

Summary
Brain metastasis is an ineffective process, and many cancer cells enter into an indolent state following extravasation in the brain. Single cell RNA sequencing of melanoma brain metastases reveals that non-proliferating brain metastatic melanoma cells exhibit a pattern of gene expression associated with inhibition of DNA methyltransferase 1 (DNMT1). The brain microenvironment, specifically the combination of reactive astrocytes and mechanically soft surroundings, suppressed DNMT1 expression in various cancer types and caused cell cycle delay. Somewhat unexpectedly, we find that DNMT1 suppression not only induces cell cycle delay but also activates pro-survival signals in brain metastatic cancer cells, including L1CAM and CRYAB. Our results demonstrate that transcriptional changes triggered by DNMT1 suppression is a key step for cancer cells to survive in the brain microenvironment and that they also restrict cancer cell proliferation. The dual consequences of DNMT1 suppression can explain the persistence of indolent cancer cells in the brain microenvironment.
Subject Areas: Biological Sciences, Cell Biology, Cancer, Transcriptomics
Graphical Abstract
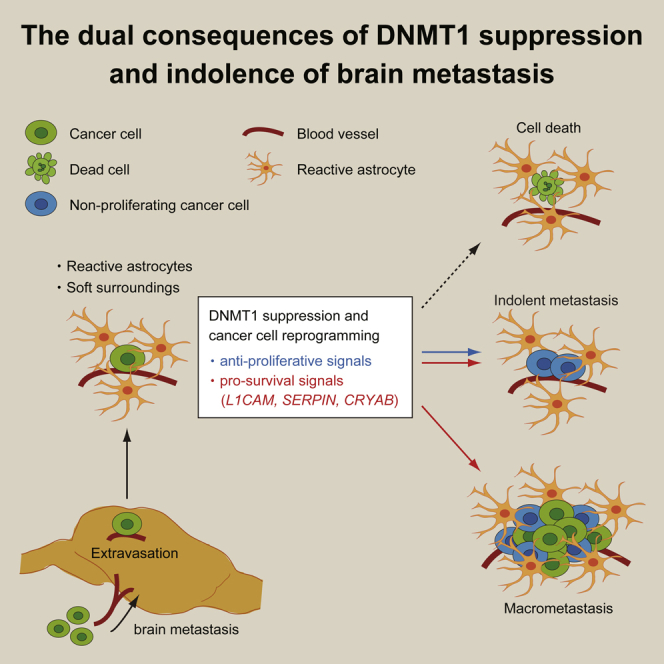
Highlights
-
•
Indolence of brain metastatic melanoma cells is associated with DNMT1 inhibition
-
•
Reactive astrocytes and mechanically soft surroundings suppress DNMT1 expression
-
•
DNMT1 suppression dually activates pro-survival and anti-proliferative signals
-
•
DNMT1-αB crystallin axis supports the survival of indolent brain metastatic cells
Biological Sciences; Cell Biology; Cancer; Transcriptomics
Introduction
Brain metastasis is a functionally devastating complication with a very poor prognosis, and the number of cases is greatly increasing in association with marked improvement of overall survival of patients with cancer (Smedby et al., 2009; Tabouret et al., 2012; Valiente et al., 2018). Melanoma, lung, and some sub-types of breast cancer have a high propensity for brain metastasis (Cagney et al., 2017). Traditional autopsy studies and recent advances in cancer genomics have revealed that brain metastasis can occur in the early stage of cancer progression, and the fate of these disseminated cancer cells is strongly dependent on the interaction with brain microenvironment (Brastianos et al., 2015; Ghajar, 2015; McAllister and Weinberg, 2014; Noltenius and Noltenius, 1985; Obenauf and Massague, 2015; Sosa et al., 2014). The brain microenvironment consists of specialized cell types such as neurons, astrocytes, oligodendrocytes, microglial cells, and brain capillary endothelial cells (Quail and Joyce, 2017; Winkler, 2015). These cell types can affect metastasis progression in a variety of ways; in particular, interaction with blood vessels is essential for cancer cells to survive in the early stage of brain metastasis (Er et al., 2018; Kienast et al., 2010; Valiente et al., 2014). The cerebrospinal fluid that permeates the brain can also influence the response of melanoma cells to therapy (Seifert et al., 2015).
The progression of brain metastasis is spatio-temporally heterogeneous among cancer types, clinical cases, or even different lesions in a patient (Cagney et al., 2017). In some cases, brain metastasis is evident soon after detection of the primary tumor, whereas in other cases, there can be a long period of remission before cancer recurrence and the development of brain metastases, a phenomenon called cancer dormancy (Sosa et al., 2014; Yeh and Ramaswamy, 2015). The causes of cancer dormancy and mechanisms of re-awakening are multi-factorial and can include differential interactions with components of the tumor microenvironment, such as endothelial cells and infiltrating leukocytes (De Cock et al., 2016; Ghajar et al., 2013; Montagner and Sahai, 2020). In the case of brain metastasis, Kienast et al. reported that VEGF-A inhibition can induce angiogenic dormancy (Kienast et al., 2010), and Malladi et al. showed that suppression of Wnt signaling by autocrine DKK1 secretion can cause immunogenic dormancy (Malladi et al., 2016). However, the mechanism of cellular dormancy in brain metastasis remains incompletely understood.
The majority of cancer cells that extravasate into the brain parenchyma die (Kienast et al., 2010; Winkler, 2015). One potential explanation is that plasmin from the reactive brain stroma kills metastatic cancer cells via conversion of a membrane-bound FasL to a soluble variant and/or direct degradation of L1CAM, and cancer cell-derived anti-plasminogen activator serpin families work as a shield against these cell death signals (Valiente et al., 2014). Other mechanisms also exist to protect disseminated cells from apoptotic cell death. For example, αB-crystallin, a small heat shock protein that directly binds to caspase-3 and inhibits apoptosis (Kamradt et al., 2002, 2005), has been linked to brain metastasis (Malin et al., 2014; Voduc et al., 2015).
In this study, we delineate the transcriptome of proliferating and indolent non-proliferating brain metastatic melanoma cells by single cell RNA sequencing (RNA-seq), and revealed that non-proliferating cells exhibit the gene expression signature of DNA methyltransferase 1 (DNMT1) inhibition. DNMT1 expression is suppressed by the brain microenvironment, and this is linked to cell cycle delay in cancer cells, suggesting a mechanism of cellular dormancy in brain metastasis. Unexpectedly, forced expression of DNMT1 did not rescue the cell cycle progression but rather reduced the number of brain metastatic lesions. We found that DNMT1 suppression induced L1CAM and SERPINs, the genes essential for brain metastasis initiation, which explained why DNMT1 overexpression derailed brain metastasis. Further functional analysis indicated that CRYAB is negatively regulated by DNMT1 and is critically required for the survival of cancer cells in the brain microenvironment. Here we suggest a scheme of brain metastasis formation—the balance of pro-survival and anti-proliferative signals induced by DNMT1 suppression determines the fate of brain metastatic cancer cells.
