

Deep sequencing identified small non-coding RNAs involved in drought stress tolerance and a microRNA-target module that enhances drought stress tolerance in Malus sieversii.
Abstract
Drought stress severely restricts crop yield and quality. Small noncoding RNAs play critical roles in plant growth, development, and stress responses by regulating target gene expression, but their roles in drought stress tolerance in apple (Malus domestica) are poorly understood. Here, we identified various small noncoding RNAs and their targets from the wild apple species Malus sieversii via high-throughput sequencing and degradome analysis. Forty known microRNAs (miRNAs) and eight new small noncoding RNAs were differentially expressed in response to 2 or 4 h of drought stress treatment. We experimentally verified the expression patterns of five selected miRNAs and their targets. We established that one miRNA, mdm-miR171i, specifically targeted and degraded SCARECROW-LIKE PROTEINS26.1 (MsSCL26.1) transcripts. Both knockout of mdm-miR171i and overexpression of MsSCL26.1 improved drought stress tolerance in the cultivated apple line ‘GL-3’ by regulating the expression of antioxidant enzyme genes, especially that of MONODEHYDROASCORBATE REDUCTASE, which functions in metabolism under drought stress. Transient expression analysis demonstrated that MsSCL26.1 activates MsMDHAR transcription by positively regulating the activity of the P1 region in its promoter. Therefore, the miR171i-SCL26.1 module enhances drought stress tolerance in apple by regulating antioxidant gene expression and ascorbic acid metabolism.
Plant growth and development are closely related to water availability. Plants continually adjust their physiological status in response to water availability (Zhu, 2016; Bechtold and Field, 2018). Apple (Malus domestica) is one of the most economically important fruit trees worldwide. Drought stress inhibits apple growth and causes premature leaf abscission, leading to declines in fruit yield and quality (Wang et al., 2012). The widespread use of rootstocks from wild apple species greatly improves drought tolerance of apple trees (Yang et al., 2011; Tan et al., 2016). Malus sieversii, an ancestral species of cultivated apple, has excellent drought stress tolerance and is a valuable wild resource for rootstock improvement (Duan et al., 2017; Wang et al., 2018a). Therefore, drought stress-related genes from M. sieversii could be identified and manipulated to improve stress tolerance of rootstocks.
Small noncoding RNAs (snRNAs) consist mainly of microRNAs (miRNAs) and small interfering RNAs (siRNAs). These snRNAs are involved in many biological processes in plants, such as growth and organ development, hormone signaling pathways, nutrient absorption and regulation, and abiotic and biotic stress responses (Khraiwesh et al., 2012; Zhang, 2015; Chen et al., 2018). In plants, MIRNA genes are transcribed into primary miRNAs via RNA polymerase II. The primary miRNAs are then processed by Dicer-like (DCL) enzymes, with DCL1 being the main effector in the miRNA pathway, to generate miRNA/miRNA* duplexes. Mature miRNA duplexes are loaded into ARGONAUTE1 (AGO1) to form RNA-induced silencing complexes that repress target gene expression by cleaving mRNAs or inhibiting translation (Achkar et al., 2016). The other main class of snRNAs, siRNAs, are mostly phased siRNAs (phasiRNAs) or heterochromatic siRNAs (het-siRNAs). The 21- or 24-nucleotide (nt) phasiRNAs mainly arise from miRNA-cleaved fragments, are processed by DCL4/5, and regulate the expression of downstream genes such as MIRNAs (Chen et al., 2018). The 24-nt het-siRNAs, produced in plants by RNA polymerase IV and DCL3, are loaded into AGO4 and inhibit gene expression transcriptionally via RNA-directed DNA methylation (Wang and Axtell, 2017).
Substantial progress has also been made in characterizing apple snRNAs (mainly miRNAs). Before the release of the apple genome, researchers from New Zealand identified seven conserved miRNA families in apple ‘Royal Gala’ based on expressed sequence tags (Gleave et al., 2008). The same group analyzed the expression patterns of 21 miRNAs in apple vascular tissue and phloem sap (Varkonyi-Gasic et al., 2010). Subsequently, conserved and apple-specific miRNA families and several phasiRNAs were identified by small RNA deep sequencing (Xia et al., 2012). Recent studies have focused on uncovering the roles of apple miRNAs in many biological processes, such as vegetative phase transition (Xing et al., 2014b; Guo et al., 2017a), shoot growth and morphogenesis (Xing et al., 2016; Song et al., 2017), and biotic stress responses (Kaja et al., 2015; Zhang et al., 2018a). However, little is known about the response patterns and regulatory networks of apple miRNAs under drought stress.
In this study, we performed high-throughput small RNA and degradome sequencing analyses to identify snRNAs and their targets in M. sieversii. We explored the expression patterns of several of these snRNAs and targets under drought stress. Based on these results, we investigated the functions of apple miR171i and its target gene, SCARECROW-LIKE PROTEINS26.1 (MsSCL26.1), in transgenic Arabidopsis (Arabidopsis thaliana) plants and in the cultivated apple line ‘GL-3’. Based on these observations, we conclude that the miR171i-SCL26.1 module improves drought stress tolerance by regulating antioxidant gene expression and ascorbic acid (AsA) metabolism.
RESULTS
Identification and Differential Expression Analysis of snRNAs in M. sieversii under Drought Stress
Although many snRNAs have been reported in apples, their roles under short-term drought stress are unclear. Here we investigated the overall changes in expression level of snRNAs of M. sieversii during short-term drought stress. The leaves of apple plants subjected to drought stress initially wilted after treating 2 to 4 h but then recovered their original phenotype by 24 h without rehydration, indicating the responses of M. sieversii to the drought stress (Fig. 1A). Pro levels increased during the stress treatment, suggesting that the plants’ physiological status was adjusted (Fig. 1B). The levels of malondialdehyde (MDA), a product of intracellular lipid peroxidation, initially increased but returned to control levels by 24 h after the onset of drought stress. The MDA time course was similar to that of the plant phenotype, indicating that the plants might respond to the stress conditions by regulating redox homeostasis (Fig. 1C). These observations suggest that the plants’ response to drought stress might involve a complex regulatory network linked to redox homeostasis.
Figure 1.

Phenotypic changes in M. sieversii under drought stress and snRNA identification. A, Phenotypic changes in M. sieversii under drought stress. The plants have been digitally extracted for comparison. Scale bar = 1 cm. Red lines mark the same position of the plant, highlighting phenotypic changes over time. B and C, Changes in leaf Proline and MDA contents in M. sieversii treated with 20% (w/v) PEG 6000. Data are presented as means ± sd from three biological replicates. Asterisks indicate significant differences in Pro and MDA contents between the treatments and controls based on Duncan’s multiple range test (*P < 0.05). D, Length distribution of new snRNAs in M. sieversii. E, Proportion of the first bases of new snRNAs in M. sieversii. FW, Fresh weight; CK, control check.
To identify potential components of this regulatory network, we constructed and sequenced 10 small RNA libraries from drought-stressed M. sieversii plants (0, 2, 4, 12, and 24 h of 20% [w/v] polyethylene glycol [PEG] 6000) using the HiSeq 2500 platform (Illumina) with two biological replicates. We obtained >164 million clean reads after implementing strict quality control steps. Bowtie software (https://bowtiesoft.com/) was used to remove known noncoding RNAs such as ribosomal RNA, transfer RNA, small nuclear RNA, and small-nucleolar RNA from the clean reads. MiRDeep2 (http://hpc.ilri.cgiar.org/mirdeep2-software) through the adjustment of parameters and the change of scoring systems was used to identify and predict snRNAs from the remaining reads mapped to the MalDomGD1.0 reference genome (BioProject: PRJNA28845). Briefly, sequencing reads were first aligned to the apple genome to obtain possible precursor sequences. Then, the snRNAs were predicted using a Bayesian model mainly based on miRNA and siRNA generation characteristics and precursor structure energy information (Yang and Li, 2011; Friedländer et al., 2012). We ultimately identified 335 potential snRNAs in M. sieversii, including 210 known miRNAs and 125 new snRNAs (Supplemental Table S1). Analysis of the length distribution and first nucleotide of the new snRNAs showed that 24 nt was the most common length (Fig. 1D) and that the most frequent first nucleotide of the 19- to 22-nt snRNAs was “U,” whereas that of the 24-nt snRNAs was “A” (Fig. 1E). These results indicated that the 19- to 22-nt snRNAs might be new miRNAs and that most of the 24-nt snRNAs are het-siRNAs because an initial “A” is a signature of het-siRNAs loaded into AGO4 (Wang and Axtell, 2017). In addition, we identified several snRNAs (miRNc1/2/9) that were located at different sites in the stem-loop structures of miR169o/k and miR319d/h precursor molecules (Supplemental Fig. S1). A comparison of the transcripts per million (TPM) reads showed that miRNc1/2/9 have much higher TPM reads than known miRNAs (Supplemental Table S2). Thus, the precursors of miR169k/o and miR319d/h might produce two types of mature miRNA, and those snRNAs might be new miRNAs or new members of known miRNA families.
To identify drought-responsive snRNA target genes, we generated a degradome library from a mixture of roots and leaves from plants subjected to a 4-h drought stress treatment. Analysis of the degradome data revealed that of the known miRNAs, 169 members of 36 miRNA families targeted 219 transcripts (Supplemental Table S3). Of the 148 genes targeted by 14 conserved miRNAs, 92 encode transcription factors, which might play important roles in regulating plant growth and development as well as in responses to various environmental stimuli.
Functional classification of known miRNA targets revealed that miR395/398/7125 cleaved the mRNAs of ion transporter proteins involved in sulfate, copper, and zinc homeostasis, indicating that these miRNAs are important regulators of intracellular ion homeostasis (Liang et al., 2010). Furthermore, genes encoding key enzymes in the miRNA pathway, such as DCL1, AGO1, and AGO2, were degraded by miR162, miR168, and miR403, respectively, further confirming the existence of a negative feedback loop in miRNA metabolism at the post-transcriptional level (Li et al., 2012; Zhang et al., 2015; Supplemental Fig. S2A).
Among the new snRNAs, 45 members of 23 snRNA families targeting 59 transcripts were identified in the degradome library (Supplemental Table S4). The functional classifications of these targets were similar to those of known miRNA targets, except that there was a larger proportion of genes of unknown function (Supplemental Fig. S2B). Consistently, the transcripts of TEOSINTE BRANCHED1, CYCLOIDEA, PCF; NAM, ATAF1/2, and CUC2; and MYB DNA-binding factors (MYB) family were identified as targets of miRNc9, miRNc11, and miRNc12, respectively. The mature sequences of these miRNAs, derived from different genomic locations, were similar to the sequences of miR319/164/159, with only one or two nucleotide differences, indicating that miRNc9/11/12 are new members of the conserved miR319/164/159 families in M. sieversii (Supplemental Table S5).
After identifying snRNAs and target genes in M. sieversii, we looked for snRNAs that were differentially expressed under drought stress based on average TPM values > 10 and |log2 (Fold Change)| ≥ 1 between the different treatments (Supplemental Table S6).
The expression levels of 40 known miRNAs and eight new snRNAs during short-term drought stress treatment are shown in Supplemental Figure S2, C and D. Changes in gene expression, both upregulation and downregulation, were concentrated in the samples treated for 2 or 4 h. These findings indicate that snRNAs respond to drought stress signals primarily in the early stages of treatment.
Gene ontology (GO) annotation showed that a large proportion of the target genes were assigned to cell part and organelle in the cellular-component category, binding and transcription regulator activity in the molecular-functions category, and metabolic and cellular process in the biological-process category (Supplemental Figs. S3A and S4A). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses revealed that more than half of the target genes were involved in signal transduction, carbohydrate metabolism, or energy metabolism (Supplemental Figs. S3B and S4B). These results suggest that the differentially expressed snRNAs participate in plant responses to drought stress signals, plant metabolism, and resistance to adverse environmental conditions.
