

Abstract
Recent advances in identification and characterization of human-specific regulatory DNA sequences set the stage for the assessment of their global impact on physiology and pathology of modern humans. Gene set enrichment analyses (GSEA) of 8405 genes linked with 35,074 human-specific neuro-regulatory single-nucleotide changes (hsSNCs) revealed numerous significant associations with morphological structures, physiological processes, and pathological conditions of modern humans. Significantly enriched traits include more than 1000 anatomically distinct regions of the adult human brain, many different types of cells and tissues, more than 200 common human disorders, and more than 1000 records of rare diseases. Thousands of genes connected with neuro-regulatory hsSNCs have been identified, which represent essential genetic elements of the autosomal inheritance and offspring survival phenotypes. A total of 1494 hsSNC-linked genes are associated with either autosomal dominant or recessive inheritance, and 2273 hsSNC-linked genes have been associated with premature death, embryonic lethality, as well as pre-, peri-, neo-, and post-natal lethality phenotypes of both complete and incomplete penetrance. Differential GSEA implemented on hsSNC-linked loci and associated genes identify a set of 7990 hsSNC-target genes linked to evolutionary distinct classes of human-specific regulatory sequences (HSRS). Notably, the expression of a majority of these genes (5389 genes; 67%) is regulated by stem cell–associated retroviral sequences (SCARS) and SCARS-regulated genes captured a dominant fraction (91%) of significant phenotypic associations linked with hsSNCs. Interrogations of the MGI database revealed readily available mouse models tailored for precise experimental definitions of functional effects of hsSNCs and SCARS on genes causally affecting thousands of mammalian phenotypes and implicated in hundreds of common and rare human disorders. These observations suggest that a preponderance of human-specific traits evolved under a combinatorial regulatory control of distinct classes of HSRS and neuro-regulatory loci harboring hsSNCs that are fixed in humans, distinct from other primates, and located in differentially accessible chromatin regions during brain development.
Electronic supplementary material
The online version of this article (10.1007/s10577-020-09639-w) contains supplementary material, which is available to authorized users.
Keywords: human phenotypic uniqueness, human-specific regulatory sequences, stem cell–associated retroviral sequences, human-specific traits, fixed neuro-regulatory human-specific single nucleotide mutations
Introduction
DNA sequences of coding genes defining the structure of macromolecules comprising the essential building blocks of life at the cellular and organismal levels remain highly conserved during the evolution of humans and other Great Apes (Chimpanzee Sequencing and Analysis Consortium 2005; Kronenberg et al. 2018). In contrast, a compendium of nearly hundred thousand candidate human-specific regulatory sequences (HSRS) has been assembled in recent years (Glinsky 2015, 2016a, b, c, 2017, 2018, 2020a; Glinsky and Barakat 2019; Kanton et al. 2019), thus providing further genetic and molecular evidence supporting the idea that unique to human phenotypes may result from human-specific changes to genomic regulatory sequences defined as “regulatory mutations” (King and Wilson 1975). Structurally, functionally, and evolutionary distinct classes of HSRS appear to cooperate in shaping developmentally and physiologically diverse human-specific genomic regulatory networks (GRNs) impacting preimplantation embryogenesis, pluripotency, and development and functions of the human brain (Glinsky 2020a). The best evidence of the exquisite degree of accuracy of the contemporary molecular definition of human-specific regulatory sequences is exemplified by the identification of 35,074 single nucleotide changes (SNCs) that are fixed in humans, distinct from other primates, and located within differentially accessible (DA) chromatin regions during the human brain development in cerebral organoids (Kanton et al. 2019). Therefore, this type of mutations could be defined as fixed neuro-regulatory human-specific single nucleotide changes (hsSNCs). However, only a small fraction of identified DA chromatin peaks (600 of 17,935 DA peaks; 3.3%) manifest associations with differential expression in human versus chimpanzee cerebral organoids model of brain development, consistent with the hypothesis that regulatory effects on gene expression of these DA chromatin regions are not restricted to the early stages of brain development. Annotation of SNCs derived and fixed in modern humans that overlap DA chromatin regions during brain development revealed that essentially all candidate neuro-regulatory human-specific SNCs are shared with the archaic humans (35,010 SNCs; 99.8%), and only 64 SNCs are unique to modern humans (Kanton et al. 2019). This remarkable conservation on the human lineage of human-specific SNCs associated with human brain development sows the seed of interest for in-depth exploration of coding gene expression of which may be affected by genetic regulatory loci harboring human-specific SNCs.
Recently, a catalogue of 59,732 genomic loci harboring evolutionary distinct candidate HSRS has been assembled which facilitated the systematic analyses of HSRS that were either inherited from extinct common ancestors (ECAs) or created de novo in human genomes (Glinsky 2020a). It has been concluded that manifestations of human-specific phenotypes are controlled by the unique-to-human mosaic of human-specific mutations and highly conserved genomic regulatory sequences inherited from extinct common ancestors (ECAs), which is supplemented with 12,486 HSRS created de novo in human genomes after divergence from ECAs (Glinsky 2020a). Notably, human-specific gene expression signatures (GES) of brain development and pluripotency phenotypes appear to manifest significant patterns of associations with HSRS assembled during evolution into human-specific regulatory pathways designed to govern various transcriptional networks in human cells. Targeted interrogations of 4433 genes encoding human virus–interacting proteins (hVIPs) revealed that 95.9% of hVIPs represent components of human-specific regulatory networks (GRNs) operating in markedly distinct types of human cells from preimplantation embryos to adult dorsolateral prefrontal cortex, dorsolateral prefrontal cortex (DLPFC) (Glinsky 2020a). It has been proposed that hVIP-encoding genes may represent a principal genomic target during evolution of human-specific GRNs, which contribute to fitness of modern humans and affect a functionally diverse spectrum of biological and cellular processes controlled by VIP-containing liquid-liquid phase-separated condensates.
Neuro-regulatory hsSNCs were discovered using the unique methodological approach combining a panel of advanced single-cell analytical techniques applied to cerebral organoid models of human and non-human primates (Kanton et al. 2019). Hence, neuro-regulatory hsSNCs represent a structurally and functionally related family of genomic regulatory sequences which are markedly distinct from previously reported and analyzed HSRS. Therefore, it was of interest to determine whether inferred regulatory actions of hsSNCs and other HSRS would likely to affect different or common sets of downstream target genes. Furthermore, of considerable importance would be to find out whether expression and functions of genes associated with hsSNCs and/or other HSRS are known to contribute to distinct and/or common physiological functions and pathological phenotypes of modern humans, in particular, aging as well as common and rare human disorders.
