

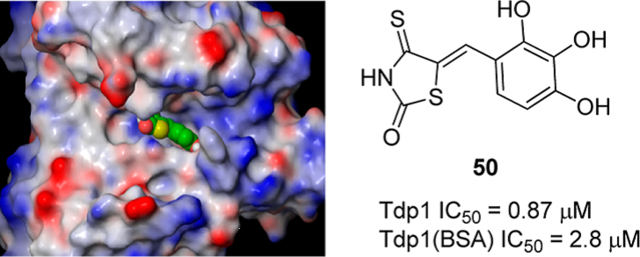
Abstract
Tyrosyl–DNA phosphodiesterase I (Tdp1) is a cellular enzyme that repairs the irreversible topoisomerase I (Top1)–DNA complexes and confers chemotherapeutic resistance to Top1 inhibitors. Inhibiting Tdp1 provides an attractive approach to potentiating clinically used Top1 inhibitors. However, despite recent efforts in studying Tdp1 as a therapeutic target, its inhibition remains poorly understood and largely underexplored. We describe herein the discovery of arylidene thioxothiazolidinone as a scaffold for potent Tdp1 inhibitors based on an initial tyrphostin lead compound 8. Through structure–activity relationship (SAR) studies we demonstrated that arylidene thioxothiazolidinones inhibit Tdp1 and identified compound 50 as a submicromolar inhibitor of Tdp1 (IC50 = 0.87 μM). Molecular modeling provided insight into key interactions essential for observed activities. Some derivatives were also active against endogenous Tdp1 in whole cell extracts. These findings contribute to advancing the understanding on Tdp1 inhibition.
Graphical Abstract
INTRODUCTION
Topoisomerase I (Top1) is an essential enzyme that catalyzes the relaxation of supercoiling and the release of torsional strains during DNA replication, transcription, recombination, and chromatin remodeling.1,2 This enzymatic function involves a dynamic equilibrium between the cleavage of the DNA substrate and the religation of the resulting transient Top1–DNA cleavage complex, which features a 3′-phosphotyrosyl bond (Figure 1). Inhibitors of Top1 stall this equilibrium by binding to and stabilizing an irreversible ternary Top1–DNA–inhibitor complex, preventing the religation of the 5′-hydroxyl group.3,4
Figure 1.

Tdp1 in the repair of the stalled Top1–DNA complex: (A) noncovalent Top1–DNA complex; (B) transient Top1–DNA complex, which is trapped in the presence of a Top1 inhibitor; (C) the stalled Top1–DNA complex (cleavage complex) is repaired by Tdp1 via a transient Tdp1–DNA covalent intermediate; (D) Top1 is released and the Tdp1–DNA covalent intermediate resolved by hydrolysis; (E) 3′-phospho–DNA–end product.
This inhibition creates a DNA lesion and underlies the use of camptothecin (CPT)-based Top1 inhibitors, such as topotecan and irinotecan, for treating ovarian, lung, and colon cancers5,6 and the development of the indenoisoquinolines as non-camptothecin Top1 inhibitors.7 Stalled Top1–DNA complexes are repaired by Tdp1, a cellular enzyme conserved from yeast to humans and which hydrolyzes the unique 3′-phosphotyrosyl linkage of the Top1–DNA cleavage complex.8 Other than Top1 inhibitors, cleavage complexes can also be generated by endogenous DNA lesions such as strand breaks, abasic sites, base mismatches, and specific oxidized or modified bases and carcinogenic DNA adducts.9–11 The repair of the Top1–DNA cleavage complex begins with a nucleophilic attack of the histidine 263 of Tdp1 onto the 3′-phosphotyrosyl bond to form a Tdp1–DNA covalent intermediate (Figure 1 C) and release Top1.12 Hydrolysis of this intermediate turns over Tdp1 and generates a DNA product with a 3′-phosphate (Figure 1 D and E). Further repair presumably involves the polynucleotide kinase phosphatase (PNKP), a bifunctional enzyme with 5′-kinase and 3′-phosphatase activities that catalyze both the hydrolysis of the 3′-phosphate and the phosphorylation of the 5′-DNA end to enable their rejoining.8 The critical role of Tdp1 in the cellular repair of Top1-mediated DNA damage has been established through three lines of evidence. First, knocking out Tdp1 in yeast13 and in vertebrate cells14,15 renders such cells hypersensitive to camptothecin. Second, Tdp1-defective SCAN1 (spinocerebellar ataxia with axonal neuropathy-1) cells, which have a Tdp1 mutation N493R and accumulate the normally transient Tdp1–DNA repair complex (Figure 1C), are highly sensitive to CPT and accumulate DNA strand breaks upon treatment with CPT.14,16 Third, overexpression of wild-type Tdp1 protects cells against CPT-induced cell death, whereas the H263A mutant does not.17 These observations provide the rationale for developing Tdp1 inhibitors for combination therapy with Top1 inhibitors.18,19
Tdp1 belongs to the phospholipase D (PLD) superfamily,20 which is defined by a conserved HKD catalytic motif21 and a signature catalytic mechanism12 featuring a covalent phosphohistidine intermediate (Figure 1C). Two such motifs are present in the N- and C-terminal domains of human Tdp1, both containing the highly conserved histidine and lysine residues (H263/K265 and H493/K495). The importance of these residues for Tdp1 catalytic activity is manifested through the observations that the mutation of H263 generates a catalytically inactive enzyme and mutating H493, K265, or K495 results in compromised enzymatic activity.20,22 Thus, Tdp1 represents a distinct member of the PLD superfamily with unique HKN motifs. Although the mechanism of Tdp1 catalysis is well established, its inhibition remains poorly understood and largely unexplored. Early studies on Tdp1 discovered molecular probes that inhibit Tdp1 at millimolar range, including aminoglycoside antibiotics and the ribosome inhibitors,23 as well as tungstates and vanadates which served as transition state mimics for crystallographic studies.24,25 Later on, a few micromolar inhibitors (Figure 2) were identified through a high-throughput screening (HTS) assay,26 among which 3 was confirmed to specifically inhibit Tdp1 through SAR.11 Recently, Cushman and co-workers reported that a subset of indenoisoquinoline derivatives potently inhibit both Top1 and Tdp1, making them the first Top1/Tdp1 dual inhibitors.27 Nevertheless, though inhibiting Tdp1 at micromolar concentrations, these compounds remain far from therapeutic development. Therefore, there is a need to continue to identify potent and specific small molecule inhibitors to further advance the understanding of Tdp1 functions and develop therapeutic candidates. Herein we report the design, synthesis, molecular modeling, and biological evaluations of thioxothiazolidinones as novel and potent Tdp1 inhibitors.
Figure 2.

Known inhibitors of Tdp1.
RESULTS AND DISCUSSION
Exploring Tdp1 as a therapeutic target was made possible by the development of a biochemical screening assay using recombinant human Tdp1.11,28 This gel-based assay is based on the cleavage by Tdp1 of the 14-mer 5′−32P-labeled 3′-phosphotyrosyl DNA substrate N14Y (5′-GATCTAAAAGACTT-pY-3′) to a 14-mer 5′−32P-labeled 3′-phosphate DNA product N14P (Figure 3, A). This assay was applied to a small in-house chemical library constituted of benzylidenemalonitriles, a class of tyrphostins. This screening led to the identification of compounds 8 and 9 as Tdp1 inhibitors with IC50 values of 12 and 18 μM, respectively (Figure 3, B and C). Notably, it was observed from this screening that only tyrphostins with a thioamide functionality (compounds 8 and 9) in the head domain inhibited Tdp1. The carboxylic acid (4), carboxamides (5 and 6), and cyano (7) analogues were completely inactive at the highest tested concentration (333 μM) (Figure 3, C).
Figure 3.

Inhibition of Tdp1 by tyrphostin derivatives. (A) Schematic representation of the Tdp1 biochemical gel-based assay. Tdp1 hydrolyzes the 3′-phosphotyrosine bond and converts the N14Y DNA substrate to a 3′-phosphate oligonucleotide product (N14P); (B) representative gel demonstrating concentration-dependent inhibition of Tdp1 by compounds 8 and 9; (C) representative compounds. The pharmacophore of lead compound 8 comprises two domains: the functional head domain and the aromatic moiety.
On the basis of these initial screening results, derivatives 10–16 of lead compound 8 were prepared to explore the impact of the aromatic domain (Table 1). While carboxylic acid analogue 13 demonstrated potent inhibitory activity against Tdp1 (IC50 = 12 μM, Table 1), all other analogues were inactive up to 333 μM. The phenolic hydroxyl group (compound 8) or the carboxylic acid functionality (compound 13) appears essential for the inhibition of Tdp1.
Table 1.
Aromatic Domain SAR of Lead Compound 8a
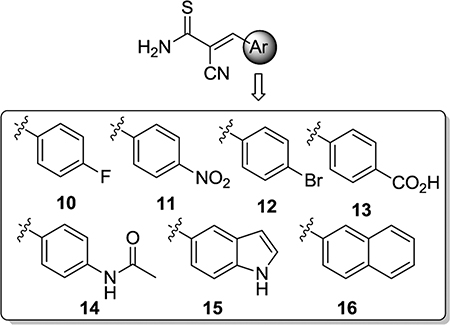 | |
|---|---|
| compound | Tdp1 IC50 (μM) |
| 10 | >333 |
| 11 | >333 |
| 12 | >333 |
| 13 | 12 |
| 14 | >333 |
| 15 | >333 |
| 16 | >333 |
| 1 | 31 ± 5.0 |
Furamidine 1 was used as a positive control,26 and its IC50 value is expressed as mean ± SD from at least three independent experiments.
At this point, we shifted our focus onto constructing heterocyclic scaffolds mimicking the head domain of lead compound 8. Notably, rhodanine benzylidene derivatives such as compound 18 have been reported as inhibitors of phosphodiesterase,29 PTP,30 and recently as broad spectrum antiviral.31 To mimic the head domain of compound 8, we chose a few rhodanine-like five-membered heterocyclic scaffolds with wide applications in medicinal chemistry (Figure 4): thiohydantoins 1932–34 and 20,35 thiazolidinedione 21,36–38 and thioxothiazolidinone 22.39,40 It must be pointed out that the medicinal chemistry value of these scaffolds is a subject of recent controversies. Many characterize these compounds as promiscuous frequent hitters that tend to produce pan-assay interference in a HTS setting.41 Others consider them privileged scaffolds42 with an extraordinary ability to engage with important biological targets as evidenced by the clinical thiazolidinedione (21, aka glitazones) antidiabetic drugs.43–47 Recent systematic studies by Klein and co-workers found no aggregation-based promiscuity or unspecific reactivity toward protein,48 arguing against the exclusion of these scaffolds in drug discovery efforts.
Figure 4.

Design of heterocyclic arylidene scaffolds 19–22 based on lead compound 8 and PTP inhibitors 18.
Synthetically, these scaffolds can be easily accessed through an aldol condensation between a heterocyclic ring (27–30) and an aromatic aldehyde (Scheme 1). The preparation of thiohydantoin intermediates 27 and 28 featured a reaction between a glycine derivative (23 and 25) and a thioisocyanate 24 or thiocyanate 26. The thioxothiazolidinone intermediate 30 was synthesized from commercial thiazolidine-2,4-dione 29 using Lawesson’s reagent. The key aldo condensation was performed under thermodynamic conditions to yield the more stable Z-isomers.49 The assignment of this stereochemistry is consistent with literature reports.49–52
Scheme 1. Synthesis of Scaffolds 19–22a.

aReagents and conditions: (a) EtOAc, reflux, 48 h, 92%; (b) EtOH, piperidine, reflux, 16 h, 41–78%; (c) 140 °C, 4 h, 72%; (d) Lawesson’s reagent, toluene, reflux, 2 h, 98%; (e) NaOAc, AcOH, reflux, 2 h, 58–90%.
