

Abstract
Development of next-generation oncolytic viruses requires the design of vectors that are potently oncolytic, immunogenic in human tumors, and well tolerated in patients. Starting with a joint-region deleted herpes simplex virus 1 (HSV-1) to create large transgene capability, we retained a single copy of the ICP34.5 gene, introduced mutations in UL37 to inhibit retrograde axonal transport, and inserted cell-type-specific microRNA (miRNA) target cassettes in HSV-1 genes essential for replication or neurovirulence. Ten miRNA candidates highly expressed in normal tissues and with low or absent expression in malignancies were selected from a comprehensive profile of 800 miRNAs with an emphasis on protection of the nervous system. Among the genes essential for viral replication identified using a small interfering RNA (siRNA) screen, we selected ICP4, ICP27, and UL8 for miRNA attenuation where a single miRNA is sufficient to potently attenuate viral replication. Additionally, a neuron-specific miRNA target cassette was introduced to control ICP34.5 expression. This vector is resistant to type I interferon compared to ICP34.5-deleted oncolytic HSVs, and in cancer cell lines, the oncolytic activity of the modified vector is equivalent to its parental virus. In vivo, this vector potently inhibits tumor growth while being well tolerated, even at high intravenous doses, compared to parental wild-type HSV-1.
Keywords: oncolytic virus, virotherapy, HSV-1, microRNA attenuation, cancer, interferon
Graphical Abstract
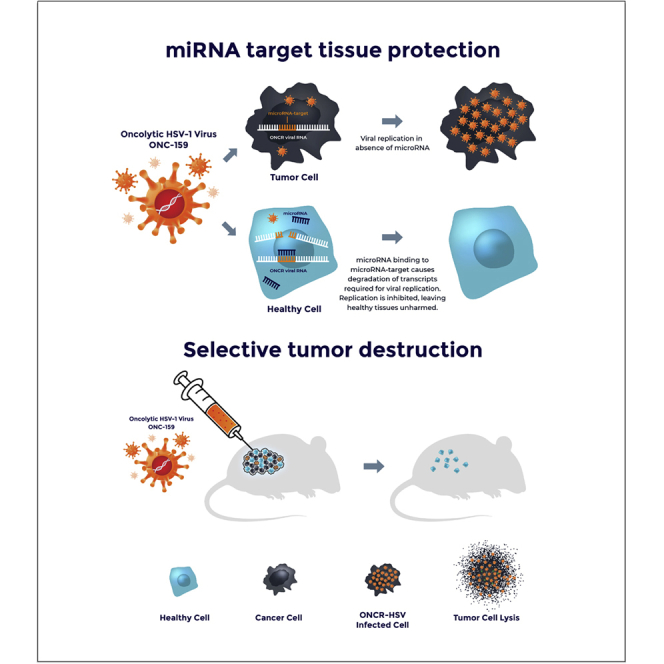
Next-generation oncolytic viral immunotherapy is designed to promote greater oncolytic potency, be highly immunogenic in human tumors, and have an improved tolerability profile among patients. This article describes the design of a highly engineered novel oncolytic HSV-1 vector, ONCR-159, demonstrates its broad therapeutic index, and confirms its potent antitumor activity.
Introduction
A central goal of next-generation oncolytic viral therapies is the design of strategies to support potent replication in cancer cells to promote oncolysis and release of tumor antigens while limiting viral replication in permissive healthy tissues. Typically, viral factors that dictate natural cellular tropism are deleted from oncolytic vectors, attenuating viral replication in most healthy tissues. These modifications can, however, limit the oncolytic activity in some tumor cells that may retain intrinsic or stromal-derived antiviral response, or may constitutively express antiviral genes, or, like cancer stem cells, may not be actively cycling.1, 2, 3, 4 Starting with HSV-1, the only oncolytic virus with a track of regulatory approval in major geographies, we sought to develop a novel vector backbone with a broadened therapeutic index and enhanced antitumor activity.
Oncolytic herpes simplex virus 1 (oHSV-1) viruses commonly employ the inactivation of the ribonucleotide reductase encoded by the ICP6 gene, or the deletion of neurovirulence factor ICP34.5, an inhibitor of the innate antiviral response mediated by type I interferon (IFN) and a mediator of eIF2 dephosphorylation downstream of EIF2AK2 (PKR), to limit replication in healthy tissue.5,6 ICP34.5 antagonizes multiple arms of the innate response, including signaling by PKR and TANK binding kinase (TBK1) signaling, and Beclin1-mediated apoptosis.7, 8, 9 ICP34.5-deleted viruses were found to be well tolerated in human clinical trials, although some viruses like H1716 and talimogene laherparepvec are described to replicate in discrete brain structures, such as the ependyma10 and myenteric neurons in immune-deficient mice.11 Another approach to enhance the tumor selective replication of oHSV vector is to express ICP34.5 under a tumor specific promoter, such as rQNestin34.5, an oHSV being developed for the treatment of glioma.12 Similarly, we hypothesized that ICP34.5 retention would stimulate viral replication in malignancies with residual type I IFN signaling or with basal IFN-dependent antiviral states.1,2,13,14 Therefore, the retention of ICP34.5 would potentially enhance oHSV replication and potency, but retaining the neurovirulence gene would require a novel strategy for robust and specific inhibition of replication in HSV-1 permissive normal tissues, including the nervous system. Ideally, this strategy would encompass conditional switches that would be redundantly engineered in HSV-1 genome to prevent inactivation by rearrangement through recombination and yet responsive to factor expressed in genetically stable normal cells and absent in tumor cells.
Here, we describe ONCR-159, a novel oHSV vector that retains one copy of the ICP34.5 gene and whose tumor selectivity is determined by the insertion of microRNA (miRNA) responsive target elements (miR-T) to control the expression of viral genes essential for replication and ICP34.5. We describe the evaluation of viral genes that were targeted with miRNA target cassettes and the expression profiling of the tissue-specific miRNAs selected for the engineering of the miR-T cassettes introduced in ONCR-159. Furthermore, we engineered an orthogonal neuron-specific safety mechanism by introducing the mutations in the UL37 tegument protein recently identified to eliminate retrograde axonal transport of the viral capsid.15
ONCR-159 benefits from additional improvements, namely the deletion of the joint region, which expands its transgene payload capacity to >20 kb, the inactivation of US12 to allow direct antigen presentation, and the gB:NT (D285N and A549T) mutant to promote infectivity.16,17 We present the preclinical characterization of ONCR-159 and demonstrate that it replicates efficiently in the presence of type I IFN challenge. ONCR-159 replication is effectively attenuated by distinct tissue miRNAs. ONCR-159 is well tolerated in vivo in the permissive BALB/c mouse at high doses following administration via multiple routes of administration, including single and multiple intravenous injections. These characteristics and the large payload capacity of this vector provide the basis for facile construction of multiple armed oncolytic viruses (OVs) that can address the unique immune suppressive drivers in the tumor microenvironment.
Results
siRNA Screen Identifies Candidate HSV-1 Genes for Attenuation
To increase the breadth of normal tissues protected from HSV-1 replication, we expanded viral attenuation mechanism to multiple essential viral genes with multiple miR-T cassettes. Ideally, the viral gene candidates would be essential for viral replication, preferably expressed at low copy number, and would be required to advance the early phases of the viral life cycle to avoid both viral early gene toxicity and progression to replication. A small interfering RNA (siRNA) screen was performed to identify optimal HSV-1 genes to be attenuated.
To model miRNA cleavage of a complementary target, we transfected pools of 3 siRNAs (Table S1) per HSV-1 gene in A253 cells, a oHSV permissive human submaxillary salivary gland cell line, that can be transfected with high efficiency (data not shown). These transfected cells were utilized to evaluate the effect of RNA-induced silencing complex (RISC)-mediated degradation of viral RNA on the inhibition of viral spread and infectious particle production (Figure 1A). Twenty-four hours post-transfection, A253 cells were infected with a reporter oHSV vector encoding green fluorescent protein (GFP) fused to the HSV-1 glycoprotein C (gC-T2A-GFP).18 GFP intensity was monitored over time as a proxy for viral spread.
Figure 1.

Identification of HSV-1 Genes Essential to Viral Replication When Targeted With RISC-Loaded siRNAs
(A) A schematic representation of the experimental design is depicted; a pooled siRNA transfection assay was performed and HSV-1 replication (GFP reporter) was measured by array scanning cytometry. (B) Endpoint (48 hours post infection) GFP intensity per infected well is shown for each transfected siRNA pool. Levels of viral replication for the siRNA pools targeting the viral genes were compared to the negative siRNA. (C) Plaque titers are shown for control miRNAs in the left panel, and siRNA pools for the 3 attenuation candidates ICP27, ICP4, and UL8 in the right panel. GFP, green fluorescent protein; miRNAs, microRNAs; Neg, negative; siRNA, small interfering RNA; PFU, plaque-forming unit. One-way ANOVA Dunnett’s multiple comparisons test was used for statistical analysis. ∗p < 0.05; ∗∗p < 0.001; ∗∗∗p < 0.005; ∗∗∗∗p < 0.0001.