In this contribution, the GREAT algorithm (McLean et al. 2010, 2011) was utilized to identify 8405 hsSNC-linked genes associated with 35,074 neuro-regulatory human-specific SNCs located in DA chromatin regions during brain development. Comprehensive gene set enrichment analyses (GSEA) of these genes revealed the large scale of significant associations with physiological processes and pathological conditions of Homo sapiens, including more than 1000 anatomically distinct regions of the adult human brain, many human tissues and cell types, more than 200 common human disorders, and more than 1000 rare diseases. It has been concluded that hsSNC-linked genes appear contributing to development and functions of the adult human brain and other components of the central nervous system; they were defined as genetic markers of many tissues across human body and were implicated in the extensive range of human physiological and pathological conditions, thus supporting the hypothesis that phenotype-altering effects of neuro-regulatory hsSNCs are not restricted to the early stages of human brain development. Differential GSEA implemented on hsSNC-linked loci and associated genes identify 7990 genes linked to evolutionary distinct classes of human-specific regulatory sequences (HSRS). Notably, the expression of a majority of this common set of genes (5389 genes; 67%) is regulated by stem cell–associated retroviral sequences (SCARS). Collectively, observations reported in this contribution indicate that structurally, functionally, and evolutionary diverse classes of HSRS, neuro-regulatory hsSNCs, and associated elite set of 7990 genes affect wide spectra of traits defining both physiology and pathology of modern humans by asserting human-specific regulatory impacts on thousands essential mammalian phenotypes.
Results
Identification and characterization of putative genetic regulatory targets associated with human-specific SNCs in DA chromatin regions during brain development
To identify and characterize human genes associated with 35,074 human-specific single nucleotide changes (SNCs) in differentially accessible (DA) chromatin regions defined by ATAC-seq during human and chimpanzee neurogenesis in cerebral organoids (Kanton et al. 2019), the GREAT algorithm (McLean et al. 2010,2011) have been employed. These analyses identified 8405 genes with putative regulatory connections to human-specific SNCs (Fig. 1) and revealed a large scale of highly significant associations with a multitude of biological processes, molecular functions, genetic and metabolic pathways, cellular compartments, and gene expression perturbations (Supplemental Table Set S1).
Fig. 1.

GREAT analysis identifies 8405 human genes associated with 35,074 neuro-regulatory human-specific single nucleotide changes (hsSNCs) identified in differentially accessible (DA) chromatin regions during human and chimpanzee brain development in cerebral organoids. a Patterns of genomic associations between neuro-regulatory hsSNCs and putative target genes defined at different single nearest gene maximum extensions. GREAT algorithm version 4.0.4. b A total of 1064 of all 35,074 SNCs (3%) are not associated with any genes in the human genome, while a total of 34,010 (97%) human-specific SNCs in DA regions appear associated with 8405 human genes. GREAT algorithm version 3.0.0
To ascertain patterns of genomic associations between neuro-regulatory human-specific SNCs and putative target genes, the GREAT analyses were performed at different proximity placement distances defined by the single nearest gene maximum extension ranging from 10 kb to 1 Mb (Fig. 1). It has been observed that from 92% of all hsSNC-linked genes are located within 200 kb distances from their putative regulatory loci (Fig. 1a). Since the size of more than 99% of topologically associating domains (TADs) in human genomes is 200 kb or more (Dixon et al. 2012), these findings indicate that a marked majority of neuro-regulatory hsSNCs and their putative target genes would be located in human genomes within the boundaries of the same TAD.
Using the GREAT algorithm, particularly large numbers of significant associations were discovered during the analyses of the following two databases:
The Human Phenotype Ontology containing over 13,000 terms describing clinical phenotypic abnormalities that have been observed in human diseases, including hereditary disorders (326 significant records with binominal FDR Q value < 0.05)
The MGI Expression Detected ontology referencing genes expressed in specific anatomical structures at specific developmental stages (Theiler stages) in the mouse (370 significant records with binominal FDR Q value < 0.05)
These observations support the hypothesis that biological functions of genes under the putative regulatory control of human-specific SNCs in DA chromatin regions during brain development are not limited to the contribution to the early stages of neuro- and corticogenesis. Collectively, findings reported in Supplemental Table Set S1 argue that genes associated with neuro-regulatory human-specific SNCs may represent a genomic dominion of putative regulatory dependency from HSRS that is likely to play an important role in a broad spectrum of physiological processes and pathological conditions of modern humans.
Identification of hsSNC-linked genes distinguishing thousands of anatomically distinct areas of the adult human brain, various regions of the central nervous system, and many different cell types and tissues in the human body
To validate and extend these observations, next, the comprehensive gene set enrichment analyses were performed employing the web-based Enrichr API bioinformatics platform (Chen et al. 2013; Kuleshov et al. 2016), which interrogated nearly 200,000 gene sets from more than 100 gene set libraries. The results of these analyses are summarized in Table 1 and reported in details in Supplemental Table Set S2. Genes that were placed during evolution under the regulatory control of ~ 35,000 human-specific SNCs demonstrate a comprehensive scale of significant associations with anatomically distinct regions of human body, a broad spectrum of cell and tissue types, a multitude of physiological processes, and a numerous pathological conditions of H. sapiens.
Table 1.
Associations with human physiological processes and pathological conditions of 8405 genes linked with 35,074 human-specific single nucleotide changes (SNC) within differentially accessible (DA) chromatin regions identified during human and chimpanzee brain development in cerebral organoids
| Database | Number of significant records* |
|---|---|
| ARCHS4 Human Tissues | 39 |
| GO Biological Process 2018 | 392 |
| GO Molecular Function 2018 | 89 |
| GO Cellular Component 2018 | 33 |
| KEGG 2019 Human | 129 |
| KEGG 2019 Mouse | 106 |
| MGI Mammalian Phenotype Level 4 2019 | 407 |
| MGI Mammalian Phenotype 2017 | 749 |
| Human Phenotype Ontology | 298 |
| GWAS Catalog 2019 | 241 |
| Rare Diseases AutoRIF Gene Lists | 1116 |
| Rare Diseases GeneRIF Gene Lists | 473 |
| Rare Diseases GeneRIF ARCHS4 Predictions | 603 |
| Rare Diseases AutoRIF ARCHS4 Predictions | 641 |
| Aging Perturbations from GEO (upregulated genes) | 34 |
| Aging Perturbations from GEO (downregulated genes) | 67 |
| Human Brain Regions: Allen Brain Atlas (upregulated genes) | 1218 |
| Human Brain Regions: Allen Brain Atlas (downregulated genes) | 1102 |
| Disease Perturbations from GEO (downregulated genes) | 240 |
| Disease Perturbations from GEO (upregulated genes) | 204 |
| Human Database of Genotypes and Phenotype (dbGaP) | 136 |
| DisGeNET database | 1313 |
| UK Biobank GWAS v1 | 357 |
GEO gene expression omnibus, GO Gene Ontology, GWAS genome-wide association studies, ARCHS4 all RNA-seq and ChIP-seq sample and signature search, KEGG Kyoto Encyclopedia of Genes and Genomes, MGI mouse genome informatics.