Four analogues of each scaffold were prepared and tested against Tdp1. The assay results are summarized in Table 2. Notably, with the exception of 35, which inhibited Tdp1 with an IC50 value of 13 μM, analogues of the first three heterocyclic scaffolds were essentially inactive against Tdp1. By contrast, the thioxothiazolidinone scaffold (compounds 43–46) consistently demonstrated significant potency. Of particular interest are the 3,4-dihydroxyl analogue 43 and the carboxylate analogue 44, which inhibited Tdp1 at low micromolar concentrations (Table 2).
Table 2.
Impact of the Head Domain on Tdp1 Inhibitiona
| compound | Tdp1 IC50 (μM) |
|---|---|
| 31 | 74 |
| 32 | >333 |
| 33 | >333 |
| 34 | 111–333 |
| 35 | 13 ± 3.0 |
| 36 | >333 |
| 37 | 111–333 |
| 38 | 111–333 |
| 39 | 111–333 |
| 40 | >111 |
| 41 | >333 |
| 42 | >111 |
| 43 | 2.6 ± 0.4 |
| 44 | 16 ± 8.8 |
| 45 | 27 ± 3.0 |
| 46 | 12 ± 2.2 |
IC50 values are expressed as mean ± SD from at least three independent experiments.
Having identified thioxothiazolidinone as the optimal substructure for the head domain, we then conducted a SAR on the aromatic domain. Toward this end, analogues with variable aromatic ring or substitution patterns were prepared based on the chemistry described in Scheme 1 (Table 3).
Table 3.
Aromatic Domain SAR of Thioxothiazolidinone Scaffold 22a
| Cpd | Ar = | Tdp1 IC50 (μM) | Cpd | Ar = | Tdp1 IC50 (μM) |
|---|---|---|---|---|---|
| 43 |  |
2.6 ± 0.4 | 61 |  |
51 ± 25 |
| 47 | 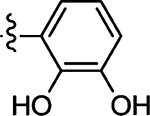 |
5.8 ± 1.1 | 45 | 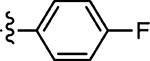 |
27 ± 3.0 |
| 48 | 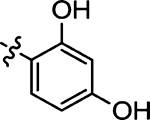 |
4.1 ± 1.0 | 62 | 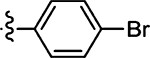 |
30 ± 12 |
| 49 | 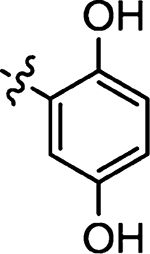 |
7.1 ± 0.50 | 63 | 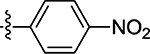 |
19 ± 6.4 |
| 50 | 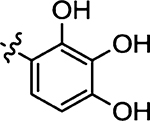 |
0.87 ± 0.07 | 64 | 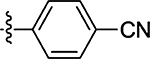 |
28 ± 7.6 |
| 51 | 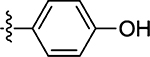 |
39 ± 9.4 | 65 | 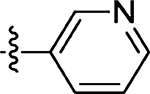 |
44 |
| 52 | 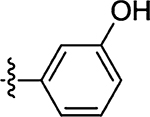 |
26 | 66 | 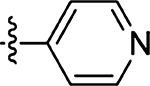 |
75 ± 29 |
| 53 | 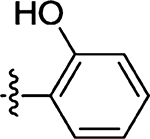 |
34 ± 13 | 67 | 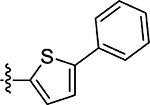 |
46 ± 15 |
| 54 | 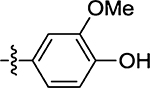 |
11 ± 4.0 | 68 | 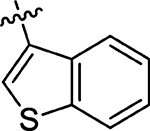 |
34 ± 7.8 |
| 55 | 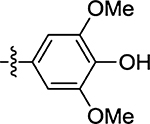 |
48 ± 16 | 46 | 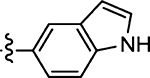 |
12 ± 2.2 |
| 44 | 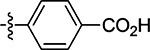 |
16 ± 8.8 | 69 | 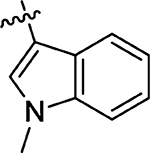 |
40 ± 11 |
| 56 | 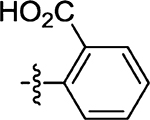 |
15 ± 1.2 | 70 | 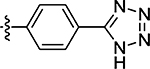 |
32 |
| 57 | 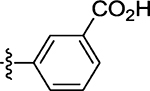 |
15 ± 4.7 | 71 | 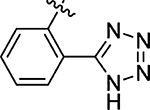 |
111 |
| 58 | 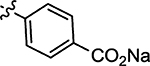 |
7.8 | 72 | 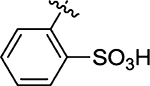 |
20 ± 3.5 |
| 59 | 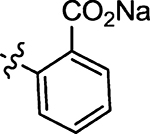 |
8.5 | 73 | 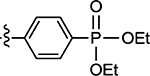 |
29 ± 6.0 |
| 60 | 6.7 ± 0.40 | 74 | 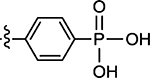 |
2.2 ± 0.5 |
IC50 values are expressed as mean ± SD from at least three independent experiments.
Analogues with two or three phenolic hydroxyl groups (43, 47–50, Table 3) were by far the best inhibitors of the series, with 50 being a submicromolar Tdp1 inhibitor (Figure 5A and B). When only one free hydroxyl group was present (compounds 51–55, Table 3, Figure 5A and B), a substantial drop in inhibitory activity was observed, suggesting that these free hydroxyl groups may provide key interactions with the enzyme possibly through hydrogen bonding.
Figure 5.

(A) Representative gel demonstrating concentration-dependent inhibition of Tdp1 by compounds 48, 50, and 51. (C) Concentration–response curves. Each point represents the mean value ± standard deviation of three independent experiments.
The polyhydroxylated phenyl moiety, particularly the pyrogallol (1,2,3-trihydroxyphenyl) functionality in compound 50, may potentially engage in excessive nonspecific protein binding. To assess the protein binding property of this functionality, 50 was tested against recombinant Tdp1 in WCE gel-based assay buffer containing bovine serum albumin (BSA, see Experimental Section) and exhibited an IC50 of 2.8 μM. Knowing that BSA does not impact Tdp1 activity (Huang, S.H. and Pommier, Y., unpublished data), the 3-fold loss of potency may reflect nonspecific BSA binding, albeit to the extent typical of small molecule inhibitors. We have also tested 50 against recombinant Tdp2 in in a gel-based assay and observed an IC50 of 17 μM, a 20-fold increase when compared to that against Tdp1 (IC50 = 0.87 μM). These studies strongly suggest that 50 is indeed a selective inhibitor of Tdp1. Efforts in bioisosteric replacement51 of catechol and pyrogallol moieties are currently underway, which could lead to more potent inhibitors with improved physicochemical properties.
The carboxylic acid analogues also demonstrated good potency, which was not affected by the position of the substitution (44, 56–57, Table 3). However, derivatization of the acid appeared to improve the activity, as the two sodium salts (58 and 59) and methyl ester 60 were all found to be approximately twice as potent as the acid analogues. Other substitutions, including halogens (45 and 62), nitro (63), and cyano (64), all led to decreased inhibitory activity. In addition, replacing the benzene ring with other heterocycles, such as pyridines (65 and 66), thiophene (67), benzothiophene (68), and indoles (46 and 69) also yielded less potent compounds. Finally, bioisosteric replacement53 of the carboxylic acid was explored, and analogues with tetrazole (70 and 71), sulfonic acid (72), phosphonate (73), and phosphonic acid (74) were prepared. The biochemical assay identified compound 74 as a potent inhibitor (IC50 = 2.2 μM) with a 7.3-fold improvement in potency over 44, demonstrating that the phosphonic acid functional group is a viable bioisostere of the carboxylic acid.
To further assess the ability of our compounds to inhibit human Tdp1 in a more complex pseudocellular environment, we performed secondary gel-based assays using the same N14Y DNA substrate while replacing recombinant Tdp1 by whole cell extracts (WCEs) as a source of endogenous Tdp1. For this purpose, the chicken Tdp1 gene in DT40 cells was knocked out and recomplemented with the human Tdp1 gene.15 Lysates were collected and used in place of recombinant Tdp1 in the gel assays.15 Compounds with a single digit micromolar IC50 from the recombinant Tdp1 assay were tested against WCE, and the results are shown in Table 4. Given the increased stringency of this assay, the observation that these compounds are generally active at micromolar concentrations in the WCE assay strongly corroborates the earlier biochemical results and confirms their inhibitory activities against endogenous Tdp1 in a pseudocellular environment. The confirmed inhibitory activity of this series of compounds, the SAR pattern and the concentration-dependent inhibition fashion, suggest that these controversial scaffolds can be valuable leads for inhibiting Tdp1.
Table 4.
Assay Results with the DT40 WCE
| Compound | Ar = | WCE Tdp1 IC50 (μM) |
|---|---|---|
| 43 | 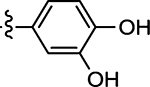 |
41 |
| 47 | 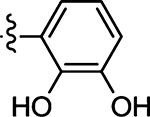 |
220 |
| 48 | 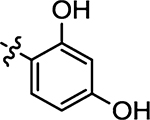 |
76 |
| 49 | 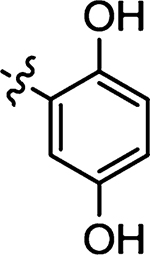 |
76 |
| 50 | 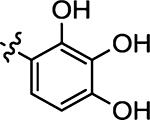 |
35 |
| 44 | 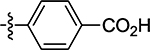 |
11 |
| 56 | 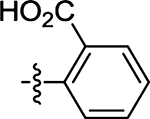 |
138 |
| 58 | 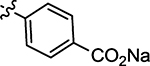 |
6.9 |
| 59 | 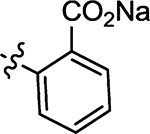 |
280 |
| 60 | 38 | |
| 74 | 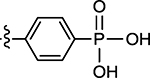 |
69 |
Binding Analysis
The binding of our inhibitors to Tdp1 was directly measured using the Surface Plasmon Resonance (SPR) technology. In this testing, Tdp1 was amine coupled to a CM5 sensor chip, and a dilution series of the representative inhibitor 50 was run through the surface. A surface without coupled Tdp1 was used as a reference. As shown in Figure 6, a concentration-dependent binding of 50 on Tdp1 was observed (A). Similarly, a 14-base oligonucleotide modified with a biotin at the 5′ end and a tyrosine at the 3′ end (biotin-GATCTAAAAGACTT-Tyr) was immobilized onto the chip surface and tested against the same dilution series of 50 (Figure 6, B). No binding was observed between 50 and the N14Y oligomer, confirming the selective binding of our inhibitors to Tdp1 under assay conditions.
Figure 6.

Binding of 50 to Tdp1 was examined using SPR spectroscopy. A concentration series of 50 (50, 25, 6.25, 3.12, 1.56. 0.78, and 0.39 μM) were injected over immobilized Tdp1 (A) or a tyrosyl-labeled oligonucleotide (GATCTAAAAGACTT-Tyr) (B). The theoretical maximal binding response to Tdp1 was 45 RU and to the oligonucleotide was 140 RU.