Our first-generation vector, ONCR-125, which encodes a miR-124 response element (miR-124T) consisting of four tandem copies of the reverse complement of mature miR-124-3p sequences in the 3′ UTR of the ICP4 gene, was used to infect A253 cells and transfected with hsa-miR-124-3p mimic to be used as our positive control for the screen. When we compared ONCR-125 in cells transfected with hsa-miR-124-3p mimic to the non-targeting control mimic, we observed a 95% reduction of viral spread and replication measured as GFP intensity (Figure 1B; p < 0.0001). Levels of viral replication in cells transfected with siRNA pools targeting various HSV-1 genes were compared to negative siRNA control. We first assessed the efficiency of knockdown for each siRNA pool with nanoString and qRT-PCR, and we observed almost complete suppression of all genes targeted with the exception of RL2 encoding ICP0 (50%), US1 encoding ICP22 (42%), UL42 (0%), and UL48 encoding VP16 (0%); therefore, these genes were not considered for miR-T inclusion (Figure S1). When siRNA pools were efficacious knocking down the expression of the HSV-1 genes, we also observed a significant reduction of viral spread and replication of 60% for UL54 encoding ICP27, 87% for RS1 encoding ICP4, 61% for UL5, 83% for UL8, 53% for UL9, 87% for UL29 encoding ICP8, 45% for UL 39/40, 79% for US1 encoding ICP22, and 79% for UL30. As expected for the HSV-1 genes where effective siRNA knockdowns were not observed, such as UL42 and VP16 (UL48), low or no inhibition of viral spread and replication was observed with these pools (Figures 1B and S1). Overall there were several genes where effective siRNA knockdown resulted in significant inhibition of viral replication, confirming that these HSV-1 genes are potential candidates for attenuation.
While sites that demonstrated significant reduction of viral replication were all considered appropriate sites to insert miR-T cassettes, we selected the 3′ UTR of UL8 for insertion as it shares a polyadenylation site with UL9, yielding to the potential repression of two HSV-1 essential gene transcripts with a single miR-T cassette.19 UL9 is the origin binding protein and UL8 is a subunit of the helicase-primase complex; these 2 proteins are essential for HSV-1 DNA replication and represent a functional block to DNA synthesis and late gene expression. To validate UL8 and the 2 other loci, ICP4 and ICP27, which were previously shown to be good candidates for viral attenuation when miRNA target sites were inserted in their 3′ UTR, we used a plaque titer assay to measure viral production after transfection with the corresponding siRNA targeting these genes.20,21 As shown in Figure 1C, almost complete inhibition of infectious virus particle production was observed when any of these 3 genes, ICP27 (UL54), ICP4 (RS1), and UL8, were silenced through repression of their transcripts.
Selection of miRNA Candidates for oHSV Attenuation
The design of the attenuation strategy for ONCR-159 required careful consideration of the tissues that are most permissive to HSV-1 replication and those that are most likely to be exposed to these viruses after intratumoral (IT) administration in tumors of interest, including squamous cell carcinoma of the head and neck (SCCHN), skin cancers, primary cancers, and metastases of the liver. Thus, our primary focus has been the protection of the central and peripheral nervous system, followed by protecting tissues including the liver, pancreas, and muscles that may be in the vicinity of tumors in indications of interest. To identify multiple miRNAs to include in miR-T cassettes to protect these tissues, we broadly expression profiled primary human normal and malignant tissues to select miRNAs that are specifically and highly expressed in healthy tissue but not in tumors.
Total RNA from snap frozen human normal (n = 110) and malignant tissues (n = 104) were prepared and profiled for miRNA expression (Tables S2–S4) using the nanoString miRNA panel v3, that allows the simultaneous measurement of 800 miRNAs from a single sample. The nanoString data was normalized, rank ordered for comparison, presented in a heat-map for several primary human tissues profiled compared to malignant tissues of various types (Figure 2A), and validated using qRT-PCR for a small panel of selected miRNA (Figure S2), demonstrating that the fold change miRNA levels in tissue measure by nanoString are similar to those measured by miRNA qRT-PCR. To assess the expression of miRNAs that could potentially protect the nervous system, we profiled brain, spinal cord, and peripheral nerves. Several miRNAs—hsa-miR-124-3p, hsa-miR-128-3p, hsa-miR-137-3p, hsa-miR-219a-5p, and hsa-miR-204-5p—were highly expressed specifically in the human brain (Figures 2B–2F). We also observed elevated levels of hsa-miR-204-5p and hsa-miR-219a-5p in spinal cord and elevated expression of hsa-miR-204-5p in nerves. In skeletal or cardiac muscle (Figure S3A), we found that hsa-miR-1-3p was highly expressed specifically in the heart, an organ potentially at risk if oHSV enters into the bloodstream. In organs with a high content of smooth muscle such as the lung and bladder, we observed high levels of hsa-miR-143-3p in these tissues; however, occasionally, a lower level signal was observed in malignant tissue presumably from contaminating margins during resection (Figure S3B).
Figure 2.

Global Expression Profiling of Human Normal and Malignant Tissue miRNAs
Tissue miRNA Profiling Identifies miRNAs for Host Tissue Protection. (A) Heatmap summary of miRNA ratios between the mean of the normal group to the malignant group was generated using the Broad Institute Morpheus software. Ratio of each candidate miRNA is normalized within its individual row to illustrate miRNA expression level for each tissue type. Counts in the red indicate high levels of miRNA present within that tissue type. Counts in blue indicate reduced levels of miRNA present in that tissue type. In (B) hsa-miR-124-3p, (C) hsa-miR-128-3p, (D) hsa-miR-137-3p, (E) hsa-miR-219a-5p, (F) hsa-miR-204-5p individual neuronal nanoString counts for each miRNA are compared to each malignant tissue profiled. Brain and spine are significantly higher than malignant tissues for all 5 neuronal miRNAs. (G) Individual nanoString miRNA counts in liver are compared to each malignant tissue profiled. Normal liver tissue is significantly higher across all malignant tissues.
Liver tissue was also a priority for profiling as image-guided repeated injection of either liver metastases or primary hepatocellular carcinoma is clinically feasible.22 We determined that hsa-miR-122-5p was highly expressed in normal liver tissue (Figure 2G) as previously shown23,24 compared to tumor tissue. Specifically, the expression level was 12,309-fold (p < 0.0001) over the average expression in colorectal cancer (CRC) samples and pancreatic cancer samples, 2 tumor histologies that are likely to metastasize to the liver. We also profiled several samples from normal human pancreas and identified hsa-miR-217-5p as being highly expressed (Figure S3C); therefore, it was also included in our cassette design to expand normal attenuation to the pancreas. In addition, hsa-miR-126-3p is known to be highly expressed by endothelial cells and has been considered in the attenuation strategy to prevent the spread of oHSV from the vasculature.25,26 Hsa-miR-126-3p is broadly expressed in healthy tissues, particularly in the heart, but was also detected in tumor tissues (Figure S3D); we reasoned that this signal likely originated from tumor margins or vasculature. For these selected miRNAs, there is substantial support for both their magnitude and tissue specific expression in the literature.27,28
Despite the fact that all these human mature miRNAs are completely conserved in the mouse (Table S5), we profiled miRNA expression in normal mouse tissue to ensure that they recapitulate both the tissue specificity and magnitude of miRNA expression found in human tissues. We performed murine nanoString miRNA panels for all 216-overlapping miRNA with 100% identical sequence (Figures S4A–S4D). To further examine miRNA enriched in each tissue type profiled, we compared human and murine miRNA expression for heart, liver, spine, and brain (Figures S5A–S5D) with all miRNA selected for inclusion into ONCR oHSV vector, and demonstrated that all these 10 miRNAs have similar tissue specificity and expression levels when comparing mouse to human tissues. In summary, our profiling data led us to select 10 miRNA candidates that demonstrated low or no expression in malignant tissue. By contrast, they were significantly highly expressed in human and murine normal tissues such as central/peripheral nervous system, heart/skeletal muscle, liver, lung, and pancreas (Table 1; Figure 2A).
Table 1.
miRNA Profiling Data Summary and Statistics
| miRNA | Tissue (Central & Peripheral Nervous System) | Mean Normala | Mean Malignantb | FC/Malignantc | p Valued |
|---|---|---|---|---|---|
| hsa-miR-124-3p | brain | 7,502.2 | 53.9 | 139.2 | 0.0001 |
| spine | 361.6 | 6.7 | 0.0611 | ||
| nerve | 4.6 | 0.1 | 0.7335 | ||
| hsa-miR-128-3p | brain | 7,505.3 | 49.8 | 150.8 | 0.0001 |
| spine | 420.6 | 8.4 | 0.0001 | ||
| nerve | 10.5 | 0.2 | 0.5145 | ||
| hsa-miR-137 | brain | 2,531.7 | 61.9 | 40.9 | 0.0001 |
| spine | 278.1 | 4.5 | 0.0265 | ||
| nerve | 43.8 | 0.7 | 0.832 | ||
| hsa-miR-204-5p | brain | 1,612.7 | 225.2 | 7.2 | 0.0001 |
| spine | 7,128.9 | 31.7 | 0.0001 | ||
| nerve | 1,762.1 | 7.8 | 0.0011 | ||
| hsa-miR-219a-5p | brain | 11,430.9 | 85 | 134.5 | 0.0001 |
| spine | 11,847.8 | 139.4 | 0.0001 | ||
| nerve | 7.9 | 0.1 | 0.6602 | ||
| miRNA | Tissue (Other Host Tissues of Interest) | Mean Normala | Mean Malignantb | FC/Malignantc | p Valued |
| hsa-miR-1-3p | oral cavity | 29,025.7 | 249 | 116.6 | 0.0001 |
| heart | 98,978.5 | 397.5 | 0.0001 | ||
| hsa-miR-122-5p | liver | 435,875.4 | 4,801.2 | 90.8 | 0.0001 |
| hsa-miR-126-3p | bladder | 6,898.4 | 10,831.8 | 0.6 | 0.2592 |
| lung | 28,685.4 | 2.6 | 0.0001 | ||
| brain | 14,516.7 | 1.3 | 0.4214 | ||
| liver | 19,348.1 | 1.8 | 0.1077 | ||
| heart | 102,967.6 | 9.5 | 0.0001 | ||
| nerve | 27,449.8 | 2.5 | 0.0119 | ||
| oral cavity | 13,779.6 | 1.3 | 0.5232 | ||
| spine | 14,236.6 | 1.3 | 0.6379 | ||
| pancreas | 3,631.8 | 0.3 | 0.1065 | ||
| vein | 12,678.6 | 1.2 | 0.756 | ||
| artery | 9,273.8 | 0.9 | 0.852 | ||
| skin | 15,504.1 | 1.4 | 0.5284 | ||
| colon | 11,710.1 | 1.1 | 0.8925 | ||
| hsa-miR-143-3p | bladder | 16,787 | 2,182 | 7.7 | 0.0001 |
| heart | 5,070.1 | 2.3 | 0.2215 | ||
| nerve | 11,945.3 | 5.5 | 0.0017 | ||
| oral cavity | 17,442.7 | 8 | 0.0124 | ||
| vein | 9,466.5 | 4.3 | 0.0017 | ||
| artery | 12,753.9 | 5.8 | 0.0045 | ||
| colon | 15,590.1 | 7.1 | 0.0003 | ||
| hsa-miR-217 | pancreas | 5,636.7 | 27.9 | 202.3 | 0.0001 |
FC, fold change.
