

Abstract
Over the last decade Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) has been developed into a potent molecular biology tool used to rapidly modify genes or their expression in a multitude of ways. In parallel, CRISPR-based screening approaches have been developed as powerful discovery platforms for dissecting the genetic basis of cellular behavior, as well as for drug target discovery. CRISPR screens can be designed in numerous ways. Here, we give a brief background to CRISPR screens and discuss the pros and cons of different design approaches, including unbiased genome-wide screens that target all known genes, as well as hypothesis-driven custom screens in which selected subsets of genes are targeted (Fig. 1). We provide several suggestions for how a custom screen can be designed, which could broadly serve as inspiration for any experiment that includes candidate gene selection. Finally, we discuss how results from CRISPR screens could be translated into drug development, as well as future trends we foresee in the rapidly evolving CRISPR screen field.
1. CRISPR
Since the first publications where CRISPR/Cas9 was used as a controlled molecular biology tool in 2012–2013 [2], [3], [4], the use of CRISPR systems has found its place as a staple in the researcher’s toolbox. These experimental CRISPR/Cas9 tools are developed from naturally occurring systems found in multiple bacterial and archaeal species [5], [6], [7], [8], [9], [10]. Many developments of the CRISPR system have been presented over the last few years, including the use of Cas9 variants from other bacteria, modified Cas9 with novel functionalities, as well as using other Cas proteins [11], [12], [13], [14], [15]. As of 2020, the CRISPR field is very active, and further exciting developments can be expected.
The basis for a standard CRISPR/Cas9 experiment intended to inactivate a gene is the formation of a complex of a guide RNA (gRNA), and the endonuclease Cas9. As the gRNA/Cas9 complex binds the specific genomic DNA sequence, dictated by the gRNA sequence, a DNA double-strand (dsDNA) break is introduced in the genomic DNA by the activity of Cas9 [2]. Cells have inherent mechanisms for rapidly repairing dsDNA breaks, but these are error-prone and commonly result in small insertions and deletions (indels) of nucleotides at the repaired site [16]. Notably, if the dsDNA break is repaired without indel formation, it is likely recut by the gRNA/Cas9 complex until indel formation will eventually preclude further recognition and recutting. If the gRNA is designed such that the double-strand break is localized in a protein-coding part of a gene, there is a high likelihood that resulting indels cause a frameshift and thus a knock out allele of that gene [17], [18]. This review will not further focus on technical details related to how to perform CRISPR experiments; excellent overviews of this can be found here [19], [20], [21].
1.1. CRISPR-based screens
In a screen, various interventions are tested in parallel, and the result on the assayed target is recorded for the different interventions. Screening approaches have historically been extensively used by the pharmaceutical industry to identify chemically synthesized small molecule drugs that affect various cellular phenotypes linked to disease [22]. Typically, such screens are performed in large scale high-throughput formats where each molecule is tested in separated wells, testing a vast number of molecules in parallel to identify those that affect the studied behavior.
In a CRISPR screen, multiple gRNA-based perturbations are introduced into a cell population, and genes that affect the studied phenotype are identified (Fig. 2). The first genome-scale CRISPR screens were published in 2014 [23], [24], [25]. Several significant developments have been presented since, showing the immense potential of CRISPR screening technologies. These potentially transformative advances involve methods using (i) single-cell RNA sequencing (scRNAseq) based readouts for CRISPR screens (which has been referred to as Perturb-seq, CRISP-seq and CROP-seq [26], [27], [28], [29], (ii) the ongoing construction of CRISPR screen databases identifying genes essential for the survival of various human cancer cells [30], [31], [32], [33], and (iii) CRISPR activation (CRISPRa) based screening using a modified Cas9 version that does not inactivate genes, but instead brings trans-activating elements to the transcription start site of a gene, thus activating gene expression [34], [35], [36]. CRISPRa share features with the CRISPR interference (CRISPRi) system, where a modified Cas9 version is used that causes transcriptional repression of the targeted gene [34], [37], [38].
Fig. 1.

Illustration highlighting the difference between genome-wide and custom screens (1).
Fig. 2.

Illustration of a CRISPR screen.
The rapid development of CRISPR screening approaches can be attributed in part to the fact that the generic setup builds on similar, previously established methods. In particular, whole genome shRNA (short-hairpin RNA) based screens share many similarities in the experimental setup and analysis to gRNA (CRISPR) based screens [39], [40], [41], [42]. Based on the different modes of action between shRNA and gRNA, with shRNAs causing degradation of gene transcripts, while gRNAs directly targeting the gene itself, one could expect differences between these approaches. Conceptually, an shRNA approach will give a range of degrees of target inhibition, while a gRNA approach will have a more binary, all or nothing pattern of inhibition on a per cell basis. Systematic comparisons between shRNA and gRNA-based screens have been performed. While gRNA-based screens are typically more effective, the two approaches likely can serve as useful complements to each other in certain situations [43], [44], [45].
A crucial development for genome-wide shRNA and subsequent CRISPR screens was the development of massively parallel sequencing technologies at the end of the 1990 s that became commercially available around 2005 [46]. This approach enabled performing large scale screens in pooled samples (referred to as pooled screens), instead of each perturbation having to be separated into individual wells (which is referred to as arrayed screens). In a pooled screen (see Fig. 3), genetic perturbations mediated by, for example, gRNAs are applied to a cell population by retroviral or lentiviral delivery, leveraging the ability of these viruses to be titrated to a concentration at which few cells will be infected by more than one virus particle and thus one gRNA, and that the virus integrate into the genome of the infected cell. After the selection of infected cells, a controlled, genetically heterogeneous cell population is thus generated, where each cell will have one gene targeted by one gRNA construct that is integrated into its genome. Standard massively parallel sequencing of PCR-amplified genomic DNA, using primers amplifying the gRNA constructs, are then used to identify the representation of different gRNAs in the studied cell population. As a readout, the pooled screen could, for example, be used to identify genes that are central for the survival of a cancer cell by comparing the gRNA representation in the cancer cell population directly after infection to a later time point when the cells have been expanding in vitro or in vivo [47], [48]. As such, gRNAs depleted from the sample taken at the later time point target genes that are essential for the expansion and/or survival of the cancer cell. Open-source software packages used to analyze gRNA representation in sequencing data, including MAGeCK [49] and pipelines such as CRISPRanalyzeR [50], have significantly facilitated the use of CRISPR screens for labs with limited bioinformatics experience.
Fig. 3.

Representation of a pooled CRISPR screen. Lenti- or retroviral particles are typically used for delivery as they can (i) be titrated to achieve a specific infection rate, (ii) will integrate into the genome of the infected cell, and (iii) infect many different cell types. The integration enables simple quantification of the gRNA representation in different cell populations by next-generation sequencing, and the subsequent identification of enriched or depleted gRNAs comparing different populations.