Modeling and Docking
Close examination of the active site identified three specific regions important for substrate binding (Figure 7A)—the hydrophobic (Y204, P461, W590), the catalytic (K265, H263, N283, H493, K495, N516), and an adjacent hydrophilic region (S400, S518, S536, E538). Both the catalytic and hydrophilic regions consist of positively charged and hydrophilic residues for the stabilization of the negatively charged phosphate backbone of the DNA (Figure 7A). The hydrophobic region is located at the top of the substrate channel that provides essential interactions to both the tyrosyl and the nucleoside group of the substrate. A docking study of the previously identified Tdp1 inhibitors (1–3) showed interactions to at least two of the three regions are required for binding (Figure 1S, Supporting Information). Docking of our first identified compounds 8 and 9 showed both inhibitors bind to the “floor” of the substrate channel at the catalytic and the hydrophilic regions (see 9 in Figure 7B). The essential interactions involved significant hydrogen bonding between the thiocarbonyl and the catalytic histidine and lysine residues as well as hydrogen bonding between the catechol and the hydrophilic residues E538 and S536 (Figure 7B). The benzyl group of 9 was easily accommodated within the tyrosyl binding site, which supports the observed similar activities between 8 and 9. Replacement of the catechol group with halobenzene or a large aromatic group significantly disrupts this observed mode of binding as the hydrophilic region energetically disfavors such substitution. For the heterocyclic head domain, placement of the catechol group directly adjacent to the thiocarbonyl is essential for maintaining the observed mode of binding. Placement of the thiocarbonyl at the 1,3 position to the catechol results in the thiocarbonyl group pointing slightly away from the catalytic lysine and histidine residues toward N283, which results in a weaker activity observed with 31 (Figure 7C). Addition of a benzyl group at the NH position of the heterocyclic ring (35–38) is prohibited as it leads to steric clashes with the interfacial region between the catalytic and hydrophilic sites. One advantage observed for the heterocyclic head is the ability to interact with P461 and Y204 within the hydrophobic region as observed in the increase in activity from 12 μM of 8 to 2.6 μM of 43. The ability to interact with all three regions enhances affinity to retain the observed mode of binding and enables SAR modification of the catechol group which would otherwise lead to significant loss of activities (10–12, 14–16). As observed in Table 3, replacement of the catechol by heteroaromatic groups and halobenzene after heterocyclization is tolerable within the hydrophilic region with only a 10-fold loss of activity. The replacement of the hydroxyl groups of catechol by negatively charged groups such as carboxylate, tetrazole, sulfonate, and phosphonate within the hydrophilic site leads to a slight shift in mode of binding, similar to that observed in 31 (Figure 7C). The observed effect depends significantly on the size, the electronegativity of the group, and the position of the aromatic substitution. Finally, addition of a third hydroxyl group to catechol (50) enables the formation of an additional hydrogen bond with E538 within the hydrophilic site that correlates directly with the improved activity of 0.87 μM (Figure 7D).
Figure 7.

Modeled structure of Human Tdp1 (PDB: 1NOP)25 with (A) Top1–DNA substrate, (B) 9, (C) 31, and (D) 50 docked within the active site. The model shows three specific regions important for substrate binding—the hydrophobic (Y204, P461, W590, labeled in white), the catalytic (K265, H263, N283, H493, K495, N516, labeled in blue), and an adjacent hydrophilic region (S400, S518, S536, E538, labeled in red). Effective binding of the hydrolytic tyrosyl–nucleosidic group of the Top1–DNA substrate involves the hydrophobic and the catalytic regions (boxed in yellow).
CONCLUSIONS
On the basis of the initial lead compound 8 identified from screening a small focused library against Tdp1, we have discovered 5-arylidene thioxothiazolidinone as a novel scaffold for low micromolar Tdp1 inhibitors. SAR studies revealed that analogues with multiple hydroxyl groups or a carboxylic acid derivative on the terminal phenyl ring consistently inhibited Tdp1 in the biochemical assay. Among these, compound 50 was identified as a submicromolar inhibitor. A modeling study identified the most likely mode of inhibition that corroborates the observed SAR. In addition, bioisosteric replacement of the carboxylic acid also led to the discovery of phosphonic acid 74 as a potent Tdp1 inhibitor. Results from the more stringent secondary WCE gel-based assay confirm the observed inhibitory activities of these compounds.
EXPERIMENTAL SECTION
Chemistry
General Procedures
All commercial chemicals were used as supplied unless otherwise indicated. Dry solvents (THF, Et2O, CH2Cl2, and DMF) were dispensed under argon from an anhydrous solvent system with two packed columns of neutral alumina or molecular sieves. Flash chromatography was performed on a Teledyne Combiflash RF-200 with RediSep columns (silica) and indicated mobile phase. All reactions were performed under an inert atmosphere of ultrapure argon with oven-dried glassware. 1H and 13C NMR spectra were recorded on a Varian 600 MHz or a Varian 400 MHz spectrometer. Mass data were acquired on an Agilent TOF II TOS/MS spectrometer capable of ESI and APCI ion sources. Analysis of sample purity was performed on a Varian Prepstar SD-1 HPLC system with a Varian Microsorb-MW 100–5 C18 column (250 mm × 4.6 mm). HPLC conditions: solvent A = H2O, solvent B = MeCN; flow rate =1.0 mL/min; compounds were eluted with a gradient of 5% MeCN/H2O to 80% MeCN for 40 min. Purity was determined by total absorbance at 254 nm. All tested compounds have a purity ≥98%. Compounds 4–9 were purchased from Axxora along with other tyrphostins as a small focused library.
General Procedure a for the Aldol Condensation
To a mixture of an aromatic aldehyde (2.0/mmol, 1 equiv) and cyanothioacetamide (2.0 mmol, 1 equiv) in ethanol (5 mL) was added one drop of N-methylmorpholine, and the resulting mixture was stirred at 35 °C for 30–60 min. The reaction mixture was evaporated to give the crude product. The obtained solid was further purified by column chromatography (SiO2, EtOAc/hexane).
(E)-2-Cyano-3-(4-fluorophenyl)prop-2-enethioamide (10)
Prepared from 4-fluorobenzaldehyde following the general procedure A. Yield 62%; mp 155–158 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.10 (s, 1H), 9.61 (s, 1H), 8.06 (s, 1H), 8.06–7.98 (m, 2H), 7.43–7.41 (m, 2H); 13C NMR (150 MHz, DMSO-d6) δ 192.9, 165.4, 146.2, 133.5, 129.2, 117.3, 112.8, 101.2; HRMS-ESI(−) m/z calcd for C10H6FN2S 205.0236 [M − H]−, found 205.0214.
(E)-2-Cyano-3-(4-nitrophenyl)prop-2-enethioamide (11)
Prepared from 4-nitrobenzaldehyde following the general procedure A. Yield 58%; mp 171–173 °C; 1H NMR (600 MHz, DMSO-d6) δ 8.24–8.26 (m, 3H), 7.81 (d, J = 7.2 Hz, 2H), 7.63 (br, 2H); 13C NMR (150 MHz, DMSO-d6) δ 194.6, 148.4, 148.0, 132.3, 131.9, 124.3, 123.7, 118.9; HRMS-ESI(−) m/z calcd for C10H6N3O2S 232.0259 [M − H]−, found 232.0181.
(E)-3-(4-Bromophenyl)-2-cyanoprop-2-enethioamide (12)
Prepared from 4-bromobenzaldehyde following the general procedure A. Yield 59%; mp 176–179 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.13 (s, 1H), 9.64 (s, 1H), 8.01 (s, 1H), 7.84 (d, J = 8.4 Hz, 2H), 7.77 (d, J = 8.4 Hz, 2H); 13C NMR (150 MHz, DMSO-d6) δ 192.7, 146.0, 133.0, 132.5, 131.8, 126.4, 116.7, 113.8; HRMS-ESI(−) m/z calcd for C10H6BrN2S 264.9435 [M − H]−, found 264.9444.
(E)-4-(3-Amino-2-cyano-3-thioxoprop-1-en-1-yl)benzoic Acid (13)
Prepared from 4-formylbenzoic acid following the general procedure A. Yield 63%; mp >300 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.12 (s, 1H), 10.06 (s, 1H), 9.51 (s, 1H), 8.18 (s, 1H), 7.98 (d, J = 8.4 Hz, 2H), 7.87 (d, J = 8.4 Hz, 2H); 13C NMR (150 MHz, DMSO-d6) δ 192.2, 167.3, 161.9, 151.8, 140.1, 138.0, 132.6, 130.2, 128.5, 115.3; HRMS-ESI(−) m/z calcd for C11H7N2O2S 231.0228 [M − H]−, found 231.0257.
(E)-N-(4-(3-Amino-2-cyano-3-thioxoprop-1-en-1-yl)phenyl)-acetamide (14)
Prepared from N-(4-formylphenyl)acetamide following the general procedure A. Yield 74%; mp 231–236 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.36 (s, 1H), 9.98 (s, 1H), 9.48 (s, 1H), 8.01 (s, 1H), 7.92 (d, J = 8.9 Hz, 2H), 7.73 (d, J = 8.9 Hz, 2H), 2.07 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 193.2, 169.7, 147.4, 143.7, 132.4, 126.8, 119.5, 117.4, 110.4, 24.8; HRMS-ESI(+) m/z calcd for C12H12N3SO 246.0701 [M + H]+, found 246.0705.
(E)-2-Cyano-3-(1H-indol-5-yl)prop-2-enethioamide (15)
Prepared from 1H-indole-5-carbaldehyde following the general procedure A. Yield 70%; mp 189–191 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.58 (s, 1H), 9.90 (s, 1H), 9.41 (s, 1H), 8.24 (d, J = 10.2 Hz, 2H), 7.81 (d, J = 8.4 Hz 1H), 7.53 (d, J = 7.8 Hz, 1H), 7.48–7.47 (m, 1H), 6.59 (s, 1H); 13C NMR (150 MHz, DMSO-d6) δ 193.7, 150.6, 138.8, 128.6, 128.3, 125.8, 123.9, 123.4, 118.1, 113.1, 108.3, 103.5; HRMS-ESI(+) m/z calcd for C12H10N3S 228.0595 [M + H]+, found 228.0581.
(E)-2-Cyano-3-(naphthalen-2-yl)prop-2-enethioamide (16)
Prepared from 2-naphthaldehyde following the general procedure A. Yield 67%; mp 141–144 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.13 (s, 1H), 9.66 (s, 1H), 8.44 (s, 1H), 8.23 (s, 1H), 8.09–8.05 (m, 2H), 8.02 (d, J = 7.8 Hz, 1H), 7.99 (d, J = 8.4 Hz, 1H), 7.66 (t, J = 7.2 Hz, 1H), 7.61 (t, J = 7.2 Hz, 1H); 13C NMR (150 MHz, DMSO-d6) δ 193.1, 147.6, 134.9, 133.1, 130.2, 129.7, 129.5, 129.3, 128.5, 127.9, 125.5, 117.1, 113.1; HRMS-ESI(+) m/z calcd for C14H11N2S 239.0643 [M + H]+, found 239.0650.
3-Phenyl-2-thioxo-imidazolidin-4-one (27).54
A suspension of methylglycinate hydrochloride (2.5 g, 20 mmol), phenylisothiocyanate (2.7 g, 20 mmol), and triethylamine (2.02 g, 2.8 mL, 20 mmol) in dry ethylacetate was refluxed (80 °C) for 2 days. Solvent was evaporated under reduced pressure. The residue was dissolved in EtOH, and crystallization afforded 27 (3.5 g, 18 mmol, 92%) as a brown solid. 1H NMR (600 MHz, CDCl3) δ 7.53–7.46 (m, 3H), 7.33–7.32 (m, 2H), 4.23 (s, 2H).
Synthesis of 1-Benzyl-2-thiohydantoin (28)
A suspension of N-benzyl glycinethylester (3.86 g, 20 mmol) and ammonium thiocyanate (4.56 g, 60 mmol) was heated to 40 °C. A clear orange solution was observed which turned into dark red. After refluxing the reaction mixture for 4 h, the solution was immediately poured into 40 mL of (EtOH:H2O). The crystalline white solid was collected by suction filtration, washed with 100 mL of (EtOH:H2O), followed by diethyl ether (20 mL), and dried under vacuum to afford 28 (2.8 g, 13.7 mmol, 70%) as a pale yellow solid. 1H NMR (600 MHz, DMSO-d6) δ 11.84 (s, 1H), 7.27–7.36 (m, 5H), 4.85 (s, 2H), 4.07 (s, 2H).