Mean miRNA counts for each relevant normal tissue for each individual miRNA.
Mean miRNA counts for all malignant tissues for each individual miRNA.
Fold change over malignant (FC/malignant) was calculated by dividing the mean normal count over the mean of all malignant tissues for each individual miRNA.
The p value was calculated using a two-tailed parametric paired t test between each normal tissue miRNA count versus all malignant for each individual miRNA.
Design of a Next-Generation miR-T Cassette
We developed the miR-T cassette design pipeline in silico (Figure S6A) with the top 10 miRNA candidates selected. First, we wanted to insert tissue-specific miRNA target sequences such that they are not repeated back-to-back to avoid RISC steric hindrance. Then, since some tissue-specific miRNAs are not co-expressed in the same cell type (i.e., mutually exclusively), we used these candidates as spacers for their neighbors’ target sequences by interleaving these targets.29 These miR-T cassettes were also designed to circumvent large repeats to avoid viral genomic recombination and to remove tumor-expressed miRNA (“OncomiR”) seed targets and canonical polyadenylation sites (Figure S6A). It is also well known that RISC is minimally effective in highly structured target regions; thus, we predicted the most appropriate structure cassettes and selected those with minimal secondary structure. These tasks were automated in silico with a script (Figure S6A, see Methods) and yielded cassettes of approximately 315 nucleotides designed for the 4 HSV-1 loci of interest, ICP4, ICP27, UL8, and ICP34.5. Each of these miR-T cassettes contains 3 miRNA cognate targets repeated 4 four times with 4 base spacers.
To validate the function of the miR-T cassette, we employed a dual luciferase assay that evaluates the ability of each individual miRNA to inhibit the expression of a renilla luciferase normalized for transfection efficiency with firefly luciferase (psiCHECK-2, Promega) in cells transfected with miRNA mimics, which are small chemically modified double-stranded mature miRNA designed to mimic endogenous miRNA.30 Each miRNA mimic inhibited luciferase expression within each miR-T cassette, ranging from 47%–75% for miR-T-UL8 (p = 0.0025), 46%–87% for miR-T-ICP4 (p = 0.0521), 34%–48% for miR-T-ICP27 (p = 0.0319), and 33%–66% for T-ICP34.5 (p = 0.103), indicating that this strategy confers potent inhibition of gene expression in vitro (Figures S6B–S6E).
Development of the Next-Generation oHSV Vectors ONCR-157 and ONCR-159
We derived ONCR-157 and ONCR-159 from a parental vector, ONCR-125. ONCR-125 is a replication-attenuated HSV-1 that differs from the KOS strain with the following modifications (shown in Figure 3A): deletion of the joint region, the introduction of a gateway recombination cassette in the UL3/UL4 intergenic region for facilitating transgene insertion, a null mutation in US12, to promote antigen presentation on MHC-class I, the mutation of amino-acids D285N and A549T in the fusogenic glycoprotein B (gB) to enhance viral entry, GFP fused to glycoprotein C (gC) via a T2A peptide sequence, and insertion of a single miR-T-124 insertion in the 3′ UTR of the essential gene ICP4.16,18,31, 32, 33 ONCR-157 and ONCR-159 were generated by sequential modification ONCR-125 as described in Figure 3A. miR-T cassettes were inserted in the 3′ UTR of ICP4, ICP27, ICP34.5, and UL8 genes. Deletion of GFP gene from gC gene restored KOS wild-type sequence. Null mutations were introduced in ICP47. To combine this modality with mutations shown to eliminate retrograde transport to the central nervous system (CNS), we produced ONCR-157 and ONCR-159, 2 oHSV-1 variants of our base vector with either miRNA attenuation alone (ONCR-157), or miRNA attenuation and UL37 mutagenesis of the R2 region as described by Richards et al.15 (ONCR-159; Figure 3A).
Figure 3.

ONCR-159 Incorporates Multiple Modifications Including Next-Generation miR-T Cassettes That Are Efficiently Attenuated by Multiple miRNAs
(A) Depicts the vector maps of the predecessors leading to ONCR-159. ONCR-159 contains 4 miR-T cassettes and the UL37 mutations. (B) Schematic representation of the overall miRNA attenuation strategy including tissue protected, miRNAs selected from each tissue group, and the HSV-1 gene of interest where the miR-T has been inserted. Green represents the CNS, orange smooth, and cardiac/skeletal muscle, and blue soft tissue (e.g., liver and pancreas) protection. (C) ONCR-159 viral replication in miRNA-mimic pretreated A253 cells is shown as a ratio of the negative control mimic (PFU/neg). The miRNA mimics is labeled on the x-axis. Statistical significance was calculated using Bonferroni-Dunn Student’s t-test, ∗p = 0.02, ∗∗∗∗p < 0.00001. CNS, central nervous system; PFU, plaque forming units; Neg, negative.
Single miRNA Expression Is Sufficient to Attenuate or Eliminate Viral Replication
To evaluate the ability of each miRNA to attenuate viral replication functionally, and to confirm our overall miRNA attenuation strategy (Figure 3B), we developed a miRNA mimic assay that reproduces physiological conditions. ONCR-159 replication was inhibited entirely in A253 cells transiently transfected with miR-124-3p, miR-143-3p, miR-219a-5p, miR-122-5p, or miR-137-3p mimics. In cells transfected with miR-1-3p, miR-128-3p, miR-217-5p, and miR-126-3p mimics, ONCR-159 replication was attenuated 95%, 80%, 90%, and 80%, respectively (p = 0.02 for miR-128-3p and p < 0.00001 for all other miRs; Figures 3C and S7). To confirm that transfection of miRNA mimics results in physiologically relevant intracellular levels, we compared A253 mimic-transfected cells to tissue levels with miRNA-specific Taqman assays. We demonstrated that the expression level of neuronal miRNAs hsa-miR-124-3p, hsa-miR-128-3p, and hsa-miR-137-3p in A253 mimic-transfected cells were below those observed in primary human tissue (Figure S8), supporting the physiological relevance of the findings with the mimic assay.
ONCR-159 Is Resistant to Type I IFN and Comparably Lytic to Their Predecessors
The purpose of developing a virus that is conditionally replicating based on miRNA expression has been to retain a copy of the ICP34.5 gene and enable replication in the presence of a residual antiviral type I IFN response. We compared the replication of our base vectors ONCR-125, ONCR-157, and ONCR-159 to an established oHSV-1 vector lacking both copies of ICP34.5 (G207) in the presence of type I IFN challenge.5 For this test, we used 2 cancer cell lines that were previously confirmed to be capable of responding to IFN-α by monitoring Stat1 phosphorylation and interferon stimulated gene 15 (ISG15) expression when pretreated with 500U IFN-α, and robust IFN signaling and ISG induction were observed (Figure 4A, left panels). Next, these cell lines were infected with either G207, ONCR-157, or ONCR-159 (ICP34.5+; Figure 4A, right panels). We observed that in NCI-H1299 cells pretreated with IFN-α, ONCR-159 replication decreased by ∼4- to 6-fold. By contrast, the replication of G207 was completely inhibited. A similar phenotype was observed in NCI-H1975 tumor cell line.
Figure 4.

The Sum of All Modifications of ONCR-159 Does Not Perturb Oncolytic Efficacy and IFN-α Resistance In Vitro
In (A), H1299 and H1975 cells are shown in the upper and lower panels, respectively. Within each upper and lower panel, IFN pretreatment western blots for p-STAT1 and ISG15 are shown on the left and normalized plaque titer yields for mock or pretreated cells are shown on the right. (B) IC50 traces for the 3 vectors are depicted in (A) for the A375, SW837, FaDU, SKMEL-28, COLO-205, and SSC25 cells. IC50 values are shown in tabular format below the traces. IFN, interferon; MOI, multiplicity of infection.
As a result of ongoing, extensive modification of these backbones, we assessed whether these modifications would impact viral replication and oncolysis. Using a cell viability assay (CellTiter-Glo Luminescent Cell Viability Assay Kit, Promega, Madison, WI, USA) in the cancer cell lines A375 (melanoma), SK-MEL-28 (melanoma), COLO-205 (CRC), SW837 (CRC), FaDu (SCCHN), and SCC25 (SCCHN), the half-maximal inhibitory concentration (IC50) was measured. The oncolytic potential of ONCR-157 or ONCR-159 compared to the starting joint deleted virus ONCR-125 was not altered (Figure 4B). Furthermore, based on these in vitro IC50 values, the UL37 R2 domain mutation did not impact oncolytic activity in vitro.