*Defined at adjusted p value < 0.05
Of particular interest is the apparent significant enrichment of human-specific SNC-associated genes among both upregulated and downregulated genes, expression of which discriminates thousands of anatomically distinct areas of the adult human brain defined in the Allen Brain Atlas. Detailed results of these analyses are depicted in the Supplemental Figure S1 and reported in Supplemental Table Set S2. Notably, genes expressed in various thalamus regions appear frequently among the top-scored anatomical areas of the human brain (Supplemental Figure S1; Supplemental Table Set S2). These findings were further corroborated by the identification of hsSNC-linked genes among genetic markers of 26 human brain regions retrieved from the Allen Human Brain Atlas database (Fig. 2). Anatomical structures implicated in consciousness and higher cognitive functions (cerebral cortex; temporal, frontal, parietal, and occipital lobes; limbic system); information integration, communication, connectivity, and processing functions (insula; claustrum; corpus callosum); and thalamic and hypothalamic structures (including thalamic and hypothalamic nuclei; subiculum; dentate gyrus) were considered among human brain regions interrogated in these analyses (Fig. 2). Notably, a significant majority of hsSNC-linked genes (6640 of 8405 genes; 79%) represents genetic markers of 26 human brain regions examined in this study (Fig. 2).
Fig. 2.


A dominant majority (6640 of 8405 genes; 79%) of genes linked to 35,074 human-specific single nucleotide changes (hsSNCs) in chromatin’s differentially accessible (DA) regions during human and chimpanzee brain development in cerebral organoids represents genetic markers of 26 human brain regions. a Number of brain regions’ marker genes linked to 35,074 neuro-regulatory hsSNCs in specified human brain regions (the normalized values calculated per 1000 region-specific marker genes are shown). Genes linked to hsSNCs were identified among genes significantly upregulated in specified human brain regions using the Allen Brain Atlas database (records manifesting increased expression at 1.5-fold cutoff were identified and selected for analyses). b Number (percent) of genes differentially expressed (DE) in human versus chimpanzee adult brains among genes linked to neuro-regulatory human-specific SNCs. Genes linked to hsSNCs were identified among genes differentially expressed in eight regions of human versus chimpanzee adult brains (Xu et al., 2018). c The relative ranking of the 26 brain regions based on the numbers of hsSNC-linked marker genes identified in each region of the human brain (normalized values per 1000 marker genes are reported)
In agreement with the hypothesis that neuro-regulatory hsSNCs may exert the human-specific regulatory effects on target genes, a notable fraction of hsSNC-linked genes (3212 genes; 38%) manifests significant expression changes in human versus chimpanzee adult brains (Fig. 2b). A dominant majority of hsSNC-linked genes manifesting differential expression in human versus chimpanzee adult brains represents genetic markers of human brain regions (2780 of 3212 genes; 87%). The fraction of hsSNC-linked genes differentially expressed in human versus chimpanzee brains is significantly higher among human brain regions’ marker genes compared with non-markers (Fig. 2b; p = 1535E−42; 2-tailed Fisher’s exact test). These observations support the hypothesis that genetic loci harboring human-specific neuro-regulatory SNCs not only contribute to the early development of human brain but also may exert regulatory effects on structural and functional features of the adult human brain, thus likely affecting both the development and functions of the central nervous system in modern humans.
Consistent with this idea, the examination of the enrichment patterns of human-specific SNC-associated genes in the ARCHS4 Human Tissues gene expression database revealed that top 10 most significantly enriched records overlapping a majority of region-specific marker genes constitute various anatomically distinct regions of the central nervous system (Supplemental Figure 1; Supplemental Table Set S2). However, the results of gene set enrichment analyses convincingly demonstrate that inferred regulatory effects of genetic loci harboring human-specific SNCs are not restricted only to the various regions of the central nervous system; they appear to affect gene expression profiles of many different cell types and tissues in the human body (Table 1; Supplemental Table Set S2).
Identification and characterization of hsSNC-linked genes manifesting altered expression during aging of humans, rats, and mice
Genes altered expression of which is implicated in the aging of various tissues and organs of humans, rats, and mice are significantly enriched among 8405 genes associated with human-specific regulatory SNCs (Supplemental Figure S2; Supplemental Table Set S2).
Aging of the hippocampus was implicated most frequently among genes manifesting increased expression with age, while among genes exhibiting aging-associated decreased expression, the hippocampus and frontal cortex were identified repeatedly (Supplemental Figure S2). Overall, twice as many significant association records were observed among aging-associated downregulated genes compared with upregulated genes (Table 1). While these observations clearly indicate that altered expression of hsSNC-linked genes may contribute to both up- and downregulation of genes in many anatomical regions of aging brains, they also suggest that diminished expression of hsSNC-linked genes in various brain regions may represent one of the important molecular determinants of human aging. Interestingly, the results of follow-up analyses have shown that the phenomenon of decreased expression of hsSNC-linked genes in human brain regions during aging appears connected with activated expression of stem cell–associated retroviral sequences (SCARS). These conclusions are supported by findings that SCARS exert the predominantly inhibitory effect on the expression of genes associated with human-specific neuro-regulatory SNCs, and SCARS-regulated genes represent a vast majority of hsSNC-linked genes manifesting significant associations with brain aging. Collectively, these observations indicate that gene changes in expression of which were associated with aging in mammals, in particular, hippocampal and frontal cortex aging, represent important elements of a genomic dominion that was placed under regulatory control of genetic loci harboring human-specific neuro-regulatory SNCs.
Identification of hsSNC-linked genes implicated in development and manifestations of hundreds physiological and pathological phenotypes and autosomal inheritance in modern humans
The Interrogations of the Human Phenotype Ontology database (298 significantly enriched records identified), the Genome-Wide Association Study (GWAS) Catalogue (241 significantly enriched records identified), and the database of Human Genotypes and Phenotypes (136 significantly enriched records identified) revealed several hundred physiological and pathological phenotypes and thousands of genes manifesting significant enrichment patterns defined at the adjusted p value < 0.05 (Supplemental Figure S3; Table 1; Supplemental Table Set S2). Interestingly, 645 and 849 genes implicated in the autosomal dominant (HP:0000006) and recessive (HP:0000007) inheritance were identified among genes associated with human-specific regulatory SNCs (Supplemental Figure S3; Supplemental Table Set S2). Notable pathological conditions among top-scored records identified in the database of Human Genotypes and Phenotypes are stroke, myocardial infarction, coronary artery disease, and heart failure (Supplemental Figure S3).