1.2. Which genes should be targeted in a CRISPR screen?
Conceptually, there exist at least three overall approaches related to the genes to be included in any screen. Here we broadly refer to these as genome-wide, restricted, and hypothesis-driven, where the latter two could be categorized as custom or focused approaches.
1. Genome-wide screens; targeting all known genes. This approach is unbiased in its design and thus has the highest potential to identify unexpected genes affecting the studied phenotype. Several genome-wide libraries currently exist and can be obtained at a relatively low cost from, for example, the non-profit plasmid repository Addgene [17], [36], [47], [48], [51], [52], [53], [54], [55], [56], [57], [58], [59], [60]. However, targeting roughly 20,000 genes and controls comes with challenges. Typically, 4–10 gRNAs are recommended to be included for each gene in such screens, which results in at least 80,000 different gRNAs in a genome-wide screen. Furthermore, for proper library representation, it is suggested that at least 500 cells per gRNA should be used, resulting in 40 million cells per experimental group in a pooled screen. In addition, a low multiplicity of infection (MOI; the ratio of viral particles to cells that are being infected) is required to assure that few cells are infected with more than one gRNA each. To achieve this, the viral library is often diluted to a level where 25–30% of the cells are infected [20]. Thus, three to four times more cells than needed for the screen must be initially transduced. In cells that are challenging to infect, achieving a 25–30% infection rate could be difficult, and an even larger starting cell population might be required. Of course, the number of cells needed is further increased by biological replicates and different interventions included in the screen design.
Minimum number of cells needed for a genome-wide CRISPR screen:
20 000 targets × 4 gRNAs/target = ~80 000 gRNAs
80 000 gRNAs × 500 cells/gRNA = ~40 million cells per group
(25% infection rate = need to start with greater than 160 million cells)
A full genome CRISPR screen thus requires a large number of cells, which could pose a problem for the specific experiment. Established cancer cell lines are typically easy to expand to unlimited numbers. In contrast, specific primary cell populations and cells from patient material can be difficult or impossible to obtain in sufficient numbers. The specific screening setup can also dictate other limitations. For example, in vivo mouse cancer experiments have limitations for the number of cancer cells that can be administered to the recipient mouse. A standard experimental protocol for subcutaneous grafting of cancer cells, subsequently studying the in vivo growth profile over time, would typically involve the administration of fewer than one million cells, which is far from the 40 million cells needed for a genome-wide screen in the administered cancer cells.
One approach that could facilitate screening in situations where the number of cells is limited is to include unique molecular identifiers (UMIs) in the CRISPR library [61], [62]. This has been shown to increase the power of the analysis by combining information about gRNA representation with the clonality of the cells, adding another layer to the analysis. Such an approach could make it feasible to screen fewer cells per gRNA with retained statistical power.
2. Restricted screens; focusing on subsets of genes that for different reasons seem relevant. For example, this could be all kinases, all transcription factors, all G-protein coupled receptors etc. This review will not further discuss such approaches in-depth. However, these approaches could be a plausible start for a screen where the total number of genes that can be targeted is limited, but where a clear hypothesis as to the regulation of a studied phenotype is lacking. Several restricted libraries are available through Addgene and commercial providers. Multiple resources for identifying subsets of genes can also be used to design restricted libraries de novo. These resources include the Human Protein Atlas, comprising a compiled database for easy identification of categories of genes to be included [63]. The Gene Ontology (GO) database is another useful resource to identify smaller and larger subsets of genes, categorized as GO-terms [64].
3. Hypothesis-driven, custom screens; targeting genes experimentally identified to potentially be involved in the studied behavior. These types of screens could start with a list of genes generated by, for example, an OMICS experiment. Many different approaches exist to generate such candidate lists of genes that potentially affect a phenotype of interest. These include RNA-based methods such as RNAseq, single-cell RNAseq [65], and spatial transcriptomics approaches [66], DNA-based methods such as ChIP-seq [67] and ATAC-seq [68], as well as protein-based approaches like mass spectrometry, phosphoproteomics [69], and different affinity and proximity-based methods like BioID [70]. The candidate gene list could also be derived from previous larger CRISPR screens or from in silico predictions.
The rest of this review will primarily focus on such hypothesis-driven screens. Here the concept is to use a CRISPR screening approach to identify which of the genes, identified by e.g. RNAseq, that are central to the studied cellular behavior, aiming to extract functional information from a descriptive OMICS data set. The overall aim of the study could, for instance, be to describe the foundations of a cellular phenotype and to identify potential drug targets that could be targeted to modify the phenotype. A custom hypothesis-driven screen could, in a simple form, target all significantly up and downregulated genes in the studied cell population with the hypothesis that differentially expressed genes are likely involved in the studied phenotype. Initial transcriptomics data could be complemented by including, for example, proteomic data, as many proteins are not primarily regulated on the transcriptional level, but on a post-transcriptional level. Independently of which method, or combination of methods, that is used to generate the candidate gene list, it is likely not going to contain all critical genes of the studied system. Different approaches to link additional genes to the initial OMICs generated candidate list will be discussed in the following section.
A custom screen design can enable discovery in cases like the in vivo mouse cancer model highlighted above, where a genome-wide CRISPR screen would not be feasible. Such an experiment could, for example, be aimed at identifying genes affecting how cancer cells survive as a drug is administered to the mouse. In a study with a similar setup, Manguso et al. elegantly solved the problem related to the limited amount of cells that could be administered to a mouse, by only including genes in the screen that (i) belonged to specific relevant categories of genes (kinase, phosphatase, cell surface, plasma membrane, antigen processing and presentation, immune system process, and chromatin remodeling based on GO-terms), and (ii) had an expression level above a pre-determined threshold [71]. Limiting the screen in such a way allowed the screen to include only around 10% of the number of genes needed in comparison to a genome-wide screen. Manguso et al. furthermore divided the gRNA library into four sub-libraries with different gRNAs, where each library contained gRNAs for all genes, but only one gRNA per gene. The four different sub-libraries were then used in parallel mouse experiments, and the results from the four different screens were combined to generate data that thus included four gRNAs per gene; in the end, generating impressive resolution of the screen despite the limitations of the assay.
In summary, several parameters need to be considered when deciding how many genes to target in a screen. Genome-wide screens have the highest possibility for unbiased discovery, although a large number of cells need to be included, which can pose a significant challenge. More targeted, restricted or hypothesis-driven screens are more straightforward, but can only identify genes that are included in the screen library. Genome-wide and different types of restricted libraries are readily accessible, whereas custom libraries need to be synthesized and cloned into the CRISPR plasmid of choice. This is more labor-intensive and typically more expensive than buying readymade libraries. However, the cost of generating custom libraries is typically not prohibitively high.