UL37 R2 Mutation Does Not Affect In Vivo Viral Spread or Anti-Tumor Activity
Mutant UL37 was associated with a small plaque-size phenotype;15 therefore, we tested whether the mutations introduced into the R2 domain of UL37 in ONCR-159 would compromise its ability to infect tumor cells in vivo and to mediate anti-tumor responses. BALB/c mice bearing oHSV-1 sensitive A20 lymphoma subcutaneous tumors were administered a single IT dose (3 × 106 plaque forming units [PFU]) of either ONCR-157 or ONCR-159. Tumors were harvested at 24, 48, and 72 h post dose, formalin fixed, and examined for HSV-1 antigen by immunohistochemistry (Figure 5A). HSV-1 positively stained cells were generally noted across samples, with the most robust immunoreactivity noted at 48 h post-dose. HSV-1 immunoreactivity was similar and not significantly different for ONCR-157 versus ONCR-159.
Figure 5.

UL37 Mutation Does Not Affect In Vivo Antigen Spread or Anti-Tumor Efficacy
(A) A single 3 × 105 PFU IT dose of ONCR-157 or ONCR-159 was administered to established A20 subcutaneous tumors. Formalin-fixed tumors were analyzed at various time points after dosing for viral antigen using a polyclonal anti-HSV-1 sera. Images represent 48 h post-dose. Insets are magnifications of positively stained regions. (B) In vivo efficacy of ONCR-157 and ONCR-159 in the A20 syngeneic tumor model. Tumor volume curves ONCR-125, ONCR-157, and ONCR-159 differed significantly from that of PBS (p ≤ 0.0001 in all cases, by two-way ANOVA). Differences between ONCR-125, ONCR-157, and ONCR-159 curves were not significant. (B) Longitudinal group mean tumor volume curves after IT administration of PBS or 3 × 105 PFU of ONCR-125, ONCR-157, or ONCR-159. Dosing occurred on days 1, 4, and 7; n = 10 mice/group. IT, intratumoral; PBS, phosphate buffered saline; PFU, plaque forming units.
Next, the anti-tumor activity of ONCR-157 and ONCR-159 was evaluated in the A20 tumor model after 3 IT doses of ONCR-125, ONCR-157, or ONCR-159 (3 × 105 PFU) administered on days 1, 4, and 7. All oHSVs were similarly efficacious compared to PBS control, resulting in significant tumor growth inhibition or tumor regressions (Figures 5B and 5C; Table 2). Similar results were obtained from 2 other mouse syngeneic tumor models (Figure S10; Table 2). Taken together, these results suggest that UL37 R2 mutation does not negatively affect in vivo antigen spread or anti-tumor activity of ONCR-159.
Table 2.
Summary of In Vivo Anti-Tumor Activity of ONCR-157 and ONCR-159
| Model | Treatment | Dose (PFU) | Anti-Tumor Efficacy |
||
|---|---|---|---|---|---|
| % RR (PR; CR) | % TGI | p Value | |||
| A20 | PBS | – | 0 (0;0) | – | – |
| ONCR-157 | 3 × 105 | 90 (1;8) | 86 | ≤0.0001 | |
| ONCR-159 | 3 × 105 | 70 (4;3) | 86 | ≤0.0001 | |
| MC38 | PBS | – | 0 (0;0) | – | – |
| ONCR-157 | 3 × 106 | 0 (0;0) | 39 | 0.04 | |
| ONCR-159 | 3 × 106 | 10 (1;0) | 71 | ≤0.0001 | |
| B16F10N1 | PBS | – | 0 (0;0) | – | – |
| ONCR-157 | 6 × 106 | 25 (2;0) | 87 | ≤0.0001 | |
| ONCR-159 | 6 × 106 | 63 (5;0) | 97 | ≤0.0001 | |
–, not assessed; CR, complete response; PBS, phosphate buffered saline; PFU, plaque forming units; PR, partial regression; RR, response rate (PR + CR); TGI, tumor growth inhibition, relative to last day most of PBS control group was still present (day 17 for A20 and MC38, and day 12 for B16F10N1). PR and CR values are based on the entire duration of the study. Animals (n = 10 per group for A20 and MC38, n = 8 per group for B16F10N1 were dosed intratumorally on days 1, 4, and 7, in which day 1 is considered the 1st day of dosing. For the statistical analysis of efficacy data, a mixed linear model was used for A20 and MC38 until day 21 and B16F10N1 until day 12. p values are in reference to the PBS control. Analyses were performed using two-way ANOVA and Tukey’s multiple comparisons post-hoc test.
Systemic Exposure of ONCR-159 Is Well Tolerated in HSV-1 Permissive Mice
ONCR-159 base vector has been primarily designed for the purpose of engineering armed oncolytic HSV-1 for the treatment of subcutaneous and visceral lesions. Repeat IT injections of subcutaneous tumors were well tolerated at dose levels up to 6 × 106 PFU/injection, as shown by the continuous body weight increase in these mice and absence of lethality (data not shown). However, the duration of these studies was short (30 days maximum) due to the observed tumor burden in the control arm group. ONCR-159 exposure was limited to the injected tumor, as shown by qPCR to detect HSV-1 genome (Figure S9). Therefore, dedicated safety studies were performed to compare the tolerability of intravenous and intrahepatic injections of ONCR-159 and its originating wild-type KOS strain. These studies were conducted using the BALB/c mouse strain that is known to be permissive to HSV-1.34,35 A single administration of 5 × 106 PFU KOS in 8 mice recapitulated the known toxicities associated with wild-type HSV-1 infection. Intravenous dosing with KOS at 5 × 106 PFU was poorly tolerated, with most animals sacrificed in a moribund state between days 8 to 10. Clinical observations included emaciation, tachypnea, tremors, dehydration, hunched posture, impaired movement, and hind limb paralysis. Multifocal gliosis, often associated with a small number of individual necrotic, possibly neuron, cells, and perivascular lymphohistiocytic inflammation in the brainstem, were observed in animals surviving until scheduled sacrifice. Since female mice were shown to be more sensitive to KOS, a follow-up study investigating ONCR-159 used only females. Intravenous administration of ONCR-159 (1 × 107 PFU) administered 3 times weekly (3 × 107 PFU in total) was shown to be well tolerated with female mice (n = 4) living without any notable clinical observation until scheduled sacrifice. No pathologically significant macroscopic or microscopic findings were observed. Intrahepatic injection in BALB/c mice were also performed to evaluate the potential for ONCR-159 vector for intralesional treatment of metastatic disease in the liver. All 4 females survived intrahepatic injection of 1 × 106 PFU KOS and 1 × 107 PFU of ONCR-159 until scheduled sacrifice on day 8, but 2 of the 4 animals dosed with KOS developed clinical symptoms, including hind limb paralysis, convulsion, and dyspnea. No clinical observation was noted in the ONCR-159 treated group. Histological findings were limited to the liver and 1 or 2 foci of hepatocellular necrosis found in close association with local inflammation, likely a consequence of the passage of the injection needle. These data indicate that in an HSV-1 permissive mouse model in which intravenous or intrahepatic KOS leads to neuropathy, the miRNA attenuation and UL37 mutations improved the tolerability of ONCR-159 and protected the mice from HSV-1 induced neuropathy.
Discussion
First generation oncolytic vectors included deletions of type I IFN resistance genes, such as ICP34.5 or genes that enable replication in quiescent cells such as ribonucleotide reductase, in order to attenuate viral replication in normal cells.36 Although groundbreaking, these vectors showed limited efficacy in the clinic, possibly from the attenuation of replication in tumors with residual intrinsic or extrinsic (from myeloid stromal cells) IFN signaling. The central assumption underlying these viral attenuation approaches was that tumor cells were generally defective for IFN response and/or that the majority of the cells within a tumor were dependent on nucleotide pools for proliferation or DNA repair. However, it is known that a majority of cells in a typical tumor, including tumor initiating cells, are quiescent and that many tumor cells express IFN or are, by default, in an antiviral state.1, 2, 3, 4 In some cases, this results from the constitutive expression of type I IFN downstream of ADAR1 for example, or in others, from the constitutive expression of ISGs.13,14 Here, we show that our vector replicates even in the presence of excessive IFN-α, whereas the well-established ICP34.5 deleted oHSV vector, G207, does not. These in vitro results suggest higher potency and may result in greater in vivo efficacy in tumor models and patients.