4-Thioxo-thiazolidin-2-one (30).55
To a solution of compound 29 (0.61 g, 5.21 mmol) in dry toluene (12 mL) was added Lawesson’s reagent (2.2 g, 5.2 mmol). The reaction mixture was refluxed for 2 h and cooled to room temperature. The solid formed was filtered off and crystallized from acetone to afford 0.64 g (98%) of 30 as yellow crystals. 1H (600 MHz, DMSO-d6) δ 13.58 (s, 1H), 4.67 (s, 2H).
General Procedure B for Aldol Condensation.56
An aromatic aldehyde (0.55 mmol, 1 equiv) and appropriate heterocyclic ring (27–29) (0.73 mmol, 1.3 equiv) were dissolved in ethanol (14 mL) which was followed by the addition of piperidine (90 μL). The reaction mixture was refluxed for 6–14 h (monitored by TLC). The ethanol was evaporated, and the residue was dissolved in ethylacetate (100 mL) and washed with water (50 mL) and brine (50 mL). The organic layer was dried over Na2SO4, filtered, and concentrated to give the crude product. The obtained solid was further purified by crystallization from ethanol or by column chromatography to yield the desired compound.
(Z)-5-(3,4-Dihydroxybenzylidene)-3-phenyl-2-thioxoimidazolidin-4-one (31)
Prepared from 3,4-dihydroxybenzaldehyde and compound 27 (0.14 g, 0.73 mmol, 1.3 equiv) following the general procedure B. Yield 51%; mp 214–220 °C; 1H NMR (600 MHz, DMSO-d6) δ 7.48–7.45 (m, 2H), 7.41–7.37 (m, 2H), 7.32 (d, J = 7.8 Hz, 2H), 7.14–7.13 (m, 1H), 6.76 (d, J = 8.4 Hz, 1H), 6.54 (s, 1H); 13C NMR (150 MHz, DMSO-d6) δ 178.7, 169.8, 146.0, 145.4, 139.9, 137.1, 129.0, 128.2, 126.7, 123.3, 117.4, 111.6; HRMS-ESI(−) m/z calcd for C16H11N2O3S 311.0490 [M − H]−, found 311.0505.
(Z)-4-((5-Oxo-1-phenyl-2-thioxoimidazolidin-4-ylidene)methyl)-benzoic Acid (32)
Prepared from 4-formylbenzoic acid and compound 27 (0.14 g, 0.73 mmol, 1.3 equiv) following the general procedure B. Yield 78%; mp > 300 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.07 (s, 1H), 12.70 (s, 1H), 7.96 (d, J = 8.4 Hz, 2H), 7.89 (d, J = 8.4 Hz, 2H), 7.52–7.46 (m, 3H), 7.38 (d, J = 8.4 Hz, 2H), 6.70 (s, 1H); 13C NMR (150 MHz, DMSO-d6) δ 179.6, 167.3, 164.4, 137.2, 133.7, 131.1, 130.6, 129.9, 129.3, 129.2, 111.3; HRMS-ESI(−) m/z calcd for C16H11N2O3S 323.0490 [M − H]−, found 323.0490.
(Z)-5-(4-Fluorobenzylidene)-3-phenyl-2-thioxoimidazolidin-4-one (33)
Prepared from 4-fluorobenzaldehyde and compound 27 (0.14 g, 0.73 mmol, 1.3 equiv) following the general procedure B. Yield: 74%. 1H NMR (600 MHz, DMSO-d6) δ 12.69 (s, 1H), 7.98 (m, 2H), 7.61 (m, 2H), 7.37–7.56 (m, 5H), 6.78 (s, 1H).
(Z)-5-((1H-Indol-5-yl)methylene)-3-phenyl-2-thioxoimidazolidin-4-one (34)
Prepared from 1H-indole-5-carbaldehyde and compound 27 (0.14 g, 0.73 mmol, 1.3 equiv) following the general procedure B. Yield 58%; mp 254–259 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.25 (s, 1H), 11.33 (s, 1H), 8.14 (s, 1H), 7.54–7.36 (m, 8 H), 6.80 (s, 1H), 6.50 (s, 1H); 13C NMR (150 MHz, DMSO-d6) δ 178.1, 164.4, 137.0, 133.9, 129.3,129.2, 129.0, 128.6, 127.3, 124.7, 124.1, 123.9, 123.7, 116.8, 112.3, 102.5; HRMS-ESI(−) m/z calcd for C18H12N3OS 318.0701 [M − H]−, found 318.0708.
(Z)-1-Benzyl-5-(3,4-dihydroxybenzylidene)-2-thioxoimidazolidin-4-one (35)
Prepared from 3,4-dihydroxybenzaldehyde and compound 28 (0.15 g, 0.73 mmol, 1.3 equiv) following the general procedure B. Yield 63%; mp 252–255 °C; 1H NMR (600 MHz, CD3OD) δ 7.64 (s, 1H), 7.26–7.22 (m, 4H), 7.18–7.13 (m, 2H), 6.60 (d, J = 8.4 Hz, 1H), 6.27 (s, 1H), 5.28 (s, 2H); 13C NMR (150 MHz, CD3OD) δ 175.9, 162.8, 147.9, 144.4, 135.6, 128.4, 127.2, 126.7, 125.0, 124.3, 123.3, 117.4, 114.5, 45.7; HRMS-ESI(−) m/z calcd for C17H13N2O3S 325.0647 [M − H]−, found 325.0637.
(Z)-4-((3-Benzyl-5-oxo-2-thioxoimidazolidin-4-ylidene)methyl)-benzoic acid (36)
Prepared from 4-formylbenzoic acid and compound 28 (0.15 g, 0.73 mmol, 1.3 equiv) following the general procedure B. Yield 64%; mp 209–211 °C; 1H NMR (600 MHz, DMSO-d6) δ 7.91 (d, J = 8.4 Hz, 2H), 7.79 (d, J = 8.4 Hz, 2H), 7.33–7.32 (m, 5H), 6.49 (s, 1H), 5.35 (s, 2H); 13C NMR (150 MHz, DMSO-d6) δ 169.6, 165.9, 137.7, 136.4, 134.7, 130.4, 130.2, 129.0, 127.7, 127.4, 118.6, 43.7; HRMS-ESI(−) m/z calcd for C18H13N2O3S 337.0647 [M − H]−, found 337.0624.
(Z)-1-Benzyl-5-(4-fluorobenzylidene)-2-thioxoimidazolidin-4-one (37)
Prepared from 4-fluorobenzaldehyde and compound 28 (0.15 g, 0.73 mmol, 1.3 equiv) following the general procedure B. Yield 47%; mp 135–138 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.62 (s, 1H), 8.02 (m, 2H), 7.24–7.34 (m, 5H), 7.20 (m, 2H), 6.64 (s, 1H), 5.32 (s, 2H); 13C NMR (150 MHz, DMSO-d6) δ 177.1, 163.2, 162.1, 135.8, 133.6, 133.5, 129.1, 128.8, 128.5, 127.9, 127.4, 119.8, 115.6, 45.7; HRMS-ESI(−) m/z calcd for C16H11N2O3S 311.0654 [M − H]−, found 311.0664.
(Z)-5-((1H-Indol-5-yl)methylene)-1-benzyl-2-thioxoimidazolidin-4-one (38)
Prepared from 1H-indole-5-carbaldehyde and compound 28 (0.15 g, 0.73 mmol, 1.3 equiv) following the general procedure B. Yield 41%; 1H NMR (600 MHz, DMSO-d6) δ 12.49 (s, 1H), 12.29 (s, 1H), 8.32 (s, 1H), 7.83 (d, 1H), 7.23–7.36 (m, 7H), 6.75 (s, 1H), 6.44 (s, 1H), 5.40 (s, 2H).
(Z)-5-(3,4-Dihydroxybenzylidene)thiazolidine-2,4-dione (39)
Prepared from 3,4-dihydroxybenzaldehyde and commercially available 2,4-thiazolidinedione 29 (0.15 g, 1.3 mmol, 1.3 equiv) following the general procedure B. Yield 65%; mp > 300 °C; 1H NMR (600 MHz, DMSO-d6) δ 9.47 (br, 2H), 7.37 (s, 1H), 6.97 (s, 1H), 6.87 (d, J = 7.8 Hz, 1H), 6.81 (d, J = 7.8 Hz, 1H); 13C NMR (150 MHz, DMSO-d6) δ 174.6, 171.9, 148.0, 146.1, 129.3, 125.8, 124.5, 123.4, 116.7, 116.5; HRMS-ESI(−) m/z calcd for C10H6NO4S 236.0018 [M − H]−, found 236.0016.
(Z)-4-((2,4-Dioxothiazolidin-5-ylidene)methyl)benzoic Acid (40)
Prepared from 4-formylbenzoic acid and commercially available 2,4-thiazolidinedione 29 (0.15 g, 1.3 mmol, 1.3 equiv) following the general procedure B. Yield 63%; mp > 300 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.17 (s, 1H), 12.68 (s, 1H), 8.02 (d, J = 8.4 Hz, 2H), 7.80 (s, 1H), 7.67 (d, J = 8.4 Hz, 2H); 13C NMR (150 MHz, DMSO-d6) δ 168.1, 167.6, 167.0, 137.4, 132.1, 130.9, 130.8, 130.7, 130.3, 126.4; HRMS-ESI(−) m/z calcd for C11H6NO4S 248.0018 [M − H]−, found 247.9998.
(Z)-5-(4-Fluorobenzylidene)thiazolidine-2,4-dione (41)
Prepared from 4-fluorobenzaldehyde and commercially available 2,4-thiazolidinedione 29 (0.15 g, 1.3 mmol, 1.3 equiv) following the general procedure B. Yield 78%; mp > 300 °C; 1H (600 MHz, DMSO-d6) δ 7.55–7.54 (m, 2H), 7.28 (s, 1H), 7.23–7.26 (m, 2H); 13C NMR (150 MHz, DMSO-d6) δ 181.8, 175.2, 162.7, 161.1, 135.2, 132.6, 131.5, 122.4, 116.3, 116.1; HRMS-ESI(−) m/z calcd for C10H5NO2SF 222.0025 [M − H]−, found 222.0026.
(Z)-5-((1H-Indol-5-yl)methylene)thiazolidine-2,4-dione (42)
Prepared from 1H-indole-5-carbaldehyde and commercially available 2,4-thiazolidinedione 29 (0.15 g, 1.3 mmol, 1.3 equiv) following the general procedure B. Yield 67%; mp > 300 °C; 1H (600 MHz, DMSO-d6) δ 12.42 (s, 1H), 11.41 (s, 1H), 7.87 (s, 1H), 7.82 (s, 1H), 7.51 (d, J = 8.4 Hz, 1H), 7.42 (s, 1H), 7.31 (d, J = 8.4 Hz, 1H); 13C NMR (150 MHz, DMSO-d6) δ 168.7, 167.9, 137.3, 134.8, 134.7, 128.6, 127.9, 127.7, 124.4, 124.2, 123.4, 119.2; HRMS-ESI(−) m/z calcd for C12H7N2O2S 243.0228 [M − H]−, found 243.0232.
General Procedure C for Aldol Condensation.55
To a mixture of aromatic aldehyde (1 equiv), 30 (1 equiv), and sodium acetate (1 equiv) was added glacial acetic acid (3 mL). The mixture was heated under reflux for 2 h and then cooled to room temperature and poured into cold water. The yellow solid precipitate was collected by filtration. The obtained solid was further purified by crystallization from ethanol or by column chromatography to yield the desired compound.