Results
Non-proliferating Brain Metastatic Cancer Cells Exhibit the Gene Expression Signature of DNMT1 Inhibition
To understand better the biology underlying the behavior of brain metastasis, we established a model using BRAF mutant WM266.4 human melanoma cells. These cells form brain metastasis within 30 days after intra-cardiac injection in nude mice (Figure 1A and Data S1). Intriguingly, there was considerable variation in the size of the metastases, with single cells still observed 30 days after injection suggesting that a subset of WM266.4 cells enter an indolent or dormant state. For subsequent analysis we classified the metastases in four classes based on their size: single cell (SC), micro-cluster (MC, a cell cluster consists of less than five cells), micro-metastasis (MM, <100 μm), and established metastasis (EM, >100 μm) (Figures 1B and 1C). We performed Ki67 staining to investigate the relationship between metastasize size and proliferative state and found that there is no simple relationship between metastasis size and the proportion of Ki67-positive cells (Figures 1D–1F). The majority of SC and MC do not contain Ki67-positive cells (Figure 1D). In contrast, EM have a relatively consistent proportion of Ki67-positive cells suggesting that they grow in an exponential manner (Figures 1E and 1F). The situation for MM is more complex: on average 45% of cells are Ki67-positive at day 20, although there is a wide spread in this value between metastases (Figure 1E). However, the average percentage of Ki67-positive cells declines to 29% at day 30 (Figure 1F). This can be explained if some of the MM lesions at day 20 with higher proportions of Ki67-positive cells transition into becoming larger EMs at day 30; this is supported by the increasing numbers of EM between day 20 and 30 in Figure 1C. In contrast, those MM with a lower proportion of Ki67-positive cells remain as MM (Figure 1F). The transition of the more proliferative metastases into the EM category can explain the drop of the average percentage of Ki67-positive cells in MM. In addition, several MM have no Ki67-positive cells at day 30, suggesting that cells can exit from a proliferative state. Furthermore, because there is little change in the total number of MM between day 20 and day 30, despite the presence of proliferative cells, it is possible that proliferation in MM may be balanced by cell death. Non-proliferating cancer cells in SC and MC could also represent either cells that have not divided since arrival in the brain or cells that have divided, but some of the resultant cells subsequently died.
Figure 1.

WM266.4 Human Melanoma Cells form Aggressive and Indolent Brain Metastatic Lesions
(A and B) Representative images of WM266.4-Luc-EGFP brain metastasis at the indicated days after intra-cardiac injection. Sagittal brain sections are stained for Ki67 and whole slices (A) and enlarged views (B) are shown. Colored arrowheads indicate four stages (SC, MC, MM, and EM) of brain metastasis progression. Green: WM266.4 cells, Blue: DAPI, Red: Ki67. Scale bar, 2.5 mm (A), 100 μm (B).
(C and D) The number of lesions per sagittal brain section (C) and the proportion of SC and MC that contain at least one Ki67-positive cell (D) at the indicated days; 30 brain slices from 8 mice (day10), 32 brain slices from 8 mice (day 20), and 28 brain slices from 8 mice (day 30) were used for quantification.
(E and F) Size of lesions (number of cells) and Ki67 index in MM and EM at day 20 (E) and day 30 (F). Av., average of Ki67-index (n = 117 [MM at day 20], 64 [EM at day 20], 123 [MM at day 30], and 82 [EM at day 30]).
Data are mean ± SEM (C and D).
A situation of low levels of ongoing proliferation and cell death would be predicted to result in the dilution of a stable cell label. Therefore, we labeled WM266.4-EGFP cells with a stable dye, CellVue Claret Far Red, just before intra-cardiac injection (Bantly et al., 2007). We extracted the cancer cells from the mouse brain at day 20 and examined the number of EGFP/CellVue Claret Far Red double-positive cells. We found that only 16 among 3,077 EGFP-positive cells were positive for the dye (positive rate = 0.52%) (Figure S1A). Thus, WM266.4 melanoma cells show diverse and interesting behaviors upon arrival in the brain. Some cells establish large metastases that grow progressively, whereas others form either micro-metastases with balanced proliferation and cell death or enter a predominantly indolent state with only rare proliferation or death. This is consistent with intravital imaging studies that also report the diverse fates of metastatic cells in the brain (Kienast et al., 2010).
To try to understand some of the mechanisms underpinning the divergent behavior of WM266.4 melanoma cells in the brain environment, we performed single cell RNA-seq of brain metastatic WM266.4 cells at day 20 using the cells to Fluidigm C1 system (Figure S1B). Principal component analysis (PCA) with ninety-nine house-keeping genes (the list provided in Table S1) selected 61 samples as unbiasedly amplified cDNA libraries (Figure S1C and Data S2). Interestingly, MKI67 expression clearly divided the cells into two groups, termed “Proliferating” and “Non-proliferating” (Figure 2A). The expression of well-established cell-cycle linked genes TK1, MCM7, TOP2A, and PCNA (Aufderklamm et al., 2012) were all higher in the “Proliferating” group (Figures 2B and 2C and Data S3). Furthermore, gene set enrichment analysis (GSEA) (Subramanian et al., 2005) showed that the genes related to cell cycle progression, DNA replication, and E2F transcription were enriched in the “Proliferating” group (Figure S1D), indicating that we can successfully separate the cells into two groups as intended.
Figure 2.

Non-proliferating WM266.4 Cells Exhibit the Gene Expression Signature of DNMT1 Inhibition
(A) Fragments per kilobase of exon per million reads mapped (FPKM) of MKI67 in the 61 single cell-derived cDNA libraries, separated into Non-proliferating (23 cells, FPKM <1) and Proliferating (38 cells, FPKM >1) group.
(B and C) FPKM of cell proliferation-related genes (TK1, MCM7, TOP2A and PCNA) in Non-proliferating (NP) and Proliferating (P) group in the single cell-derived cDNA libraries.
(D) GSEA results with the indicated gene sets between Non-proliferating (NP) and Proliferating (P) groups.
(E) Representative images of WM266.4 brain metastasis, stained for DNMT1. Green: WM266.4 cells, Blue: DAPI, Red: DNMT1. Scale bar, 100 μm.
(F) Size of lesions (number of cells) and DNMT1-positive ratio in SC, MC, MM, and EM at day 30. Av., average of DNMT1-positive ratio (n = 37 [SC], 32 [MC], 39 [MM], and 21 [EM]). Data are mean (B and C).