A total of 241 significantly enriched records (Table 1) were documented by gene set enrichment analyses of the GWAS catalogue (2019), among which a highly diverse spectrum of pathological conditions linked to genes associated with human-specific regulatory SNCs was identified, including obesity, type 2 diabetes, amyotrophic lateral sclerosis, autism spectrum disorders, attention deficit hyperactivity disorder, bipolar disorder, major depressive disorder, schizophrenia, Alzheimer’s disease, malignant melanoma, diverticular disease, asthma, coronary artery disease, glaucoma, as well as breast, prostate, and colorectal cancers (Supplemental Figure S3; Supplemental Table Set S2). These observations indicate that thousands of genes putatively associated with genetic regulatory loci harboring human-specific SNCs affect the risk of developing numerous pathological conditions in modern humans.
Identification of hsSNC-linked genes manifesting altered expression in several hundred common human disorders
Gene set enrichment analysis–guided interrogation of the Gene Expression Omnibus (GEO) database revealed the highly diverse spectrum of human diseases with the etiologic origins in multiple organs and tissues and highly heterogeneous pathophysiological trajectories of their pathogenesis (Supplemental Figure S4; Supplemental Table Set S2). Overlapping gene sets between disease-associated genes and human-specific SNC-linked genes compose of hundreds of genes that were either upregulated (204 significant disease records) or downregulated (240 significant disease records) in specific pathological conditions, including schizophrenia, bipolar disorder, various types of malignant tumors, Crohn’s disease, ulcerative colitis, Down syndrome, Alzheimer’s disease, spinal muscular atrophy, multiple sclerosis, autism spectrum disorders, type 2 diabetes mellitus, morbid obesity, and cardiomyopathy (Supplemental Figure S4; Supplemental Table Set S2). These observations demonstrate that thousands of genes manifesting altered expression in a myriad of human diseases appear associated with genetic regulatory loci harboring human-specific SNCs.
Viral infections caused by persisting and novel viruses and manifesting sporadic, endemic, epidemic, or pandemic patterns of disease in human populations are regarded as a major contemporary public health threat. Clinical dynamics and outcomes of virus-host encounters are determined to a significant extend by interactions of viral and host proteins. Importantly, genes encoding virus-interacting proteins (VIPs) represent one of important regulatory targets of different families of HSRS, which collectively may exert regulatory effects on more than 95% of all known VIP-encoding genes in human genome (Glinsky 2020a). These findings suggest that human-specific genomic regulatory networks (GRNs) play important roles in governing virus-host interactions and affecting the outcomes of viral infections. It was of interest to evaluate the validity of this concept in the context of the current COVID-19 pandemic caused by the novel coronavirus SARS-CoV-2.
A recent proteomics study of the SARS-CoV-2 interactome in human cells identified 332 high-confidence human protein targets of the 27 SARS-CoV-2 viral proteins (Gordon et al. 2020). This knowledge has been exploited for identification of medicinal substances manifesting genomic profiles of candidate pandemic mitigation agents (Glinsky 2020b). In agreement with the concept of regulatory effects of HSRS on VIP-encoding genes, 229 of 332 genes (69%) encoding SARS-CoV-2 prey proteins in human cells represent regulatory targets of HSRS (Supplemental Table Set 6). Similarly, nearly half of human genes (162 of 332 genes; 49%) encoding prey proteins for SARS-CoV-2 coronavirus are genes associated with neuro-regulatory hsSNCs. In total, a significant majority of genes encoding protein targets of novel SARS-CoV-2 coronavirus in human cells (250 of 332; 75%) appears associated with HSRS and/or neuro-regulatory hsSNCs (Supplemental Table Set 6). Collectively, observations reported in present study appear consistent with the hypothesis that genes linked to human-specific neuro-regulatory SNCs represent a network of essential genetic loci implicated in a broad spectrum of physiological and pathological traits of modern humans, including genes implicated in pathogenesis of viral infections.
Identification of hsSNC-linked genes implicated in more than 1000 records classified as human rare diseases
Present analyses demonstrate that thousands of genes associated with human-specific regulatory SNCs have been previously identified as genetic elements affecting the likelihood of development a multitude of common human disorders. Similarly, thousands of genes with altered expression during development and manifestation of multiple common human disorders appear linked to genetic regulatory loci harboring human-specific SNCs. Interestingly, interrogations of the Enrichr’s libraries of genes associated with modern humans’ rare diseases identified 473, 603, 641, and 1116 significantly enriched records of various rare disorders employing the Rare Diseases GeneRIF gene lists library, the Rare Diseases GeneRIF ARCHS4 predictions library, the Rare Diseases AutoRIF ARCHS4 predictions library, and the Rare Diseases AutoRIF Gene lists library, respectively (Supplemental Figure S5; Supplemental Table Set S2). Taken together, these observations demonstrate that thousands of genes associated with hundreds of human rare disorders appear linked with human-specific regulatory SNCs.
Gene ontology analyses of putative regulatory targets of genetic loci harboring human-specific SNCs
Gene Ontology (GO) analyses identified a constellation of biological processes (GO Biological Process 308 significant records) supplemented with a multitude of molecular functions (GO Molecular Function 81 significant records) that appear under the regulatory control of human-specific SNCs (Supplemental Figure S6; Supplemental Table Set 2). Consistently, both databases identified frequently the components of transcriptional regulation and protein kinase activities among most significant records. Other significantly enriched records of interest are regulation of apoptosis, cell proliferation, migration, and various binding properties (cadherin binding; sequence-specific DNA binding; protein-kinase binding; amyloid-beta binding; actin binding; tubulin binding; microtubule binding; PDZ domain binding) which are often supplemented by references to the corresponding activity among the enriched records, for example, enriched records of both binding and activity of protein kinases.
Interrogation of GO Cellular Component database identified 29 significantly enriched records, among which nuclear chromatin as well as various cytoskeleton and membrane components appear noteworthy (Supplemental Figure S6). Both GO Biological Process and GO Cellular Component databases identified significantly enriched records associated with the central nervous system development and functions such as axonogenesis and axon guidance; generation of neurons, neuron differentiation, and neuron projection morphogenesis; cellular components of dendrites and dendrite’s membrane; and ionotropic glutamate receptor complex. In several instances, biologically highly consistent enrichment records have been identified in different GO databases: cadherin binding (GO Molecular Function) and catenin complex (GO Cellular Component); actin binding (GO Molecular Function) and actin cytoskeleton, cortical actin cytoskeleton, and actin-based cell projections (GO Cellular Component); microtubule motor activity, tubulin binding, and microtubule binding (GO Molecular Function); and microtubule organizing center and microtubule cytoskeleton (GO Cellular Component).