2. How to design a hypothesis-driven custom screen
In a hypothesis-driven custom CRISPR screen, we suggest starting with a list of genes experimentally identified in the studied system. Differentially expressed genes identified by RNAseq comparing a tumor treated with a drug or control is an example to which we return. Here, the hypothesis would be that the differentially regulated genes are involved in the biological activity of the drug and that a screen could identify which of these genes are central to the phenotype.
In the following section, a number of different analytical tools that can be used to expand the list of genes to include in a custom screen, are introduced. This is not a comprehensive list but focuses on simple and freely available tools we find helpful. Table 1 below provides more information and links to the suggested resources.
Table 1.
Online tools and databases.
| Tool | Location | Comment |
|---|---|---|
| MAGeCK | https://sourceforge.net/p/mageck/wiki/Home/ | An open source computational tool for CRISPR screen analysis. |
| CRISPRanalyzeR | http://crispr-analyzer.dkfz.de | Web based tool for CRISPR screen analysis. |
| Addgene | https://www.addgene.org/crispr/ | A non-profit plasmid repository where many CRISPR relevant plasmids, and pooled gRNA libraries can be obtained. |
| The Human Protein Atlas | https://www.proteinatlas.org/humanproteome/proteinclasses | Database including protein class categorization (including drug targets), as well as extensive expression information from human cells and tissues. |
| Gene Ontology (GO) database | http://geneontology.org/ | Extensive categorization of genes into GO-terms. |
| Gene Set Enrichment Analysis (GSEA) | https://www.gsea-msigdb.org/gsea/index.jsp | Pathway analysis tool for RNAseq data. |
| g:GOSt of g:Profiler | https://biit.cs.ut.ee/gprofiler/gost | Pathway analysis tool based on lists of manually input genes. |
| Mouse Genome Informatics (MGI) Gene Ontology Browser | http://www.informatics.jax.org/vocab/gene_ontology/ | Simple tool to search GO-terms. |
| g:Converter of g:Profiler | https://biit.cs.ut.ee/gprofiler/convert | Tool that can be used to convert Gene Ids, and to extract genes from GO-terms, KEGG pathways etc. |
| Pathway Commons | https://www.pathwaycommons.org/ | Analyses lists of genes and shows interactions and enriched pathways. |
| GeneMANIA | https://genemania.org/ | Analyses lists of genes and shows interactions and enriched pathways (plugin for Cytoscape also exists). |
| Harmonizome | https://amp.pharm.mssm.edu/Harmonizome/ | Database that extracts information from multiple other sources and integrates it into a search feature |
| Geneshot | https://amp.pharm.mssm.edu/geneshot/ | Literature mining tools providing lists of genes linked to the search term(s). |
| Green Listed tool | http://greenlisted.cmm.ki.se/ | Rapid gRNA design tool for custom CRISPR screens. Can also be used to extract non-targeting and intergenic control gRNAs (select Zhang/GeCKOv2 or Wang/Lander/Sabatini and press “Detailed Information”). |
| Depmap portal | https://depmap.org/portal | Cancer dependencies analytical and visualization tools, which e.g. can be used to identify essential genes. |
| g:Orth of g:Profiler | https://biit.cs.ut.ee/gprofiler/orth | Translates gene identifiers between organisms. |
| MGI batch query | http://www.informatics.jax.org/batch | Tool that can identify alternative names of genes. |
| Drug Gene Interaction | http://www.dgidb.org/druggable_gene_categories | Database of drug targets. |
| Probe Miner | https://probeminer.icr.ac.uk/#/ | Database of small molecule drugs and their targets. |
2.1. Identifying genes linked to your data set
A common starting point for evaluating an OMICs dataset is a pathway analysis, and several tools exist to perform such. Genes identified by the pathway analysis could be included in the subsequent screen library, complementing the original OMICs dataset. In such a way, the screen is expanded by thoroughly exploring the involvement of pathways linked to the original data set. An excellent recent protocol by Reimand et al. describes some approaches that can be used to perform pathway analysis [72].
One of the most advanced freely available analysis tools for pathway analysis of RNAseq data is the Gene Set Enrichment Analysis (GSEA) [73]. In the standard setup, GSEA takes the expression levels of all genes in the expression data set and compares them to defined gene sets to identify pathways linked to the expression profile.
Another useful pathway analysis tool is g:GOSt of g:Profiler [74]. In contrast to GSEA, g:GOSs is not restricted to uploading a complete expression data set. Instead, the researcher enters a list of genes, and the tool identifies pathways (e.g. GO, KEGG, Reactome and WikiPathways) in which the entered genes are enriched. Additionally, information on transcription factors and microRNAs that could be involved in regulating the gene set can be obtained. Importantly, in contrast to GSEA, the design of this tool allows for the analysis of lists of genes identified based on methods beyond RNA expression.
The Gene Ontology (GO) consortium [75] is one of the most popular resources for classifying genes into categories. The tool performs GO-term enrichment analysis and extracts lists of genes linked to the identified GO-terms (e.g. “neutrophil migration” = GO:1990266, which includes 118 genes). The Mouse Genome Informatics (MGI) Gene Ontology Browser is another simple tool for browsing mouse GO-terms. g:Converter is an additional convenient tool to extract lists of genes linked to different GO-terms, but also from e.g. KEGG, Reactome, and WikiPathways.
Pathway commons is a web-based tool that analyzes a gene set integrating data related to e.g. biological pathways and physical interactions from various publicly available databases [76] such as Reactome, and PANTHER. Pathway commons allows the user to identify related pathways and suggests potential interactions between genes in the entered gene set.
A very useful tool that shares functionalities with Pathway Commons is GeneMANIA [77]. A plugin version for Cytoscape of GeneMANIA [78] also exists and is the primary tool we currently use to identify genes to include in our custom screens. The GeneMANIA plugin can quickly analyze large lists of genes and generate graphical representations of gene interactions. Importantly, GeneMANIA can also suggest genes that are linked to a gene or a list of genes. The tool links genes based on factors such as physical interactions, co-localization, and co-expression. It is also possible to define which organism the data should be related to. Fig. 4A shows the graphical output of GeneMANIA when the Hoxb8 gene was entered, including 15 linked genes suggested by the software. The size of the grey circles (nodes) represents link strength, and the color of the connecting lines (edges) represents different categories of interactions.
Fig. 4.
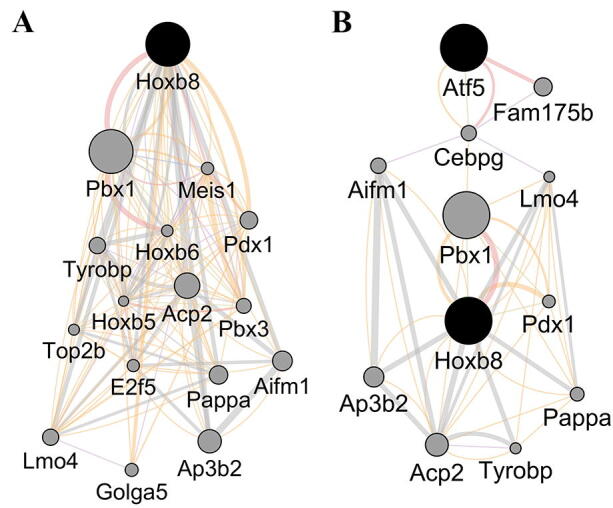
Analysis using the geneMANIA plugin for Cytoscape. (A) Genes identified to be linked to HoxB8. (B). Genes identified to connect Atf5 and HoxB8.