Intriguingly, in this analysis, the top-ranked gene sets up-regulated and down-regulated in “Non-proliferating” group were the genes induced and suppressed by decitabine treatment, respectively (Figure 2D, Data S4 and S5) (Missiaglia et al., 2005). Decitabine, a cytidine analog, is a DNA methyltransferase inhibitor and known to suppress cell cycle progression in both a DNA demethylation-dependent and -independent manner (Seelan et al., 2018; Tsai et al., 2012). Because DNA methylation is mainly maintained by DNA methyltransferase-1 (DNMT1) in cancer cells (Zhang and Xu, 2017) and WM266.4 cells express no or little DNMT3A, 3B, and 3L (Figure S2A), we examined the expression of DNMT1 in brain metastatic WM266.4 cells. Immunostaining shows that most of SC and MC do not express DNMT1, whereas about 27% and 42% of the cells in MM and EM at day 30 expressed DNMT1, respectively (Figures 2E and 2F). Interestingly, quantitative analysis revealed that the expression levels of DNMT1 in WM266.4 cells distribute far more widely in vivo than in vitro (Figure S2B), suggesting that DNMT1 expression is highly influenced in brain. We also found that the expression of Ki67 and DNMT1 clearly overlapped (Figure S2C). These data raise the possibility that DNMT1 down-regulation by brain microenvironment might be the cause of cell-cycle arrest in indolent brain metastasis.
Brain Microenvironment Suppresses DNMT1 and Cell Cycle Progression in Various Types of Cancer Cells
DNMT1 ablation is well known to suppress cell cycle progression in many cell types; therefore, we sought to investigate the relationship between DNMT1 and proliferation in the context of brain metastasis. We found that DNMT1 knockdown using four independent siRNA suppressed WM266.4 cell cycle progression in vitro (Figures 3A–3G). More importantly, silencing of DNMT1 affected systemic metastasis progression (Figures 3H and 3I) including brain metastasis (Figures 3J and 3K). Of particular note, the number of larger metastases (MM + EM) was greatly reduced, whereas the number of indolent metastatic lesions (SC + MC) was not altered (Figure 3K), indicating that reduced DNMT1 levels seem to prevent the progression of metastatic lesions.
Figure 3.

DNMT1 Suppression Induces Cell Cycle Delay in WM266.4 Cells
(A) The time course of siRNA experiments. WM266.4 cells were transfected with control siRNA or siRNA against DNMT1 at days 0 and 3, and further experiments were started at day 7.
(B) Immunoblotting at day 7 showed effective ablation of DNMT1 in WM266.4 cells treated with four different siRNAs (siDNMT1 #1–4).
(C) Cell proliferation analysis of WM266.4 cells treated with siCtrl (#1) or siDNMT1 (#1–4).
(D) Immunoblotting at day 7 showed effective ablation of DNMT1 in WM266.4 cells treated with siCtrl smart pool (siCtrl-SP) or siDNMT1 smart pool (siDNMT1-SP).
(E and F) Cell cycle analysis of WM266.4 cells treated with siCtrl-SP or siDNMT1-SP. Representative FACS images with cell cycle profiling by EdU incorporation (y-axis) and DNA content (x-axis, DAPI staining) are shown in (E) and the percentages of the cells in G0/1, S and G2/M phase are shown in (F) (n = 3).
(G) CpG methylation analysis at transcription start site of the indicated genes by pyrosequence. LINE-1 is used as a marker of global DNA methylation (average of three CpG sites, n = 2) and EBF3 as a representative highly methylated region in melanoma cells (n = 2).
(H and I) WM266.4 cells pre-treated with siCtrl-SP or siDNMT1-SP were injected into mouse hearts and induced brain metastasis. Representative images of bioluminescence detection (H) and total flux from the whole body (photons/s) at day 0 (after injection) and day 29 (siCtrl-SP; n = 5, siDNMT1-SP; n = 6) (I) are shown.
(J and K) Representative images of sagittal brain sections stained for Ki67 (J) and the number of lesions per sagittal brain section at day 29 (K) are shown. A total of 10 brain slices from 5 mice (siCtrl-SP) and 12 brain slices from 6 mice (siDNMT1-SP) were used for quantification. Green: WM266.4 cells pre-treated with siCtrl-SP or siDNMT1-SP, Blue: DAPI, Red: Ki67. Scale bar, 2.5 mm (large images), 100 μm (small panels).
Data are mean (I), mean ± SD (C, F, and G), or mean ± SEM (K).
Having established the link between low DNMT1 levels and lack of proliferation in the brain microenvironment, we investigated if there were any factors that might down-regulate DNMT1 in cancer cells arriving in the brain. The presence of reactive astrocytes (recognized as glial fibrillary acidic protein [GFAP]-positive cells) around WM266.4 cells even at early stages of brain metastasis led us to test whether astrocytes might modulate DNMT1 expression (Figure 4A). We also found that microglial cells (recognized as ionized calcium binding adapter molecule-1 [Iba-1]-positive cells) are recruited to the metastatic lesions; however, the accumulation is not as prominent as reactive astrocytes, especially around SC and MC (Figure S3A). We established primary astrocytes (Figure 4B) and microglial cells (Figure S3B) in culture and tested the effect of media conditioned by these two cell types. As a control, we used media conditioned by melanoma-associated fibroblasts (MAFs) (Hirata et al., 2015). Remarkably, conditioned media from astrocytes, but not microglial cells or MAFs, suppressed DNMT1 expression in WM266.4 cells (Figures 4C, 4D, S3C, and S3D). Furthermore, because the brain is very soft (Young's modulus ~0.2 kPa) compared with most primary cancer tissues (3–10 kPa) (Barnes et al., 2017), we also studied the effect of differential mechanical properties of the brain microenvironment. To this end, we designed two “quasi microenvironments” reflecting primary tumors where cancer cells were cultured on a hard substrate (3 kPa) with conditioned media from MAFs or the brain with cells cultured on a soft substrate (0.2 kPa) with conditioned media from astrocytes (Figure 4E). We found that culture on soft substrates suppressed DNMT1 expression in WM266.4 cells (Figure 4F) and the effect was further enhanced by conditioned media from astrocytes (Figures 4G and 4H). Cell culture on a soft substrate with astrocyte-conditioned media significantly suppressed cell cycle progression compared with cell culture on a hard substrate with MAF-conditioned media, which was partially rescued by DNMT1 over-expression in WM266.4 cells (Figure 4I). To determine whether this suppression of DNMT1 by the combination of astrocyte-derived factors and soft ECM was restricted to WM266.4 melanoma cells, we also investigated cancer models capable of brain metastasis. We obtained the similar results with MDA231 human breast cancer cells and PC9 human lung cancer cells, where DNMT1 knockdown suppressed cell cycle progression (Figures S4A–S4D and S5A–S5D) and cell culture on a soft substrate with astrocyte-conditioned media suppressed DNMT1 expression and cell cycle progression (Figures S4E–S4H and S5E–S5H). Taken together, these data demonstrate the down-regulation of DNMT1 by the brain microenvironment and establish its functional role in altered cell cycle progression.
Figure 4.

Brain Microenvironment Suppresses DNMT1 and Cell Cycle Progression
(A) A representative image of MC at day 10, stained for glial fibrillary acidic protein (GFAP). Green: WM266.4 cells, Blue: DAPI, Red: GFAP. Scale bar, 100 μm.
(B) Representative images of primary astrocytes established from C57BL/6 neonatal mouse brains. Cells were stained for GFAP and confirmed that more than 90% of the cells were positive for this astrocyte marker. Green: GFAP, Magenta: DAPI. Scale bar, 100 μm.
(C) Representative images of immunoblotting for DNMT1 in WM266.4 cells cultured in DMEM/10% FBS or conditioned media prepared from the indicated cell cultures.