Analyses of human and mouse databases of the Kyoto Encyclopedia of Genes and Genomes (KEGG; Supplemental Figure S7) identified more than 100 significantly enriched records in each database (KEGG 2019 Human 129 significant records; KEGG 2019 Mouse 106 significant records). Genes associated with human-specific regulatory SNCs were implicated in a diverse spectrum of signaling pathways ranging from pathways regulating the pluripotency of stem cells to cell type-specific morphogenesis and differentiation pathways, for example, melanogenesis and adrenergic signaling in cardiomyocytes (Supplemental Figure S7). Genes under putative regulatory control of human-specific SNCs include hundreds of genes contributing to specific functions of specialized differentiated cells (gastric acid secretion; insulin secretion; aldosterone synthesis and secretion), multiple receptor/ligand-specific signaling pathways, as well as genetic constituents of pathways commonly deregulated in cancer and linked to the organ-specific malignancies, for example, breast, colorectal, and small cell lung cancers (Supplemental Figure S7). Other notable entries among most significantly enriched records include pathways of the axon guidance; dopaminergic, glutamatergic, and cholinergic synapses; neuroactive receptor-ligand interactions; and AGE-RAGE signaling pathway in diabetic complications (Supplemental Figure S7; Supplemental Table Set 2).
Identification of 2273 genes associated with human-specific SNCs and implicated in premature death and embryonic, prenatal, perinatal, neonatal, and postnatal lethality phenotypes
Interrogation of MGI Mammalian Phenotype databases revealed several hundred mammalian phenotypes affected by thousands of genes associated with genomic regulatory regions harboring human-specific SNCs: the MGI Mammalian Phenotype (2017) database identified 749 significant enrichment records, while the MGI Mammalian Phenotype Level 4 (2019) database identified 407 significant enrichment records (Table 2; Supplemental Figure S8; Supplemental Table Set S2). Notably, among the records of mammalian phenotypes identified by the gene set enrichment analyses of neuro-regulatory hsSNC-linked genes, there are 2273 genes mutations of which result in phenotypes of premature death, embryonic lethality, as well as prenatal, perinatal, neonatal, and postnatal lethality of both complete and incomplete penetrance. A significant fraction of these 2273 genes, which collectively could be defined based on patterns of phenotypes caused by their mutations as an offspring survival genomic dominion, was implicated in the autosomal dominant (389 genes) and recessive (426 genes) inheritance in modern humans. Based on these observations, it has been concluded that thousands of genes within the genomic dominions of putative regulatory dependencies from human-specific SNCs represent the essential genetic elements of the mammalian offspring survival phenotypes.
Table 2.
Gene set enrichment analyses of the MGI Mammalian Phenotype Level 4 (2019) database identify mammalian phenotypes manifesting significant associations with neuro-regulatory human-specific SNC-linked genes. Top 40 of the 407 significant records are reported
| Mammalian phenotype | Overlap | p value | Adjusted p value |
|---|---|---|---|
| MP:0011087_neonatal_lethality,_complete_penetrance | 315/517 | 1.55E-18 | 8.15E-15 |
| MP:0001262_decreased_body_weight | 773/1471 | 2.04E-17 | 5.36E-14 |
| MP:0001405_impaired_coordination | 247/405 | 6.90E-15 | 1.21E-11 |
| MP:0011086_postnatal_lethality,_incomplete_penetrance | 362/643 | 9.65E-14 | 1.27E-10 |
| MP:0001463_abnormal_spatial_learning | 120/172 | 1.46E-13 | 1.54E-10 |
| MP:0002169_no_abnormal_phenotype_detected | 958/1944 | 6.87E-12 | 6.02E-09 |
| MP:0004811_abnormal_neuron_physiology | 78/107 | 8.69E-11 | 6.53E-08 |
| MP:0002206_abnormal_CNS_synaptic_transmission | 75/103 | 2.18E-10 | 1.27E-07 |
| MP:0000267_abnormal_heart_development | 111/168 | 2.43E-10 | 1.28E-07 |
| MP:0011085_postnatal_lethality,_complete_penetrance | 246/432 | 2.04E-10 | 1.34E-07 |
| MP:0011110_preweaning_lethality,_incomplete_penetrance | 360/669 | 2.90E-10 | 1.39E-07 |
| MP:0011109_lethality_throughout_fetal_growth_and_development,_incomplete_penetrance | 122/191 | 8.21E-10 | 3.60E-07 |
| MP:0001899_absent_long_term_depression | 27/28 | 1.11E-09 | 4.50E-07 |
| MP:0001732_postnatal_growth_retardation | 360/677 | 1.85E-09 | 6.48E-07 |
| MP:0001698_decreased_embryo_size | 293/537 | 2.09E-09 | 6.88E-07 |
| MP:0002152_abnormal_brain_morphology | 119/187 | 1.84E-09 | 6.92E-07 |
| MP:0011090_perinatal_lethality,_incomplete_penetrance | 154/256 | 3.23E-09 | 9.44E-07 |
| MP:0002741_small_olfactory_bulb | 35/40 | 3.07E-09 | 9.51E-07 |
| MP:0001469_abnormal_contextual_conditioning_behavior | 48/61 | 5.45E-09 | 1.43E-06 |
| MP:0001473_reduced_long_term_potentiation | 84/124 | 6.05E-09 | 1.45E-06 |
| MP:0000788_abnormal_cerebral_cortex_morphology | 104/161 | 5.79E-09 | 1.45E-06 |
| MP:0011088_neonatal_lethality,_incomplete_penetrance | 172/293 | 5.33E-09 | 1.48E-06 |
| MP:0001954_respiratory_distress | 121/194 | 7.90E-09 | 1.81E-06 |
| MP:0001575_cyanosis | 129/210 | 1.00E-08 | 2.19E-06 |
| MP:0001953_respiratory_failure | 103/161 | 1.47E-08 | 3.09E-06 |
| MP:0002906_increased_susceptibility_to_pharmacologically_induced_seizures | 70/101 | 2.60E-08 | 5.25E-06 |
| MP:0002910_abnormal_excitatory_postsynaptic_currents | 59/83 | 7.68E-08 | 1.50E-05 |
| MP:0002083_premature_death | 499/997 | 9.80E-08 | 1.84E-05 |
| MP:0002066_abnormal_motor_capabilities/coordination/movement | 102/164 | 1.41E-07 | 2.55E-05 |
| MP:0006254_thin_cerebral_cortex | 53/74 | 2.36E-07 | 4.14E-05 |