(Z)-5-(3,4-Dihydroxybenzylidene)-4-thioxothiazolidin-2-one (43)
Prepared from 3,4-dihydroxybenzaldehyde following the general procedure C. Yield 63%; mp 210–214 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.62 (s, 1H), 7.92 (s, 1H), 7.05–7.03 (m, 2H), 6.86 (d, J = 8.4 Hz, 1H); 13C NMR (150 MHz, DMSO-d6) δ 195.2, 171.1, 150.1, 146.5, 137.9, 125.9, 125.3, 117.0, 116.9; HRMS-ESI(−) m/z calcd for C10H6NO3S2 251.9789 [M − H]−, found 251.9803.
(Z)-4-((2-Oxo-4-thioxothiazolidin-5-ylidene)methyl)benzoic Acid (44)
Prepared from 4-formylbenzoic acid following the general procedure C. Yield 67%; mp > 300 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.22 (s, 1H), 8.08 (s, 1H), 8.02 (d, J = 8.4 Hz, 2H), 7.75 (d, J = 8.4 Hz, 2H); 13C NMR (150 MHz, DMSO-d6) δ 195.8, 170.8, 166.9, 137.6, 134.6, 132.4, 132.1, 130.8, 130.5; HRMS-ESI(−) m/z calcd for C11H6NO3S2 263.9789 [M − H]−, found 263.9808.
(Z)-5-(4-Fluorobenzylidene)-4-thioxothiazolidin-2-one (45)
Prepared from 4-fluorobenzaldehyde following the general procedure C. Yield 53%; mp 192–194 °C; 1H NMR (600 MHz, CD3OD) δ 8.01 (s, 1H), 7.57–7.55 (m, 2H), 7.16–7.13 (m, 2H); 13C NMR (150 MHz, CD3OD) δ 195.5, 170.2, 164.5, 162.8, 134.3, 132.5, 132.4, 130.4, 116.1, 115.9; HRMS-ESI(−) m/z calcd for C12H7N2OS2 237.9797 [M − H]−, found 237.9827.
(Z)-5-((1H-Indol-5-yl)methylene)-4-thioxothiazolidin-2-one (46)
Prepared from 1H-indole-5-carbaldehyde following the general procedure C. Yield 49%; mp 192–194 °C; 1H NMR (600 MHz, CD3OD) δ 8.24 (s, 1H), 7.81 (s, 1H), 7.41 (d, J = 8.4 Hz, 1H), 7.41 (d, J = 8.4 Hz, 1H), 7.24 (d, J = 3.1 Hz, 1H), 6.49 (d, J = 3.1 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 196.2, 172.8, 139.2, 137.6, 128.7, 126.5, 125.8, 125.2, 124.5, 123.7, 111.8, 102.4; HRMS-ESI(−) m/z calcd for C12H7N2OS2 259.0000 [M − H]−, found 259.0007.
(Z)-5-(2,3-Dihydroxybenzylidene)-4-thioxothiazolidin-2-one (47)
Prepared from 2,3-dihydroxybenzaldehyde following the general procedure C. Yield 69%; mp 188–192 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.89 (s, 1H), 9.99 (s, 1H), 9.76 (s, 1H), 8.49 (s, 1H), 6.92 (d, J = 6.6 Hz, 1H), 6.88 (d, J = 6.6 Hz, 1H), 6.76 (t, J = 8.4 Hz, 1H); 13C NMR (150 MHz, DMSO-d6) δ 195.8, 171.7, 161.6, 147.6, 146.3, 132.7, 128.5, 121.7, 120.1, 118.6; HRMS-ESI(−) m/z calcd for C10H6NO3S2 251.9789 [M − H]−, found 251.9763.
(Z)-5-(2,4-Dihydroxybenzylidene)-4-thioxothiazolidin-2-one (48)
Prepared from 2,4-dihydroxybenzaldehyde following the general procedure C. Yield 62%; mp 238–241 °C; 1H NMR (600 MHz, CD3OD) δ 8.53 (s, 1H), 7.24 (d, J = 8.4 Hz, 1H), 6.31 (d, J = 8.4 Hz, 1H), 6.26 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 194.3, 170.4, 161.7, 159.9, 132.1, 129.1, 112.8, 107.1, 101.2; HRMS-ESI(−) m/z calcd for C10H6NO3S2 251.9789 [M − H]−, found 251.9776.
(Z)-5-(2,5-Dihydroxybenzylidene)-4-thioxothiazolidin-2-one (49)
Prepared from 2,5-dihydroxybenzaldehyde following the general procedure C. Yield 62%; mp 226–229 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.7 (s, 1H), 9.49 (s, 1H), 9.18 (s, 1H), 8.41 (s, 1H), 6.82–6.80 (m, 3H); 13C NMR (150 MHz, DMSO-d6) δ 195.7, 175.1, 152.2, 150.6, 132.3, 128.0, 121.4, 121.0, 117.6, 113.2; HRMS-ESI(−) m/z calcd for C10H6NO3S2 251.9789 [M − H]−, found 251.9781.
(Z)-4-Thioxo-5-(2,3,4-trihydroxybenzylidene)thiazolidin-2-one (50)
Prepared from 2,3,4-trihydroxybenzaldehyde following the general procedure C. Yield 60%; mp 227–231 °C; 1H NMR (600 MHz, CD3OD) δ 8.54 (s, 1H), 6.81 (d, J = 8.4 Hz, 1H), 6.36 (d, J = 8.4 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 195.1, 171.4, 149.8, 148.6, 133.2, 132.9, 124.9, 119.9, 114.3, 107.5; HRMS-ESI(−) m/z calcd for C10H6NO4S2 267.9738 [M − H]−, found 267.9759.
(Z)-5-(4-Hydroxybenzylidene)-4-thioxothiazolidin-2-one (51)
Prepared from 4-hydroxybenzaldehyde following the general procedure C. Yield 69%; mp 241–244 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.66 (s, 1H), 10.50 (s, 1H), 8.03 (s, 1H), 7.54 (d, J = 8.9 Hz, 2H), 6.92 (d, J = 8.9 Hz, 2H); 13C NMR (150 MHz, DMSO-d6) δ 195.3, 171.1, 161.2, 137.5, 137.4, 133.8, 133.7, 126.2, 124.9, 117.2; HRMSESI(−) m/z calcd for C10H6NO2S2 235.9840 [M − H]−, found 235.9858.
(Z)-5-(3-Hydroxybenzylidene)-4-thioxothiazolidin-2-one (52)
Prepared from 3-hydroxybenzaldehyde following the general procedure C. Yield 67%; mp 222–225 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.86 (s, 1H), 9.86 (s, 1H), 7.97 (s, 1H), 7.32–7.31 (m, 1H), 7.10 (d, J = 6.0 Hz, 1H), 7.02 (s, 1H), 6.91 (d, J = 7.8 Hz, 1H); 13C NMR (150 MHz, DMSO-d6) δ 195.8, 175.0, 171.0, 158.4, 136.5, 134.9, 130.9, 122.4, 118.8, 116.5; HRMS-ESI(−) m/z calcd for C10H6NO2S2 235.9840 [M − H]−, found 235.9860.
(Z)-5-(2-Hydroxybenzylidene)-4-thioxothiazolidin-2-one (53)
Prepared from 2-hydroxybenzaldehyde following the general procedure C. Yield 63%; mp 210–212 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.73 (s, 1H), 10.62 (s, 1H), 8.45 (s, 1H), 7.40 (d, J = 7.2 Hz, 1H), 7.35–7.32 (m, 1H), 6.97–6.92 (m, 2H); 13C NMR (150 MHz, DMSO-d6) δ 195.8, 186.0, 175.0, 171.2, 158.7, 132.2, 128.6, 126.0, 120.4; HRMS-ESI(−) m/z calcd for C10H6NO2S2 235.9840 [M − H]−, found 235.9855.
(Z)-5-(4-Hydroxy-3-methoxybenzylidene)-4-thioxothiazolidin-2-one (54)
Prepared from 4-hydroxy-3-methoxybenzaldehyde following the general procedure C. Yield 83%; mp 195–198 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.66 (s, 1H), 10.1 (s, 1H), 8.04 (s, 1H), 7.22 (s, 1H), 7.16 (d, J = 8.4 Hz, 1H), 6.93 (d, J = 8.4 Hz, 1H), 2.47 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 195.2, 171.0, 150.8, 148.6, 137.8, 126.4, 125.6, 123.3, 116.8, 115.2, 56.5; HRMS-ESI(−) m/z calcd for C11H8NO3S2 265.9946 [M − H]−, found 265.9937.
(Z)-5-(4-Hydroxy-3,5-dimethoxybenzylidene)-4-thioxothiazolidin-2-one (55)
Prepared from 4-hydroxy-3,5-dimethoxybenzaldehyde following the general procedure C. Yield 72%; mp 222–226 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.63 (s, 1H), 8.01 (s, 1H), 6.93 (s, 1H), 3.81 (s, 6H); 13C NMR (150 MHz, DMSO-d6) δ 195.0, 175.0, 171.0, 148.7, 140.1, 138.1, 126.6, 124.2, 109.3, 108.9, 56.6, 56.3; HRMS-ESI(−) m/z calcd for C12H10NO4S2 296.0051 [M − H]−, found 296.0069.
(Z)-2-((2-Oxo-4-thioxothiazolidin-5-ylidene)methyl)benzoic Acid (56)
Prepared from 2-formylbenzoic acid following the general procedure C. Yield 58%; mp 206–209 °C; 1H NMR (600 MHz, CD3OD) δ 8.71 (s, 1H), 8.09 (d, J = 7.8 Hz, 1H), 7.67–7.65 (m, 1H), 7.62 (d, J = 7.2 Hz, 1H), 7.55–7.53 (m, 1H); 13C NMR (150 MHz, CD3OD) δ 195.6, 171.2, 167.9, 137.0, 135.3, 133.1, 132.0, 131.9, 131.5, 130.7, 128.6; HRMS-ESI(−) m/z calcd for C11H6NO3S2 263.9789 [M − H]−, found 263.9808.
(Z)-3-((2-Oxo-4-thioxothiazolidin-5-ylidene)methyl)benzoic Acid (57)
Prepared from 3-formylbenzoic acid following the general procedure C. Yield 58%; mp 242–244 °C; 1H NMR (600 MHz, DMSO-d6) δ 8.15 (s, 1H), 8.07 (s, 1H), 8.00 (d, J = 7.2 Hz, 1H), 7.74 (d, J = 7.2 Hz, 1H), 7.52 (t, J = 8.4 Hz, 1H); 13C NMR (150 MHz, DMSO-d6) δ 195.7, 170.7, 166.9, 135.0, 134.2, 132.3, 131.5, 131.2, 130.3, 126.7; HRMS-ESI(−) m/z calcd for C11H6NO3S2 263.9789 [M − H]−, found 263.9807.
Sodium (Z)-4-((2-Oxo-4-thioxothiazolidin-5-ylidene)methyl)-benzoate (58)
Compound 44 (43 mg, 0.162 mmol) and NaOH (6.5 mg, 0.162 mmol) were dissolved in 0.7 mL (H2O:MeOH/3:2) and stirred at room temperature for 2 h. The solution was evaporated and dried to afford 58 (43.8 mg, 94%) as a yellow colored compound; mp > 300 °C; 1H NMR (600 MHz, DMSO-d6) δ 7.99 (d, 2H), 7.97 (s, 1H), 7.55 (d, J = 7.8 Hz, 2H); 13C NMR (150 MHz, DMSO-d6) δ 210.5, 180.2, 170.1, 140.6, 140.5, 136.2, 131.4, 129.9, 128.8, 128.6, 117.1; HRMS-ESI(−) m/z calcd for C11H6NO3S2 263.9789 [M − Na]−, found 263.9838.