(D) Relative expression of DNMT1/β-tubulin in WM266.4 cells cultured in DMEM/10% FBS or conditioned media from astrocytes was quantified (n = 3).
(E) A cartoon of quasi microenvironment experiment on poly-acrylic acid (PAA) gels with defined stiffness.
(F) DNMT1 expression in WM266.4 cells cultured on different stiffness. Y axis indicates normalized intensity of DNMT1/DAPI (n = 166–305).
(G and H) Representative images of WM266.4 cells cultured in the indicated conditions and stained for DNMT1 (G) and normalized intensity of DNMT1/DAPI was quantified (n = 227–338) (H). Scale bar, 100 μm.
(I) EdU incorporation by WM266.4 cells and WM266.4 cells overexpressing DNMT1 (WM266.4-DNMT1) in the indicated culture conditions (n = 6).
(J and K) WM266.4 or WM266.4-DNMT1 cells were injected into mouse hearts and induced brain metastasis. Representative images of bioluminescence detection in vivo (day 0) and ex vivo (day 28) (J) and total flux from the brain (photon/sec) (n = 5 in each group) (K) are shown.
(L and M) Representative images of sagittal brain sections stained for Ki67 (L) and the number of lesions per sagittal brain section at day 28 (M) are shown. A total of 20 brain slices from 5 mice (WM266.4) and 24 brain slices from 6 mice (WM266.4-DNMT1) were used for quantification. Green: WM266.4 or WM266.4-DNMT1 cells, Blue: DAPI, Red: Ki67. Scale bar, 2.5 mm (large images), 100 μm (small panels).
(N) WM266.4 cells labeled with mCherry and WM266.4-DNMT1 cells labeled with mEGFP were mixed at 1:1 ratio and injected into mouse hearts. The number of lesions per sagittal brain section at day 10 are quantified. A total of 36 brain slices from 6 mice were used for quantification.
Data are mean (K), mean ± SD (D, F, H, and I) or mean ± SEM (M and N).
Forced Expression of DNMT1 Unexpectedly Reduced the Number of Brain Metastatic Lesions
The data above suggest that forced DNMT1 expression might circumvent cell-cycle arrest/delay and enable rapid outgrowth of brain metastasis. However, somewhat surprisingly, DNMT1-overexpressing WM266.4 cells failed to form brain metastasis (Figures 4J and 4K), although the growth rate was similar to the parental cells in vitro (Figures S6A and S6B). Detailed histopathological analysis revealed that there was a significant decrease even in single cell metastases (Figures 4L and 4M), suggesting that the reduced number of metastases might reflect a defect in the ability of cells to survive in the brain. The few remaining metastases had a similar range of sizes to control WM266.4 cells. We also found that similar results were observed following DNMT1 over-expression in MDA231 cells (Figures S4I–S4L). Thus, both DNMT1 suppression and over-expression reduce the overall metastatic burden, but they do so in different ways with high DNMT1 expression hindering survival upon arrival in the brain and low/absent DNMT1 expression precluding outgrowth of micrometastases. To confirm more precisely whether DNMT1 overexpression hinders survival of the cells in the early stage of brain metastasis, we labeled WM266.4 and WM266.4-DNMT1 cells with different fluorescent proteins (mCherry and mEGFP, respectively), mixed the cells in a 1:1 ratio before intra-cardiac injection to seed brain metastasis. Histopathological analysis at day 10 clearly showed that the number of lesions is greatly reduced by DNMT1 overexpression (Figure 4N). We further investigated the small number of SC and MC in mice injected with DNMT1-overexpressing WM266.4 cells. Intriguingly, DNMT1 staining of the limited number of brain metastatic lesions induced by intra-cardiac injection of DNMT1-overexpressing WM266.4 cells revealed that SC and MC do not express DNMT1 (Figure S6C), suggesting that these cells might have silenced the construct driving DNMT1 over-expression or reduced its levels post-transcriptionally. These results further strengthen the argument that DNMT1-low cells and/or the cells in which DNMT1 is strongly suppressed by the brain microenvironment form indolent brain metastasis. Moreover, although the number is limited, MM and EM express high levels of DNMT1 (Figure S6C). Taken all together, these data support a model in which low levels of DNMT1 are advantageous for both the initial survival of metastatic cells in the brain and their persistence as indolent single cell or micro-cluster metastases. In contrast, the transition to large growing EM is driven by elevated DNMT1 levels.
DNMT1 Suppression Activates Pro-survival Signals Essential for Brain Metastasis Initiation
The results described above evoked the idea that DNMT1 suppression by brain microenvironment might simultaneously activate pro-survival (pro-tumor) and anti-proliferative (anti-tumor) signals in brain metastatic cancer cells. Hence, low DNMT1 expression would be associated with persistent but indolent metastases. To explore this idea and seek pro-survival factors, we performed RNA-seq to examine the effect of DNMT1 ablation on gene expression in vitro. The 262 genes suppressed by DNMT1 included L1CAM, SERPIND1, and E2, the genes reported to be essential for brain metastasis initiation (Valiente et al., 2014) (Figures 5A and 5B and Data S6). Technical constraints precluded us from directly analyzing CpG promoter methylation in single cell metastases; we therefore investigated if DNMT1 targets might be differentially expressed in melanoma cells selected for efficient brain metastasis and if this correlated with altered DNA methylation. To this end, we established WM266.4 cells with enhanced brain metastatic ability by repetition of intra-cardiac injection and cancer cell collection from the mouse brain (WM266.4-BrM1, 2, and 3) (Figures S7A–S7G and Data S1 and S7) (Bos et al., 2009). We found that WM266.4-BrM3 expressed higher levels of the DNMT1 target, L1CAM (Data S7). As might be expected for a gene regulated by DNMT1, the promoter CpG site of L1CAM was extensively demethylated in WM266.4-BrM3 cells, whereas that of LINE-1 was not affected (Figure 5C). Intriguingly, total DNMT1 levels were not altered in WM266.4-BrM3 cells (Figure 5D), suggesting that its activity may be regulated by additional mechanisms besides protein expression. Together, these data support the idea that DNA demethylation of pro-survival genes favors successful brain metastasis in BrM3 cells.
Figure 5.

Brain Microenvironment Activates Pro-survival Signals via DNMT1 Suppression
(A) Transcriptome analysis of WM266.4 cells treated with siCtrl-SP or siDNMT1-SP. Genes up-regulated by DNMT1 knockdown are shown with fold change and p value (n = 3). Please also refer to Data S6.
(B) Relative expression of the indicated genes in WM266.4 cells treated with siDNMT1-#1 or -#2 compared with siCtrl-#1 were quantified by quantitative RT-PCR.
(C) CpG methylation analysis of the indicated genes in WM266.4 (parent) and its derivatives (BrM1-3) by pyrosequence. The methylation of L1CAM is the average of five CpG sites in the promoter region (n = 2), and LINE-1 is used as a marker of global DNA methylation (average of three CpG sites, n = 2).