| MP:0011098_embryonic_lethality_during_organogenesis,_complete_penetrance | 339/656 | 2.60E-07 | 4.28E-05 |
| MP:0000807_abnormal_hippocampus_morphology | 63/92 | 2.56E-07 | 4.34E-05 |
| MP:0006009_abnormal_neuronal_migration | 59/85 | 2.94E-07 | 4.69E-05 |
| MP:0011108_embryonic_lethality_during_organogenesis,_incomplete_penetrance | 143/247 | 3.20E-07 | 4.95E-05 |
| MP:0000031_abnormal_cochlea_morphology | 44/59 | 3.84E-07 | 5.77E-05 |
| MP:0000852_small_cerebellum | 58/84 | 4.91E-07 | 7.17E-05 |
| MP:0002063_abnormal_learning/memory/conditioning | 42/56 | 5.43E-07 | 7.52E-05 |
| MP:0003633_abnormal_nervous_system_physiology | 79/123 | 5.36E-07 | 7.62E-05 |
| MP:0010025_decreased_total_body_fat_amount | 263/498 | 5.94E-07 | 8.01E-05 |
| MP:0009937_abnormal_neuron_differentiation | 75/116 | 7.01E-07 | 9.22E-05 |
Gene set enrichment analyses were performed employing the Enrichr bioinformatics platform (see “Methods”). All analyzed records are reported in the Supplemental Table Set S2. Overlap refers to the number of hsSNC-linked genes and mammalian phenotype–associated genes in corresponding categories. Italicized records highlight the classification categories defined as offspring survival phenotypes
Identification of the experimentally tractable models for molecular definitions of regulatory effects of human-specific SNCs on expression of genes associated with thousands of mammalian phenotypes and human diseases
To identify all genes linked with human-specific neuro-regulatory SNCs that are associated with defined mammalian phenotypes and human diseases with one or more mouse models, the analyses have been carried out utilizing the Mouse Genome Informatics (MGI) database (http://www.informatics.jax.org/). These analyses identified 125,938 Mammalian Phenotype Ontology records and 1807 Human Disease Ontology records associated with 5730 and 1162 human-specific regulatory SNC-linked genes, respectively (Supplemental Table Sets S4 and S5). Significantly, genes linked with human-specific regulatory SNCs have been associated with a majority (61%) of all human diseases with one or more mouse models (967 of 1584 human disease ontology terms; Supplemental Table Set S4). Similarly, human-specific SNC-linked genes have been associated with 71% of all Mammalian Phenotype Ontology terms (9190 of 12,936 records; Supplemental Table Set S5). These observations identify readily available mouse models for experimental interrogations of regulatory effects of human-specific SNCs and other types of HSRS on genes causally affecting thousands of defined mammalian phenotypes and hundreds of common and rare human disorders.
Structurally, functionally, and evolutionary distinct classes of HSRS share the relatively restricted elite set of common genetic targets
It has been suggested that unified activities of thousands candidate HSRS comprising a coherent compendium of genomic regulatory elements markedly distinct in their structure, function, and evolutionary origin may have contributed to development and manifestation of human-specific phenotypic traits (Glinsky 2020a). It was of interest to determine whether genes previously linked to other classes of HSRS, which were identified without considerations of neuro-regulatory human-specific SNCs, overlap with genes associated in this contribution with genomic regulatory loci harboring human-specific SNCs. To this end, genes associated with different classes of HSRS were identified using the GREAT algorithm, subjected to the GSEA, and compared with the set of 8405 genes linked with neuro-regulatory hsSNCs. Notably, all classes of HSRS appear to share common sub-sets of putative genetic regulatory targets with neuro-regulatory hsSNCs (Table 3; Fig. 3; Supplemental Figure S9; Supplemental Table Set S3). GSEA of genes linked with different classes of HSRS revealed apparently similar patterns of associations with human phenotypic traits (Supplemental Notes 1 and 2), which recapitulate, in part, the patterns of phenotypic associations of neuro-regulatory hsSNC-linked genes.
Table 3.
Structurally, functionally, and evolutionary distinct families of human-specific regulatory sequences (HSRS) manifest common enrichment patterns of associations with 8405 hsSNC-linked genes
| Classification category/reference database | Number of records (hg19) | Associated genes | Common with all 8405 hsSNC-linked genes | Percent, hsSNCs genes | Percent, HSRS genes |
|---|---|---|---|---|---|
| Fixed human-specific insertions. | 11,878 | 7979 | 5290 | 62.94 | 66.30 |
| Human-specific TE loci expressed in human dorsolateral prefrontal cortex | 4637 | 4051 | 2719 | 32.35 | 67.12 |
| Set of duplicated regions in GRCh38 space | 7599 | 6618 | 3654 | 43.47 | 55.21 |
| Fixed human-specific deletions | 5883 | 5489 | 3835 | 45.63 | 69.87 |
| Human-specific STR expansions | 4875 | 4844 | 3354 | 39.90 | 69.24 |
| hsTFBS | 3803 | 1087 | 750 | 8.92 | 69.00 |
| ace-DHS | 3538 | 3445 | 2553 | 30.37 | 74.11 |
| FHSRR | 4249 | 2810 | 1899 | 22.59 | 67.58 |
| Human-specific STR contractions | 1279 | 973 | 554 | 6.59 | 56.94 |
| hESC_FHSRR_DHS | 1932 | 1458 | 1096 | 13.04 | 75.17 |
| DHS_FHSRR (non-hESC) | 2118 | 552 | 307 | 3.65 | 55.62 |
| Human accelerated regions (HARs) | 2745 | 2281 | 1890 | 22.49 | 82.86 |
| haDHS | 524 | 747 | 659 | 7.84 | 88.22 |
| Human-biased CNCC enhances | 1000 | 1439 | 1110 | 13.21 | 77.14 |
| Chimp-biased CNCC enhances | 1000 | 1445 | 1106 | 13.16 | 76.54 |
| H3K4me3 peaks with human-specific enrichment in prefrontal neurons | 410 | 578 | 308 | 3.66 | 53.29 |
| Human-specific hESC functional enhancers | 1619 | 1214 | 816 | 9.71 | 67.22 |
| All HSRS | 59,089 | 13,824 | 7406 | 88.11 | 53.57 |
Fig. 3.