To increase tumor-selective viral replication, we took advantage of the nearly general loss of expression of tissue-specific miRNA in cancer and inserted miR-T elements that are complementary to highly expressed and RISC-loaded cell-type-specific miRNA to restrict viral replication to cancer cells. This strategy has been successfully applied to gene therapy vectors or first generation oHSVs.18,20,37 However, these vectors typically contained few miRNAs or were well tolerated in their unmodified state. Here, we have expanded this paradigm to include many tissues and cell types specific miR-T sequences in the context of a more potent vector that we have shown to be IFN-α resistant. Specifically, we extensively profiled miRNA and selected 10 miRNA candidates with high differential expression in tumor and normal tissue. Further, we have developed an in silico design pipeline to produce miR-Ts that are highly susceptible to Ago2 cleavage resulting from efforts to reduce RNA secondary structure, and we have eliminated seed targets for known miRNAs that are expressed in tumor cells (OncomiRs). These cassettes are highly effective in vitro (Figure 3), and any of the miRNAs included in these cassettes are capable of strongly, if not completely, inhibiting viral production assessed by plaque titer. If we consider the case of neuronal tissues, miR-124-3p, miR-128-3p, and miR-137-3p are expected to be highly expressed in all differentiated neurons and loaded in the RISC prior to the infection; any one of these miRNAs would be repressing ONCR-159 at the onset of infection. Considering this possibility, the conservative interpretation of the strong attenuation mediated by single mimics (Figure 3C) suggests that this outcome might be an underestimate of the actual repression that would be observed in primary cortical/peripheral neurons where multiple RISC loaded miRNAs (miR-124-3p, miR-128-3p, miR-137-3p) would be repressing multiple essential HSV-1 genes simultaneously. Overall, in addition to the focus on the nervous system, ONCR-159 was engineered to respond to miRNAs specific for smooth/cardiac/skeletal muscle and liver to protect those tissues from potentially high viral production during tumor lysis. Data from high dose IV infusion shows that this strategy protects normal tissues from undesired viral replication, as is evidenced by survival relative to HSV-1 KOS and the lack of toxicity. This vector is also well tolerated when directly injected in the liver, expanding the potential clinical applications to hepatic lesions.
Escape from the mechanisms driving tumor versus normal tissue selectivity is a concern for any replication competent vector. In consideration of this, we engineered tissue-specific redundancy into the miR-T platform to minimize the probability of selecting oHSV mutants that may have mutations in these cassettes. We have built redundancy into each cassette with multiple elements of each miR-T per cassette. For the attenuation specifically in the nervous system miR-Ts for miRNAs expressed in neurons were built into all 3 essential gene cassettes and in ICP34.5 (Figure 3B). ONCR-159 cassettes were stable during the production of this viral vector as assessed by next-generation sequencing, and no evidence of loss or mutagenesis during scaling or production of an armed vector derived from ONCR-159 was observed.
The retrograde transport from the periphery into the CNS is a general concern with HSV-1 vectors, especially when they are delivered at a high intratumorally dose. We made use of the series of mutations in the HSV-1 UL37 gene described by Richards et al.15 that completely ablates retrograde transport into the CNS in the context of a corneal scratch model and incorporated these mutations to enhance the safety strategy of ONCR-159. This additional modification adds further redundancy to the miRNA attenuation platform and are expected to prevent migration of ONCR-159 into the CNS.
Despite the extensive modification of our oHSV vector, we observed efficient oncolysis in vitro in a panel of human cancer cell lines (Figure 4B), indicating that these modifications did not disrupt essential aspects of the viral life cycle or its oncolytic activity. This data is reinforced by antigen spread and antitumor activity observed in vivo after IT administration in tumors established from murine tumor cell lines ranging in HSV-1 sensitivity from completely sensitive (A20, Figures 5B and 5C) to refractory (B16F10N1, Figure S10B). The antitumor efficacy in combination with the fact that this vector is well tolerated at high doses when administered via multiple routes, warrants the development of this vector for clinical use. However, in studies with contralateral tumors, we observed modest activity at best, which serves as a rationale for the selection and inclusion of the immunomodulatory payloads. This vector can accommodate >20 kb of payload cassettes, differentiating this oncolytic viral vector from other viral vectors currently in development. This large cargo capacity permits this vector to be armed by multiple transgenes, far beyond the capacity observed for other HSV-1 oncolytic viruses, or other smaller RNA viruses. The selection of immunomodulatory payloads is currently underway and will provide a vehicle for their expression to enhance the immune stimulatory potential of the virus. In conclusion, ONCR-159 and vectors that are fully armed with immunomodulatory payloads will provide potent therapeutics to improve outcomes for patients with advanced disease.
Materials and Methods
Cell Lines and Oncolytic Viruses
HEK293T (CRL-3216, ATCC Manassas, VA, USA), A253 (HTB-41, ATCC), Vero (CCL-81, ATCC), A375 (CRL 1619, ATCC), and SKMEL-28 (HTB-72, ATCC) cells were grown in high-glucose DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37°C, 100% humidity, and 5% CO2. COLO 205 (CCL-222, ATCC), NCI-H1299, NCI-1975, and MC38 cells were grown in RPMI-1640 supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C, 100% humidity, and 5% CO2. SW837 cells (CCL-235, ATCC) were grown in Leibovitz L-15 supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C, 100% humidity, and 5% CO2. FaDu (HTB-43, ATCC) cells were grown in EMEM supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C, 100% humidity, and 5% CO2. SCC25 (CRL-1628) cells were grown in DMEM+F12 supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C, 100% humidity, and 5% CO2. A20 (TIB-208, ATCC) cells were maintained in vitro as a suspension culture in RPMI-1640 medium supplemented with 10% heat-inactivated FBS, 1% penicillin/streptomycin, and 0.05 mM β-mercapto-ethanol. Cell cultures were incubated in a humidified atmosphere with 5% CO2 at 37°C and sub-cultured once cell density reached 1 × 106 cells/mL. B16F10 (CRL-6475, ATCC) cells were stably expressed to include HSV-1 entry receptor Nectin 1 (B16F10N1) and grown in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C, 0.75 μg/mL puromycin at 100% humidity, and 5% CO2.
All genetic alterations leading to ONCR-157, and mONCR-159 were conducted on their respective full-length genome cloned in bacterial artificial chromosome (BAC). All modifications were confirmed by Sanger sequencing. The excision of the BAC vector sequence including the ori-replication and the antibiotic selection marker, was achieved by Cre loxP recombination and confirmed by X-gal staining of ONCR-157- or ONCR-159-infected cells. BAC-removed viruses were plaque purified in the Vero-SF (ATCC CCL-81.5) cell line prior to expansion of the viruses in Vero-SF cells, purification by centrifugation, determination of titer, and vialing.
Transient Transfection for siRNA Screen on MiniMax
A253 cells were seeded at 4 × 104 cells/well in 48-well plates and incubated for 24 h in a CO2 incubator. On target plus siRNA (GE Healthcare Dharmacon, Lafayette, CO, USA) were transfected using RNAiMax (Invitrogen, Thermo Fisher, Waltham, MA) according to manufacturer’s protocol at a final concentration of 6.7 nM in biological and technical triplicate. Each experiment also included a negative siRNA control #1 (GE #D-001810-01-05), a non-targeting sequence molecule that serves as a baseline for evaluation of the control and experimental siRNA on target gene expression.
Transient Transfection for Dual-Luciferase Assay
HEK293T cells were seeded at 1 × 104 cells/well in opaque 96-well plates and incubated for 24 h at 37°C in a CO2 incubator. mirVana miRNA mimics were co-transfected in triplicate with psiCHECK-2 (Promega, C8021) vectors using Lipofectamine 2000 (Invitrogen, Thermo Fisher, 11668-019) according to manufacturer’s protocol at the indicated final concentration for the mimic and 50 ng of plasmid DNA for the vector containing the miR-T cassette, in biological and technical triplicate. Each experiment also included a negative mirVana mimic miRNA control #1 (Invitrogen, Thermo Fisher, 4464058), a random sequence of miRNA mimic molecule that serves as a baseline for evaluation of the transcriptional regulation control and experimental miRNA mimic on target gene expression.38 A control negative (Neg) mimic with little or no off-target effect was provided by the manufacturer.
Dual-Luciferase Assays
To control for transfection efficiency, we used firefly luciferase (fluc) expression to normalize for Renilla luciferase (rluc) expression. The dual-luciferase assay was performed with the SpectraMax DuoLuc Reporter Assay Kit (Molecular Devices, San Jose, CA, USA, R8361). All reagents were prepared as described by the manufacturer. Twenty microliters of 1X passive lysis buffer was added to each well for 15 min at 250 rpm on a plate shaker. Using the SpectaMax i3x Dual-Luc injector cartridge, automated injection of 100 μL of the fluc reagent was added to each test sample, with a 2 s equilibration time and measurement of luminescence with a 5 s integration time, followed by addition of 100 μL of the rluc reagent and firefly quenching, 2 s equilibration time, and measurement of luminescence with a 5 s integration time. The data are represented as the ratio of firefly to Renilla luciferase activity (fluc/rluc). Fluc expression is controlled by the miR-T cassette, and rluc serves as the control for transfection efficiency. The data were normalized to the negative control (miR-T vector and negative mirVana mimic miRNA control #1, Invitrogen, Thermo Fisher, 4464058).
Transient Transfection of miRNA Mimics for Virus Infection
A253 cells were seeded at 4 × 104 cells/well in 48-well plates and incubated for 24 h in a CO2 incubator. mirVanamiRNA mimics were transfected using RNAiMax (Invitrogen, Thermo Fisher) according to the manufacturer’s protocol at a final concentration of 6.7 nM in biological and technical duplicate. Each experiment also included a negative mirVana mimic miRNA control #1 (Invitrogen, Thermo Fisher, 4464058), a random sequence miRNA mimic molecule that serves as a baseline for evaluation of the control and experimental miRNA mimic on target gene expression.38 This mimic with a random sequence has been tested in human cell lines and does not produce any off-target effects. All mimic sequences are listed in Table S6.
Virus Infection
After 24 h post transfection, A253 cells were enumerated and infected with ONCR-159 (Lot 18-159-058) with a multiplicity of infection (MOI) of 0.1 in a final volume of 250 μL (in DMEM). Infection was carried out in serum-free conditions with an absorption period of 1 h on a rocking platform. Plates were then incubated for a period of 3 days at 37°C in a CO2 incubator. To measure plaque titer for each condition, we froze plates at −20°C and freeze/thawed them 3 times. After the final thaw, each supernatant was transferred to 1.5 mL Eppendorf tubes and briefly vortexed, followed by centrifugation at 3,500 RPM for 5 min to pellet cell debris. Ninety-six well S blocks (QIAGEN, Germantown, MD, USA) were used to serially dilute each condition with serum-free media across a −1 to −6 log dilution range.