(D) Immunoblotting for DNMT1 in WM266.4 (parent) and its derivatives with enhanced brain metastasis (BrM1-3).
(E) Immunoblotting for DNMT1 in WM266.4-BrM3 cells and WM266.4-BrM3 cells overexpressing DNMT1 (WM266.4-BrM3-DNMT1).
(F) Cell proliferation analysis of WM266.4-BrM3 and WM266.4-BrM3-DNMT1 cells (n = 3).
(G and H) WM266.4-BrM3 or WM266.4-BrM3-DNMT1 cells were injected into mouse hearts and induced brain metastasis. Representative images of bioluminescence detection in vivo (day 0) and ex vivo (day 21) (G) and total flux from the brain (photons/s) (WM266.4-BrM3; n = 4, WM266.4-BrM3-DNMT1; n = 5) (H) are shown.
(I and J) Representative images of sagittal brain sections stained for Ki67 (I) and the number of lesions per sagittal brain section at day 25 (J) are shown. A total of 12 brain slices from 6 mice were used for quantification in each group. Green: WM266.4-BrM3 or WM266.4-BrM3-DNMT1 cells, Blue: DAPI, Red: Ki67. Scale bar, 2.5 mm (large images), 100 μm (small panels).
Data are mean (H), mean ± SD (B, C, and F), or mean ± SEM (J).
Interestingly, we also found that forced expression of DNMT1 in WM266.4-BrM3 cells canceled their enhanced ability to form brain metastases without affecting cell growth in vitro (Figures 5E–5J). To test the universality of this observation, we generated PC9 cells with enhanced brain metastatic ability using the same method as for WM266.4 cells. Reassuringly, similar results were obtained with PC9 and PC9-BrM4 cells (Figures S7H–S7M and S8A–S8G and Data S1 and S8). Taken all together, these results reinforce the view that the relationship between DNMT1 and metastasis is complex. More specifically, efficient metastasis requires an ability to temporarily down-regulate DNMT1 upon arrival in the brain and subsequently re-activate it to outgrow. Cells selected for efficient brain metastases do not have different levels of DNMT1 but show increased expression of some DNMT1-regulated genes.
αB-Crystallin Is Required for the Survival of Non-proliferating Brain Metastatic Cancer Cells
To further identify players in the survival of dormant brain metastases, we interrogated the results of single cell RNA-seq again for pro-survival genes up-regulated in the indolent cells. The volcano plot showed that CRYAB was the most highly expressed gene in the “Non-proliferating” group (Figures 6A and 6B and Data S3). Moreover, we found that αB-crystallin (encoded by CRYAB) expression was strongly induced by DNMT1 suppression (Figures 6C–6F). αB-crystallin is a molecular chaperon protein that suppresses apoptosis by inhibiting caspase-3 activation (Shin et al., 2009) and is related to breast cancer brain metastasis (Malin et al., 2014; Voduc et al., 2015). Consistent with low DNMT1 levels being linked to elevated levels of CRYAB, we found that the expression of αB-crystallin was restricted to a very small population in MM and EM (Figure 6G) and is not related to enhanced brain metastasis (Figures 6H and 6I), suggesting that this protein might be required for the survival of SC and MC. Crucially, we found that knockout (KO) of αB-crystallin did not affect cell growth in vitro (Figures 6J and 6K) but strongly suppressed brain metastasis (Figures 6L and 6M). Similar to forced DNMT1 expression, although the overall number of metastases was greatly reduced, the probability of single cell metastases progressing to macro-metastases was not affected by CRYAB deletion (Figures 6N and 6O). These results demonstrate that DNMT1-αB-crystallin axis plays a pivotal role for the survival of SC and MC brain metastases.
Figure 6.

DNMT1-αB-Crystallin Axis Plays a Pivotal Role for the Survival of Non-proliferating Brain Metastatic WM266.4 Cells
(A) A volcano plot showing average gene expression and p value in Proliferating and Non-proliferating groups.
(B) FPKM of CRYAB in Proliferating (P) and Non-proliferating (NP) groups. Bars indicate mean.
(C) Transcriptome analysis of WM266.4 cells treated with siCtrl-SP or siDNMT1-SP shows that CRYAB is up-regulated by DNMT1 knockdown (n = 3).
(D) Relative expression of CRYAB mRNA in WM266.4 cells treated with siDNMT1-#1 or -#2 compared with siCtrl-#1 were quantified by quantitative RT-PCR.
(E and F) Representative images of immunoblotting for αB-crystallin in WM266.4 cells treated with siCtrl-SP or siDNMT1-SP (E) and relative expression of αB-crystallin/β-tubulin are quantified (F) (n = 3).
(G) Representative images of WM266.4 brain metastasis, stained for αB-crystallin. Green: WM266.4 cells, Blue: DAPI, Red: αB-crystallin. Scale bar, 100 μm.
(H and I) Representative images of immunoblotting for αB-crystallin in WM266.4 (Parent) and its derivatives with enhanced brain metastasis (BrM1-3) (H) and relative expression of αB-crystallin/β-tubulin are shown (I) (n = 3).
(J) Immunoblotting for αB-crystallin in WM266.4 cells and WM266.4 cells knocked out for αB-crystallin (WM266.4-CRYAB-KO).
(K) Cell proliferation analysis of WM266.4 and WM266.4-CRYAB-KO cells.
(L and M) WM266.4 or WM266.4-CRYAB-KO cells were injected into mouse hearts and induced brain metastasis. Representative images of bioluminescence detection in vivo (day 0) and ex vivo (day 30) (L) and total flux from the brain (photon/sec) (n = 5 in each group) (M) are shown.
(N and O) Representative images of sagittal brain sections stained for Ki67 (N) and the number of lesions per sagittal brain section at day 30 (O) are shown. A total of 16 brain slices from 4 mice (WM266.4) and 10 brain slices from 5 mice (WM266.4-CRYAB-KO) were used for the quantification. Green: WM266.4, Blue: DAPI, Red: Ki67. Scale bar, 2.5 mm.
Data are mean (B and M), mean ± SD (D, F, I, and K), or mean ± SEM (O).