We used reverse transcription quantitative PCR (RT-qPCR) to verify the tissue expression patterns of several miRNAs and target genes during drought stress, including miR171i and miR477b, two known miRNAs that were the most significantly downregulated at 4 h of drought stress treatment (Supplemental Fig. S2C). RT-qPCR analysis showed that the expression level of miR171i and miR477b precursors was significantly lower in roots, and higher in leaves and stems, during drought stress (Fig. 2, A and D). In roots, we hypothesize that miR171i and miR477b play major roles in the response to drought stress, given their relatively high expression under normal and drought stress conditions (Fig. 2, B and E). Although miR171i/477b and their target genes had similar V-shaped expression patterns in the root in response to drought stress (Fig. 2, C and F), the increase in target gene transcript abundance lagged behind the significant decrease in miR171i/477b expression. Thus, the miRNA-mediated repression of target gene expression might be delayed under drought conditions.
Figure 2.
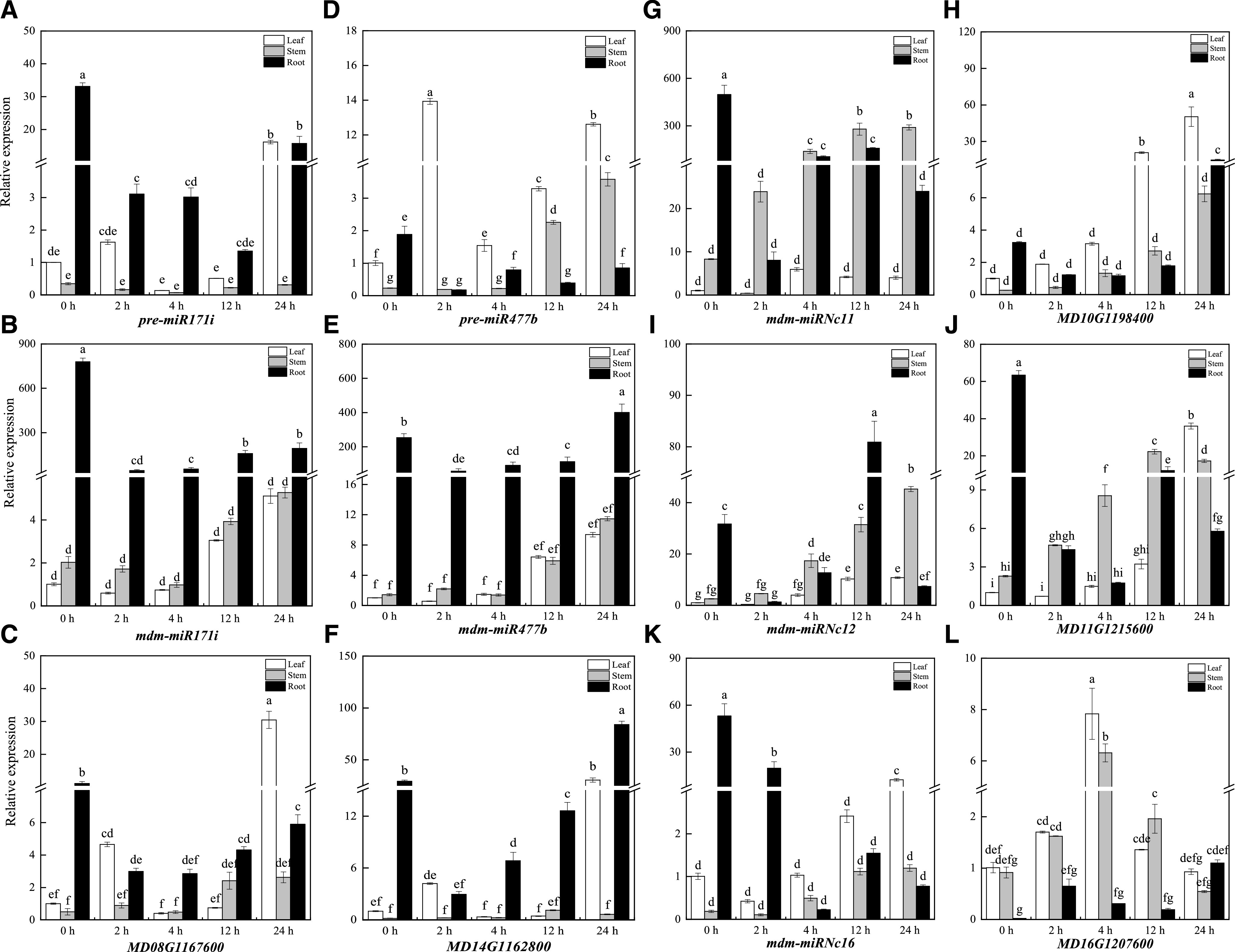
Expression patterns of two known and three new snRNAs and their targets in M. sieversii under drought stress. A to C, Expression patterns of mdm-miR171i precursors, mature sequences, and target genes in M. sieversii. D to F, Expression patterns of mdm-miR477b precursors, mature sequences, and target genes in M. sieversii. G and H, Expression patterns of mdm-miRNc11 mature sequences and target genes in M. sieversii. I and J, Expression patterns of mdm-miRNc12 mature sequences and target genes in M. sieversii. K and L, Expression patterns of mdm-miRNc16 mature sequences and target genes in M. sieversii. MdActin was used as an internal control. The values are means ± sd from three biological replicates. Lowercase letters indicate significance differences in gene expression among tissues based on Duncan’s multiple range test (P < 0.05).
We also examined the expression patterns of miRNc11/12/16 and their target genes in response to drought stress. MiRNc11/12/16 and miR171i/477b had similar expression profiles, and we inferred that this expression pattern might be associated with the responses of M. sieversii to drought stress. MiRNc11/12 accumulated mainly in stems and roots after 12 or 24 h of drought stress treatment, and their target genes were induced mostly in leaves (Fig. 2, G–J). miRNc16 expression gradually decreased in roots during drought stress treatment, whereas it increased mainly in leaves during the later stages of drought stress (Fig. 2K). There was a corresponding gradual increase in the expression of the miRNc16 target gene in roots, whereas the target gene’s expression in leaves and stems was induced during the early stages of drought stress and then decreased during the later stages (Fig. 2L).
Thus, we identified a set of known miRNAs and previously unidentified snRNAs that were differentially expressed during drought stress in apple.
Identification of mdm-miR171i and Its Target Gene MsSCL26.1
Based on the expression patterns of the differentially expressed miRNAs (Supplemental Fig. S2), we selected (M. domestica) mdm-miR171i for functional analysis in both Arabidopsis and apple. We constructed a phylogenetic tree using the sequences of 149 miR171 precursors from 17 plant species (data from miRBase 22.1). The phylogenetic tree is divided into four clades, with two to six apple miR171 family members per clade (Supplemental Fig. S5A). The number of miR171 family members varies widely among plant species, ranging from 2 to 21 (Supplemental Fig. S5B), indicating that miR171s are conserved throughout the plant kingdom. Soybean (Glycine max) contains a relatively large number (21) of miR171 family members, which might be related to the role of GAI-RGA-SCR (GRAS) transcription factors in signal transduction during nodulation (Hirsch et al., 2009). In M. domestica, the 17 miR171 family members identified by Xia et al. (2012) and Kaja et al. (2015) were divided into eight types based on the degree of similarity of their mature sequences (Supplemental Fig. S5C). The mature sequence of mdm-miR171i is slightly different from that of the other family members, with two nucleotide differences in the 8- to 9-nt sequence at the 5′ end, perhaps explaining the different expression pattern of mdm-miR171i under drought stress.
To further confirm the unique expression pattern of mdm-miR171i, we investigated the expression levels of all miR171 family members in M. sieversii treated with 20% (w/v) PEG 6000. All family members exhibited similar expression patterns, except that mdm-miR171i was expressed at a lower level in roots after 4 h of treatment (Supplemental Fig. S6). Alignment of the mature miR171 sequence with the target gene recognition sites revealed that mdm-miR171i target genes were different from those of other miR171 members due to nucleotide differences (Supplemental Fig. S7A). Phylogenetic analysis indicated that two mdm-miR171i target mRNAs, MD08G1167600 and MD15G1353200, shared high sequence similarity with AtSCL26 (Supplemental Fig. S7B). MD08G1167600 was named MdSCL26.1 and MD15G1353200 was named MdSCL26.2 based on the amino acid sequence similarity. Under normal conditions, MsSCL26.1 expression was significantly higher than that of MsSCL26.2 in both leaves and roots of M. sieversii (Supplemental Fig. S7C). Therefore, future studies should investigate the functions of mdm-miR171i and its target gene, MsSCL26.1.
To verify the interaction between mdm-miR171i and MsSCL26.1, we performed a dual luciferase-based miRNA sensor assay to qualitatively and quantitatively evaluate the cleavage of MsSCL26.1 by mdm-miR171i. The potential mdm-miR171i cleavage sites are located in the open-reading frame (ORF) of MsSCL26.1. When premdm-miR171i was coexpressed in Nicotiana benthamiana with MsSCL26.1 cleavage sites fused to the ORF of the FIREFLY LUCIFERASE (LUC) gene, there was significantly less LUC transcript abundance and enzyme activity compared to the control. However, there was no decrease in LUC expression and enzyme activity when premdm-miR171i was coexpressed with LUC fused to MsSCL26.1 cleavage sites harboring synonymous mutations (Fig. 3A).
Figure 3.

The interactional mode between mdm-miR171i and MsSCL26.1 and characterization of the GRAS transcription factor MsSCL26.1. A, Dual LUC reporter assay to qualitatively and quantitatively evaluate mdm-miR171i-mediated repression of the target gene MsSCL26.1. (MsCL26.1 is a synonymous mutation of MsSCL26.1 in mdm-miR171i cleavage sites.) Renilla luciferase was used as a positive control in the pGreen-dualluc-ORF-sensor vector. Data are presented as means ± sd from three biological replicates. Asterisks indicate significant differences between the test group and control group based on Duncan’s multiple range test (*P < 0.05). B, mdm-miR171i-directed cleavage site in MsSCL26.1 identified by 5′-RACE. The arrow indicates the mdm-miR171i cleavage site in the MsSCL26.1 transcript. The numbers represent the frequency of clones used to validate the cleavage sites of the target mRNAs. C, Four typical GRAS protein domains in MsSCL26.1. D, Transactivation activity assay of MsSCL26.1 in yeast strain AH109. MsDREB6.2 was used as a positive control. E, Subcellular localization of MsSCL26.1. GFP (Control) and the MsSCL26.1-GFP fusion gene driven by the CaMV 35S promoter were separately transiently expressed in N. benthamiana leaf epidermal cells. DAPI (4’,6-diamidino-2-phenylindole) was used as a nuclear marker.
We also performed 5′-RACE (rapid amplification of cDNA ends) to identify the cleavage site in MsSCL26.1. Multiple sequence alignment showed that the mdm-miR171i/MsSCL26.1 duplex is cleaved between the 10th and 11th nucleotide from the 5′ end of mdm-miR171i (Fig. 3B).
MsSCL26.1 is predicted to have a typical GRAS domain, including Leu repeat I, the VHIID motif, Leu repeat II, and the PFYRE motif (Fig. 3C). To determine whether MsSCL26.1 functions as a transcription factor, we performed a transcriptional activation activity assay in yeast. Fusion of the full-length MsSCL26.1 sequence to the DNA-binding domain of GAL4, similar to a previous study of MsDREB6.2 (Liao et al., 2017), strongly induced the expression of the reporter genes, indicating that MsSCL26.1 has transcriptional activation activity (Fig. 3D). We also analyzed the subcellular location of MsSCL26.1 expressed in N. benthamiana leaf epidermal cells. Fluorescence from MsSCL26.1 fused with GFP was observed in the nucleus (Fig. 3E). These results suggest that MsSCL26.1 is a GRAS transcription factor.