Sodium (Z)-2-((2-Oxo-4-thioxothiazolidin-5-ylidene)methyl)-benzoate (59)
Prepared from 56 following the procedure preparation of 58. Yield 92%; mp 252–256 °C; 1H NMR (600 MHz, DMSO-d6) δ 8.68 (s, 1H), 7.62 (d, J = 7.8 Hz, 1H), 7.54 (d, J = 7.8 Hz, 1H), 7.32 (t, 1H), 7.26 (t, 1H); 13C NMR (150 MHz, DMSO-d6) δ 210.5, 181.1, 172.5, 144.6, 140.4, 134.7, 131.4, 128.9, 127.3, 117.2; HRMS-ESI(−) m/z calcd for C11H6NO3S2 263.9789 [M − Na]−, found 263.9829.
(Z)-Methyl 4-((2-oxo-4-thioxothiazolidin-5-ylidene)methyl)-benzoate (60)
Prepared from methyl 4-formylbenzoate following the general procedure C. Yield 90%; mp 187–191 °C; 1H NMR (600 MHz, DMSO-d6) δ 8.08 (s, 1H), 8.05 (d, J = 8.4 Hz, 2H), 7.79 (d, J = 8.4 Hz, 2H), 3.86 (s, 3 H); 13C NMR (150 MHz, DMSO-d6) δ 195.8, 170.8, 165.9, 138.1, 134.3, 130.9, 130.4, 52.8; HRMS-ESI(−) m/z calcd for C12H8NO3S2 277.9946 [M − H]−, found 277.9965.
(Z)-Methyl 3-((2-oxo-4-thioxothiazolidin-5-ylidene)methyl)-benzoate (61)
Prepared from methyl 3-formylbenzoate following the general procedure C. Yield 79%; mp 200–203 °C; 1H NMR (600 MHz, DMSO-d6) δ 8.19 (s, 1H), 8.12 (s, 1H), 8.04 (d, J = 7.2 Hz, 1H), 7.93 (d, J = 7.2 Hz, 1H), 7.68 (t, J = 7.8 Hz, 1H), 3.88 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 195.7, 170.6, 165.8, 135.2, 134.7, 134.3, 131.3, 131.0, 130.7, 130.4, 52.5; HRMS-ESI(−) m/z calcd for C12H8NO3S2 277.9946 [M − H]−, found 277.9951.
(Z)-5-(2-Bromobenzylidene)-4-thioxothiazolidin-2-one (62)
Prepared from 4-bromobenzaldehyde following the general procedure C. Yield 40%; mp 165–168 °C; 1H NMR (600 MHz, CD3OD) δ 8.28 (s, 1H), 7.64 (d, J = 8.4 Hz, 1H), 7.52 (d, J = 8.4 Hz, 1H), 7.38 (d, J = 7.8 Hz, 1H), 7.25 (d, J = 7.8 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 195.2, 179.7, 141.0, 134.2, 133.8, 133.4, 131.4, 128.7, 127.8, 125.9; HRMS-ESI(−) m/z calcd for C10H5NBrOS2 297.8996 [M − H]−, found 297.8992.
(Z)-5-(4-Nitrobenzylidene)-4-thioxothiazolidin-2-one (63)
Prepared from 4-nitrobenzaldehyde following the general procedure C. Yield 58%; mp 192–194 °C; 1H NMR (600 MHz, DMSO-d6) δ 8.32 (d, J = 8.4 Hz, 2H), 8.11 (s, 1H), 7.91 (d, J = 8.7 Hz, 2H); 13C NMR (150 MHz, DMSO-d6) δ 195.3, 172.9, 147.9, 139.9, 131.9, 130.6, 123.8; HRMS-ESI(−) m/z calcd for C10H5N2O3S2 264.9742 [M − H]−, found 264.9757.
(Z)-4-((2-Oxo-4-thioxothiazolidin-5-ylidene)methyl)benzonitrile (64)
Prepared from 4-formylbenzonitrile following the general procedure C. Yield 61%; mp 178–181 °C; 1H NMR (600 MHz, DMSO-d6) δ 8.03 (s, 1H), 7.75 (d, J = 8.4 Hz, 2H), 7.68 (d, 1H, J = 8.4 Hz, 2H); 13C NMR (150 MHz, DMSO-d6) δ 195.7, 170.6, 138.1, 133.5, 133.2, 131.2, 118.8, 112.7; HRMS-ESI(−) m/z calcd for C11H5N2OS2 244.9843 [M − H]−, found 244.9861.
(Z)-5-(Pyridin-3-ylmethylene)-4-thioxothiazolidin-2-one (65)
Prepared from nicotinaldehyde following the general procedure C. Yield 84%; mp 255–258 °C; 1H NMR (600 MHz, DMSO-d6) δ 8.80 (s, 1H), 8.64–8.63 (m, 1H), 8.07 (s, 1H), 8.03 (d, J = 8.4 Hz, 1H), 7.56 (dd, J = 6.0 Hz, J = 7.8 Hz, 1H); 13C NMR (150 MHz, DMSO-d6) δ 195.7, 170.7, 152.1, 151.1, 136.7, 132.6, 132.2, 130.1, 124.7; HRMS-ESI(−) m/z calcd for C9H5N2OS2 220.9843 [M − H]−, found 220.9863.
(Z)-5-(Pyridin-4-ylmethylene)-4-thioxothiazolidin-2-one (66)
Prepared from isonicotinaldehyde following the general procedure C. Yield 67%; mp 252–254 °C; 1H NMR (600 MHz, DMSO-d6) δ 8.71 (d, J = 5.1 Hz, 2H), 7.97 (s, 1H), 7.59 (d, J = 5.4 Hz, 2H); 13C NMR (150 MHz, DMSO-d6) δ 198.2, 172.4, 153.8, 153.1, 126.1, 124.4, 111.8; HRMS-ESI(−) m/z calcd for C9H5N2OS2 220.9843 [M − H]−, found 220.9860.
(Z)-5-((5-Phenylthiophen-2-yl)methylene)-4-thioxothiazolidin-2-one (67)
Prepared from 5-phenylthiophene-2-carbaldehyde following the general procedure C. Yield 65%; mp 250–255 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.76 (s, 1H), 8.27 (s, 1H), 7.86 (d, J = 3.6 Hz, 1H), 7.78 (d, J = 7.8 Hz, 2H), 7.75 (d, J = 4.2 Hz, 1H), 7.47 (t, J = 7.8 Hz, 2H), 7.41 (d, J = 7.2 Hz, 1H); 13C NMR (150 MHz, DMSO-d6) δ 194.3, 170.0, 151.9, 138.9, 137.6, 132.9, 129.7, 129.2, 127.5, 126.4, 126.5; HRMS-ESI(−) m/z calcd for C14H8NOS3 301.9768 [M − H]−, found 301.9779.
(Z)-5-(Benzo[b]thiophen-3-ylmethylene)-4-thioxothiazolidin-2-one (68)
Prepared from benzo[b]thiophene-3-carbaldehyde following the general procedure C. Yield 73%; mp 218–221 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.83 (s, 1H), 8.35 (s, 1H), 8.24 (s, 1H), 8.10 (d, J = 7.8 Hz, 1H), 8.01 (d, J = 7.8 Hz, 1H), 7.55 (t, J = 7.2 Hz, 1H), 7.50 (t, J = 7.8 Hz, 1H); 13C NMR (150 MHz, DMSO-d6) δ 195.2, 170.7, 139.5, 138.4, 132.8, 132.4, 131.1, 130.0, 125.4, 125.6, 123.8, 121.8; HRMS-ESI(−) m/z calcd for C12H6NOS3 275.9612 [M − H]−, found 275.9628.
(Z)-5-((1-Methyl-1H-indol-3-yl)methylene)-4-thioxothiazolidin-2-one (69)
Prepared from 1-methyl-1H-indole-3-carbaldehyde following the general procedure C. Yield 59%; mp 224–228 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.38 (s, 1H), 8.39 (s, 1H), 7.97 (s, 1H), 7.84 (d, J = 8.4 Hz, 1H), 7.58 (d, J = 7.8 Hz, 1H), 7.33 (t, J = 7.2 Hz, 1H), 7.28 (t, J = 7.8 Hz, 1H), 3.93 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 193.3, 175.0, 170.5, 137.5, 134.2, 128.9, 128.1, 124.1, 123.5, 122.5, 118.7, 111.6, 34.0; HRMS-ESI(−) m/z calcd for C13H9N2OS2 273.0156 [M − H]−, found 273.0171.
(Z)-5-(4-(1H-Tetrazol-5-yl)benzylidene)-4-thioxothiazolidin-2-one (70)
Prepared from 4-(1H-tetrazol-5-yl)benzaldehyde31 following the general procedure C. Yield 84%; 1H NMR (600 MHz, DMSO-d6) δ 13.9 (s, 1H), 8.16 (d, J = 8.4 Hz, 2H), 8.10 (s, 1H), 7.88 (d, J = 8.4 Hz, 2H); 13C NMR (150 MHz, DMSO-d6) δ 195.7, 170.8, 134.1, 131.7, 128.2, 117.4.
(Z)-5-(2-(1H-Tetrazol-5-yl)benzylidene)-4-thioxothiazolidin-2-one (71)
Prepared from 2-(1H-tetrazol-5-yl)benzaldehyde57 following the general procedure C. Yield 41%; mp 206–210 °C; 1H NMR (600 MHz, CD3OD) δ 8.26 (s, 1H), 7.79 (d, J = 7.8 Hz, 1H), 7.70 (d, J = 7.8 Hz, 1H), 7.62 (t, J = 7.2 Hz, 1H), 7.56 (t, J = 7.2 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 195.1, 170.9, 133.7, 133.2, 131.0, 130.1, 130.0, 128.2, 112.2, 109.9; HRMS-ESI(−) m/z calcd for C11H6N5OS2 288.0014 [M − H]−, found 288.0026.
(Z)-2-((2-Oxo-4-thioxothiazolidin-5-ylidene)methyl)-benzenesulfonic Acid (72)
Prepared from 2-formylbenzenesulfonic acid sodium salt following the general procedure C. Yield 27%; mp 218–221 °C; 1H NMR (600 MHz, CD3OD) δ 9.02 (s, 1H), 8.04 (d, J = 7.8 Hz, 1H), 7.63 (d, J = 7.8 Hz, 1H), 7.38 (t, J = 7.8 Hz, 1H), 7.25 (t, J = 7.8 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 192.5, 176.7, 144.8, 134.6, 132.8, 130.2, 129.0, 128.1, 127.5, 127.0; HRMS-ESI(−) m/z calcd for C10H6NO4S3 299.9459 [M − H]−, found 299.9445.
(Z)-Diethyl (4-((2-Oxo-4-thioxothiazolidin-5-ylidene)methyl)-phenyl)phosphonate (73)
Prepared from diethyl (4-formylphenyl)-phosphonate58 following the general procedure C. Yield 51%; mp 151–154 °C; 1H NMR (600 MHz, CD3OD) δ 8.05 (s, 1H), 7.81–7.77 (m, 2H), 7.68–7.66 (m, 2H), 4.04 (q, J = 6.6 Hz, J = 14.4 Hz, 4H), 1.24 (t, J = 7.2 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 195.3, 138.1, 133.3, 132.3, 132.0, 131.9, 129.9, 129.8, 62.6, 15.2; HRMSESI(−) m/z calcd for C14H15NO4PS2 356.0180 [M − H]−, found 356.0213.