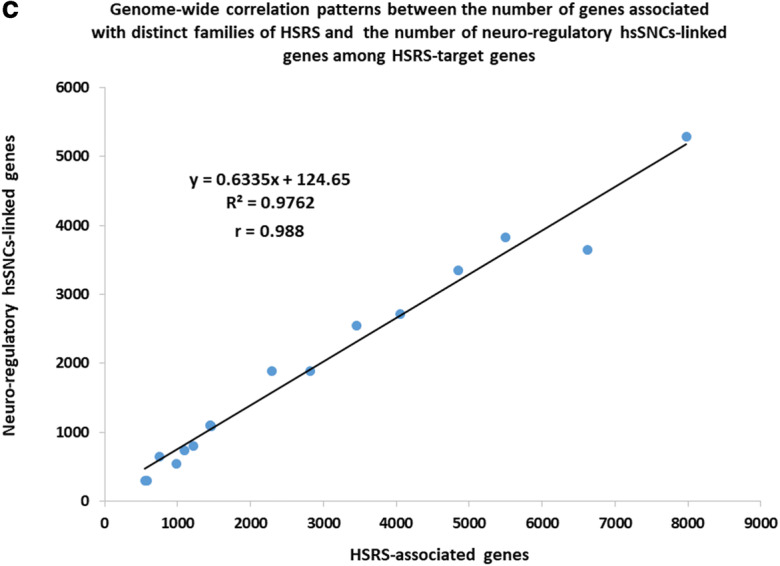
Distinct families of regulatory DNA sequences comprising a compendium of 59,089 human-specific regulatory sequences (HSRS) manifest common enrichment patterns of associations with sub-sets of 8405 neuro-regulatory hsSNC-linked genes. a Number of genes identified by the GREAT algorithm as putative regulatory targets of distinct families of HSRS. b Number of hsSNC-linked genes among genes comprising putative regulatory targets of distinct families of HSRS. c Genome-wide correlation patterns between the number of genes comprising the putative regulatory targets of distinct families of HSRS and the number of hsSNC-linked genes among the HSRS-target genes
To determine whether the patterns of significant phenotypic associations observed for genes linked with HSRS are specific and not related to the size effects of relatively large gene sets subjected to the GSEA, 42,847 human genes not linked by the GREAT algorithm with HSRS were randomly split into 21 control gene sets of various sizes ranging from 2847 to 6847 genes and subjected to the GSEA (Supplemental Notes 1 and 2). Importantly, no significant phenotypic associations were observed for 21 control gene sets, consistent with the conclusion that significant phenotypic associations documented for genes linked with HSRS and neuro-regulatory hsSNCs are not likely due to non-specific size effects captured by the GSEA.
In agreement with this conclusion, it was observed that the common gene set of putative regulatory targets shared by HSRS and neuro-regulatory hsSNCs composes of 7406 coding genes (88% of all human-specific SNC-associated genes), indicating that structurally and functionally diverse HSRS, the evolutionary origin of which has been driven by mechanistically distinct processes, appear to favor the genomic regulatory alignment with the relatively restricted elite set of genetic targets (Fig. 3; Table 3; Supplemental Figure S9; Supplemental Table Set S3). The estimated timeline of the evolutionary origin of 59,089 HSRS is likely to encompass many thousands, perhaps, hundred thousand years. Therefore, it seems likely to have biologically meaningful implications that the patterns of their genomic placement appear uniformly associated with sub-sets of genes comprising the putative regulatory targets of human-specific neuro-regulatory SNCs (Fig. 3; Table 3; Supplemental Table Set S3).
Previous studies have identified stem cell–associated retroviral sequences (SCARS) encoded by human endogenous retroviruses LTR7/HERVH and LTR5_Hs/HERVK as one of the significant sources of the evolutionary origin of HSRS (Glinsky 2015,2016a,b,c,2017,2018,2020a,b), including human-specific transcription factor–binding sites (TFBS) for NANOG, OCT4, and CTCF (Glinsky 2015,2016a,b,c).
Next, the common sets of genetic regulatory targets were identified for SCARS-regulated genes and genes associated in this study with human-specific regulatory SNCs (Supplemental Figure S9). It has been determined that each of the structurally distinct families of SCARS appears to share a common set of genetic regulatory targets with human-specific SNCs (Supplemental Figure S8). Overall, the expression of nearly two-thirds (5389 genes; 64%) of all genes identified as putative regulatory targets of human-specific SNCs is regulated by SCARS (Supplemental Figure S9; Supplemental Table Set S3). Consistent with the idea that structurally diverse HSRS may favor the relatively restricted elite set of genetic targets, the combined gene set of regulatory targets for HSRS, SCARS, and SNCs composes of 7990 coding genes or 95% of all genes associated in this contribution with human-specific neuro-regulatory SNCs (Supplemental Figure S9; Supplemental Table Set S3).
To gain insights into mechanisms of SCARS-mediated effects on expression of 5389 genes linked to human-specific regulatory SNCs, the numbers of genes manifesting either activated (downregulated following SCARS silencing) or inhibited (upregulated following SCARS silencing) expression have been determined. It was observed that SCARS exert the predominantly inhibitory effect on expression of genes associated with human-specific regulatory SNCs, which is exemplified by activated expression of as many as 87% of genes affected by SCARS silencing (Supplemental Figure S9; Supplemental Table Set S3). These findings indicate that when SCARS-associated networks are active during the human preimplantation embryogenesis, they exert a dominant effect on gene expression, whereas when SCARS are silenced during the postimplantation embryonic development and in the adulthood, regulatory impact of human-specific neuro-regulatory SNCs may be prevalent.
Genes linked with neuro-regulatory hsSNCs represent intrinsic genetic elements of developmentally and physiologically distinct human-specific GRNs
Since distinct families of HSRS, including neuro-regulatory hsSNCs, share common sets of genetic targets, it was of interest to determine whether hsSNC-linked genes are represented among genes previously identified as components of human-specific GRNs operating in developmentally and physiologically distinct human tissues and cells. Importantly, human-specific GRNs selected for these analyses were defined employing vastly different experimental, analytical, and computational approaches that were applied within the broad range of experimental settings (Glinsky, 2019). Specifically, the interrogated human-specific GRNs include the following datasets: (i) Great Apes’ whole-genome sequencing–guided human-specific insertions and deletions (Kronenberg et al. 2018); (ii) genome-wide analysis of retrotransposon’s transcriptome in postmortem samples of human dorsolateral prefrontal cortex (Guffanti et al. 2018); (iii) shRNA-mediated silencing of LTR7/HERVH retrovirus–derived long non-coding RNAs in human embryonic stem cell (hESC) (Wang et al. 2014); (iv) single-cell expression profiling analyses of human preimplantation embryos (Glinsky et al. 2018); (v) network of genes associated with regulatory transposable elements (TE) operating in naïve and primed hESC (Theunissen et al. 2016; Pontis et al. 2019; Barakat et al. 2018; Glinsky and Barakat 2019); (vi) pluripotency-related network of genes manifesting concordant expression changes in human fetal brain and adult neocortex (Glinsky 2017); (vii) network of genes governing human neurogenesis in vivo (Nowakowski et al. 2017); (viii) network of genes differentially expressed during human corticogenesis in vitro (van de Leemput et al. 2014). Thus, selected for these analyses, human-specific GRNs appear to function in a developmentally and physiologically diverse spectrum of human cells that are biologically and anatomically highly relevant to manifestations of human-specific phenotypes ranging from preimplantation embryos to adult dorsolateral prefrontal cortex (Table 4; Supplemental Table Set S3).
Table 4.