Discussion
Brain tissue presents a very different microenvironment from the other sites of metastases. Following successful extravasation, cancer cells that arrive in the brain will encounter reactive astrocytes and mechanically soft environment. The combination of these factors strongly suppresses DNMT1 expression in cancer cells. Multiple factors are likely to contribute to DNMT1 down-regulation. Schnepp et al. have reported that glial cells can suppress DNMT1 in breast cancer cells via clusterin secretion (Schnepp et al., 2017). Furthermore, Ras-c-Jun signaling pathway is reported to be the major inducer of DNMT1 (Bigey et al., 2000), and this pathway is strongly suppressed by reduction of cell adhesion-mediated signals on soft substrate (Hirata et al., 2015; Paszek et al., 2005). The end result of DNMT1 suppression by brain microenvironments is simultaneous activation of pro-survival and growth-suppressive signals. Cancer cells that fail to activate the pro-survival reprograming become sensitive to apoptosis. This is evidenced by the failure of cancer cells with continuously forced high expression of DNMT1 to survive, even as single cell metastases. In contrast, cells with suppressed DNMT1 have no defect in survival as single cells but are compromised in their ability to outgrow and form larger metastases. Altogether, our analyses indicate that an ability for DNMT1 to be dynamically modulated is likely to be optimal for efficient brain metastases. This could explain the observation that selecting for increased brain metastatic capability does not lead to a change in overall DNMT1 levels (Figure 5D). Another possibility is that the cell culture between the rounds of in vivo selection imposes a selective pressure against low DNMT1 cells; thus, any selection occurring in vivo is counteracted by that occurring during the in vitro phases of the experiment. Interestingly, although the regulation of DNMT1 during brain metastasis formation is dynamic and complex, many DNMT1-regulated genes, such as L1CAM, are stably increased in cells with enhanced brain metastatic ability (Data S6, S7, and S8). This could be the result of either a more specific down-regulation of DNMT1 activity at the L1CAM promoter or the action of other DNA methylases and de-methylases. In addition, although the mechanisms of regulation of DNMT1 that we document are inherently brain specific because they involve astrocyte-derived factors, the DNMT1-knockdown experiments suggest that differential regulation of DNMT1 might affect metastasis progression even in other organs (Figures 3H and 3I and discuss further below).
We also identified αB-crystallin as a key mediator of cancer cell survival downstream of DNMT1 suppression. Although it is required for the survival of single cell brain metastases, elevated αB-crystallin expression was not observed in either advanced brain metastasis (MM and EM) (Figure 6G) or cells with enhanced brain metastasis (BrM1-BrM3) (Figures 6H and 6I). These data are consistent with CRYAB being dynamically regulated by DNMT1; however, they also imply that αB-crystallin has a context-specific effect on cell survival. Intriguingly, a recent report by Montagner et al. shows that CRYAB is much higher in indolent breast cancer cells (D2.0R cells) than in the aggressive counterparts (D2A1 cells), although it does not appear to be regulated by the lung microenvironment (Figure S9). However, in these cells, CRYAA seems to be the regulated isoform of crystallin-α and this is inversely correlated with DNMT1 levels (Figure S9) (Montagner et al., 2020). These results suggest that diverse cues in different organs might be able to regulate DNMT1 and the expression of crystallins and are consistent with the effects of DNMT1 manipulation on extra-cranial metastases. We propose that single cell metastases and micro-clusters experience particularly high levels of cellular and metabolic stress and therefore are particularly dependent on crystallins, which are reported to suppress stress-induced apoptosis (Hamann et al., 2013; Mao et al., 2004). In cases where metastatic cells transition to active growth, their apparent dependence on αB-crystallin decreases. Thus, in addition to the possibility of blood-brain barrier-permeable decitabine for the treatment of growing brain metastasis (Chabot et al., 1983; Riccadonna et al., 2016; Tawbi et al., 2013), targeting αB-crystallin and other pro-survival factors expressed in dormant cells may represent an attractive strategy for eliminating indolent single cells and micro-clusters. Furthermore, we can also hypothesize from our results that only the cells that undergo reprograming for cell survival and also circumvent anti-proliferative effects might progress to macrometastasis. Multiple events may enable the circumvention of growth arrest (Liddelow and Barres, 2017; Wasilewski et al., 2017); excessive activation of astrocytes may increase the mechanical properties of the metastatic microenvironment and promote the re-expression of DNMT1 and cell cycle entry.
In conclusion, we identify how brain microenvironment-induced reprogramming via DNMT1 suppression determines the fate of brain metastatic cancer cells. If cells fail to down-regulate DNMT1 in the brain microenvironment, then they will lack survival molecules required in single metastatic cells. If they stably down-regulate DNMT1, then they will survive but with minimal ability to actively resume growth leading to an indolent state. Only if DNMT1 can be dynamically modulated can the cells efficiently form macroscopic metastases. The initial triggers for DNMT1 down-regulation are astrocytes and the soft brain environment. In the future, it will be interesting to determine the subsequent triggers for DNMT1 re-expression and metastatic outgrowth.
Limitation of the Study
We established WM266.4 brain metastasis models and re-derived experimental models of brain metastasis that are widely used in previous studies (Bos et al., 2009; Priego et al., 2018; Valiente et al., 2014) and could successfully analyze the process of brain metastasis progression at single cell levels. To further verify the epigenetic reprogramming by the brain microenvironment, DNA methylation analysis at single cell levels (Farlik et al., 2015) should be very useful; however, this remains technically challenging to set up. Therefore, we investigated CpG demethylation in the cells that survived and proliferated in brain microenvironment and could speculate that selective DNA demethylation of pro-survival genes favors successful brain metastasis in these cells. Another challenge should be more detailed and prolonged observation of brain metastatic cancer cells in vivo—we could not know in this study whether indolent cancer cells are capable of regrowing by some triggers. In addition, we could only obtain indirect evidence about the time frame of brain metastasis progression and DNMT1 regulation. To this end, real-time intra-vital imaging with dynamic visualization of DNMT1 expression should be powerful approach in the future. Syngeneic mouse models should also help to totally describe complicated cancer-stroma interactions in these experiments.
In this study, we have only considered mechanisms that might control indolence in the brain. However, indolent metastases can be found in many organs and the brain is the only organ with astrocytes, implying that the mechanism we describe would not apply universally to all organs. Interestingly, examination of RNA-seq of indolent breast cancer cells in the lung environment reveals apparent convergence on DNMT1 and crystallin regulation.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Eishu Hirata (ehirata@staff.kanazawa-u.ac.jp).
Materials Availability
Brain metastatic cancer cells generated in this study will be made available on reasonable requests with a completed Materials Transfer Agreement.
Data and Code Availability
All the RNA sequence data in this study are available at the National Center for Biotechnology Information's Gene Expression Omnibus database with accession numbers GSE150556, GSE150557, and GSE150560.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank M. Matsuda and T. Akagi for the plasmids. T. Yu (AS ONE corporation, Osaka, JAPAN), S. Yamagishi, K. Taniguchi, A. Nakayama, and B. Spencer-Dene are also to be thanked for their technical assistance. We are grateful to the members of the Hirata, Sahai, and Kiyokawa Laboratory for their helpful discussions. E.H. was supported by the Japan Society for the Promotion of Science (JSPS) Kakenhi Grant [17K07181] and [20H03510], The Uehara Memorial Foundation, Japan Brain Foundation, Extramural Collaborative Research Grant of Cancer Research Institute of Kanazawa University, The Grant for Collaborative Research from Kanazawa Medical University [S2016-16], Hokkoku Cancer Foundation, Takeda Science Foundation, The Naito Foundation, Kanae Foundation for the Promotion of Medical Science, Astellas Foundation for Research on Metabolic Disorders, The Tokyo Biomedical Research Foundation, MSD Life Science Foundation, and the P-CREATE (Project for Cancer Research And Therapeutic Evolution) grant from The Japan Agency for Medical Research and Development (AMED) [17cm0106224h0002] and [19cm0106261h0001]. E.S. was funded by the Francis Crick Institute, which receives its core funding from Cancer Research UK (FC001144), the UK Medical Research Council (FC001144), and the Wellcome Trust (FC001144).
