. Sci. USA. 2012;109:5934–5941. doi: 10.1073/pnas.1202490109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rad R, et al. A conditional piggyBac transposition system for genetic screening in mice identifies oncogenic networks in pancreatic cancer. Nat. Genet. 2015;47:47–56. doi: 10.1038/ng.3164. [DOI] [PubMed] [Google Scholar]
- 12.Witkiewicz AK, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015;6:6744. doi: 10.1038/ncomms7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mueller S, et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature. 2018;554:62–68. doi: 10.1038/nature25459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lange S, et al. Analysis pipelines for cancer genome sequencing in mice. Nat. Protoc. 2020;15:266–315. doi: 10.1038/s41596-019-0234-7. [DOI] [PubMed] [Google Scholar]
- 15.Friedrich MJ, et al. Genome-wide transposon screening and quantitative insertion site sequencing for cancer gene discovery in mice. Nat. Protoc. 2017;12:289–309. doi: 10.1038/nprot.2016.164. [DOI] [PubMed] [Google Scholar]
- 16.Kuilman T, et al. CopywriteR: DNA copy number detection from off-target sequence data. Genome Biol. 2015;16:49. doi: 10.1186/s13059-015-0617-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
NGS and aCGH data from Niknafs et al.1 is available from the National Center for Biotechnology Information Sequence Read Archive (NCBI SRA) using study accession PRJNA546566 and from the Gene Expression Omnibus (GEO) database using the accession GSE132235. WES and aCGH data of our KC cohort, described by Mueller et al.13 is available from the European Nucleotide Archive using study accession PRJEB23787 and from the GEO database using accession GSE107458. Nanopore sequencing, WES and aCGH data of our KPC cohort is available from the European Nucleotide Archive using study accession PRJEB39427, PRJEB39429 and from the GEO database using accession GSE154537, respectively. aCGH data from Maser et al.2, Foijer et al.3, Foijer et al.4, and Cutler et al.6 is available from the GEO database using study accession GSE7615, GSE57334, GSE63686, and GSE9186, respectively. Human pancreatic cancer WES data of Witkiewicz et al.12 is available from NCBI SRA using study accession PRJNA278883.
The source code for WES analyses pipelines is available at https://github.com/roland-rad-lab/MoCaSeq.