The miR171i-SCL26.1 Module Enhances Tolerance to Drought Stress
Like miR156 and miR172, the miR171 sequence is conserved among plant species, suggesting that it might be involved in regulating basic developmental and metabolic processes (Gu et al., 2012). Sequence alignment between mdm-miR171i and Arabidopsis miR171 family members revealed only four nucleotide mismatches, which caused the same target genes to be predicted in Arabidopsis and apple (Supplemental Fig. S8). Heterologous expression of mdm-miR171i in Arabidopsis resulted in delayed shoot apical meristem formation at the seedling stage (Fig. 4, A and B), and the transgenic Arabidopsis plants were more sensitive to drought stress than the wild type at the adult stage (Fig. 4C).
Figure 4.
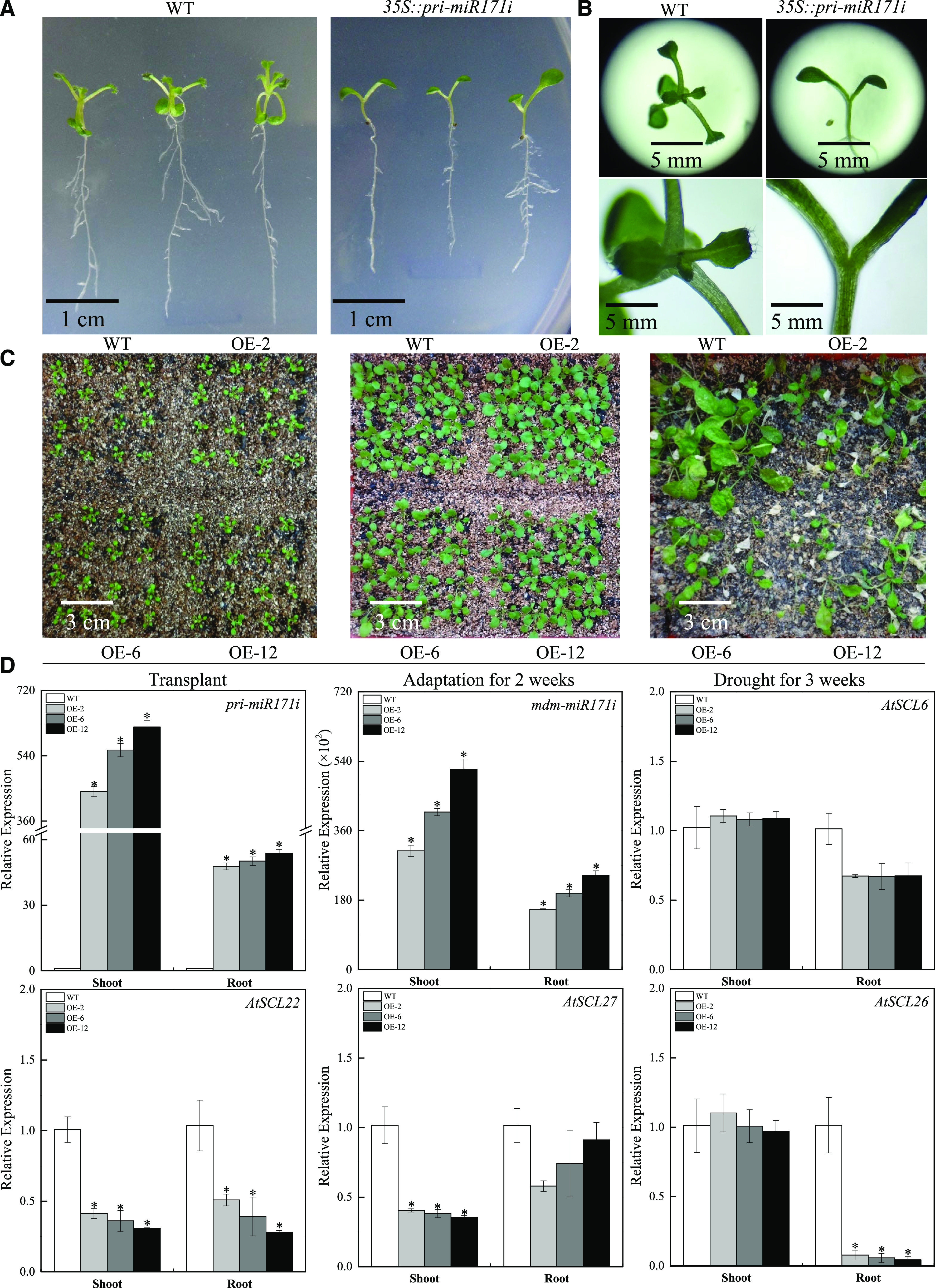
The function of mdm-miR171i in Arabidopsis. A and B, Phenotypic differences between Arabidopsis plants heterologously expressing mdm-miR171i and the wild type (WT). C, mdm-miR171i-expressing Arabidopsis show reduced drought tolerance. D, Transcriptional levels of the precursor of mdm-miR171i (pri-miR171i), mdm-miR171i, and predicted target genes in transgenic plants. The data are presented as means ± sd from three biological replicates. Asterisks indicate significant differences between the transgenic lines and wild type based on Duncan’s multiple range test (*P < 0.05).
AtSCL22 and AtSCL27 are GRAS transcription factors encoded by LOST MERISTEMS (LOM) genes that promote cell differentiation at the periphery of shoot meristems. Hence, the lack of shoot apical meristem formation at the seedling stage might be related to the inhibition of AtSCL22 and AtSCL27 expression (Fig. 4D). RT-qPCR analysis also revealed that the expression of AtSCL26, the homolog of the mdm-miR171i target gene in apple, was sharply reduced in roots, probably due to similarities in the recognition sites of these genes (Fig. 4D). The expression changes of these downstream genes in the transgenic Arabidopsis plants might account for the delayed shoot apical meristem formation and increased drought sensitivity.
To verify the function of mdm-miR171i and its target gene MsSCL26.1 in apple, we used clustered regularly interspaced short palindromic repeats/CRISPR-associated protein9 (CRISPR/Cas9) and Agrobacterium tumefaciens-mediated transformation of apple leaf discs to generate mdm-miR171i knockout plants and plants overexpressing MsSCL26.1 with synonymous mutations in the mdm-miR171i recognition sites. CRISPR/Cas9 has been used to manipulate miRNA levels in rice (Oryza sativa) and tomato (Solanum lycopersicum; Zhou et al., 2017; Damodharan et al., 2018; Basso et al., 2019). First, we edited the phytoene desaturase (PDS) gene in apple to confirm the efficiency of our system and obtained 0.27% editing rates (Supplemental Table S7). The transformed plants exhibited an albino phenotype, as would be expected for PDS-deficient mutants, and had nucleotide deletions in the fifth and seventh exons of PDS (Fig. 5, A–D). Next, we selected three independent miR171i knockout lines (CRI-171i-7, CRI-171i-10, and CRI-171i-12) for further functional analysis by identifying pri-miR171i editing types in two target sites (Fig. 5E), and the editing rate was 1.2% (Supplemental Table S7). mdm-miR171i expression was significantly reduced in the three knockout lines. By contrast, MsSCL26.1 was expressed at significantly higher levels in the miR171i-knockout lines and MsSCL26.1-overexpression lines (OE-MsSCL26.1-3, OE-MsSCL26.1-5, and OE-MsSCL26.1-8) than in nontransgenic apple plants (Fig. 6A).
Figure 5.
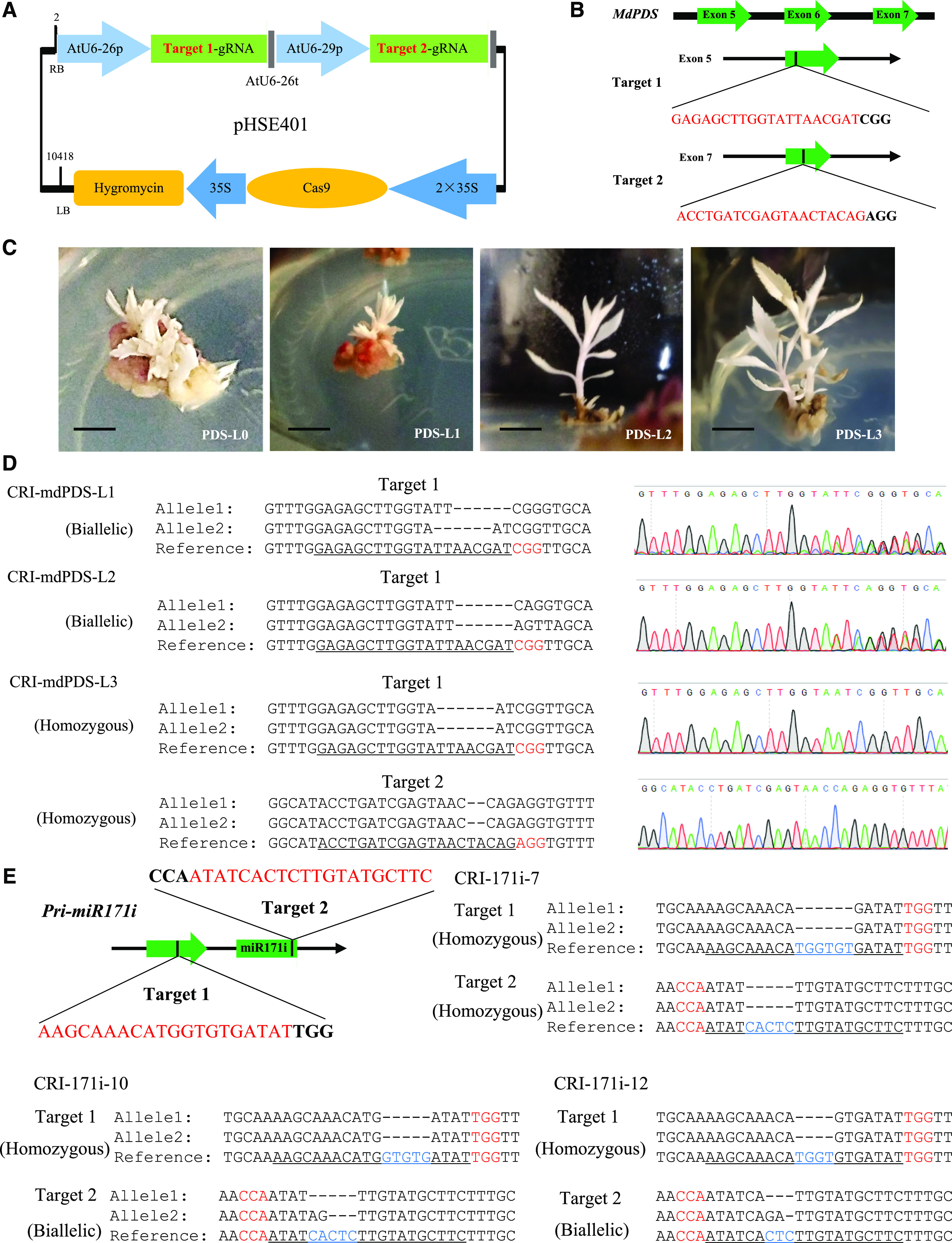
CRISPR/Cas9-mediated genome editing of apple line ‘GL-3’ A, Schematic diagram of the CRISPR/Cas9 vector pHSE401 with two target sites. B, Two target site sequences of MdPDS in the fifth and seventh exons. Green boxes indicate exons. C and D, Albino phenotype of three CRI-PDS lines (C) and mutant alleles (D) identified in ‘GL-3’ apple plants transformed with the CRISPR/Cas9-PDS vector. Scale bars = 1 cm. E, Sequences of target sites in the mdm-miR171i precursor and mutant alleles identified in the three independent transgenic lines.
Figure 6.