GeneMANIA can also identify suggested connections between two, or more, genes that are not directly interacting, as exemplified in Fig. 4B by searching Hoxb8 and Atf5, where Cebpg, Lmo4, Aifm1, and Pbx1 are suggested as connecting nodes.
Harmonizome [79] is yet another impressive example of a database extracting information from multiple sources. This tool can, for example, identify transcription factor binding to the promoter of an entered gene, pathways the gene is involved in, as well as the expression level and importance of the selected gene for the survival of different cell lines.
Finally, Geneshot [80] is a literature mining tool identifying genes often mentioned in context with the query gene in the literature.
2.2. Control genes
Another vital part of designing a custom screen is to include positive and negative control gRNAs. The purpose of these are to establish a baseline for the screen, defined by the negative controls, and for evaluation of the efficiency of the screen, shown by the strength of the enrichment/depletion of positive controls. Including 5–10% of the total gRNAs as controls in a screen is a suggested setup. However, in smaller, hypothesis-driven screens, including a higher proportion of negative controls should be considered. The readout of a CRISPR screen is enrichment and depletion of gRNAs comparing experimental groups. In a genome-wide screen, many of the included genes will not affect the studied phenotype, and as a consequence, gRNAs targeting these genes will act as a baseline facilitating the identification of enriched/depleted gRNAs. In contrast, a smaller hypothesis-driven screen would likely be designed to have a higher proportion of the included genes that affect the phenotype, and fewer that act as a baseline. As a consequence, the enrichment/depletion of gRNAs might not be as noticeable depending on the readout used. Therefore, in very small screens, the use of up to 50% negative controls could be considered.
2.2.1. Negative controls
Negative controls are gRNAs that are expected to not affect the studied phenotype. Two types of negative controls are typically considered; (i) gRNAs that do not bind anywhere in the studied genome, so-called non-targeting controls (NTCs), and (ii) gRNAs that bind intergenic regions that are predicted to not affect any protein-coding gene. The rationale for using “intergenic controls” is that they do not affect any known gene, but still control for the DNA damage response that is triggered when the gRNA/Cas9 complex cuts the genome, which is something the NTC does not control for. In this context, it is worth noting that the DNA damage response induced in a CRISPR experiment can have a negative impact on cell survival and proliferation, thereby introducing noise in a screen [81], [82]. Importantly, a gRNA that binds to multiple genomic regions, having a high off-target activity, will cause an increased DNA damage and non-specific toxicity to the cell [83]. Combining NTC and intergenic controls can be useful if possible. The negative control population should theoretically neither be enriched nor depleted when comparing experimental groups. In practice, NTC sets are commonly slightly enriched, due to the absence of DNA damage induced by the NTC in contrast to targeting gRNAs. A resource to extract lists of negative control gRNAs for mouse and human can be found using the ‘Green Listed’ tool [84]. Pressing “detailed information” in the Zhang/GeCKOv2 or Wang/Lander/Sabatini reference libraries provides more information. Intergenic controls targeting well-defined loci like AAVS1 could also be considered [4].
2.2.2. Positive controls
Positive controls that are known or expected to affect the studied phenotype should always be included, if available. Typically, essential genes that affect cell survival could be considered standard positive controls. As such, gRNAs targeting essential genes are expected to be depleted from the cell population over time. Examples of lists of essential genes can be found in e.g. Hart et al. (Supplemental Table 2 in [58]). Another useful resource for identifying essential genes is the Depmap portal, where e.g. result from the Project Achilles can be accessed [85], containing genome-wide gene essentiality data for more than 600 human cell lines. For other screening readouts, such as those based on FACS sorting to isolate cells with different phenotypes, the inclusion of gRNAs against genes coding for the proteins that the FACS antibodies bind to is advisable as positive controls. These gRNAs are expected to be depleted from the sorted population. Similarly, in screens studying the response to a ligand, including a gRNA targeting known receptors for the ligand is a good strategy, as is including gRNAs against GFP in screens using GFP reporters as a readout.
2.3. Converting gene identifiers
A common challenge in handling OMICS data are inconsistencies in gene identifiers, either within a species or between different species. There is no simple general approach to solve this problem. We typically use two websites to identify alternative names: The g:Convert Gene ID conversion tool or the g:Orth Orthology search of g:Profiler, as well as the Batch Query Tool of the MGI webpage. As a last resort, alternative names need to be searched manually.
2.4. Generating lists of gRNAs from a list of genes
After generating a list of genes and controls to include in a custom screen, we typically use the free web-based ‘Green Listed’ tool for rapid gRNA design, something we have successfully used for several published and unpublished custom screens [84], [86]. Extensive information about how to work with the tool can be found on the webpage, as well as in [1]. Briefly, a Reference Library is selected, a list of genes to be targeted is entered, and adapter sequences for downstream cloning are provided by the user, and Green Listed produces a list of gRNAs to order.
3. Trends and future development in CRISPR-based screening
The CRISPR field is rapidly changing, and significant developments can be expected. Here follow some concepts we anticipate will be further developed going forward.
3.1. Using different CRISPR systems for screening
Multiple diverse CRISPR systems have been used for genome-wide screening, including systems based on nuclease deactivated Cas9 (dCas9) [34], [35], [36]. Still, there are additional CRISPR-based systems that have features that make them potentially attractive from a large scale screening point-of-view, for example, base editors [87], Cas13 [88] and different epigenetic modifying dCas9 versions [89]. A bottleneck establishing novel CRISPR-based screening platforms is the development of reliable gRNA prediction tools for the specific setup, allowing for large scale design of gRNAs without the need to validate individual gRNAs [17], [18], [90]. Thus, reliable gRNA prediction algorithms need to be developed for the specific system to enable genome-wide screens.
Alternative gRNA design approaches could potentially be considered, for example, where gRNA sequences are generated from mRNA or DNA isolated from the cells to be screened [91], [92]. Such approaches would, however, likely demand a significantly larger amount of cells to be included in the screen as the activity of the generated gRNAs as a population must be expected to have low activity.