Virus Plaque Titer Assay
Vero cells were seeded in a 24-well format at 1 × 105 cells/well. After 24 h, infectious particles recovered from supernatants collected from the virus infection were added to these cells. Each condition was analyzed in biological triplicate and technical duplicate in serum-free conditions with a 100 μL volume for an absorption period of 1 h on a rocking platform. Post-absorption, 400 μL of complete media was added to a final volume of 500 μL/well. After 20 h, each well was aspirated and replenished with 500 μL complete media containing 2% human serum. The assay was conducted for a period of 8 days. Wells were aspirated and stained with crystal violet solution. Discrete plaque forming colonies were counted manually ≥5/well to determine ONCR-159 titer.
Tissue RNA Extraction
Human tissue samples were obtained from Cooperative Human Tissue Network (University of Pennsylvania, Vanderbilt University, The Ohio State University, University of Virginia, Duke University and Nationwide Children’s Hospital). Additional tissue samples were obtained from BioIVT (Westbury, NY, USA) and DxBio (San Diego, CA, USA). Tissue metadata is listed in Table S3. Tissues were rapidly snap frozen after harvest and were histologically characterized with respect to tissue composition.39 Subcellular distribution of these miRNAs in tissue has been characterized utilizing miRNA in situ hybridization, and references for these studies have been included in the appendix (Table S7). Twenty milligram sections were cut by scalpel and placed in a 1.5 mL Eppendorf tube and stored at −80°C.
For RNA isolation, each tissue sample was thawed on ice. Qiazol (QIAGEN) and chloroform were chilled on ice prior to use. Qiazol (700 μL) was added to each tube containing a tissue section, and disposable RNase-free pestles were used to homogenize the tissue. Subsequently, 140 μL of chloroform was added and centrifuged at maximum speed for 10 min. The upper aqueous phase (300 μL) was transferred to a new tube, and 450 μL of 100% ethanol at room temperature was then added and mixed. Approximately 700 μL of this mixture was transferred to a QIAGEN miRneasy column. The remaining steps in this process were performed according to the manufacturer’s protocol (QIAGEN miRneasy Kit, 217004). Each RNA sample in this report was analyzed by a NanoDrop spectrophotometer (Thermo Fisher Scientific, Wilmington, MA, USA) to confirm sample quality as recommended by nCounter miRNA Expression Assay User Manual.
nanoString Human miRNA Profiling
miRNA profiling using nanoString (NanoString Technologies, Seattle, WA, USA) was performed exactly as per the manufacturer protocol (nCounter miRNA Expression Assay User Manual). Each total RNA sample was analyzed in a separate multiplexed reaction. Briefly, unique oligonucleotide tags are ligated onto miRNAs, allowing short RNAs to be detected without amplification. Multiplexed hybridization of specific tags to their target miRNA, with a biotin capture probe and a unique reporter probe forming a Target-Probe complex being hybridized in solution. After hybridization, the hybridization mixture containing the target/probe complexes binds to magnetic beads complementary to sequences on the capture probe. Excess probes without a complementary target were removed using a two-step magnetic bead-based purification. Final purified target/probe complexes are eluted off the beads, immobilized, and aligned to the cartridge. Barcodes were read on a digital analyzer in a high-throughput automated fashion for up to 800 individual miRNAs per sample. Quantification was performed on a nanoString SPRINT instrument with the nanoString Human miRNA Panel 3.0.
All individual tissue samples were analyzed with the human miRv3 panel on the nanoString Sprint instrument. Sample preparation was performed as per the manufacturer recommendations, and the raw data quality control was conducted. Raw data were checked using the nSolver 4.0 software to ensure that (1) binding density was appropriate, (2) ligation controls were in the appropriate range, and (3) positive hybridization controls were correct. These analyses were performed with nSolver 4.0, and raw data were exported.
Datasets were normalized in nSolver by background thresholding the geometric mean across the negative controls, any samples outside of the 0.3 to 3 normalization factor range were omitted. Subsequently, normalization to the geometric mean of the positive controls was conducted to account for hybridization efficiency and was scaled to all endogenous and housekeeping genes to 1,000,000 total counts of the lane. Any samples for which the normalization factors were outside of the 0.1 to 10 range were omitted.
Lastly, every miRNA below 0.1% was excluded from analysis as it has been established in the field.40 Ratios between the mean of the normal group to the malignant group were calculated. Individual candidate miRNAs were then compared sample-by-sample, and an unpaired two-tailed t test was applied. A heatmap summary of the miRNA ratios was generated using Broad Institute’s https://software.broadinstitute.org/morpheus/ Morpheus software.
Cell Lines and Viral Infection for Interferon Sensitivity
Cells (1.0 × 105) in complete media supplemented with 5,000 IU/mL of recombinant human IFNα-2a per well were seeded onto 24-well format multiwell tissue culture plates.41 Each cell type was also seeded in complete media without IFNα-2a. On the following day, total media was aspirated out and substituted with 100 μL of OptiMEM containing ONCR-177 and G207 (each 0.03 MOI). The cells were treated with either mock or ONCR-177 or G207. Viruses were allowed to adsorb for a period of 60 min followed by substitution with 400 μL of complete media with and without IFNα-2a at 5,000 IU/mL 48 h post infection. Cells and conditioned media were collected and frozen at −80°C, until the viral titer was determined by virus plaque titer assay.
Western Blot Protocol
Cells plated at 2 × 105cells/well/2 mL media were plated in a 6-well plate the night before IFN-α treatment as described. After 30 min, media were removed, and cells were lysed and collected using 150 μL 1X NuPAGE LDS Sample Buffer (Invitrogen, Thermo Fisher). Lysates were stored at −80°C. Lysate samples were directly sonicated with a microtip at 20% amplitude 3X for 5 s before being incubated at 95°C for 5 min. Lysate samples (10 μL) were electrophoresed on a Nu-PAGE 4%–12% Bis-Tris gel with MOPS buffer at 150 V for 70 min and transferred to a membrane using an iBlot2 gel transfer system. The membrane was blocked with 5% milk in PBS-T, incubated with primary antibodies overnight at 1:1,000 dilution at 4°C, followed by secondary antibody incubations at 1:10,000 dilution for 60 min at 25°C. Antibody binding was detected using Immobilon Western Chemiluminescent HRP Substrate and imaged using Protein Simple Fluor Chem R. The following antibodies were used for western blot analysis: Stat1 (#14994), Phospho-Stat1 Tyr701 (#9167), ISG15 (#2758) from Cell Signaling Technologies (Danvers, MA, USA), and beta actin HRP (#ab20272) from Abcam (Cambridge, MA, USA).
Oncolysis and In Vitro Viability Assays
The oncolytic activity of ONCR vectors was determined by calculating the IC50 value for each virus in each cell line. The IC50 is determined by the MOI, achieving the killing of 50% of the input cells 72 h after infection as compared to mock-treated cells and as determined by CellTiter-Glo assay. CellTiter-Glo (#G9242) was obtained from Promega Corporation (Madison, WI, USA). Annexin V red reagent (#4641) was purchased from Essen Bioscience (Sartorius, Bohemia, NY, USA).
Briefly, 10,000 cells were seeded in 96-well tissue culture plates in their recommended complete media followed by overnight incubation at 37°C, 5% CO2. On the following day, culture medium was aspirated and replaced with 25 μL freshly prepared serial diluted (1:3) virus (ONCR vectors) in OptiMEM-reduced serum media, starting from MOI = 30 (9 points of dilution and a blank control in quadruplicates). An adsorption step was carried out for 1 h at 37°C, 5% CO2, by gentle rotating the multi-well plate on a shaker. After 1 h, 75 μL of complete culture media was added. The next day media containing viral inoculum were aspirated and substituted with 100 μL pre-warmed complete media.
In vitro viability assay was performed at 72 h post-infection by adding 100 μL of CellTiter-Glo 2.0 reagent and equilibrating (shaking) the plate at room temperature for 30 min. Total luminescence (RLU) was measured in a plate reader (Molecular Devices, Spectramax i3X minimax imaging cytometer), and data were acquired by Softmax Pro v7.0.2 from Molecular Devices (San Jose, CA, USA).
Raw data acquired by Softmax Pro v7.0.2 were reduced to numbers with 3 decimal points only. The values were then converted to percentage survival relative to mock (100%).
The values were graphed in GraphPad Software Prism 7.0 (San Diego, CA, USA) and analyzed using a non-linear sigmoidal plot with variable slope (asymmetric four-point linear regression) to generate IC50 values.
In Vivo Tumor Studies
Animal Care
Female BALB/c and C57BL/6 mice (Charles River Laboratories), 5 to 8 weeks of age, were cared for in accordance with the “Guide for the Care and Use of Laboratory Animals.” All protocols were approved by an Institutional Animal Care and Use Committee. Animals had ad libitum access to sterile pelleted feed and reverse osmosis-purified water and were maintained on a 12:12-h light:dark cycle with access to environmental enrichment opportunities.