MiR171i and SCL26.1 expression levels and phenotypes of transgenic apple plants treated with salt-, osmotic-, and drought stress. A, miR171i and SCL26.1 expression levels in transgenic apple plants. The data are presented as means ± sd from three biological replicates. Asterisks indicate significant differences between the transgenic lines and ‘GL-3’ based on Duncan’s multiple range test (*P < 0.05). B, Phenotypic comparison of miR171i knockout and MsSCL26.1-overexpressing apple plants under salt and osmotic stress in vitro. Thirty-day-old nontransgenic and transgenic apple plants were grown for 10 d under 200 mm of NaCl or 300 mm of mannitol treatment. Scale bars = 1 cm. Plants were digitally extracted for comparison. C, Phenotypic changes in soil-grown MsSCL26.1-overexpressing apple plants under drought stress. Three-month–old nontransgenic and transgenic apple plants were grown for 15 to 20 d under drought stress in 48-cm × 20-cm × 14-cm pots. Plants were digitally extracted for comparison.
Although there were no distinct phenotypic differences between the transgenic lines and nontransgenic ‘GL-3’ apple plants under normal conditions, the transgenic plants were more tolerant than nontransgenic plants to salt, osmotic, and drought stresses both in vitro and in soil. In nontransgenic plants, salt stress was much more damaging to leaves than mannitol stress. These stresses were less damaging to the transgenic plants (Fig. 6B). Similarly, in soil-grown plants subjected to reductions in soil moisture, leaf wilting occurred 5 d later in transgenic plants overexpressing MsSCL26.1 compared to nontransgenic plants under the same conditions (Fig. 6C; Supplemental Fig. S9A). We examined several physiological indicators related to drought stress in the plants. The leaves of the transgenic plants had higher levels of Pro, lower levels of MDA and hydrogen peroxide (H2O2), and higher superoxide dismutase (SOD) and catalase (CAT) activity than the leaves of the nontransgenic plants. The 3,3′-diaminobenzidine (DAB) and nitrotetrazolium blue chloride (NBT) staining revealed lower levels of H2O2 and O2– in the leaves of transgenic versus nontransgenic plants (Fig. 7A; Supplemental Fig. S9B). These observations indicate that the miR171i-SCL26.1 module enhances tolerance to drought stress in transgenic Arabidopsis and apple.
Figure 7.
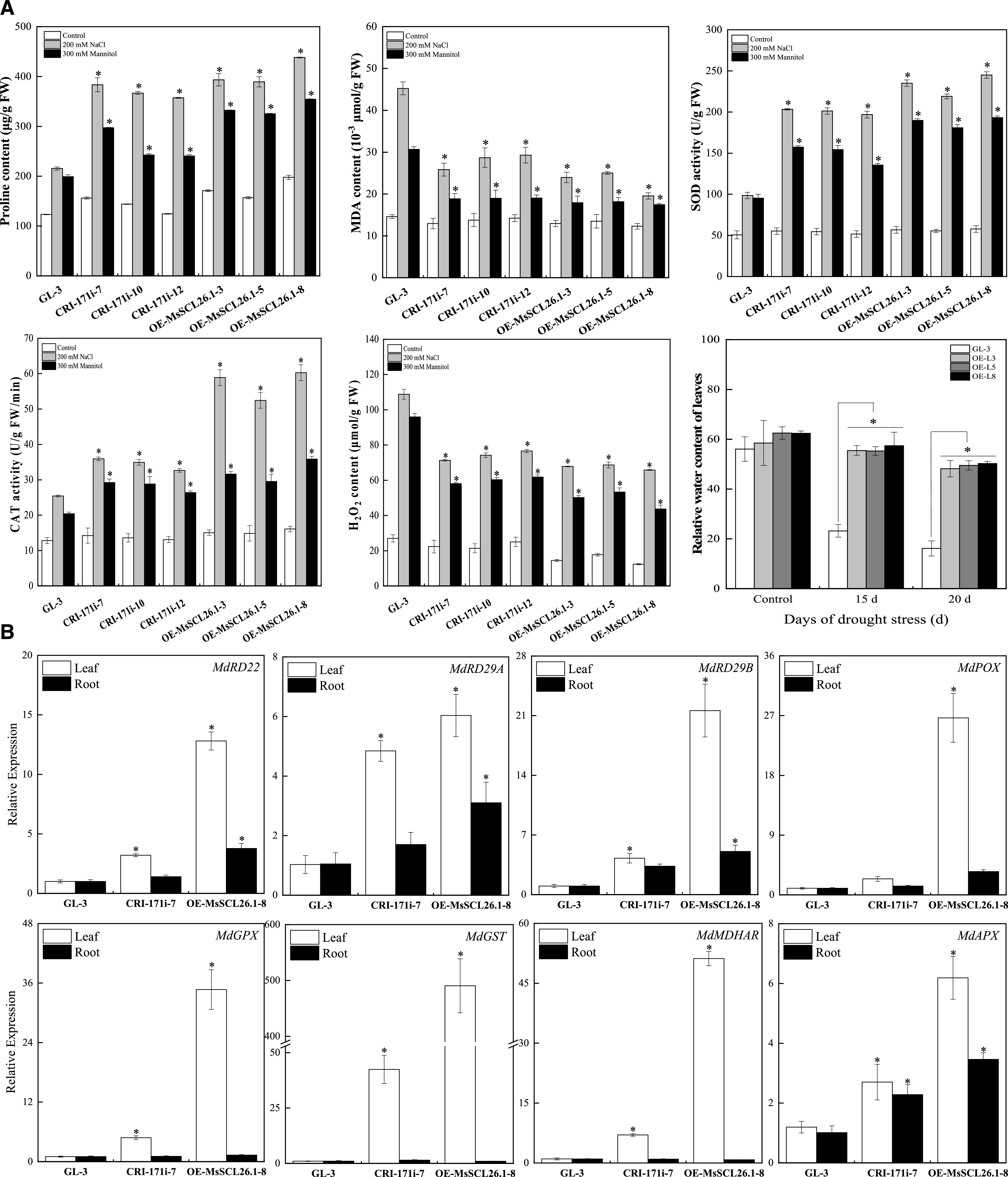
Physiological changes in transgenic apple plants under abiotic stress and the expression level of stress-related downstream genes in transgenic lines under normal conditions. A, Pro and MDA concentration, SOD, and CAT activity, H2O2 concentration of transgenic and nontransgenic apple plants subjected to salt and osmotic stress, and changes in relative water content in leaves of transgenic and nontransgenic apple plants under drought stress. B, Expression levels of stress-related downstream genes in transgenic apple plants. The data are presented as means ± sd from three biological replicates. Asterisks indicate significant differences between the transgenic lines and ‘GL-3’ based on Duncan’s multiple range test (*P < 0.05). FW, fresh weight.
MsSCL26.1 Promotes the Expression of Antioxidant Genes, Especially MsMDHAR
To understand the mechanism by which miR171i-SCL26.1 enhanced tolerance to drought stress, we analyzed the transcript levels of the stress-responsive genes MdRD22, MdRD29A, and MdRD29B and of various reactive oxygen species (ROS) scavenging enzyme genes based on published transcriptome data (Supplemental Table S8), including those encoding peroxidase superfamily protein (POX), glutathione peroxidase, GST family protein, ascorbate peroxidase (APX), and monodehydroascorbate reductase (MDHAR). Transcript levels were measured in CRI-171i-7, OE-MsSCL26.1-8, and nontransgenic plants under normal conditions. The stress-responsive genes and MdAPX were highly induced in the leaves and roots of transgenic lines compared to nontransgenic plants, whereas the other downstream genes were significantly upregulated mainly in the leaves of the transgenic lines. These differences in the tissue specificity of expression might be related to the functions of the genes. Most of the genes were expressed at much higher levels in OE-MsSCL26.1-8 than in CRI-171i-7 plants (Fig. 7B). These findings suggest that MsSCL26.1 regulates the expression of stress-responsive genes and genes encoding antioxidant enzymes involved in ROS scavenging.
To investigate whether MsSCL26.1 binds to the promoter regions of the antioxidant enzyme genes, we performed a dual LUC assay by coexpressing MsSCL26.1 in N. benthamiana leaves along with constructs in which the promoter sequences of the antioxidant enzyme genes were fused to the LUC reporter gene. When coexpressed with MsSCL26.1, LUC fused to the promoter region of MsMDHAR, MsPOX, or MsAPX produced significantly higher LUC activity than the controls without the 35S:MsSCL26.1 effector plasmid, with the highest activity detected using the MsMDHAR promoter (Fig. 8A).
Figure 8.
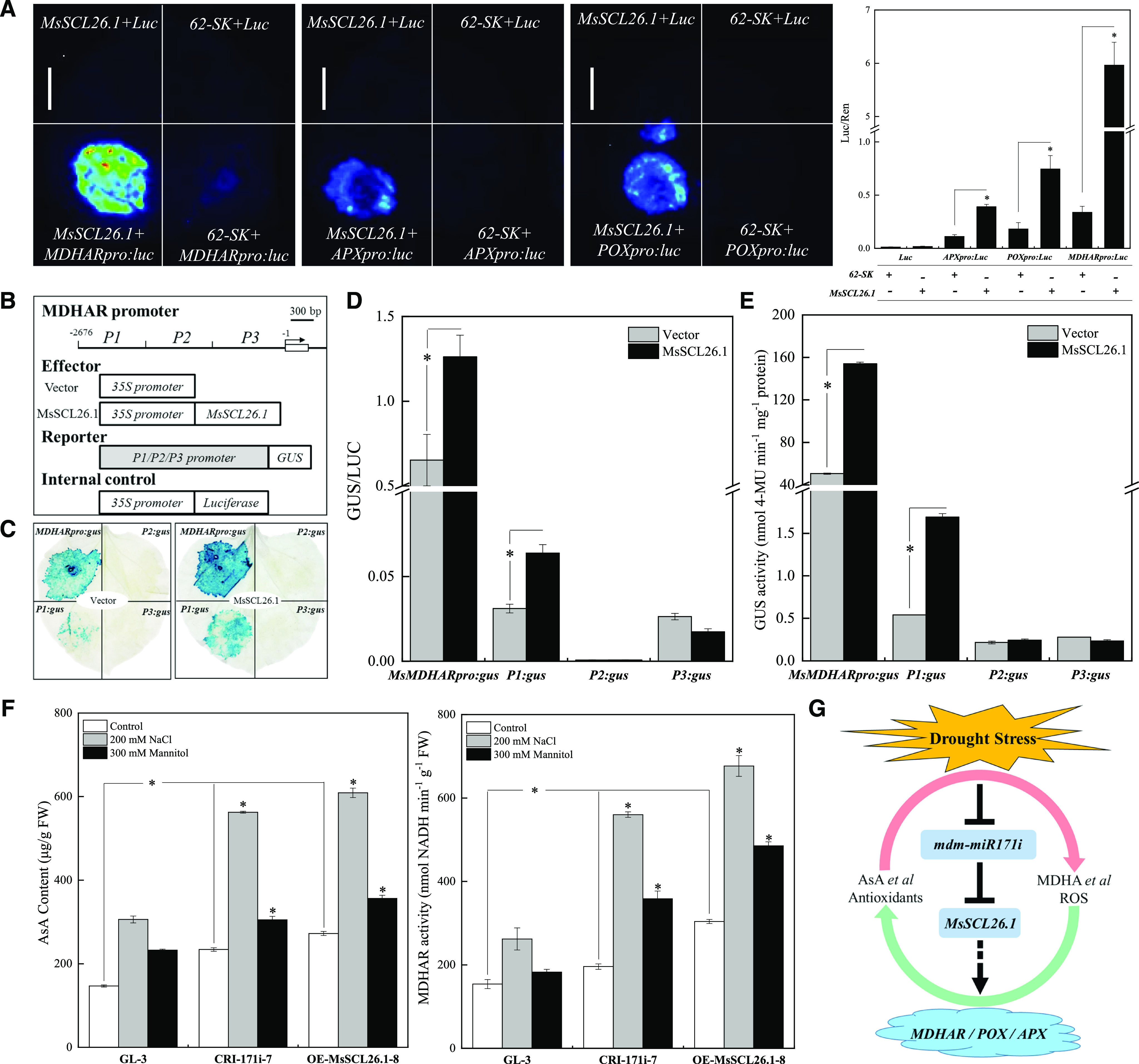
The effect of MsSCL26.1 on the transcription of genes encoding ROS scavenging system-related enzymes. A, Qualitative and quantitative evaluation of MsSCL26.1-mediated activation of antioxidant enzyme genes in a dual LUC reporter system. The data are presented as means ± sd from three biological replicates. Asterisks indicate significant differences between treatments and controls based on Duncan’s multiple range test (*P < 0.05). Scale bar = 1 cm. B to E, Transient expression assays of MDHAR promoter activity modulated by MsSCL26.1 in N. benthamiana leaves. B, Effector and reporter vectors used in the transient expression system. C, GUS staining of N. benthamiana leaves from the transient expression assays. D and E, Relative GUS activity (GUS/LUC) and quantitative analysis of GUS activity from coinjected N. benthamiana leaves. F, MDHAR activity and AsA content in transgenic and nontransgenic apple plants subjected to salt and osmotic stress in vitro for 10 d. The data are presented as means ± sd from three biological replicates. Asterisks indicate significant differences between treatments and controls based on Duncan’s multiple range test (*P < 0.05). G, Model of the mdm-miR171i-MsSCL26.1-mediated drought-stress–responsive network in apple. Antioxidants, such as AsA, are oxidized to MDHA under drought stress, which is accompanied by the release of a large number of ROS. The accumulation of high levels of ROS and other oxidation products, such as MDHA, seriously threaten the survival of the plant. Meanwhile, the transcript level of mdm-miR171i is significantly inhibited by drought stress, which reduces the repression of MsSCL26.1 in plants, especially in the roots, and might partly contribute to the upregulation of MsSCL26.1. MsSCL26.1 activates the expression of antioxidant enzyme genes, such as MsMDHAR. MsMDHAR catalyzes the reduction of MDHA into AsA, thus maintaining the homeostasis of AsA metabolism in plant cells under drought stress. Arrows, positive relationship; T-bar, negative (inhibitory) relationship.