3.2. Screens identifying pathways involving genes with overlapping functionality
A standard CRISPR screen setup, where each cell is targeted by one gRNA, has a good chance of identifying genes that have a non-redundant activity in the studied behavior. However, if redundancies exist in an involved pathway, for example, if several genes have overlapping functionality, these genes will typically not be identified in the screen. For drug target discovery, this does not pose a significant problem, as a drug target optimally should have a non-redundant functionality in the pathway the drug is intended to interfere with. Still, from a biological point of view, identifying pathways involved in a studied behavior is an important development, even if functionally overlapping genes are involved. Several approaches could be considered going forward with a project focused on detailed understanding of the involved biology. For example, performing the screen in cells that have been generated to lack genes thought to contribute to redundancies in a pathway of interest. Another approach is screening with combinations of more than one gRNA in each cell. Such screens have been elegantly performed by using orthogonal Cas9 version [93], [94]. Importantly, controlled combinatorial screens rapidly become prohibitively large as the number of included genes increase. For feasibility, such screen approaches need to be more limited, targeting subsets of genes. Finally, performing screens based on both inactivation of genes (such as traditional Cas9-based screens or CRISPRi) with screens based on activation of gene expression, CRISPRa, could significantly increase the biological understanding of a system. Taken together, we foresee more intricate CRISPR screen designs allowing for dissecting of complex biological pathways [93].
3.3. The DNA damage response challenge
Standard CRISPR experiments, where dsDNA breaks are formed by the gRNA/Cas9 complex, results in a DNA damage response in cells, which introduce non-specific noise into screens [81], [82], [95], [96]. The type of cells used will likely influence this problem, for example, related to the specific status of TP53 in the cell, central to the DNA-damage response. Different approaches to limit the negative impact of the DNA damage response will likely be an integrated part of future CRISPR screen projects, for example, by performing screens in TP53 KO cells, by applying transient TP53 inactivation, or by using CRISPR versions that do not induce a DNA damage response, such as CRISPRa- and CRISPRi-based screens.
3.4. Performing CRISPR screens in more complex biological settings
Large scale screening efforts have identified common and specific patterns related to the survival of cancer cell lines cultured in well-defined in vitro conditions [32], [97], [98]. Importantly, the result of a screen will be linked to the selection pressure applied by the screen setup. Therefore, further discoveries can likely be obtained by introducing layers of relevant complexity in the screen design. For example, performing in vivo screens in the context of animal disease models, or in more complex co-culture settings, including different organoid designs, have the potential to identify prospective drug targets that are not found in a standard in vitro survival screen setup [71], [99], [100], [101], [102], [103]. Performing screens with primary patient material, where the unique genetic and epigenetic state of the patient contributes to the complexity, could also be highly relevant for discovery. A somewhat similar but potentially more feasible setup is to generate cell lines with patient-specific mutations and perform screens in these, aiming to identify how the specific mutation is affecting the cellular behavior.
All of these more complex screen settings are limited by multiple factors, including the number of cells that can successfully be infected with the CRISPR library, how long the studied cells survive, the possibility of developing relevant readouts that the cells are separated based on in the screen, etc. We argue that using custom CRISPR screen approaches, as discussed above, could be a powerful way to approach these more complex screen setups and still retain discovery potential.
3.5. Translating results from screens into drug development
The identification of candidate gene and protein targets linked to disease often serves as an initial step in contemporary drug development. In this regard, a CRISPR screen can identify genes that are central to a studied pathway, for example related to T cell activation [104]. Conceptually, the phenotype resulting from efficient targeting of a gene with an inhibitory drug should recapitulate, at least in part, the phenotype, as a result of a knockout of the same gene. A CRISPR screen could thus serve as a rapid platform to prioritize drug target candidates.
However, only 5–10% of the protein-coding genes are considered to be “druggable” with small molecule drugs. This is based on structural characteristics, where druggable proteins have a three-dimensional structure that allows for a small molecule to dock into a unique pocket and thereby affect the function of the protein. Commonly, such a pocket is the active site of an enzyme. Prediction algorithms have suggested genes referred to as the druggable genome [105], [106], [107]. Lists of validated and potential drug targets can be found, for instance, through the Human Protein Atlas [63] and the Drug Gene Interaction Database [108]. Already developed small molecule drugs targeting different proteins can also be found, for example, through the ‘Probe Miner’ tool [109].
The development of biological drugs, like antibodies and soluble receptors, adds another mode of action to affect cellular behavior [110], [111]. Secreted proteins and proteins exposed on the cell surface are apparent candidates for biological drugs. Nevertheless, the observation that current drug modalities cannot target most human proteins still holds true also when including biological drugs, and translating results from a CRISPR screen to drug development is therefore not necessarily straight forward.
3.5.1. RNA interference and antisense oligonucleotides
A less explored therapeutic approach, which is not limited by specific features of the target’s protein structure, is based on RNA interference (RNAi) and antisense oligonucleotide (ASO) technologies that can be designed to inhibit any coding or noncoding RNA [112], [113].
RNAi is based on different small RNA oligonucleotides, including shRNAs mentioned above, that are complementary in sequence to their target RNA, thus forming the basis of their specificity and ability to target the specific RNA. The RNAi molecules interfere with their target RNA and cause its degradation through the activity of the RNA-induced silencing complex (RISC) [114], [115]. ASOs, on the other hand, are composed of a DNA-based oligonucleotide that comprises a chemically modified backbone and modified bases that aid in stability and activity [116]. Similar to RNAi, ASOs are designed to be complementary to the target RNA sequence. The binding of the ASO to its target RNA results in an ASO-RNA complex that recruits the RNAse H enzyme and the subsequent degradation of the RNA [117].
RNAi and ASO-based drugs could thus be designed to target any gene identified in a CRISPR screen, independently of whether the target is "druggable" or not. Nonetheless, there are significant challenges to these types of drugs, including the difficulty of delivering the drug to the correct cell and the potentially higher cost compared to traditional small molecule drugs. Different approaches have been explored to solve these challenges e.g. using lipid carriers aiming for more specific delivery to the cells of interest [118], [119]. The carriers can be further functionalized by, for example, introducing ligands for specific endocytic receptors, facilitating uptake into cells of interest [120], [121], [122], [123]. Another possible approach is to deliver the RNAi/ASO molecules specifically to the site where the action is intended. For example, in the case of rheumatoid arthritis, RNAi/ASOs could be injected directly into the affected joint as a therapy [124], [125]. Similar concepts could be considered in other contexts where features of the disease are localized, for instance delivery into the cerebrospinal fluid of patients with multiple sclerosis or neurodegenerative diseases, as well as delivery into a tumor or draining lymph node of a cancer patient to trigger a stronger immune cell activation against the cancer.