Author Contributions
E.H. designed the study and conducted most of the experiments with help of K.I. S. Kohsaka and S. Kojima carried out RNA-seq and data analysis. K.S. carried out pyrosequencing and DNA methylation analysis. Y.K., H.M., S.Y., E.K., and E.S. supervised the project and experiments. E.H. and E.S. wrote the paper with assistance from the other authors.
Declaration of Interests
S.Y. obtained commercial research grants from AstraZeneca, Chugai Pharm, and Boehringer-Ingelheim and has received speaking honoraria from AstraZeneca, Chugai Pharma, and Boehringer-Ingelheim. Other authors declare no competing interests.
Published: September 25, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101480.
Supplemental Information
References
- Aufderklamm S., Todenhofer T., Gakis G., Kruck S., Hennenlotter J., Stenzl A., Schwentner C. Thymidine kinase and cancer monitoring. Cancer Lett. 2012;316:6–10. doi: 10.1016/j.canlet.2011.10.025. [DOI] [PubMed] [Google Scholar]
- Bantly A.D., Gray B.D., Breslin E., Weinstein E.G., Muirhead K.A., Ohlsson-Wilhelm B.M., Moore J.S. CellVue Claret, a new far-red dye, facilitates polychromatic assessment of immune cell proliferation. Immunol. Invest. 2007;36:581–605. doi: 10.1080/08820130701712461. [DOI] [PubMed] [Google Scholar]
- Barnes J.M., Przybyla L., Weaver V.M. Tissue mechanics regulate brain development, homeostasis and disease. J. Cell Sci. 2017;130:71–82. doi: 10.1242/jcs.191742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigey P., Ramchandani S., Theberge J., Araujo F.D., Szyf M. Transcriptional regulation of the human DNA Methyltransferase (dnmt1) gene. Gene. 2000;242:407–418. doi: 10.1016/s0378-1119(99)00501-6. [DOI] [PubMed] [Google Scholar]
- Bos P.D., Zhang X.H., Nadal C., Shu W., Gomis R.R., Nguyen D.X., Minn A.J., van de Vijver M.J., Gerald W.L., Foekens J.A. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–1009. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brastianos P.K., Carter S.L., Santagata S., Cahill D.P., Taylor-Weiner A., Jones R.T., Van Allen E.M., Lawrence M.S., Horowitz P.M., Cibulskis K. Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov. 2015;5:1164–1177. doi: 10.1158/2159-8290.CD-15-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagney D.N., Martin A.M., Catalano P.J., Redig A.J., Lin N.U., Lee E.Q., Wen P.Y., Dunn I.F., Bi W.L., Weiss S.E. Incidence and prognosis of patients with brain metastases at diagnosis of systemic malignancy: a population-based study. Neuro Oncol. 2017;19:1511–1521. doi: 10.1093/neuonc/nox077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabot G.G., Rivard G.E., Momparler R.L. Plasma and cerebrospinal fluid pharmacokinetics of 5-Aza-2'-deoxycytidine in rabbits and dogs. Cancer Res. 1983;43:592–597. [PubMed] [Google Scholar]
- De Cock J.M., Shibue T., Dongre A., Keckesova Z., Reinhardt F., Weinberg R.A. Inflammation triggers zeb1-dependent escape from tumor latency. Cancer Res. 2016;76:6778–6784. doi: 10.1158/0008-5472.CAN-16-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Er E.E., Valiente M., Ganesh K., Zou Y., Agrawal S., Hu J., Griscom B., Rosenblum M., Boire A., Brogi E. Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat. Cell Biol. 2018;20:966–978. doi: 10.1038/s41556-018-0138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlik M., Sheffield N.C., Nuzzo A., Datlinger P., Schonegger A., Klughammer J., Bock C. Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Rep. 2015;10:1386–1397. doi: 10.1016/j.celrep.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghajar C.M. Metastasis prevention by targeting the dormant niche. Nat. Rev. Cancer. 2015;15:238–247. doi: 10.1038/nrc3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghajar C.M., Peinado H., Mori H., Matei I.R., Evason K.J., Brazier H., Almeida D., Koller A., Hajjar K.A., Stainier D.Y. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013;15:807–817. doi: 10.1038/ncb2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann S., Metrailler S., Schorderet D.F., Cottet S. Analysis of the cytoprotective role of alpha-crystallins in cell survival and implication of the alphaA-crystallin C-terminal extension domain in preventing Bax-induced apoptosis. PLoS One. 2013;8:e55372. doi: 10.1371/journal.pone.0055372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata E., Girotti M.R., Viros A., Hooper S., Spencer-Dene B., Matsuda M., Larkin J., Marais R., Sahai E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin beta1/FAK signaling. Cancer Cell. 2015;27:574–588. doi: 10.1016/j.ccell.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamradt M.C., Chen F., Sam S., Cryns V.L. The small heat shock protein alpha B-crystallin negatively regulates apoptosis during myogenic differentiation by inhibiting caspase-3 activation. J. Biol. Chem. 2002;277:38731–38736. doi: 10.1074/jbc.M201770200. [DOI] [PubMed] [Google Scholar]
- Kamradt M.C., Lu M., Werner M.E., Kwan T., Chen F., Strohecker A., Oshita S., Wilkinson J.C., Yu C., Oliver P.G. The small heat shock protein alpha B-crystallin is a novel inhibitor of TRAIL-induced apoptosis that suppresses the activation of caspase-3. J. Biol. Chem. 2005;280:11059–11066. doi: 10.1074/jbc.M413382200. [DOI] [PubMed] [Google Scholar]
- Kienast Y., von Baumgarten L., Fuhrmann M., Klinkert W.E., Goldbrunner R., Herms J., Winkler F. Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med. 2010;16:116–122. doi: 10.1038/nm.2072. [DOI] [PubMed] [Google Scholar]
- Liddelow S.A., Barres B.A. Reactive astrocytes: production, function, and therapeutic potential. Immunity. 2017;46:957–967. doi: 10.1016/j.immuni.2017.06.006. [DOI] [PubMed] [Google Scholar]
- Malin D., Strekalova E., Petrovic V., Deal A.M., Al Ahmad A., Adamo B., Miller C.R., Ugolkov A., Livasy C., Fritchie K. alphaB-crystallin: a novel regulator of breast cancer metastasis to the brain. Clin. Cancer Res. 2014;20:56–67. doi: 10.1158/1078-0432.CCR-13-1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malladi S., Macalinao D.G., Jin X., He L., Basnet H., Zou Y., de Stanchina E., Massague J. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell. 2016;165:45–60. doi: 10.1016/j.cell.2016.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y.W., Liu J.P., Xiang H., Li D.W. Human alphaA- and alphaB-crystallins bind to Bax and Bcl-X(S) to sequester their translocation during staurosporine-induced apoptosis. Cell Death Differ. 2004;11:512–526. doi: 10.1038/sj.cdd.4401384. [DOI] [PubMed] [Google Scholar]