To identify the potential MsSCL26.1-binding regions in the MsMDHAR promoter, we divided the 2.6-kb MsMDHAR promoter into three fragments and fused them to the β-glucuronidase (GUS) reporter gene. We transiently coexpressed an effector vector of 35S:MsSCL26.1 with the GUS reporter constructs in N. benthamiana leaves (Fig. 8B). GUS staining and quantitative analysis of GUS activity suggested that MsSCL26.1 significantly increased the activity of the P1 region in the MsMDHAR promoter at position −1,682 to −2,676 bp relative to the ATG start codon (Fig. 8, C–E). MsMDHAR mediates AsA metabolism (Sultana et al., 2012), and accumulation of AsA enhances abiotic stress tolerance in plants (Akram et al., 2017; Yu et al., 2019). Correspondingly, MDHAR activity and AsA contents were much higher in the leaves of transgenic apple plants than in those of nontransgenic plants under both normal and drought stress conditions (Fig. 8F).
Taken together, these observations indicate that the miR171i-SCL26.1 pathway improves plant tolerance to drought stress in apple by regulating the expression of genes encoding antioxidant enzymes in the ROS scavenging system. This regulation is especially strong in the case of MsMDHAR, which is involved in maintaining AsA homeostasis.
DISCUSSION
Identification of snRNAs and Target Genes in M. sieversii
Even though 38,589 miRNAs have already been deposited in miRBase 22.1, miRNAs continue to be identified in various plant species via next-generation sequencing. Plants contain numerous conserved miRNAs that regulate their growth, development, and environmental responses (Song et al., 2019). In addition, because of differences in habitat and evolutionary history, various plants possess species-specific miRNAs, which add to the number of miRNAs being discovered.
In this study, we identified 210 known miRNAs and 125 candidate snRNAs in the wild apple species M. sieversii via high-throughput sequencing. In 2012, Xia et al. (2012) identified 207 miRNAs in apple, 204 of which were also identified in this study. Three years later, Kaja et al. (2015) discovered 102 apple miRNAs, only six of which were identified in this study. M. sieversii was identified as an ancestor of cultivated apple in previous studies (Duan et al., 2017). Many reported apple miRNAs were not detected in this study, suggesting that they might have been derived from other ancestral species of cultivated apple or arose more recently than the evolutionary split between M. sieversii and cultivated apple (Nozawa et al., 2012). In addition, we identified three previously unidentified snRNA family members, miRNc1, miRNc2, and miRNc9h/i, located in the precursor sequences of known miRNAs, suggesting that these precursors produce at least two mature miRNAs. The ability of a precursor to produce multiple mature miRNAs has been observed in Arabidopsis (Voinnet, 2009; Fahlgren et al., 2010).
We also identified 278 target genes for 169 known and 45 new snRNAs using degradome analysis (Supplemental Table S9). By comparing our data with five available miRNA degradome data sets from apple, we identified additional target genes that are cleaved by known miRNAs. Specifically, miR172, miR159, and miR396 degrade the transcripts of WRKY51 (except AP2), MADS-box (except MYB DNA-binding factors), and bHLH (except GRF), respectively (Supplemental Table S3). The new miRNA family members (miRNc9/11/12) target the same genes due to the high similarity of their mature sequences to those of miR319/164/159, suggesting that additional known apple miRNAs might be identified in M. sieversii. The identification of apple snRNAs and target genes in M. sieversii lays the foundation for understanding the regulation of gene expression in this important crop.
snRNAs that Are Differentially Expressed in Response to Drought Stress in M. sieversii
These findings show that most of the known miRNAs and newly identified snRNAs were upregulated or downregulated after short-term drought stress of only 2 or 4 h (Supplemental Fig. S2), indicating that snRNAs quickly respond to environmental stimuli and regulate gene expression during the early stages of drought stress. For instance, three known miRNAs (miR395, miR398, and miR7125) with target genes involved in sulfate, copper, and zinc transport, respectively, were downregulated after 2 h of drought stress, indicating that ion transport and metabolism were regulated to maintain a high osmotic potential in the cell. Although miR398 expression is repressed in tomato (Candar-Cakir et al., 2016) and cotton (Gossypium hirsutum; Xie et al., 2015) in response to drought stress, the target gene encodes a copper/zinc SOD, in contrast to the target gene of apple miR398. Therefore, the same miRNAs from different species might participate in different drought-related regulatory networks. GO and KEGG pathway analysis revealed that these genes are involved in multiple biological processes, including signal transduction, transcriptional regulation, and multiple metabolic processes (Supplemental Figs. S3 and S4).
We selected two known and three new snRNAs to verify the expression patterns under drought stress in M. sieversii. These snRNA levels were significantly higher in roots than in other tissues under normal conditions. Furthermore, the abundance in roots declined to their lowest levels after 2 or 4 h of drought stress, and the concomitant increase in shoots did not compensate for the decline in roots, showing that drought stress causes a global decrease in the transcription of miRNAs and their targets due to general physiological changes. Hence, the expression patterns of selected snRNAs were largely consistent with the results of small RNA sequencing (Supplemental Fig. S2).
On the other hand, we established that miR171i/477b and miRNc11/12 and their target genes exhibited V-shaped expression patterns in the roots, which might be because the negative association between miRNAs and their targets is reduced under drought stress (Fig. 2, A‒J). Firstly, we speculated that the V-shaped expression trends in the roots might be associated with the responsive phenotype of M. sieversii during drought stress (Fig. 1A). Except for miRNc16 and MD16G1207600, the abundance of other miRNAs in roots reached the lowest levels at 2 h of drought stress, whereas their target genes began to be significantly upregulated at 4 h or 12 h, further suggesting that the delay in miRNA regulation might desynchronize their negative association.
Secondly, the reduced expression of target genes, with the exception of MD16G1207600, in roots under 4 or 12 h of drought stress compared to unstressed plants might be related to the tissue-specific expression of the genes. MD16G1207600 encodes a phloem filament protein, which accumulated in stems and leaves under normal and drought stress conditions. This protein might facilitate the transport of nutrients and osmotic regulators from the leaves and stems to the roots under drought stress, relieving stress damage in the roots. Indeed, previous studies have demonstrated that genes can have different functions under drought stress and normal conditions, due to tissue-specific regulatory mechanisms (Campo et al., 2014; Liao et al., 2017; Martignago et al., 2020; Gupta et al., 2020). The tissue-specific regulatory mechanisms that control drought-responsive miRNAs remain to be elucidated (Candar-Cakir et al., 2016; Guo et al., 2017b; Pekmezci et al., 2017).
Therefore, M. sieversii adjusts its physiological status and responds to complex environmental changes by rapidly regulating the abundance of miRNAs and their target genes under the short-term drought stress.
The MiR171i-SCL26.1 Module Improves Drought Stress Tolerance by Maintaining AsA Homeostasis
Highly conserved miRNA families in plants play conserved roles in growth and development, as well as other diverse roles that depend on genomic differences among plant species (Gramzow and Theißen, 2019). The miR171 family is one of the best conserved miRNA families, but the number and sequences of mature miR171s differ markedly between Arabidopsis and other plants (Zhu et al., 2015), suggesting that miR171s play diverse roles in different plant species. For example, miR171 was induced by abiotic stress in embryogenic callus tissues from Larix leptolepis (Zhang et al., 2010), but in potato (Solanum tuberosum), the abundance of stu-miR171 decreased, followed by an increase, upon air-drying and 15% (w/v) PEG 6000 treatment (Hwang et al., 2011). In rice, osa-miR171c accumulated substantially under abiotic stress and exogenous abscisic acid treatment, and transgenic lines overexpressing osa-miR171c displayed increased sensitivity to salt stress (Yang et al., 2017). In addition, plant miR171s have been implicated in tolerance to long-term boron toxicity (Huang et al., 2019), chlorophyll biosynthesis (Ma et al., 2014), and arbuscular mycorrhizal and root nodule symbiosis (Hofferek et al., 2014). However, the functions of apple miR171s are unclear.
Furthermore, miR171-targeted GRAS proteins play important roles in regulating plant growth and development as well as biotic and abiotic stress resistance (Hirsch and Oldroyd, 2009). Heterologous expression of PeSCL7 from Populus euphratica (Ma et al., 2010) and VaPAT1 from Vitis amurensis (Yuan et al., 2016) in Arabidopsis and overexpression of OsGRAS23 in rice (Xu et al., 2015a) markedly enhanced plant tolerance to drought and salt stress by upregulating a series of stress-responsive genes. Although additional abiotic stress-related GRAS transcription factors have been identified in grapevine (Vitis vinifera; Grimplet et al., 2016), tea plant (Camellia sinensis; Wang et al., 2018b), cotton (Zhang et al., 2018b), and Tamarix hispida (Wang et al., 2019), it is not known whether GRAS transcription factors play similar roles in apple.
In this study, the target gene of mdm-miR171i shares high levels of sequence similarity with AtSCL26 at the amino acid level. Heterologous expression of mdm-miR171i repressed AtSCL22 and AtSCL27 expression in Arabidopsis, which led to delayed shoot apical meristem formation at the seedling stage. In addition, AtSCL26 expression was significantly inhibited in the roots of the transgenic plants, resulting in lower tolerance to drought stress than in the wild type (Fig. 4). These results indicate that mdm-miR171i is involved in the formation of the shoot apical meristem and drought tolerance in Arabidopsis.