3.5.2. Using CRISPR as a drug
Yet another potential therapeutic alternative could be delivering CRISPR constructs directly into the patient to modify genes identified to be linked to a disease [126]. However, several complicating factors exist, including the fact that CRISPR systems are derived from prokaryotes, which trigger the activation of the immune system [127], [128]. Delivery of the CRISPR constructs to the correct cells or tissues is also a significant challenge. One alternative approach to administering the CRISPR construct directly into a patient is to extract relevant cells from the patient, modify them ex vivo, and reintroduce them back to the patient. Such approaches are currently in clinical trials, both aiming to correct inherited genetic modifications affecting hematopoietic stem cells that result in severe disease related to the hematopoietic system [129], as well as to generate aggressive tumor-targeting T cells that can be given to cancer patients [130].
In summary, over the last decade, CRISPR and CRISPR-based screens have been developed into powerful discovery tools used by the research community. Designing a screen can be done in several ways, where the number of genes included in the screen should be considered. The more genes that are included, the more unbiased result can be expected, but as a consequence, more cells need to be used, which can become a significant technical bottleneck for the screen. We propose using hypothesis-based, custom screens as a rational and straightforward alternative approach and have, in detail, discussed concepts to design such screens using, for example, an RNAseq experiment as a starting point. In this scenario, we aim to transform a descriptive expression dataset into biological understanding. As these smaller screens are much less demanding than genome-wide screens, performing a series of custom screens where the hypothesis is refined for each screen can be a practical discovery approach. Finally, using CRISPR to identify genes that affect a cellular behavior linked to disease can serve as a starting point for drug development. However, since only a limited number of proteins/genes can be targeted with traditional drugs, novel approaches likely need to be explored to develop drugs targeting identified genes. Combining CRISPR screening approaches for discovery with the development of RNAi or ASO based drugs could serve as a foundation for future precision medicine (Fig. 5).
Fig. 5.

Summary of proposed discovery process.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We are thankful for thoughtful suggestions from Professor AT Phan (School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore), Aaron Winkler (Pfizer, Cambridge, MA, US), as well as Laura Plant, Marek Skupinski and Mats Ceder. This review was based on concepts developed partly funded by grants from the Swedish Research Council, the Swedish Cancer Society, the Karolinska Institutet, The Swedish Rheumatism Association, King Gustaf V’s 80-year fund, Åke Wiberg Foundation, Stiftelsen Professor Nanna Svartz Fond, The Jeansson Foundations, Alex and Eva Wallström Foundation for scientific research and education (to F.W.), The Wenner-Gren Foundations (to F.W., S.K.P., and S.V.B.), the Börje Dahlin fund (to S.K.P, L.J, and Y.S.), the China Scholarship Council (to L.J. and Y.S), and the Nanyang Technological University - Karolinska Institutet Joint Ph.D. Programme (to V.S.I). This work has received support from the EU/EFPIA Innovative Medicines Initiative Joint Undertaking (ULTRADD grant n° 115766) (to S.K.P. and Z.K.). This work has received support from the EU/EFPIA Innovative Medicines Initiative 2 Joint Undertaking (RTCure grant n° 777357) (to F.W.).
References
- 1.F. Wermeling (2017) Custom CRISPR Screens & the Green Listed Software. (https://blog.addgene.org/custom-crispr-screens-the-green-listed-software).
- 2.Jinek M. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cong L. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mali P. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marraffini L.A., Sontheimer E.J. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008;322:1843–1845. doi: 10.1126/science.1165771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horvath P., Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 7.Brouns S.J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mojica F.J.M., Montoliu L. On the Origin of CRISPR-Cas Technology: From Prokaryotes to Mammals. Trends Microbiol. 2016;24:811–820. doi: 10.1016/j.tim.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 9.Makarova K.S. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol. 2020;18:67–83. doi: 10.1038/s41579-019-0299-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishino Y., Krupovic M., Forterre P. History of CRISPR-Cas from Encounter with a Mysterious Repeated Sequence to Genome Editing Technology. J Bacteriol. 2018;200 doi: 10.1128/JB.00580-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun. 2018;9:1911. doi: 10.1038/s41467-018-04252-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pickar-Oliver A., Gersbach C.A. The next generation of CRISPR-Cas technologies and applications. Nat Rev Mol Cell Biol. 2019;20:490–507. doi: 10.1038/s41580-019-0131-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knott G.J., Doudna J.A. CRISPR-Cas guides the future of genetic engineering. Science. 2018;361:866–869. doi: 10.1126/science.aat5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gibson G.J., Yang M. What rheumatologists need to know about CRISPR/Cas9. Nat Rev Rheumatol. 2017;13:205–216. doi: 10.1038/nrrheum.2017.6. [DOI] [PubMed] [Google Scholar]
- 15.Li Y., Li S., Wang J., Liu G. CRISPR/Cas Systems towards Next-Generation Biosensing. Trends Biotechnol. 2019;37:730–743. doi: 10.1016/j.tibtech.2018.12.005. [DOI] [PubMed] [Google Scholar]
- 16.Hsu P.D., Lander E.S., Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doench J.G. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chari R., Mali P., Moosburner M., Church G.M. Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat Methods. 2015;12:823–826. doi: 10.1038/nmeth.3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ran F.A. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doench J.G. Am I ready for CRISPR? A user's guide to genetic screens. Nat Rev Genet. 2018;19:67–80. doi: 10.1038/nrg.2017.97. [DOI] [PubMed] [Google Scholar]
- 21.Bak R.O., Dever D.P., Porteus M.H. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat Protoc. 2018;13:358–376. doi: 10.1038/nprot.2017.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Janzen W.P. Screening technologies for small molecule discovery: the state of the art. Chem Biol. 2014;21:1162–1170. doi: 10.1016/j.chembiol.2014.07.015. [DOI] [PubMed] [Google Scholar]
- 23.Shalem O. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang T., Wei J.J., Sabatini D.M., Lander E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koike-Yusa H., Li Y., Tan E.P., Velasco-Herrera Mdel C., Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol. 2014;32:267–273. doi: 10.1038/nbt.2800. [DOI] [PubMed] [Google Scholar]
- 26.Adamson B. A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell. 2016;167:1867–1882 e1821. doi: 10.1016/j.cell.2016.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dixit A., Perturb-Seq Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell. 2016;167:1853–1866 e1817. doi: 10.1016/j.cell.2016.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Datlinger P. Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods. 2017;14:297–301. doi: 10.1038/nmeth.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jaitin D.A. Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell. 2016;167:1883–1896 e1815. doi: 10.1016/j.cell.2016.11.039. [DOI] [PubMed] [Google Scholar]
- 30.Dempster J.M. Agreement between two large pan-cancer CRISPR-Cas9 gene dependency data sets. Nat Commun. 2019;10:5817. doi: 10.1038/s41467-019-13805-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi J. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol. 2015;33:661–667. doi: 10.1038/nbt.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Behan F.M. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature. 2019;568:511–516. doi: 10.1038/s41586-019-1103-9. [DOI] [PubMed] [Google Scholar]
- 33.Chan E.M. WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature. 2019;568:551–556. doi: 10.1038/s41586-019-1102-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gilbert L.A. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Konermann S. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanson K.R. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat Commun. 2018;9:5416. doi: 10.1038/s41467-018-07901-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gilbert L.A. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qi L.S. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berns K. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004;428:431–437. doi: 10.1038/nature02371. [DOI] [PubMed] [Google Scholar]
- 40.Paddison P.J. A resource for large-scale RNA-interference-based screens in mammals. Nature. 2004;428:427–431. doi: 10.1038/nature02370. [DOI] [PubMed] [Google Scholar]
- 41.Kamath R.S. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- 42.Moffat J. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 43.Evers B. CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat Biotechnol. 2016;34:631–633. doi: 10.1038/nbt.3536. [DOI] [PubMed] [Google Scholar]
- 44.Morgens D.W., Deans R.M., Li A., Bassik M.C. Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat Biotechnol. 2016;34:634–636. doi: 10.1038/nbt.3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taylor J., Woodcock S. A Perspective on the Future of High-Throughput RNAi Screening: Will CRISPR Cut Out the Competition or Can RNAi Help Guide the Way? J Biomol Screen. 2015;20:1040–1051. doi: 10.1177/1087057115590069. [DOI] [PubMed] [Google Scholar]
- 46.Metzker M.L. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 47.Tzelepis K. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. 2016;17:1193–1205. doi: 10.1016/j.celrep.2016.09.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hart T. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell. 2015;163:1515–1526. doi: 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 49.Li W. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014;15:554. doi: 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.J. Winter et al., CRISPRAnalyzeR: Interactive analysis, annotation and documentation of pooled CRISPR screens. bioRxiv, 109967 (2017).