(Z)-(4-((2-Oxo-4-thioxothiazolidin-5-ylidene)methyl)phenyl)-phosphonic Acid (74).59
To a solution of compound 73 (0.05 g, 0.14 mmol) in dry DCM (3 mL) was added TMSBr (0.3 g, 1.9 mmol) and stirred at room temperature for 16 h. The solvent was evaporated under vacuum and coevaporated with methanol. The reaction mixture was poured into water, and the solid separated was filtered off and washed with water to afford 74 (0.035 g, 84%). Mp 225–228 °C; 1H NMR (600 MHz, CD3OD) δ 8.14 (s, 1H), 7.91–7.88 (m, 2H), 7.71–7.61 (m, 2H); 13C NMR (150 MHz, CD3OD) δ 195.4, 170.2, 136.8, 133.9, 133.9, 131.6, 131.3, 131.2, 129.6, 129.5; HRMS-ESI(−) m/z calcd for C10H7NO4PS2 299.9554 [M − H]−, found 299.9580.
Biology
Tdp1 Gel-Based Assay.11
5′−32P-labeled DNA substrate (1 nM; N14Y; 5′-GATCTAAAAGACTT-pY-3′) was incubated with 0.02 nM Tdp1 in the absence or presence of inhibitor for 15 min at room temperature in an assay buffer containing PBS 1X, pH 7.4, 80 mM KCl, and 0.01% Tween-20. Reactions were terminated by the addition of 1 volume of gel loading buffer [99.5% (v/v) formamide, 5 mM EDTA, 0.01% (w/v) xylene cyanol, and 0.01% (w/v) bromophenol blue]. Samples were subjected to a 16% denaturing PAGE, and gels were exposed after drying to a PhosphorImager screen (GE Healthcare). Gel images were scanned using a Typhoon 8600 (GE Healthcare), and densitometric analyses were performed using the ImageQuant software (GE Healthcare).
WCE Tdp1 Gel-Based Assay
An amount of 1 ×107 DT40 knockout cells for chicken Tdp1 and complemented with human Tdp1 were collected and washed once with PBS. The supernatant was removed, and the pellet was resuspended with 50 μL of CellLytic M Cell Lysis Reagent (SIGMA C2978), pipetting several times over ice. After 15 min, the lysate was centrifuged at 12 000g for 10 min, and the protein concentration of the supernatant was determined using the Protein Assay Dye Reagent Concentrate (Bio-Rad #500–0006). 5′−32P-labeled DNA substrate (1 nM; N14Y; 5′-GATCTAAAAGACTT-pY-3′) was incubated with 1 μg/mL of whole cell extract in the absence or presence of inhibitor for 15 min at room temperature in an assay buffer containing 25 mM Tris HCl, pH 7.5, 80 mM KCl, 2 mM EDTA, 1 mM DTT, 40 μg/mL of BSA, and 0.01% Tween-20. Reactions were terminated by the addition of 1 volume of gel loading buffer [99.5% (v/v) formamide, 5 mM EDTA, 0.01% (w/v) xylene cyanol, and 0.01% (w/v) bromophenol blue]. Samples were subjected to a 16% denaturing PAGE, and gels were exposed after drying to a PhosphorImager screen (GE Healthcare). Gel images were scanned using a Typhoon 8600 (GE Healthcare), and densitometric analyses were performed using the ImageQuant software (GE Healthcare).
Surface Plasmon Resonance Analysis.
Materials
Coupling reagents N-ethyl-N′-(3dimethylaminopropyl)carbodiimide (EDC), N-hydroxysuccinimide (NHS), and ethanolamine were purchased from GE Heathcare (Piscatawy, NJ). Neutravidin was obtained from Pierce.
Binding experiments were performed on a Biacore T200 instrument (GE, Piscatawy, NJ). Tdp1 was amine coupled to a CM5 sensor chip (GE Healthcare, Piscatawy, NJ). The amine groups in the active site of Tdp1 were protected from modification by binding a 14 base oligonucleotide to Tdp1 during the coupling step. Specifically 1 μM Tdp1 was incubated with 2 μM of a 14 base oligonucleotide containing a phosphate group at the 3′ end (GATCTAAAAGACTT) in 10 mM sodium acetate pH 4.5 for 20 min. The CM5 chip surface was activated for 7 min with 0.1 M NHS and 0.4 M EDC at a flow rate of 20 μL/min, and the Tdp1–oligonucleotide mixture was injected until approximately 6000 RUs were attached. Activated amine groups were quenched with an injection of 1 M solution of ethanolamine pH 8.0 for 7 min. Any bound oligonucleotide was removed by washing the surface with 1 M NaCl. A reference surface was prepared in the same manner without coupling of Tdp1. Using the same approach, 8000 RUs of Neutravidin were amine coupled to flow cells 3 and 4. A 14 base oligonucleotide modified with a biotin at the 5′ end and a tyrosine at the 3′ end (biotin-GATCTAAAAGACTT-Tyr) was captured in the flow cell 4. A dilution series of 50 (50, 25, 6.25, 3.12, 1.56. 0.78, and 0.39 μM) was prepared in running buffer [50 mM Tris/HCl, 80 mM KCl, 0.01% tween 20 (v/v), 5% DMSO (v/v), 2 mM EDTA, 1 mM DTT, 40 μg/mL of BSA pH 7.5] and injected over all flow cells at 30 μL/min at 25 °C. Following compound injections, the surface was regenerated with a 30 s injection of 1 M NaCl, a 30 s injection of 50% DMSO (v/v), and a 30 s running buffer injection. Each cycle of compound injection was followed by a buffer cycle for referencing purposes. A DMSO calibration curve was included to correct for refractive index mismatches between the running buffer and compound dilution series.
Modeling and Docking
The study was carried out using the Schrodinger modeling suite.60 The X-ray crystallographic structure of human Tdp1 in complex with vanadate, DNA, and human Top1 derived peptide (PDB: 1NOP)25 was used as the starting point of the study. The crystallographic vanadate ion was replaced with a pyrophosphate linker to generate the complete Top1–DNA covalent intermediate. All missing hydrogen atoms were added based on ionization states determined by PROPKA.61 Restraint energy was minimized with the implicit solvent model using the OPLS 2005 force field.62 All Tdp1 inhibitors were built and prepared with Ligprep to enumerate all possible ionization states, tautomers, and low ring energy conformers. The docking studies were carried out using Glide at standard precision without any constraint within the catalytic binding site of Tdp1. The van der Waals radii of nonpolar atoms for each of the ligands were scaled by a factor of 0.8 to account for structure variability to specific ligand binding. Visual inspection was employed to identify the potential mode of binding that best corroborates with our observed SAR study.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the Center for Drug Design at the University of Minnesota and by the Center for Cancer Research, Intramural Program of the National Cancer Institute, National Institutes of Health. We thank the University of Minnesota Supercomputing Institute for providing the computational resources.
ABBREVIATIONS USED
- Tdp1
tyrosyl–DNA phosphodiesterase
- Top1
topoisomerase I
- SAR
structure–activity relationship
- CPT
camptothecin
- PLD
phospholipase D
- HTS
high-throughput screening
- PTK
protein tyrosine kinase
- PTP
protein tyrosine phosphatase
- WCE
whole cell extracts
- SPR
Surface Plasmon Resonance
Footnotes
The authors declare no competing financial interest.
Supporting Information
Docking of previously reported inhibitors (1–3). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Champoux JJ DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem 2001, 70, 369–413. [DOI] [PubMed] [Google Scholar]
- (2).Pommier Y; Pourquier P; Fan Y; Strumberg D Mechanism of action of eukaryotic DNA topoisomerase I and drugs targeted to the enzyme. Biochim. Biophys. Acta, Gene Struct. Expression 1998, 1400, 83–105. [DOI] [PubMed] [Google Scholar]
- (3).Pommier Y DNA topoisomerase I inhibitors: chemistry, biology, and interfacial inhibition. Chem. Rev 2009, 109, 2894–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Pommier Y; Marchand C Interfacial inhibitors: targeting macromolecular complexes. Nat. Rev. Drug Discovery 2012, 11, 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Pommier Y Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [DOI] [PubMed] [Google Scholar]
- (6).Pourquier P; Pommier Y Topoisomerase I-mediated DNA damage. Adv. Cancer Res 2001, 80, 189–216. [DOI] [PubMed] [Google Scholar]
- (7).Pommier Y; Cushman M The indenoisoquinoline non-camptothecin topoisomerase I inhibitors: update and perspectives. Mol. Cancer Ther 2009, 8, 1008–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Yang S.-w.; Burgin AB Jr.; Huizenga BN; Robertson CA; Yao KC; Nash HA A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc. Natl. Acad. Sci. U.S.A 1996, 93, 11534–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pommier Y; Barcelo JM; Rao VA; Sordet O; Jobson AG; Thibaut L; Miao ZH; Seiler JA; Zhang H; Marchand C; Agama K; Nitiss JL; Redon C Repair of topoisomerase I-mediated DNA damage. Prog. Nucleic Acid Res. Mol. Biol 2006, 81, 179–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Pommier Y; Redon C; Rao VA; Seiler JA; Sordet O; Takemura H; Antony S; Meng L; Liao Z; Kohlhagen G; Zhang H; Kohn KW Repair of and checkpoint response to topoisomerase I-mediated DNA damage. Mutat. Res 2003, 532, 173–203. [DOI] [PubMed] [Google Scholar]
- (11).Dexheimer TS; Gediya LK; Stephen AG; Weidlich I; Antony S; Marchand C; Interthal H; Nicklaus M; Fisher RJ; Njar VC; Pommier Y 4-Pregnen-21-ol-3,20-dione-21-(4-bromobenzenesufonate) (NSC 88915) and Related Novel Steroid Derivatives as Tyrosyl-DNA Phosphodiesterase (Tdp1) Inhibitors. J. Med. Chem 2009, 52, 7122–7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gottlin EB; Rudolph AE; Zhao Y; Matthews HR; Dixon JE Catalytic mechanism of the phospholipase D superfamily proceeds via a covalent phosphohistidine intermediate. Proc. Natl. Acad. Sci. U.S.A 1998, 95, 9202–9207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Liu C; Pouliot JJ; Nash HA The role of TDP1 from budding yeast in the repair of DNA damage. DNA Repair 2004, 3, 593–601. [DOI] [PubMed] [Google Scholar]
- (14).Interthal H; Chen HJ; Kehl-Fie TE; Zotzmann J; Leppard JB; Champoux JJ SCAN1 mutant Tdp1 accumulates the enzyme-DNA intermediate and causes camptothecin hypersensitivity. EMBO J. 2005, 24, 2224–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Murai J; Huang SY; Das BB; Dexheimer TS; Takeda S; Pommier Y Tyrosyl-DNA phosphodiesterase 1 (TDP1) repairs DNA damage induced by topoisomerases I and II and base alkylation in vertebrate cells. J. Biol. Chem 2012, 287, 12848–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Miao Z-H; Agama K; Sordet O; Povirk L; Kohn KW; Pommier Y Hereditary ataxia SCAN1 cells are defective for the repair of transcription-dependent topoisomerase I cleavage complexes. DNA Repair 2006, 5, 1489–1494. [DOI] [PubMed] [Google Scholar]