A20 Syngeneic Tumor Model
A20 tumor cells were maintained in vitro as a suspension culture in RPMI-1640 medium supplemented with 10% heat-inactivated FBS, 1% penicillin/streptomycin, and 0.05 mM β-mercapto-ethanol. Cell cultures were incubated in a humidified atmosphere with 5% CO2 at 37°C and sub-cultured once cell density reached 1 × 106 cells/mL. BALB/c female mice that were 8 weeks of age weighing approximately 18 to 22 lbs were ordered under Oncorus Animal Protocol 001-2017. Animals were housed up to 5 mice per cage and received food and water ad libidum throughout the course of this study. Prior to tumor cell implantation, all mice were identified by ear-tag and tumor implantation sites were shaved. Each animal was inoculated subcutaneously with ≥90% viable 5 × 106 A20 cells (in 0.1 mL in serum-free Dulbecco's Phosphate Buffered Saline [DPBS] per implant) on the flank (0.1 mL in serum-free DPBS) for tumor development. For HSV-1 antigen spread studies, BALB/c mice with ∼50 mm3 A20 subcutaneous tumors were administered either ONCR157 or ONCR-159 intratumorally at 3 × 106 PFU. Tumors were harvested at 24, 48, or 72 h post-treatment, formalin-fixed, paraffin embedded, and sectioned into slides. Slides were stained for HSV-1 expression using an anti-HSV-1 rabbit polyclonal antibody APA 3027 AAK (Biocare Medical, Pacheco, CA, USA).
MC38 Syngeneic Tumor Model
MC38 tumor cells were cultured as a monolayer in vitro in DMEM supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin and maintained in a humidified atmosphere with 5% CO2 at 37°C. Cells were sub-cultured at 80% confluency. C57BL/6 female mice that were 8 weeks of age weighing approximately 18 to 22 lbs were ordered under Oncorus Animal Protocol 001-2017. Animals were housed up to 5 mice per cage and received food and water ad libidum throughout the course of these studies. Prior to tumor cell implantation, all mice were identified by a unique ear tag and tumor implantation sites shaved. Each animal was inoculated subcutaneously with ≥90% viable 5 × 105 MC38 cells (in 0.1 mL in serum-free DPBS per implant) on the flank (0.1 mL in serum-free DPBS) for tumor development.
B16F10 Tumor Model
To generate a B16F10 cell line that stably expresses HSV-1 entry receptor Nectin 1 (B16F10N1), we transduced B16F10 cells with lentivirus-pCDH-Nectin1 and clones selected in the presence of 0.75 μg/mL puromycin dihydrochloride. B16F10N1 tumor cells were maintained in vitro as a monolayer cell culture in DMEM medium supplemented with 10% heat-inactivated FBS, 1% penicillin/streptomycin, and 0.75 μg/mL puromycin. Cell cultures were incubated in a humidified atmosphere with 5% CO2 at 37°C and sub-cultured once at 80% confluency. C57BL/6 female mice that were 8 weeks of age weighing approximately 18 to 22 lbs were ordered under Oncorus Animal Protocol 001-2017. Animals were housed up to 5 mice per cage and received food and water ad libidum throughout the course of this study. Prior to tumor cell implantation, all mice were identified by a unique ear tag and tumor implantation sites were shaved. Each animal was inoculated subcutaneously with ≥90% viable 5 × 105 B16F10N1 cells (in 0.1 mL in serum-free DPBS per implant) on the flank (0.1 mL in serum-free DPBS) for tumor development.
In all in vivo studies, mice were lightly anesthetized with 5% isoflurane before implantation to ensure subcutaneous delivery of cells. Any animals harboring tumors growing intradermally (ID) or intramuscularly (i.m.) or with ulceration/necrosis were not enrolled into the study. Randomization, enrollment, and initiation of treatment began when an adequate number of mice with properly sized tumors were identified (median right flank tumor volume of approximately 100 ± 25 mm3 for studies in A20 and M38 tumor models and median right flank tumor volume of approximately 80 ± 30 mm3 for a study in B16F10N1 melanoma model). Animals were matched based on tumor volume size and randomly placed into groups. Doses were formulated according to the following formula:
where VS is volume of virus stock solution (μL), V is volume of virus preparation being prepared for dosing (μL), D is dose in PFU, CS is the concentration of the virus stock, and VI is volume of injection.
All viruses were formulated immediately before dosing and kept on ice until ready to be injected. A pre-specified volume of virus aliquot (VS) was diluted in a volume of sterile DPBS (VDPBS) and gently mixed. The injection of virus into the tumor tissue was performed using a sterile 31G insulin needle (bevel up) attached to a sterile syringe. Doses were administered as a single injection of 25 μL per tumor per dosing date. Mice received 3 IT injections of either PBS, ONCR-157, or ONCR-159 on days 1, 4, and 7.
Animals had ad libitum access to sterile pelleted feed and reverse-osmosis-purified water and were maintained on a 12:12-h light:dark cycle with access to environmental enrichment. Throughout the course of the efficacy study, the animals were monitored daily for adverse events and body weight was recorded twice per week. Tumor volumes, measured at least 2 times per week, were calculated using the following formula: TV = a × b2/c, where “a” is tumor length and “b” is tumor width. Mice were humanely euthanized once the combined tumor burden reached the endpoint value of >2,000 mm3. All animal protocols were approved by the Oncorus Institutional Animal Care and Use Committee (IACUC) and were performed in accordance with IACUC regulations.
Safety Studies
All safety studies used 5- to 8-week-old BALB/c mice (Charles River Laboratories, Wilmington, MA, USA). In one study, animals (4 per sex) were given a single intravenous dose of KOS at 5 × 106 or 1 × 107 PFU. Animals were thereafter followed or evaluated for clinical observations, body weight, and food consumption until study takedown on days 8 or 29 post-dose, upon which tissues (brain, liver, spleen, lungs, kidneys, and heart) were evaluated for pathology. In another study, repeat intravenous doses (on days 1, 8, and 15) of ONCR-159 at 1 × 107 PFU were administered. Animals were followed as described for the previous study, and take downs were on days 8 or 30, upon which tissues (brain, liver, spleen, lungs, kidneys, and heart) were evaluated for pathology. Finally, a cohort (4 female BALB/c mice per treatment group) was administered a single dose of vehicle control, KOS (1 × 106 PFU), or ONCR-159 (1 × 107 PFU) into the left lobe of the liver (intra-hepatic [IH]) and thereafter followed or evaluated for clinical observations, clinical chemistry, body weight, and food consumption, until study takedown on days 8. Tissues (brain, liver, spleen, lung, kidney, heart, and trigeminal ganglia) were subsequently evaluated for pathology.
Statistical Analysis
The in vitro data are presented as the mean ± standard deviation (SD). The in vivo data are presented as the mean ± standard error of the mean (SEM) The statistical tests for data are justified and the data meets the assumptions of the test and was performed using GraphPad Prism version 8.01 for Windows (GraphPad Software, La Jolla, CA, USA; https://www.graphpad.com/). Significance between specific datasets is described in the respective figure legends.
Author Contributions
Conceptualization, E.M.K., M.H.F., J.C.G., B.B.H., C.Q., and L.L.; Methodology, E.M.K., T.F., P.G., J.L., A.D., J.H., J.B., P.B., C.G., D.W.; Software, E.M.K.; Validation, E.M.K., T.F., and L.L.; Formal Analysis, E.M.K., T.F., A.D., A.D.A., J.J., D.D., B.B.H., J.B.R., C.Q., L.L.; Writing – Review & Editing, E.M.K., L.L., B.B.H., and C.Q.; Supervision, E.M.K., B.B.H., L.L, and CQ.; Project Administration, E.M.K., L.L., B.B.H., and C.Q.
Conflicts of Interest
E.M.K., T.F., P.G., J.L., A.D., J.H., J.B., P.B. (at the time the study was conducted), C.G. (at the time the study was conducted), D.W., A.D.A., J.J., M.H.F. (at the time the study was conducted), B.B.H., C.Q., and L.L. are all employees of Oncorus. D.D. and J.B.R. are paid consultants for Oncorus, and J.C.G. is Oncorus cofounder, paid consultant, and shareholder. All other authors declare no competing interests.
Acknowledgments
This study was sponsored by Oncorus, Inc. The authors would like to acknowledge Sofie Denies for expert statistical analysis and Michael Cristini and Jim Markert for providing G207 for comparison. The authors also wish to thank Chastity Bradley, PhD, of FBL ClinWriters for her editorial review of the manuscript and assistance with manuscript submission.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.08.004.