- McAllister S.S., Weinberg R.A. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat. Cell Biol. 2014;16:717–727. doi: 10.1038/ncb3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missiaglia E., Donadelli M., Palmieri M., Crnogorac-Jurcevic T., Scarpa A., Lemoine N.R. Growth delay of human pancreatic cancer cells by methylase inhibitor 5-aza-2'-deoxycytidine treatment is associated with activation of the interferon signalling pathway. Oncogene. 2005;24:199–211. doi: 10.1038/sj.onc.1208018. [DOI] [PubMed] [Google Scholar]
- Montagner M., Bhome R., Hooper S., Chakravarty P., Qin X., Sufi J., Bhargava A., Ratcliffe C.D.H., Naito Y., Pocaterra A. Crosstalk with lung epithelial cells regulates Sfrp2-mediated latency in breast cancer dissemination. Nat. Cell Biol. 2020;22:289–296. doi: 10.1038/s41556-020-0474-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagner M., Sahai E. In vitro models of breast cancer metastatic dormancy. Front. Cell Dev. Biol. 2020;8:37. doi: 10.3389/fcell.2020.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noltenius C., Noltenius H. Dormant tumor cells in liver and brain. An autopsy study on metastasizing tumors. Pathol. Res. Pract. 1985;179:504–511. doi: 10.1016/S0344-0338(85)80191-6. [DOI] [PubMed] [Google Scholar]
- Obenauf A.C., Massague J. Surviving at a distance: organ specific metastasis. Trends Cancer. 2015;1:76–91. doi: 10.1016/j.trecan.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek M.J., Zahir N., Johnson K.R., Lakins J.N., Rozenberg G.I., Gefen A., Reinhart-King C.A., Margulies S.S., Dembo M., Boettiger D. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Priego N., Zhu L., Monteiro C., Mulders M., Wasilewski D., Bindeman W., Doglio L., Martinez L., Martinez-Saez E., Cajal S.R.Y. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat. Med. 2018;24:1024–1035. doi: 10.1038/s41591-018-0044-4. [DOI] [PubMed] [Google Scholar]
- Quail D.F., Joyce J.A. The microenvironmental landscape of brain tumors. Cancer cell. 2017;31:326–341. doi: 10.1016/j.ccell.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccadonna C., Yacoub Maroun C., Vuillefroy de Silly R., Boehler M., Calvo Tardon M., Jueliger S., Taverna P., Barba L., Marinari E., Pellegatta S. Decitabine treatment of glioma-initiating cells enhances immune recognition and killing. PLoS One. 2016;11:e0162105. doi: 10.1371/journal.pone.0162105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnepp P.M., Lee D.D., Guldner I.H., O'Tighearnaigh T.K., Howe E.N., Palakurthi B., Eckert K.E., Toni T.A., Ashfeld B.L., Zhang S. GAD1 upregulation programs aggressive features of cancer cell metabolism in the brain metastatic microenvironment. Cancer Res. 2017;77:2844–2856. doi: 10.1158/0008-5472.CAN-16-2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelan R.S., Mukhopadhyay P., Pisano M.M., Greene R.M. Effects of 5-Aza-2'-deoxycytidine (decitabine) on gene expression. Drug Metab. Rev. 2018;50:193–207. doi: 10.1080/03602532.2018.1437446. [DOI] [PubMed] [Google Scholar]
- Seifert H., Hirata E., Gore M., Khabra K., Messiou C., Larkin J., Sahai E. Extrinsic factors can mediate resistance to BRAF inhibition in central nervous system melanoma metastases. Pigment Cell Melanoma Res. 2015;29:92–100. doi: 10.1111/pcmr.12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J.H., Kim S.W., Lim C.M., Jeong J.Y., Piao C.S., Lee J.K. alphaB-crystallin suppresses oxidative stress-induced astrocyte apoptosis by inhibiting caspase-3 activation. Neurosci. Res. 2009;64:355–361. doi: 10.1016/j.neures.2009.04.006. [DOI] [PubMed] [Google Scholar]
- Smedby K.E., Brandt L., Backlund M.L., Blomqvist P. Brain metastases admissions in Sweden between 1987 and 2006. Br. J. Cancer. 2009;101:1919–1924. doi: 10.1038/sj.bjc.6605373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa M.S., Bragado P., Aguirre-Ghiso J.A. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat. Rev. Cancer. 2014;14:611–622. doi: 10.1038/nrc3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabouret E., Chinot O., Metellus P., Tallet A., Viens P., Goncalves A. Recent trends in epidemiology of brain metastases: an overview. Anticancer Res. 2012;32:4655–4662. [PubMed] [Google Scholar]
- Tawbi H.A., Beumer J.H., Tarhini A.A., Moschos S., Buch S.C., Egorin M.J., Lin Y., Christner S., Kirkwood J.M. Safety and efficacy of decitabine in combination with temozolomide in metastatic melanoma: a phase I/II study and pharmacokinetic analysis. Ann. Oncol. 2013;24:1112–1119. doi: 10.1093/annonc/mds591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai H.C., Li H., Van Neste L., Cai Y., Robert C., Rassool F.V., Shin J.J., Harbom K.M., Beaty R., Pappou E. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21:430–446. doi: 10.1016/j.ccr.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiente M., Ahluwalia M.S., Boire A., Brastianos P.K., Goldberg S.B., Lee E.Q., Le Rhun E., Preusser M., Winkler F., Soffietti R. The evolving landscape of brain metastasis. Trends Cancer. 2018;4:176–196. doi: 10.1016/j.trecan.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiente M., Obenauf A.C., Jin X., Chen Q., Zhang X.H., Lee D.J., Chaft J.E., Kris M.G., Huse J.T., Brogi E. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell. 2014;156:1002–1016. doi: 10.1016/j.cell.2014.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voduc K.D., Nielsen T.O., Perou C.M., Harrell J.C., Fan C., Kennecke H., Minn A.J., Cryns V.L., Cheang M.C. alphaB-crystallin expression in breast cancer is associated with brain metastasis. NPJ Breast Cancer. 2015;1:15014. doi: 10.1038/npjbcancer.2015.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasilewski D., Priego N., Fustero-Torre C., Valiente M. Reactive astrocytes in brain metastasis. Front. Oncol. 2017;7:298. doi: 10.3389/fonc.2017.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler F. The brain metastatic niche. J. Mol. Med. (Berl.) 2015;93:1213–1220. doi: 10.1007/s00109-015-1357-0. [DOI] [PubMed] [Google Scholar]
- Yeh A.C., Ramaswamy S. Mechanisms of cancer cell dormancy-another hallmark of cancer? Cancer Res. 2015;75:5014–5022. doi: 10.1158/0008-5472.CAN-15-1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Xu J. DNA methyltransferases and their roles in tumorigenesis. Biomark. Res. 2017;5:1. doi: 10.1186/s40364-017-0081-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the RNA sequence data in this study are available at the National Center for Biotechnology Information's Gene Expression Omnibus database with accession numbers GSE150556, GSE150557, and GSE150560.