- 51.Wang T. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–1101. doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morgens D.W. Genome-scale measurement of off-target activity using Cas9 toxicity in high-throughput screens. Nat Commun. 2017;8:15178. doi: 10.1038/ncomms15178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Horlbeck M.A. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife. 2016;5 doi: 10.7554/eLife.19760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma H. A CRISPR-Based Screen Identifies Genes Essential for West-Nile-Virus-Induced Cell Death. Cell Rep. 2015;12:673–683. doi: 10.1016/j.celrep.2015.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Henriksson J. Genome-wide CRISPR Screens in T Helper Cells Reveal Pervasive Crosstalk between Activation and Differentiation. Cell. 2019;176:882–896 e818. doi: 10.1016/j.cell.2018.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Joung J. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc. 2017;12:828–863. doi: 10.1038/nprot.2017.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hart T. Evaluation and Design of Genome-Wide CRISPR/SpCas9 Knockout Screens. G3 (Bethesda) 2017;7:2719–2727. doi: 10.1534/g3.117.041277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mair B. Essential Gene Profiles for Human Pluripotent Stem Cells Identify Uncharacterized Genes and Substrate Dependencies. Cell Rep. 2019;27:599–615 e512. doi: 10.1016/j.celrep.2019.02.041. [DOI] [PubMed] [Google Scholar]
- 60.Wang T. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell. 2017;168:890–903 e815. doi: 10.1016/j.cell.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Michlits G. CRISPR-UMI: single-cell lineage tracing of pooled CRISPR-Cas9 screens. Nat Methods. 2017;14:1191–1197. doi: 10.1038/nmeth.4466. [DOI] [PubMed] [Google Scholar]
- 62.Schmierer B. CRISPR/Cas9 screening using unique molecular identifiers. Mol Syst Biol. 2017;13:945. doi: 10.15252/msb.20177834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Uhlen M. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 64.C The Gene Ontology, The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019;47:D330–D338. doi: 10.1093/nar/gky1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sandberg R. Entering the era of single-cell transcriptomics in biology and medicine. Nat Methods. 2014;11:22–24. doi: 10.1038/nmeth.2764. [DOI] [PubMed] [Google Scholar]
- 66.Stahl P.L. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353:78–82. doi: 10.1126/science.aaf2403. [DOI] [PubMed] [Google Scholar]
- 67.Johnson D.S., Mortazavi A., Myers R.M., Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 68.Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu Y., Chance M.R. Integrating phosphoproteomics in systems biology. Comput Struct Biotechnol J. 2014;10:90–97. doi: 10.1016/j.csbj.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.May D.G., Roux K.J. BioID: A Method to Generate a History of Protein Associations. Methods Mol Biol. 2019;2008:83–95. doi: 10.1007/978-1-4939-9537-0_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Manguso R.T. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017;547:413–418. doi: 10.1038/nature23270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reimand J. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA Cytoscape and EnrichmentMap. Nat Protoc. 2019;14:482–517. doi: 10.1038/s41596-018-0103-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Subramanian A. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Raudvere U. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Res. 2019;47:W191–W198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ashburner M. Gene ontology: tool for the unification of biology The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rodchenkov I. Pathway Commons 2019 Update: integration, analysis and exploration of pathway data. Nucleic Acids Res. 2020;48:D489–D497. doi: 10.1093/nar/gkz946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Franz M. GeneMANIA update 2018. Nucleic Acids Res. 2018;46:W60–W64. doi: 10.1093/nar/gky311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Montojo J. GeneMANIA Cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics. 2010;26:2927–2928. doi: 10.1093/bioinformatics/btq562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rouillard A.D. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database (Oxford) 2016;2016 doi: 10.1093/database/baw100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lachmann A. Geneshot: search engine for ranking genes from arbitrary text queries. Nucleic Acids Res. 2019;47:W571–W577. doi: 10.1093/nar/gkz393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Haapaniemi E., Botla S., Persson J., Schmierer B., Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med. 2018;24:927–930. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 82.Ihry R.J. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med. 2018;24:939–946. doi: 10.1038/s41591-018-0050-6. [DOI] [PubMed] [Google Scholar]
- 83.Schiroli G. Precise Gene Editing Preserves Hematopoietic Stem Cell Function following Transient p53-Mediated DNA Damage Response. Cell Stem Cell. 2019;24:551–565 e558. doi: 10.1016/j.stem.2019.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Panda S.K. Green listed-a CRISPR screen tool. Bioinformatics. 2017;33:1099–1100. doi: 10.1093/bioinformatics/btw739. [DOI] [PubMed] [Google Scholar]
- 85.J. M. Dempster et al., Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. bioRxiv, 720243 (2019).