Supplemental Information
References
- 1.Diallo J.S., Le Boeuf F., Lai F., Cox J., Vaha-Koskela M., Abdelbary H., MacTavish H., Waite K., Falls T., Wang J. A high-throughput pharmacoviral approach identifies novel oncolytic virus sensitizers. Mol. Ther. 2010;18:1123–1129. doi: 10.1038/mt.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Felt S.A., Droby G.N., Grdzelishvili V.Z. Ruxolitinib and Polycation Combination Treatment Overcomes Multiple Mechanisms of Resistance of Pancreatic Cancer Cells to Oncolytic Vesicular Stomatitis Virus. J. Virol. 2017;91 doi: 10.1128/JVI.00461-17. e00461-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Y.P., Suksanpaisan L., Steele M.B., Russell S.J., Peng K.W. Induction of antiviral genes by the tumor microenvironment confers resistance to virotherapy. Sci. Rep. 2013;3:2375. doi: 10.1038/srep02375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moerdyk-Schauwecker M., Shah N.R., Murphy A.M., Hastie E., Mukherjee P., Grdzelishvili V.Z. Resistance of pancreatic cancer cells to oncolytic vesicular stomatitis virus: role of type I interferon signaling. Virology. 2013;436:221–234. doi: 10.1016/j.virol.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mineta T., Rabkin S.D., Yazaki T., Hunter W.D., Martuza R.L. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat. Med. 1995;1:938–943. doi: 10.1038/nm0995-938. [DOI] [PubMed] [Google Scholar]
- 6.Liu B.L., Robinson M., Han Z.Q., Branston R.H., English C., Reay P., McGrath Y., Thomas S.K., Thornton M., Bullock P. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10:292–303. doi: 10.1038/sj.gt.3301885. [DOI] [PubMed] [Google Scholar]
- 7.He B., Gross M., Roizman B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA. 1997;94:843–848. doi: 10.1073/pnas.94.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manivanh R., Mehrbach J., Knipe D.M., Leib D.A. Role of Herpes Simplex Virus 1 γ34.5 in the Regulation of IRF3 Signaling. J. Virol. 2017;91 doi: 10.1128/JVI.01156-17. e01156-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orvedahl A., Alexander D., Tallóczy Z., Sun Q., Wei Y., Zhang W., Burns D., Leib D.A., Levine B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 10.Kesari S., Lasner T.M., Balsara K.R., Randazzo B.P., Lee V.M., Trojanowski J.Q., Fraser N.W. A neuroattenuated ICP34.5-deficient herpes simplex virus type 1 replicates in ependymal cells of the murine central nervous system. J. Gen. Virol. 1998;79:525–536. doi: 10.1099/0022-1317-79-3-525. [DOI] [PubMed] [Google Scholar]
- 11.Therapeutic Goods Administration Australian Government Australian Public Assessment Report for Talimogene Laherparepvec. 2016. https://www.tga.gov.au/sites/default/files/auspar-talimogene-laherparepvec-160531.pdf
- 12.Kambara H., Saeki Y., Chiocca E.A. Cyclophosphamide allows for in vivo dose reduction of a potent oncolytic virus. Cancer Res. 2005;65:11255–11258. doi: 10.1158/0008-5472.CAN-05-2278. [DOI] [PubMed] [Google Scholar]
- 13.Gannon H.S., Zou T., Kiessling M.K., Gao G.F., Cai D., Choi P.S., Ivan A.P., Buchumenski I., Berger A.C., Goldstein J.T. Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nat. Commun. 2018;9:5450. doi: 10.1038/s41467-018-07824-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishizuka J.J., Manguso R.T., Cheruiyot C.K., Bi K., Panda A., Iracheta-Vellve A., Miller B.C., Du P.P., Yates K.B., Dubrot J. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature. 2019;565:43–48. doi: 10.1038/s41586-018-0768-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richards A.L., Sollars P.J., Pitts J.D., Stults A.M., Heldwein E.E., Pickard G.E., Smith G.A. The pUL37 tegument protein guides alpha-herpesvirus retrograde axonal transport to promote neuroinvasion. PLoS Pathog. 2017;13:e1006741. doi: 10.1371/journal.ppat.1006741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.York I.A., Roop C., Andrews D.W., Riddell S.R., Graham F.L., Johnson D.C. A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell. 1994;77:525–535. doi: 10.1016/0092-8674(94)90215-1. [DOI] [PubMed] [Google Scholar]
- 17.Uchida H., Chan J., Shrivastava I., Reinhart B., Grandi P., Glorioso J.C., Cohen J.B. Novel mutations in gB and gH circumvent the requirement for known gD Receptors in herpes simplex virus 1 entry and cell-to-cell spread. J. Virol. 2013;87:1430–1442. doi: 10.1128/JVI.02804-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mazzacurati L., Marzulli M., Reinhart B., Miyagawa Y., Uchida H., Goins W.F., Li A., Kaur B., Caligiuri M., Cripe T. Use of miRNA response sequences to block off-target replication and increase the safety of an unattenuated, glioblastoma-targeted oncolytic HSV. Mol. Ther. 2015;23:99–107. doi: 10.1038/mt.2014.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baradaran K., Dabrowski C.E., Schaffer P.A. Transcriptional analysis of the region of the herpes simplex virus type 1 genome containing the UL8, UL9, and UL10 genes and identification of a novel delayed-early gene product, OBPC. J. Virol. 1994;68:4251–4261. doi: 10.1128/jvi.68.7.4251-4261.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee C.Y., Rennie P.S., Jia W.W. MicroRNA regulation of oncolytic herpes simplex virus-1 for selective killing of prostate cancer cells. Clin. Cancer Res. 2009;15:5126–5135. doi: 10.1158/1078-0432.CCR-09-0051. [DOI] [PubMed] [Google Scholar]
- 21.Li J.M., Kao K.C., Li L.F., Yang T.M., Wu C.P., Horng Y.M., Jia W.W., Yang C.T. MicroRNA-145 regulates oncolytic herpes simplex virus-1 for selective killing of human non-small cell lung cancer cells. Virol. J. 2013;10:241. doi: 10.1186/1743-422X-10-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marabelle A., Kohrt H., Caux C., Levy R. Intratumoral immunization: a new paradigm for cancer therapy. Clin. Cancer Res. 2014;20:1747–1756. doi: 10.1158/1078-0432.CCR-13-2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fornari F., Gramantieri L., Giovannini C., Veronese A., Ferracin M., Sabbioni S., Calin G.A., Grazi G.L., Croce C.M., Tavolari S. MiR-122/cyclin G1 interaction modulates p53 activity and affects doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res. 2009;69:5761–5767. doi: 10.1158/0008-5472.CAN-08-4797. [DOI] [PubMed] [Google Scholar]
- 24.Gramantieri L., Ferracin M., Fornari F., Veronese A., Sabbioni S., Liu C.G., Calin G.A., Giovannini C., Ferrazzi E., Grazi G.L. Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res. 2007;67:6092–6099. doi: 10.1158/0008-5472.CAN-06-4607. [DOI] [PubMed] [Google Scholar]
- 25.Fish J.E., Santoro M.M., Morton S.U., Yu S., Yeh R.F., Wythe J.D., Ivey K.N., Bruneau B.G., Stainier D.Y., Srivastava D. miR-126 regulates angiogenic signaling and vascular integrity. Dev. Cell. 2008;15:272–284. doi: 10.1016/j.devcel.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang S., Aurora A.B., Johnson B.A., Qi X., McAnally J., Hill J.A., Richardson J.A., Bassel-Duby R., Olson E.N. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev. Cell. 2008;15:261–271. doi: 10.1016/j.devcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sempere L.F., Freemantle S., Pitha-Rowe I., Moss E., Dmitrovsky E., Ambros V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004;5:R13. doi: 10.1186/gb-2004-5-3-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lagos-Quintana M., Rauhut R., Yalcin A., Meyer J., Lendeckel W., Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 29.Ameres S.L., Martinez J., Schroeder R. Molecular basis for target RNA recognition and cleavage by human RISC. Cell. 2007;130:101–112. doi: 10.1016/j.cell.2007.04.037. [DOI] [PubMed] [Google Scholar]
- 30.Wang Z. The guideline of the design and validation of MiRNA mimics. Methods Mol. Biol. 2011;676:211–223. doi: 10.1007/978-1-60761-863-8_15. [DOI] [PubMed] [Google Scholar]
- 31.Früh K., Ahn K., Djaballah H., Sempé P., van Endert P.M., Tampé R., Peterson P.A., Yang Y. A viral inhibitor of peptide transporters for antigen presentation. Nature. 1995;375:415–418. doi: 10.1038/375415a0. [DOI] [PubMed] [Google Scholar]
- 32.Todo T., Martuza R.L., Rabkin S.D., Johnson P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA. 2001;98:6396–6401. doi: 10.1073/pnas.101136398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uchida H., Chan J., Goins W.F., Grandi P., Kumagai I., Cohen J.B., Glorioso J.C. A double mutation in glycoprotein gB compensates for ineffective gD-dependent initiation of herpes simplex virus type 1 infection. J. Virol. 2010;84:12200–12209. doi: 10.1128/JVI.01633-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujioka N., Akazawa R., Ohashi K., Fujii M., Ikeda M., Kurimoto M. Interleukin-18 protects mice against acute herpes simplex virus type 1 infection. J. Virol. 1999;73:2401–2409. doi: 10.1128/jvi.73.3.2401-2409.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sundaresan P., Hunter W.D., Martuza R.L., Rabkin S.D. Attenuated, replication-competent herpes simplex virus type 1 mutant G207: safety evaluation in mice. J. Virol. 2000;74:3832–3841. doi: 10.1128/jvi.74.8.3832-3841.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bommareddy P.K., Peters C., Saha D., Rabkin S.D., Kaufman H.L. Oncolytic Herpes Simplex Viruses as a Paradigm for the Treatment of Cancer. Annu. Rev. Cancer Biol. 2018;2:155–173. [Google Scholar]
- 37.Geisler A., Fechner H. MicroRNA-regulated viral vectors for gene therapy. World J. Exp. Med. 2016;6:37–54. doi: 10.5493/wjem.v6.i2.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang S., Li J., Li J., Yang Y., Kang X., Li Y., Wu X., Zhu Q., Zhou Y., Hu Y. Up-regulation of microRNA-203 in influenza A virus infection inhibits viral replication by targeting DR1. Sci. Rep. 2018;8:6797. doi: 10.1038/s41598-018-25073-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grizzle W.E., Gunter E.W., Sexton K.C., Bell W.C. Quality management of biorepositories. Biopreserv. Biobank. 2015;13:183–194. doi: 10.1089/bio.2014.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mullokandov G., Baccarini A., Ruzo A., Jayaprakash A.D., Tung N., Israelow B., Evans M.J., Sachidanandam R., Brown B.D. High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nat. Methods. 2012;9:840–846. doi: 10.1038/nmeth.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chee A.V., Roizman B. Herpes simplex virus 1 gene products occlude the interferon signaling pathway at multiple sites. J. Virol. 2004;78:4185–4196. doi: 10.1128/JVI.78.8.4185-4196.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.