- (17).Barthelmes HU; Habermeyer M; Christensen MO; Mielke C; Interthal H; Pouliot JJ; Boege F; Marko D TDP1 Overexpression in Human Cells Counteracts DNA Damage Mediated by Topoisomerases I and II. J. Biol. Chem 2004, 279, 55618–55625. [DOI] [PubMed] [Google Scholar]
- (18).Beretta GL; Cossa G; Gatti L; Zunino F; Perego P Tyrosyl-DNA phosphodiesterase 1 targeting for modulation of camptothecin-based treatment. Curr. Med. Chem 2010, 17, 1500–1508. [DOI] [PubMed] [Google Scholar]
- (19).Dexheimer TS; Antony S; Marchand C; Pommier Y Tyrosyl-DNA phosphodiesterase as a target for anticancer therapy. Anti-Cancer Agents Med. Chem 2008, 8, 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Interthal H; Pouliot JJ; Champoux JJ The tyrosyl-DNA phosphodiesterase Tdp1 is a member of the phospholipase D superfamily. Proc. Natl. Acad. Sci. U.S.A 2001, 98, 12009–12014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Waite M The PLD superfamily: insights into catalysis. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 1999, 1439, 187–197. [DOI] [PubMed] [Google Scholar]
- (22).Raymond AC; Rideout MC; Staker B; Hjerrild K; Burgin AB Jr. Analysis of Human Tyrosyl-DNA Phosphodiesterase I Catalytic Residues. J. Mol. Biol 2004, 338, 895–906. [DOI] [PubMed] [Google Scholar]
- (23).Liao Z; Thibaut L; Jobson A; Pommier Y Inhibition of human tyrosyl-DNA phosphodiesterase by aminoglycoside antibiotics and ribosome inhibitors. Mol. Pharmacol 2006, 70, 366–372. [DOI] [PubMed] [Google Scholar]
- (24).Davies DR; Interthal H; Champoux JJ; Hol WGJ The crystal structure of human tyrosyl-DNA phosphodiesterase, Tdp1. Structure 2002, 10, 237–248. [DOI] [PubMed] [Google Scholar]
- (25).Davies DR; Interthal H; Champoux JJ; Hol WGJ Crystal structure of a transition state mimic for Tdp1 assembled from vanadate, DNA, and a topoisomerase I-derived peptide. Chem. Biol 2003, 10, 139–147. [DOI] [PubMed] [Google Scholar]
- (26).Antony S; Marchand C; Stephen AG; Thibaut L; Agama BK; Fisher RJ; Pommier Y Novel high-throughput electro-chemiluminescent assay for identification of human tyrosyl-DNA phosphodiesterase (Tdp1) inhibitors and characterization of furamidine (NSC 305831) as an inhibitor of Tdp1. Nucleic Acids Res. 2007, 35, 4474–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Nguyen TX; Morrell A; Conda-Sheridan M; Marchand C; Agama K; Bermingam A; Stephen AG; Chergui A; Naumova A; Fisher R; O’Keefe BR; Pommier Y; Cushman M Synthesis and Biological Evaluation of the First Dual Tyrosyl-DNA Phosphodiesterase I (Tdp1)-Topoisomerase I (Top1) Inhibitors. J. Med. Chem 2012, 55, 4457–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Marchand C; Lea WA; Jadhav A; Dexheimer TS; Austin CP; Inglese J; Pommier Y; Simeonov A Identification of phosphotyrosine mimetic inhibitors of human tyrosyl-DNA phosphodiesterase I by a novel AlphaScreen high-throughput assay. Mol. Cancer Ther 2009, 8, 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Irvine MW; Patrick GL; Kewney J; Hastings SF; MacKenzie SJ Rhodanine derivatives as novel inhibitors of PDE4. Bioorg. Med. Chem. Lett 2008, 18, 2032–7. [DOI] [PubMed] [Google Scholar]
- (30).Stuible M; Zhao L; Aubry I; Schmidt-Arras D; Bohmer FD; Li CJ; Tremblay ML Cellular inhibition of protein tyrosine phosphatase 1B by uncharged thioxothiazolidinone derivatives. ChemBioChem 2007, 8, 179–86. [DOI] [PubMed] [Google Scholar]
- (31).Wolf MC; Freiberg AN; Zhang T; Akyol-Ataman Z; Grock A; Hong PW; Li J; Watson NF; Fang AQ; Aguilar HC; Porotto M; Honko AN; Damoiseaux R; Miller JP; Woodson SE; Chantasirivisal S; Fontanes V; Negrete OA; Krogstad P; Dasgupta A; Moscona A; Hensley LE; Whelan SP; Faull KF; Holbrook MR; Jung ME; Lee B A broad-spectrum antiviral targeting entry of enveloped viruses. Proc. Natl. Acad. Sci. U.S.A 2010, 107, 3157–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Korohoda MJ; Kleinrok Z; Przegalinski E A search for new anticonvulsant compounds among 3-aryl-5-benzylidene derivatives of D. thiohydantoin. Pol. J. Pharmacol. Pharm 1976, 28, 329–33. [PubMed] [Google Scholar]
- (33).Marton J; Enisz J; Hosztafi S; Timar T Preparation and fungicidal activity of 5-substituted hydantoins and their 2-thio analogs. J. Agric. Food Chem 1993, 41, 148–52. [Google Scholar]
- (34).Ryczek J; Sermak W; Kleinrok Z; Przegalinski E Anticonvulsive drugs, ortho and meta isomers of 3-alkoxyphenyl-5-benzylidene-2-thiohydantoins. Acta Pharm. Jugosl 1979, 29, 131–7. [Google Scholar]
- (35).Machii D; Hagihara K; Asai A; Arai H; Yamashita Y; Chin AC; Piatyszek MA Preparation of 4-oxo-2-thioxoimidazolidine derivatives as telomerase inhibitors. 2002053155, 20011228, 2002. [Google Scholar]
- (36).Balschmidt P; Olsen HB; Kaarsholm NC; Madsen P; Jakobsen P; Ludvigsen S; Schluckebier G; Steensgaard DB; Petersen AK Preparation of high-affinity ligands for crystalline formulations of NPH-insulin. 2004080481, 20040312, 2004. [Google Scholar]
- (37).Esswein A; Schaefer W; Tsaklakidis C; Honold K; Kaluza K Preparation of thiazolidinones for the treatment and prevention of metabolic bone disorders. 2000018746, 19990930, 2000. [Google Scholar]
- (38).Fan Y-H; Chen H; Natarajan A; Guo Y; Harbinski F; Iyasere J; Christ W; Aktas H; Halperin JA Structure-activity requirements for the antiproliferative effect of troglitazone derivatives mediated by depletion of intracellular calcium. Bioorg. Med. Chem. Lett 2004, 14, 2547–2550. [DOI] [PubMed] [Google Scholar]
- (39).Cheung A; Fu Z Methods for screening for antibiotic compounds. 20110158911, 20101215, 2011. [Google Scholar]
- (40).Cheung A; Fu Z Compositions for treating bacterial infections. 2011002951, 20100701, 2011. [Google Scholar]
- (41).Baell JB; Holloway GA New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem 2010, 53, 2719–40. [DOI] [PubMed] [Google Scholar]
- (42).Tomasic T; Masic LP Rhodanine as a Privileged Scaffold in Drug Discovery. Curr. Med. Chem 2009, 16, 1596–1629. [DOI] [PubMed] [Google Scholar]
- (43).McGuire DK; Inzucchi SE New drugs for the treatment of diabetes mellitus: part I: Thiazolidinediones and their evolving cardiovascular implications. Circulation 2008, 117, 440–9. [DOI] [PubMed] [Google Scholar]
- (44).Pastromas S; Koulouris S Thiazolidinediones: antidiabetic drugs with cardiovascular effects. Hellenic J. Cardiol 2006, 47, 352–60. [PubMed] [Google Scholar]
- (45).Naito Y; Yoshikawa T Thiazolidinediones: a new class of drugs for the therapy of ischemia-reperfusion injury. Drugs Today 2004, 40, 423–30. [DOI] [PubMed] [Google Scholar]
- (46).O’Moore-Sullivan TM; Prins JB Thiazolidinediones and type 2 diabetes: new drugs for an old disease. Med. J. Aust 2002, 176, 381–6. [DOI] [PubMed] [Google Scholar]
- (47).Day C Thiazolidinediones: a new class of antidiabetic drugs. Diabet. Med 1999, 16, 179–92. [DOI] [PubMed] [Google Scholar]
- (48).Mendgen T; Steuer C; Klein CD Privileged Scaffolds or Promiscuous Binders: A Comparative Study on Rhodanines and Related Heterocycles in Medicinal Chemistry. J. Med. Chem 2012, 55, 743–753. [DOI] [PubMed] [Google Scholar]
- (49).Gouveia FL; de Oliveira RMB; de Oliveira TB; da Silva IM; do Nascimento SC; de Sena KXFR; de Albuquerque JFC Synthesis, antimicrobial and cytotoxic activities of some 5-arylidene-4-thioxo-thiazolidine-2-ones. Eur. J. Med. Chem 2009, 44, 2038–2043. [DOI] [PubMed] [Google Scholar]
- (50).Atamanyuk D; Zimenkovsky B; Lesyk R Synthesis and anticancer activity of novel thiopyrano[2,3-d]thiazole-based compounds containing norbornane moiety. J. Sulfur Chem 2008, 29, 151–162. [Google Scholar]
- (51).Jeong TS; Kim JR; Kim KS; Cho KH; Bae KH; Lee WS Inhibitory effects of multi-substituted benzylidenethiazolidine-2,4-diones on LDL oxidation. Bioorg. Med. Chem. Lett 2004, 12, 4017–4023. [DOI] [PubMed] [Google Scholar]
- (52).Seyfried MS; Linden A; Mloston G; Heimgartner H Chemoselectivity of the Reactions of Diazomethanes with 5-Benzylidene-3-phenylrhodanine. Helv. Chim. Acta 2009, 92, 1800–1816. [Google Scholar]
- (53).Meanwell NA Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem 2011, 54, 2529–91. [DOI] [PubMed] [Google Scholar]
- (54).Renault S; Bertrand S; Carreaux F; Bazureau JP Parallel solution-phase synthesis of 2-alkylthio-5-arylidene-3,5-dihydro-4Himidazol-4-one by one-pot. three-component domino reaction. J. Comb. Chem 2007, 9, 935–942. [DOI] [PubMed] [Google Scholar]
- (55).Metwally NH Synthesis of some new fused thiopyrano[2,3-d]thiazoles and their derivatives. J. Sulfur Chem 2007, 28, 275–284. [Google Scholar]
- (56).Abou El-Regal MK; Abdalha AA; El-Kassaby MA; Ali AT Synthesis of new thiohydantoin derivatives under phase transfer catalysis. Phosphorus, Sulfur Silicon Relat. Elem 2007, 182, 845–851. [Google Scholar]
- (57).Das B; Reddy CR; Kumar DN; Krishnaiah M; Narender R A Simple, Advantageous Synthesis of 5-Substituted 1H-Tetrazoles. Synlett 2010, 391–394. [Google Scholar]
- (58).Hirao T; Masunaga T; Ohshiro Y; Agawa T A Novel Synthesis of Dialkyl Arenephosphonates. Synthesis-Stuttgart 1981, 56–57. [Google Scholar]
- (59).Dang Q; Liu Y; Cashion DK; Kasibhatla SR; Jiang T; Taplin F; Jacintho JD; Li HQ; Sun ZL; Fan Y; Dare J; Tian F; Li WY; Gibson T; Lemus R; van Poelje PD; Potter SC; Erion MD Discovery of a Series of Phosphonic Acid-Containing Thiazoles and Orally Bioavailable Diamide Prodrugs That Lower Glucose in Diabetic Animals Through Inhibition of Fructose-1,6-Bisphosphatase. J. Med. Chem 2011, 54, 153–165. [DOI] [PubMed] [Google Scholar]
- (60).Maestro9.3, Macromodel9.9, Glide5.8, LigPrep4.0; Schrodinger, LLC: New York, 2012. [Google Scholar]
- (61).Li H; Robertson AD; Jensen JH Very fast empirical prediction and rationalization of protein pK(a) values. Proteins: Struct., Funct., Bioinf 2005, 61, 704–721. [DOI] [PubMed] [Google Scholar]
- (62).Jorgensen WL; Maxwell DS; Tirado-Rives J Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc 1996, 118, 11225–11236. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.