- 86.Panda S.K. IL-4 controls activated neutrophil FcgammaR2b expression and migration into inflamed joints. Proc Natl Acad Sci U S A. 2020 doi: 10.1073/pnas.1914186117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Abudayyeh O.O. RNA targeting with CRISPR-Cas13. Nature. 2017;550:280–284. doi: 10.1038/nature24049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hilton I.B. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33:510–517. doi: 10.1038/nbt.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wessels H.H. Massively parallel Cas13 screens reveal principles for guide RNA design. Nat Biotechnol. 2020;38:722–727. doi: 10.1038/s41587-020-0456-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Arakawa H. A method to convert mRNA into a gRNA library for CRISPR/Cas9 editing of any organism. Sci Adv. 2016;2 doi: 10.1126/sciadv.1600699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Koferle A. CORALINA: a universal method for the generation of gRNA libraries for CRISPR-based screening. BMC Genomics. 2016;17:917. doi: 10.1186/s12864-016-3268-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Najm F.J. Orthologous CRISPR-Cas9 enzymes for combinatorial genetic screens. Nat Biotechnol. 2018;36:179–189. doi: 10.1038/nbt.4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.DeWeirdt P.C. Genetic screens in isogenic mammalian cell lines without single cell cloning. Nat Commun. 2020;11:752. doi: 10.1038/s41467-020-14620-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Brown K.R., Mair B., Soste M., Moffat J. CRISPR screens are feasible in TP53 wild-type cells. Mol Syst Biol. 2019;15 doi: 10.15252/msb.20188679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Haapaniemi E., Botla S., Persson J., Schmierer B., Taipale J. Reply to “CRISPR screens are feasible in TP53 wild-type cells. Mol Syst Biol. 2019;15 doi: 10.15252/msb.20199059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hahn W.C. A CRISPR Way to Identify Cancer Targets. N Engl J Med. 2019;380:2475–2477. doi: 10.1056/NEJMcibr1905048. [DOI] [PubMed] [Google Scholar]
- 98.Ghandi M. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019;569:503–508. doi: 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ishizuka J.J. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature. 2019;565:43–48. doi: 10.1038/s41586-018-0768-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wei J. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature. 2019;576:471–476. doi: 10.1038/s41586-019-1821-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dong M.B. Systematic Immunotherapy Target Discovery Using Genome-Scale In Vivo CRISPR Screens in CD8 T Cells. Cell. 2019;178:1189–1204 e1123. doi: 10.1016/j.cell.2019.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen S. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell. 2015;160:1246–1260. doi: 10.1016/j.cell.2015.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chow R.D. AAV-mediated direct in vivo CRISPR screen identifies functional suppressors in glioblastoma. Nat Neurosci. 2017;20:1329–1341. doi: 10.1038/nn.4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shifrut E. Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell. 2018;175:1958–1971 e1915. doi: 10.1016/j.cell.2018.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.A. H. Wagner et al., DGIdb 2.0: mining clinically relevant drug-gene interactions. Nucleic Acids Res 44, D1036-1044 (2016). [DOI] [PMC free article] [PubMed]
- 106.Russ A.P., Lampel S. The druggable genome: an update. Drug Discov Today. 2005;10:1607–1610. doi: 10.1016/S1359-6446(05)03666-4. [DOI] [PubMed] [Google Scholar]
- 107.Hopkins A.L., Groom C.R. The druggable genome. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 108.Cotto K.C. DGIdb 3.0: a redesign and expansion of the drug-gene interaction database. Nucleic Acids Res. 2018;46:D1068–D1073. doi: 10.1093/nar/gkx1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Antolin A.A., Objective Quantitative, Data-Driven Assessment of Chemical Probes. Cell. Chem Biol. 2018;25:194–205 e195. doi: 10.1016/j.chembiol.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Elliott M.J. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344:1105–1110. doi: 10.1016/s0140-6736(94)90628-9. [DOI] [PubMed] [Google Scholar]
- 111.Hodi F.S. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kole R., Krainer A.R., Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov. 2012;11:125–140. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Watts J.K., Corey D.R. Silencing disease genes in the laboratory and the clinic. J Pathol. 2012;226:365–379. doi: 10.1002/path.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sen G.L., Blau H.M. A brief history of RNAi: the silence of the genes. FASEB J. 2006;20:1293–1299. doi: 10.1096/fj.06-6014rev. [DOI] [PubMed] [Google Scholar]
- 115.Weiss B., Davidkova G., Zhou L.W. Antisense RNA gene therapy for studying and modulating biological processes. Cell Mol Life Sci. 1999;55:334–358. doi: 10.1007/s000180050296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Potaczek D.P., Garn H., Unger S.D., Renz H. Antisense molecules: A new class of drugs. J Allergy Clin Immunol. 2016;137:1334–1346. doi: 10.1016/j.jaci.2015.12.1344. [DOI] [PubMed] [Google Scholar]
- 117.Crooke S.T. Molecular Mechanisms of Antisense Oligonucleotides. Nucleic Acid Ther. 2017;27:70–77. doi: 10.1089/nat.2016.0656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Miller A.D. Delivery of RNAi therapeutics: work in progress. Expert Rev Med Devices. 2013;10:781–811. doi: 10.1586/17434440.2013.855471. [DOI] [PubMed] [Google Scholar]
- 119.Castanotto D., Rossi J.J. The promises and pitfalls of RNA-interference-based therapeutics. Nature. 2009;457:426–433. doi: 10.1038/nature07758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bareford L.M., Swaan P.W. Endocytic mechanisms for targeted drug delivery. Adv Drug Deliv Rev. 2007;59:748–758. doi: 10.1016/j.addr.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Muro S. Challenges in design and characterization of ligand-targeted drug delivery systems. J Control Release. 2012;164:125–137. doi: 10.1016/j.jconrel.2012.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Prakash T.P. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014;42:8796–8807. doi: 10.1093/nar/gku531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nair J.K. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J Am Chem Soc. 2014;136:16958–16961. doi: 10.1021/ja505986a. [DOI] [PubMed] [Google Scholar]
- 124.Khoury M., Jorgensen C., Apparailly F. RNAi in arthritis: prospects of a future antisense therapy in inflammation. Curr Opin Mol Ther. 2007;9:483–489. [PubMed] [Google Scholar]
- 125.Morita Y. Antisense oligonucleotides targeting c-fos mRNA inhibit rheumatoid synovial fibroblast proliferation. Ann Rheum Dis. 1998;57:122–124. doi: 10.1136/ard.57.2.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Porteus M.H. A New Class of Medicines through DNA Editing. N Engl J Med. 2019;380:947–959. doi: 10.1056/NEJMra1800729. [DOI] [PubMed] [Google Scholar]
- 127.Charlesworth C.T. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat Med. 2019;25:249–254. doi: 10.1038/s41591-018-0326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Crudele J.M., Chamberlain J.S. Cas9 immunity creates challenges for CRISPR gene editing therapies. Nat Commun. 2018;9:3497. doi: 10.1038/s41467-018-05843-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Booth C., Romano R., Roncarolo M.G., Thrasher A.J. Gene therapy for primary immunodeficiency. Hum Mol Genet. 2019;28:R15–R23. doi: 10.1093/hmg/ddz170. [DOI] [PubMed] [Google Scholar]
- 130.Stadtmauer E.A. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020 doi: 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]