On the other hand, knockout of mdm-miR171i and overexpression of MsSCL26.1 improved tolerance to salt, osmotic, and drought stress compared to nontransgenic plants (Fig. 6). Furthermore, the expression of LOM genes (MdSCL6/22/27) and of other miR171 members was not dramatically changed in the knockout lines (Supplemental Fig. S10), and MdSCL26.1 transcript abundance was significantly increased in the knockout lines (Fig. 6A), as revealed by RT-qPCR. These data suggest that miR171i specifically regulates MdSCL26.1 and not the other LOM genes in apple plants. Taken together, the phenotypic differences between the transgenic Arabidopsis and ‘GL-3’ apple plants might result from the differential expression of miR171i target genes.
Under drought stress, plants accumulate massive amounts of ROS, which causes lipid peroxidation, protein dysfunction, and DNA damage (Selote and Khanna-Chopra, 2010). Plant cells have both enzymatic and nonenzymatic pathways that can scavenge ROS to maintain antioxidant homeostasis. Enzymatic scavenging systems include all types of antioxidant enzymes, whereas nonenzymatic systems primarily consists of AsA or glutathione (Xu et al., 2015b). Overexpression of antioxidant enzyme genes and exogenous application of antioxidants (mainly AsA) confer increased abiotic stress tolerance in plants (Kavitha et al., 2010; Terzi et al., 2015; Hafez and Gharib, 2016). Heterologous expression or overexpression of MdATG18a, the apple homolog of a gene critical for autophagy in Arabidopsis, enhanced drought stress tolerance in tomato and apple plants by increasing antioxidant levels (Sun et al., 2018). Nonetheless, much remains to be learned about the ROS metabolic regulatory networks in cultivated apple under drought stress.
AtSCL26 is dramatically induced in Arabidopsis roots under drought stress, but its role is largely unclear (Guo et al., 2009). MiR171i-targeted MsSCL26.1, the homolog of AtSCL26 in M. sieversii, significantly enhances drought stress tolerance in apple by activating the expression of several genes encoding key enzymes involved in ROS scavenging. Among these, MDHAR, POX, and APX were markedly upregulated by MsSCL26.1 (Figs. 7B and 8A). MDHAR mediates the regeneration of AsA from monodehydroascorbate (MDHA), the oxidation product of AsA in the chloroplasts of plants in response to environmental stimuli, maintaining proper ROS levels in plant cells during drought stress (Gallie, 2013). In this study, heterologous expression of MsSCL26.1 significantly increased GUS activity in the leaves of N. benthamiana harboring GUS driven by the P1 region of the MsMDHAR promoter (Fig. 8, B‒E), and MDHAR activity and AsA levels were substantially increased in the transgenic apple leaves under normal conditions and also under salt and osmotic stress (Fig. 8F).
These observations further confirm that MsSCL26.1 specifically regulates MsMDHAR expression and thereby increases MDHAR activity and AsA content in apple leaves under drought stress. Thus, the miR171i-SCL26.1 module enhances tolerance to drought stress in apple by functioning in the AsA cycle and regeneration process (Fig. 8G).
MATERIALS AND METHODS
Plant Materials and Treatments
Malus sieversii plants were grown in vitro for 30 d and transferred to rooting medium (Murashige and Skoog medium with 0.5 mg L−1 of indole-3-butyric acid). After 20 d, the rooted plants were grown in half-strength Hoagland nutrient solution for 2 weeks, followed by full-strength Hoagland nutrient solution before drought stress treatment. The plants were grown at 23°C and 40% relative humidity under a 16-h/8-h (day/night) photoperiod. When the plants reached a height of 20 to 30 cm, they were subjected to drought stress. The plants were immersed in a solution of 20% (w/v) PEG 6000 for various periods of time. Leaves and roots were harvested and mixed (at equal weight) for small RNA deep sequencing, and the remaining samples were separately harvested for RNA isolation and expression analysis.
Tissue-cultured plants of apple (Malus domestica) line ‘GL-3’ from the laboratory of Dr. Zhihong Zhang (Liaoning, China) were used for genetic transformation and stress treatments as described in Chen et al., (2019).
Small RNA and Degradome Sequencing
Mixed samples were sent to BioMarker Technology. Two biological replicates were performed per treatment. Small RNA libraries were constructed following standard methods as described in Yin et al. (2016). Briefly, 1.5 μg of RNA was used as input material for RNA sample preparation. A NanoDrop Spectrophotometer (Thermo Fisher Scientific), a Qubit 2.0 Fluorometer (Thermo Fisher Scientific), and a 2100 Bioanalyzer (Agilent) were used to detect the purity, concentration, and integrity of the RNA samples, respectively. Sequencing libraries were generated using a NEBNext Ultra Small RNA Library Prep Kit for Illumina (New England BioLabs) following the manufacturer’s recommendations. The libraries were sequenced on the HiSeq 2500 platform (Illumina). All downstream analyses were based on high quality clean data. RNAfold tools (http://unafold.rna.albany.edu/?q=mfold/RNA-Folding-Form2.3) were used to predict the secondary structures of miRNAs. After removing known functional RNAs, the clean reads were mapped to the M. domestica reference genome (BioProject ID: PRJNA28845). The tool MiRDeep2 (http://hpc.ilri.cgiar.org/mirdeep2-software) was used to identify known miRNAs and to predict miRNAs (Supplemental Table S10). Target genes were predicted using the tool TargetFinder (http://www.bioit.org.cn/ao/targetfinder.htm).
Samples from plants after 4 h of treatment with 20% (w/v) PEG 6000 were used to construct a degradome library as described in Addo-Quaye et al. (2009), with some modifications. Briefly, degraded fragments captured by magnetic beads containing 5′-monophosphates were ligated to a 5′ adaptor with T4 RNA ligase and used to generate first-strand complementary DNA in the presence of biotinylated random primers. After performing PCR amplification, sequencing was performed on a HiSeq 2500 (Illumina) at LC-Bio. The degraded fragments were matched to the apple genome. The raw data have been deposited in the National Center for Biotechnology Information Sequence Read Archive (https://www.ncbi.nlm.nih.gov/Traces/sra_sub/sub.cgi) and the accession number was PRJNA645498.
RNA Extraction and RT-qPCR
Total RNA was extracted from Arabidopsis (Arabidopsis thaliana) with TRIzol Reagent (CWBIO), and small RNA was extracted from apple plants using an EASYspin Plant microRNA Extract Kit (Aidlab). The RNA samples were used to generate complementary DNA with oligo dT primers and stem-loop reverse transcription primers, respectively. RT-qPCR was performed in a 20-μL reaction system containing 10 μL of 2× UltraSYBR Mixture (CWBIO) and 0.4 μm of forward and reverse primers to assess the expression of miRNAs and their targets. The reactions were incubated in a Rotor-Gene Q Machine (Qiagen) for 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 34 s at 60°C. AtActin2 and MdActin were used as internal controls for Arabidopsis and apple, respectively. Relative expression levels were calculated using the 2–ΔΔCt method (Livak and Schmittgen, 2001). All primers and accession numbers are listed in Supplemental Table S11.
Phylogenetic Analysis of miR171 and GRAS Families
A total of 149 miR171 precursor sequences from 17 plant species and the GRAS amino acid sequences from Arabidopsis and apple were used as queries in BLAST searches (http://www.mirbase.org/ftp.shtml) of miRBase 22.1 (http://www.mirbase.org/), The Arabidopsis Information Resource, and the Genome Database for Rosaceae, respectively. The phylogenetic analyses were conducted using MEGA v.6 and the neighbor-joining method with 1,000 bootstrap replicates. The phylogenetic trees were modified using the tool Evolview v3 (Subramanian et al., 2019).
Dual LUC Reporter Assay in Nicotiana benthamiana and 5′-RACE
Two different dual LUC reporter systems were used in this study: one system was used to confirm the interaction between mdm-miR171i and MsSCL26.1, and the other was used to screen for downstream genes of MsSCL26.1.
In the first system, potential mdm-miR171i cleavage sites from MsSCL26.1 and its synonymous mutation sequences (as a negative control) were inserted into the AvrII/AgeI sites of the pGreen-dualluc-ORF-sensor vector as described in Liu et al. (2014). The precursor of mdm-miR171i was driven by the CaMV35S promoter in the pGreenII 0029 62-SK vector. The pGreen-dualluc-ORF-sensor vector carrying the Renilla luciferase gene was used as a positive control. Transformation and infiltration were performed as described in Wei et al. (2017). In the second system, pGreenII 0029 62-SK, containing the coding sequences of MsSCL26.1, and pGreenII 0800-LUC, containing the promoter sequences of the candidate genes, were separately transformed into Agrobacterium tumefaciens strain GV3101 harboring the pSoup plasmids. Infiltration and promoter activity analysis were performed as described in Hellens et al. (2005).
The cleaved products of MsSCL26.1 were obtained as described in Zhang et al. (2018a) using a modified 5′-RACE method.
Transcriptional Activity Analysis and Subcellular Localization of MsSCL26.1
The full-length MsSCL26.1 sequence containing NdeI/EcoRI sites was fused to the pGBKT7 vector 78 bp downstream of the GAL4 DNA-binding region. The recombination plasmid was transformed into yeast strain AH109. The transformants were screened and MsDREB6.2 was used as a positive control.
To investigate the subcellular localization of MsSCL26.1, the full-length MsSCL26.1 sequence without the stop codon was cloned from M. sieversii and inserted into the PacI/KpnI sites of the pMDC83 vector, which enabled MsSCL26.1 to be fused with GFP. Transformation and GFP fluorescence observation were performed as described in Liu et al. (2010).
Plasmid Construction and Genetic Transformation
For mdm-miR171i overexpression, the 895-bp sequence from M. sieversii containing the miR171i stem-loop structure was amplified and inserted into the NcoI/BglII sites of pCAMBIA1302. To knock out mdm-miR171i, a CRISPR/Cas9 toolkit was used as described in Xing et al. (2014a). The PDS gene was used as a positive control to validate the CRISPR/Cas9 system. The target sites for gene editing were screened using CRISPR Primer Designer (Yan et al., 2015). The coding sequence of MsSCL26.1 containing the synonymous mutation of sites cleaved by mdm-miR171i was cloned using overlapping PCR and inserted into the PacI/KpnI sites of the pMDC83 vector. These vectors were transformed into A. tumefaciens strain EHA105. Arabidopsis and apple ‘GL-3′ were genetically transformed as described in Clough and Bent (1998) and Dai et al. (2013).
Measurement of Physiological Indicators and NBT and DAB Staining
Pro, MDA, AsA, and H2O2 levels and SOD, CAT, and MDHAR activity in leaves were measured as described in Velikova et al. (2000) and Eltayeb et al. (2007). DAB and NBT staining of leaves under stress treatments was performed using a modified method as described in Kumar et al. (2014).
GUS Reporter Gene Assay in Transiently Transformed N. benthamiana leaves
Different promoter regions of MsMDHAR were fused to the GUS gene in the pCAMBIA1301 vector. The 35S:MsSCL26.1 construct was used as an effector, and 35S:LUC was used as an internal control. The constructs were transformed into A. tumefaciens strain GV3101, and infiltration and GUS activity analysis were performed as described in Liu et al. (2016).
Statistical Analyses
Statistical analyses were performed using the one-way ANOVA followed by Duncan’s multiple range test (SPSS v.22.0 for Windows; SPSS). All the data were presented as the mean ± sd from three biological replicates; P < 0.05 was considered as statistically significant.
Accession Numbers
Sequence data from this article can be found in the Genome Database for Rosaceae Web site (https://www.rosaceae.org/) and The Arabidopsis Information Resource database (https://www.arabidopsis.org/index.jsp) under accession numbers MdSCL26.1 (MD08G1167600), MdSCL26.2 (MD15G1353200), MdPDS (MD04G1023800), MdRD22 (MD15G1098800), MdRD29A (MD01G1201000), MdRD29B (MD07G1268800), MdPOD (MD03G1059000), MdGPX6 (MD06G1081300), MdGST (MD04G1111600), MdAPX6 (MD08G1150400), MdMDHAR (MD14G1018700), MdSCL6 (MD02G1210200), MdSCL22.1 (MD07G1115800), MdSCL22.2 (MD04G1088100), MdSCL27 (MD00G1043300), AtSCL27 (AT2G45160), AtSCL22 (AT3G60630), AtSCL6 (AT4G00150), AtSCL26 (AT4G08250), and AtACT2 (AT3G18780).
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. Several new and candidate miRNAs are located in the precursors of known miRNAs.
Supplemental Figure S2. Identification of the functions of target genes and differentially expressed snRNAs in M. sieversii under drought stress.
Supplemental Figure S3. GO annotation and KEGG pathway analysis of target genes of differentially expressed known miRNAs.
Supplemental Figure S4. GO annotation and KEGG pathway analysis of target genes of differentially expressed newly identified snRNAs.
Supplemental Figure S5. Phylogenetic analysis of miR171 families in 17 plant species and sequence classification of miR171 family members from M. sieversii and Arabidopsis.
Supplemental Figure S6. The expression patterns of all mature miR171 family members in M. sieversii treated with 20% PEG 6000.
Supplemental Figure S7. Alignment analysis of mdm-miR171 family members and their targets.
Supplemental Figure S8. Comparative analysis of the targets of mdm-miR171i and ath-miR171.
Supplemental Figure S9. Changes of soil water content and the height and basal diameter of transgenic apple plants during drought stress, and leaf staining of transgenic apple plants under salt and osmotic stress.
Supplemental Figure S10. Expression levels of MdSCL6/22/27 and other miR171 members except miR171i in transgenic apple plants.
Supplemental Table S1. The 210 known miRNAs and 125 candidate snRNAs in M. sieversii.
Supplemental Table S2. Several new snRNAs with mature sequences that are located in known miRNA precursors.
Supplemental Table S3. The target genes of known miRNAs, as revealed by degradome analysis.
Supplemental Table S4. The target genes of new snRNAs, as revealed by degradome analysis.
Supplemental Table S5. Comparative analysis between miRNc9, miRNc11, and miRNc12 and miR319, miR164, and miR159. Underlined bases represent differences between miRNAs.
Supplemental Table S6. Differential expression analysis of snRNAs in M. sieversii.
Supplemental Table S7. Transformation rates of CRISPR/Cas9-mediated gene editing in T0 transgenic apple plants.
Supplemental Table S8. Information about genes encoding five crucial enzymes in the ROS scavenging system. Transcriptome data from BioProject: PRJNA379354.
Supplemental Table S9. Forty-five new snRNAs in M. sieversii that target at least one transcript, as revealed by degradome analysis.
Supplemental Table S10. TPM read counts of known miRNAs and new snRNAs in M. sieversii.
Supplemental Table S11. Primers of each gene used in this study.
Acknowledgments
We thank Qijun Chen (China Agricultural University) for providing the CRISPR/Cas9 vectors and Zhihong Zhang (Shenyang Agricultural University) for providing the ‘GL-3’ apple plants. We thank Dr. Kathleen Farquharson and Bingbing Li (China Agricultural University) for their critical review of the article.
Footnotes
This work was supported by the National Key R&D Program of China (grant no. 2019YFD1000102–02), the National Natural Science Foundation of China (grant nos. 31672119 and 31972390), and the Construction of Beijing Science and Technology Innovation and Service Capacity in Top Subjects (grant no. CEFF–PXM2019_014207_000032).
References
- Achkar NP, Cambiagno DA, Manavella PA(2016) miRNA biogenesis: A dynamic pathway. Trends Plant Sci 21: 1034–1044 [DOI] [PubMed] [Google Scholar]
- Addo-Quaye C, Miller W, Axtell MJ(2009) CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 25: 130–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akram NA, Shafiq F, Ashraf M(2017) Ascorbic acid: A potential oxidant scavenger and its role in plant development and abiotic stress tolerance. Front Plant Sci 8: 613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso MF, Ferreira PCG, Kobayashi AK, Harmon FG, Nepomuceno AL, Molinari HBC, Grossi-de-Sa MF(2019) MicroRNAs and new biotechnological tools for its modulation and improving stress tolerance in plants. Plant Biotechnol J 17: 1482–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechtold U, Field B(2018) Molecular mechanisms controlling plant growth during abiotic stress. J Exp Bot 69: 2753–2758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campo S, Baldrich P, Messeguer J, Lalanne E, Coca M, San Segundo B(2014) Overexpression of a calcium-dependent protein kinase confers salt and drought tolerance in rice by preventing membrane lipid peroxidation. Plant Physiol 165: 688–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candar-Cakir B, Arican E, Zhang B(2016) Small RNA and degradome deep sequencing reveals drought-and tissue-specific microRNAs and their important roles in drought-sensitive and drought-tolerant tomato genotypes. Plant Biotechnol J 14: 1727–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Zeng Z, Liu Z, Xia R(2018) Small RNAs, emerging regulators critical for the development of horticultural traits. Hortic Res 5: 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Song M, Guo Y, Liu L, Xue H, Dai H, Zhang Z(2019) MdMYB46 could enhance salt and osmotic stress tolerance in apple by directly activating stress-responsive signals. Plant Biotechnol J 17: 2341–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clough SJ, Bent AF(1998) Floral dip: A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16: 735–743 [DOI] [PubMed] [Google Scholar]
- Dai H, Li W, Han G, Yang Y, Ma Y, Li H, Zhang Z(2013) Development of a seedling clone with high regeneration capacity and susceptibility to Agrobacterium in apple. Sci Hortic (Amsterdam) 164: 202–208 [Google Scholar]
- Damodharan S, Corem S, Gupta SK, Arazi T(2018) Tuning of SlARF10A dosage by sly-miR160a is critical for auxin-mediated compound leaf and flower development. Plant J 96: 855–868 [DOI] [PubMed] [Google Scholar]
- Duan N, Bai Y, Sun H, Wang N, Ma Y, Li M, Wang X, Jiao C, Legall N, Mao L, et al. (2017) Genome re-sequencing reveals the history of apple and supports a two-stage model for fruit enlargement. Nat Commun 8: 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltayeb AE, Kawano N, Badawi GH, Kaminaka H, Sanekata T, Shibahara T, Inanaga S, Tanaka K(2007) Overexpression of monodehydroascorbate reductase in transgenic tobacco confers enhanced tolerance to ozone, salt and polyethylene glycol stresses. Planta 225: 1255–1264 [DOI] [PubMed] [Google Scholar]
- Fahlgren N, Jogdeo S, Kasschau KD, Sullivan CM, Chapman EJ, Laubinger S, Smith LM, Dasenko M, Givan SA, et al. (2010) MicroRNA gene evolution in Arabidopsis lyrata and Arabidopsis thaliana. Plant Cell 22: 1074–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedländer MR, Mackowiak SD, Li N, Chen W, Rajewsky N(2012) miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 40: 37–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallie DR.(2013) l-ascorbic acid: A multifunctional molecule supporting plant growth and development. Scientifica (Cairo) 2013: 795964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleave AP, Ampomah-Dwamena C, Berthold S, Dejnoprat S, Karunairetnam S, Nain B, Wang Y-Y, Crowhurst RN, MacDiarmid RM(2008) Identification and characterisation of primary microRNAs from apple (Malus domestica cv. Royal Gala) expressed sequence tags. Tree Genet Genomes 4: 343–358 [Google Scholar]
- Gramzow L, Theißen G(2019) Plant miRNA Conservation and Evolution In de Folter S, ed, Plant MicroRNAs Methods in Molecular Biology, Vol Vol 1932 Humana Press, New York, NY: [DOI] [PubMed] [Google Scholar]
- Grimplet J, Agudelo-Romero P, Teixeira RT, Martinez-Zapater JM, Fortes AM(2016) Structural and functional analysis of the GRAS gene family in grapevine indicates a role of GRAS proteins in the control of development and stress responses. Front Plant Sci 7: 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Wang X, Zhai C, Xie X, Zhou T(2012) Selection on synonymous sites for increased accessibility around miRNA binding sites in plants. Mol Biol Evol 29: 3037–3044 [DOI] [PubMed] [Google Scholar]
- Guo H, Jiao Y, Di C, Yao D, Zhang G, Zheng X, Liu L, Zhang Q, Guo A, Su Z(2009) Discovery of Arabidopsis GRAS family genes in response to osmotic and drought stresses. Zhiwu Xuebao 44: 290–299 [Google Scholar]
- Guo X, Ma Z, Zhang Z, Cheng L, Zhang X, Li T(2017a) Small RNA-sequencing links physiological changes and RdDM process to vegetative-to-floral transition in apple. Front Plant Sci 8: 873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Zhao S, Zhu C, Chang X, Yue C, Wang Z, Lin Y, Lai Z(2017b) Identification of drought-responsive miRNAs and physiological characterization of tea plant (Camellia sinensis L.) under drought stress. BMC Plant Biol 17: 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Rico-Medina A, Caño-Delgado AI(2020) The physiology of plant responses to drought. Science 368: 266–269 [DOI] [PubMed] [Google Scholar]
- Hafez EM, Gharib HS(2016) Effect of exogenous application of ascorbic acid on physiological and biochemical characteristics of wheat under water stress. Int J Plant Prod 10: 579–596 [Google Scholar]
- Hellens RP, Allan AC, Friel EN, Bolitho K, Grafton K, Templeton MD, Karunairetnam S, Gleave AP, Laing WA(2005) Transient expression vectors for functional genomics, quantification of promoter activity and RNA silencing in plants. Plant Methods 1: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch S, Kim J, Muñoz A, Heckmann AB, Downie JA, Oldroyd GED(2009) GRAS proteins form a DNA binding complex to induce gene expression during nodulation signaling in Medicago truncatula. Plant Cell 21: 545–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch S, Oldroyd GED(2009) GRAS-domain transcription factors that regulate plant development. Plant Signal Behav 4: 698–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofferek V, Mendrinna A, Gaude N, Krajinski F, Devers EA(2014) MiR171h restricts root symbioses and shows like its target NSP2 a complex transcriptional regulation in Medicago truncatula. BMC Plant Biol 14: 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J-H, Lin X-J, Zhang L-Y, Wang X-D, Fan G-C, Chen L-S(2019) MicroRNA sequencing revealed Citrus adaptation to long-term boron toxicity through modulation of root development by miR319 and miR171. Int J Mol Sci 20: 1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang E-W, Shin S-J, Yu B-K, Byun M-O, Kwon H-B(2011) miR171 family members are involved in drought response in Solanum tuberosum. J Plant Biol 54: 43–48 [Google Scholar]
- Kaja E, Szcześniak MW, Jensen PJ, Axtell MJ, McNellis T, Makałowska I(2015) Identification of apple miRNAs and their potential role in fire blight resistance. Tree Genet Genomes 11: 812 [Google Scholar]
- Kavitha K, George S, Venkataraman G, Parida A(2010) A salt-inducible chloroplastic monodehydroascorbate reductase from halophyte Avicennia marina confers salt stress tolerance on transgenic plants. Biochimie 92: 1321–1329 [DOI] [PubMed] [Google Scholar]
- Khraiwesh B, Zhu J-K, Zhu J(2012) Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim Biophys Acta 1819: 137–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D, Yusuf M, Singh P, Sardar M, Sarin N(2014) Histochemical detection of superoxide and H2O2 accumulation in Brassica juncea seedlings. Bio Protoc 4: e1108 [Google Scholar]
- Li W, Cui X, Meng Z, Huang X, Xie Q, Wu H, Jin H, Zhang D, Liang W(2012) Transcriptional regulation of Arabidopsis MIR168a and argonaute1 homeostasis in abscisic acid and abiotic stress responses. Plant Physiol 158: 1279–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G, Yang F, Yu D(2010) MicroRNA395 mediates regulation of sulfate accumulation and allocation in Arabidopsis thaliana. Plant J 62: 1046–1057 [DOI] [PubMed] [Google Scholar]
- Liao X, Guo X, Wang Q, Wang Y, Zhao D, Yao L, Wang S, Liu G, Li T(2017) Overexpression of MsDREB6.2 results in cytokinin-deficient developmental phenotypes and enhances drought tolerance in transgenic apple plants. Plant J 89: 510–526 [DOI] [PubMed] [Google Scholar]
- Liu L, Zhang Y, Tang S, Zhao Q, Zhang Z, Zhang H, Dong L, Guo H, Xie Q(2010) An efficient system to detect protein ubiquitination by agroinfiltration in Nicotiana benthamiana. Plant J 61: 893–903 [DOI] [PubMed] [Google Scholar]
- Liu Q, Wang F, Axtell MJ(2014) Analysis of complementarity requirements for plant microRNA targeting using a Nicotiana benthamiana quantitative transient assay. Plant Cell 26: 741–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Hu P, Huang M, Tang Y, Li Y, Li L, Hou X(2016) The NF-YC-RGL2 module integrates GA and ABA signalling to regulate seed germination in Arabidopsis. Nat Commun 7: 12768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD(2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC(T) method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- Ma H-S, Liang D, Shuai P, Xia X-L, Yin W-L(2010) The salt- and drought-inducible poplar GRAS protein SCL7 confers salt and drought tolerance in Arabidopsis thaliana. J Exp Bot 61: 4011–4019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Hu X, Cai W, Huang W, Zhou X, Luo Q, Yang H, Wang J, Huang J(2014) Arabidopsis miR171-targeted scarecrow-like proteins bind to GT cis-elements and mediate gibberellin-regulated chlorophyll biosynthesis under light conditions. PLoS Genet 10: e1004519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martignago D, Rico-Medina A, Blasco-Escámez D, Fontanet-Manzaneque JB, Caño-Delgado AI(2020) Drought resistance by engineering plant tissue-specific responses. Front Plant Sci 10: 1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa M, Miura S, Nei M(2012) Origins and evolution of microRNA genes in plant species. Genome Biol Evol 4: 230–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekmezci AK, Karakülah G, Unver T(2017) Discovery of drought-responsive transposable element-related peach miRNAs. bioRxiv 143115 [Google Scholar]
- Selote DS, Khanna-Chopra R(2010) Antioxidant response of wheat roots to drought acclimation. Protoplasma 245: 153–163 [DOI] [PubMed] [Google Scholar]
- Song C, Zhang D, Zheng L, Zhang J, Zhang B, Luo W, Li Y, Li G, Ma J, Han M(2017) miRNA and degradome sequencing reveal miRNA and their target genes that may mediate shoot growth in spur type mutant “Yanfu 6.”. Front Plant Sci 8: 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Li Y, Cao X, Qi Y(2019) MicroRNAs and their regulatory roles in plant-environment interactions. Annu Rev Plant Biol 70: 489–525 [DOI] [PubMed] [Google Scholar]
- Subramanian B, Gao S, Lercher MJ, Hu S, Chen W-H(2019) Evolview v3: A webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res 47(W1): W270–W275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana S, Khew C-Y, Morshed MM, Namasivayam P, Napis S, Ho C-L(2012) Overexpression of monodehydroascorbate reductase from a mangrove plant (AeMDHAR) confers salt tolerance on rice. J Plant Physiol 169: 311–318 [DOI] [PubMed] [Google Scholar]
- Sun X, Wang P, Jia X, Huo L, Che R, Ma F(2018) Improvement of drought tolerance by overexpressing MdATG18a is mediated by modified antioxidant system and activated autophagy in transgenic apple. Plant Biotechnol J 16: 545–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Wei X, Wang P, Sun X, Li M, Ma F(2016) A phytocystatin gene from Malus prunifolia (Willd.) Borkh., MpCYS5, confers salt stress tolerance and functions in endoplasmic reticulum stress response in Arabidopsis. Plant Mol Biol Report 34: 62–75 [Google Scholar]
- Terzi R, Kalaycıoglu E, Demiralay M, Saglam A, Kadioglu A(2015) Exogenous ascorbic acid mitigates accumulation of abscisic acid, proline and polyamine under osmotic stress in maize leaves. Acta Physiol Plant 37: 43 [Google Scholar]
- Varkonyi-Gasic E, Gould N, Sandanayaka M, Sutherland P, MacDiarmid RM(2010) Characterisation of microRNAs from apple (Malus domestica ‘Royal Gala’) vascular tissue and phloem sap. BMC Plant Biol 10: 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velikova V, Yordanov I, Edreva A(2000) Oxidative stress and some antioxidant systems in acid rain-treated bean plants Protective role of exogenous polyamines. Plant Sci 151: 59–66 [Google Scholar]
- Voinnet O.(2009) Origin, biogenesis, and activity of plant microRNAs. Cell 136: 669–687 [DOI] [PubMed] [Google Scholar]
- Wang F, Axtell MJ(2017) AGO4 is specifically required for heterochromatic siRNA accumulation at Pol V-dependent loci in Arabidopsis thaliana. Plant J 90: 37–47 [DOI] [PubMed] [Google Scholar]
- Wang N, Jiang S, Zhang Z, Fang H, Xu H, Wang Y, Chen X(2018a) Malus sieversii: The origin, flavonoid synthesis mechanism, and breeding of red-skinned and red-fleshed apples. Hortic Res 5: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Wang L, Liu Z, Zhang T, Wang Y, Li Y, Gao C(2019) Molecular characterization and expression profiles of GRAS genes in response to abiotic stress and hormone treatment in Tamarix hispida. Trees (Berl) 33: 213–225 [Google Scholar]
- Wang S, Liang D, Li C, Hao Y, Ma F, Shu H(2012) Influence of drought stress on the cellular ultrastructure and antioxidant system in leaves of drought-tolerant and drought-sensitive apple rootstocks. Plant Physiol Biochem 51: 81–89 [DOI] [PubMed] [Google Scholar]
- Wang Y-X, Liu Z-W, Wu Z-J, Li H, Wang W-L, Cui X, Zhuang J(2018b) Genome-wide identification and expression analysis of GRAS family transcription factors in tea plant (Camellia sinensis). Sci Rep 8: 3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q, Ma C, Xu Y, Wang T, Chen Y, Lü J, Zhang L, Jiang C-Z, Hong B, Gao J(2017) Control of chrysanthemum flowering through integration with an aging pathway. Nat Commun 8: 829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia R, Zhu H, An YQ, Beers EP, Liu Z(2012) Apple miRNAs and tasiRNAs with novel regulatory networks. Genome Biol 13: R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie F, Wang Q, Sun R, Zhang B(2015) Deep sequencing reveals important roles of microRNAs in response to drought and salinity stress in cotton. J Exp Bot 66: 789–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing H-L, Dong L, Wang Z-P, Zhang H-Y, Han C-Y, Liu B, Wang X-C, Chen Q-J(2014a) A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol 14: 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing L, Zhang D, Li Y, Zhao C, Zhang S, Shen Y, An N, Han M(2014b) Genome-wide identification of vegetative phase transition-associated microRNAs and target predictions using degradome sequencing in Malus hupehensis. BMC Genomics 15: 1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing L, Zhang D, Zhao C, Li Y, Ma J, An N, Han M(2016) Shoot bending promotes flower bud formation by miRNA-mediated regulation in apple (Malus domestica Borkh.). Plant Biotechnol J 14: 749–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Chen S, Li T, Ma X, Liang X, Ding X, Liu H, Luo L(2015a) OsGRAS23, a rice GRAS transcription factor gene, is involved in drought stress response through regulating expression of stress-responsive genes. BMC Plant Biol 15: 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Xu Q, Huang B(2015b) Ascorbic acid mitigation of water stress-inhibition of root growth in association with oxidative defense in tall fescue (Festuca arundinacea Schreb.). Front Plant Sci 6: 807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M, Zhou S-R, Xue H-W(2015) CRISPR Primer Designer: Design primers for knockout and chromosome imaging CRISPR-Cas system. J Integr Plant Biol 57: 613–617 [DOI] [PubMed] [Google Scholar]
- Yang W, Fan T, Hu X, Cheng T, Zhang M(2017) Overexpressing osa-miR171c decreases salt stress tolerance in rice. J Plant Biol 60: 485–492 [Google Scholar]
- Yang W, Liu X-D, Chi X-J, Wu C-A, Li Y-Z, Song L-L, Liu X-M, Wang Y-F, Wang F-W, Zhang C, et al. (2011) Dwarf apple MbDREB1 enhances plant tolerance to low temperature, drought, and salt stress via both ABA-dependent and ABA-independent pathways. Planta 233: 219–229 [DOI] [PubMed] [Google Scholar]
- Yang X, Li L(2011) miRDeep-P: A computational tool for analyzing the microRNA transcriptome in plants. Bioinformatics 27: 2614–2615 [DOI] [PubMed] [Google Scholar]
- Yin H, Fan Z, Li X, Wang J, Liu W, Wu B, Ying Z, Liu L, Liu Z, Li J(2016) Phylogenetic tree-informed microRNAome analysis uncovers conserved and lineage-specific miRNAs in Camellia during floral organ development. J Exp Bot 67: 2641–2653 [DOI] [PubMed] [Google Scholar]
- Yu Y, Wang J, Li S, Kakan X, Zhou Y, Miao Y, Wang F, Qin H, Huang R(2019) Ascorbic acid integrates the antagonistic modulation of ethylene and abscisic acid in the accumulation of reactive oxygen species. Plant Physiol 179: 1861–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Fang L, Karungo SK, Zhang L, Gao Y, Li S, Xin H(2016) Overexpression of VaPAT1, a GRAS transcription factor from Vitis amurensis, confers abiotic stress tolerance in Arabidopsis. Plant Cell Rep 35: 655–666 [DOI] [PubMed] [Google Scholar]
- Zhang B.(2015) MicroRNA: A new target for improving plant tolerance to abiotic stress. J Exp Bot 66: 1749–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Liu J, Yang ZE, Chen EY, Zhang CJ, Zhang XY, Li FG(2018b) Genome-wide analysis of GRAS transcription factor gene family in Gossypium hirsutum L. BMC Genomics 19: 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Xian Z, Huang W, Li Z(2015) Evidence for the biological function of mir403 in tomato development. Sci Hortic (Amsterdam) 197: 619–626 [Google Scholar]
- Zhang Q, Ma C, Zhang Y, Gu Z, Li W, Duan X, Wang S, Hao L, Wang Y, Wang S, et al. (2018a) A single-nucleotide polymorphism in the promoter of a hairpin RNA contributes to Alternaria alternata leaf spot resistance in apple (Malus × domestica). Plant Cell 30: 1924–1942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhou J, Han S, Yang W, Li W, Wei H, Li X, Qi L(2010) Four abiotic stress-induced miRNA families differentially regulated in the embryogenic and non-embryogenic callus tissues of Larix leptolepis. Biochem Biophys Res Commun 398: 355–360 [DOI] [PubMed] [Google Scholar]
- Zhou J, Deng K, Cheng Y, Zhong Z, Tian L, Tang X, Tang A, Zheng X, Zhang T, Qi Y, et al. (2017) CRISPR-Cas9 based genome editing reveals new insights into microRNA function and regulation in rice. Front Plant Sci 8: 1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J-K.(2016) Abiotic stress signaling and responses in plants. Cell 167: 313–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Leng X, Sun X, Mu Q, Wang B, Li X, Wang C, Fang J(2015) Discovery of conservation and diversification of miR171 genes by phylogenetic analysis based on global genomes. Plant Genome 8: 1–11 [DOI] [PubMed] [Google Scholar]