Enrichment within human-specific genomic regulatory networks (GRNs) of 8405 genes associated with human-specific neuro-regulatory single nucleotide changes (hsSNCs)
| Classification category | Number of genes | Genes associated with hsSNCs |
|---|---|---|
| Networks of genes associated with expression of transposable elements (TE) in human dorsolateral prefrontal cortex | ||
| Human genome | 63,677 | 8405 |
| Networks of genes associated with human DLPFC-expressed TE | 22,863 | 6547 |
| Percent | 35.9 | 77.89 |
| Enrichment** | 1 | 2.17 |
| p value* | 0 | |
| GES of the multi-lineage markers expressing (MLME) cells of human preimplantation embryo | ||
| Human genome | 63,677 | 8405 |
| GES of the MLME cells of human preimplantation embryo | 12,735 | 5218 |
| Percent | 20 | 62.08 |
| Enrichment** | 1 | 3.10 |
| p value* | 0 | |
| Regulatory networks of genes associated with human-specific structural variants*** | ||
| Human genome | 63,677 | 8405 |
| Genes associated with human-specific deletions and insertions | 10,992 | 3056 |
| Percent | 17.26 | 36.36 |
| Enrichment** | 1 | 2.11 |
| p value* | 0 | |
| Gene expression signature of the HERVH/LBP9 network in hESC | ||
| Human genome | 63,677 | 8405 |
| Genes associated with the HERVH/LBP9 pathway in hESC | 11,507 | 4073 |
| Percent | 18.07 | 48.46 |
| Enrichment** | 1 | 2.68 |
| p value* | 0 | |
| Network of genes associated with regulatory TE in naïve and primed hESC | ||
| Human genome | 63,677 | 8405 |
| Genes associated with regulatory TE in naïve and primed hESC | 6148 | 2787 |
| Percent | 9.65 | 33.16 |
| Enrichment** | 1 | 3.44 |
| p value* | 0 | |
| Network of genes differentially expressed in human fetal brain and adult neocortex | ||
| Human genome | 63,677 | 8405 |
| Human fetal brain/adult neocortex signature genes | 4764 | 2448 |
| Percent | 7.48 | 29.13 |
| Enrichment** | 1 | 3.89 |
| p value* | 0 | |
| Human neurogenesis in vivo network^ | ||
| Human genome | 63,677 | 8405 |
| Gene expression signatures of human neurogenesis in vivo | 11,911 | 5467 |
| Percent | 18.71 | 65.04 |
| Enrichment** | 1 | 3.48 |
| p value* | 0 | |
| Human corticogenesis in vitro network | ||
| Human genome | 63,677 | 8405 |
| Gene expression signatures of human corticogenesis in vitro | 12,334 | 5253 |
| Percent | 19.37 | 62.50 |
| Enrichment** | 1 | 3.23 |
| p value* | 0 | |
Human-specific genomic regulatory networks (GRNs) were defined previously (Glinsky, 2020a, see text for details) based on the following primary contributions:
Networks of genes associated with expression of transposable elements (TE) in human dorsolateral prefrontal cortex: Guffanti et al. (2018)
Gene expression signature (GES) of the multi-lineage markers expressing (MLME) cells of human preimplantation embryo: Glinsky et al. (2018)
Regulatory networks of genes associated with human-specific structural variants: Kronenberg et al. (2018)
Gene expression signature of the HERVH/LBP9 network in hESC: Wang et al. (2014)
Network of genes associated with regulatory TE in naïve and primed hESC: Theunissen et al. (2016)
Network of genes differentially expressed in human fetal brain and adult neocortex: Glinsky (2017). doi: 10.1101/022913
Human neurogenesis in vivo network: Nowakowski et al. (2017)
Human corticogenesis in vitro network: van de Leemput et al. (2014)
TE transposable genetic elements, hESC human embryonic stem cell, DLPFC dorsolateral prefrontal cortex, MLME multi lineage markers expression
*p values were estimate using the hypergeometric distribution test
**Expected values were estimated based on the number of genes in the human genome (63,677) and the number of genes in the corresponding category of human-specific regulatory networks
***This category of genes was reported in Kronenberg et al. (2018)
^This category of genes was reported in Nowakowski et al. (2017)
Importantly, in all instances, a highly significant enrichment of hsSNC-linked genes has been observed (Table 4; Supplemental Table Set S3). These observations are consistent with the hypothesis that neuro-regulatory hsSNCs and associated genes represent principal components of the exceptionally broad range of human-specific GRNs operating in the wide spectra of developmental and physiological contexts reflecting species-defining human-specific phenotypes.
SCARS-regulated genes linked with neuro-regulatory human-specific SNCs manifest dominant patterns of significant associations to physiological and pathological phenotypes of modern humans
A putative regulatory nexus of human-specific neuro-regulatory SNCs and target genes has been inferred based on their genomic proximity to each other taking into account genome-wide folding patterns of linear chromatin fibers in human cells (Fig. 1). This approach narrows the list of putative regulatory targets to 8405 genes manifesting marked enrichment patterns to thousands of physiological and pathological phenotypes of modern humans. However, this relatively large set of hsSNC-linked genes appears to represent a highly diverse biologically and structurally collection of genetic loci, which makes design and execution of follow-up functional validation experiments very challenging. Therefore, it was of interest to ascertain whether the elite list of high priority genetic targets for follow-up functional validation studies could be refined further by performing additional differential GSEA on smaller sets of genes segregated from a parent set of 8405 hsSNC-linked genes.
Present analyses identified 5389 hsSNC-linked genes, the expression of which in hESC is regulated by stem cell–associated retroviral sequences (SCARS). It was of interest to ascertain the contribution of SCARS-regulated genes to a global scale of significant associations of hsSNC-linked genes to phenotypes of modern humans. To this end, the set of 8405 hsSNC-regulated genes was segregated into a set of 5389 hsSNC-linked SCARS-regulated genes and a set of 3016 hsSNC-linked non-SCARS-regulated genes. Next, the differential GSEA were carried separately on these two sets of genes using 29 genomic databases and numbers of significantly enriched records were recorded for each interrogated database (Fig. 4). It has been observed that SCARS-regulated genes consistently scored markedly higher numbers of significantly enriched records compared with non-SCARS-regulated genes (Fig. 4). Overall, SCARS-regulated hsSNC-linked genes scored 9.8-fold greater number of significantly enriched records compared with hsSNC-linked genes that are not regulated by SCARS (10,645 versus 1089 records, respectfully; Fig. 4). Two exceptions form this pattern have been noted: the ARCHS4 Human Tissues database and the Allen Brain Atlas database of upregulated genes. In these instances, numbers of identified significantly enriched records were similar for both SCARS-regulated and non-SCARS-regulated hsSNC-linked genes (Fig. 4). Consistently, differential GSEA of multiple databases attributed all statistically significant phenotypic associations to SCARS-regulated genes (Fig. 4).
Fig. 4.
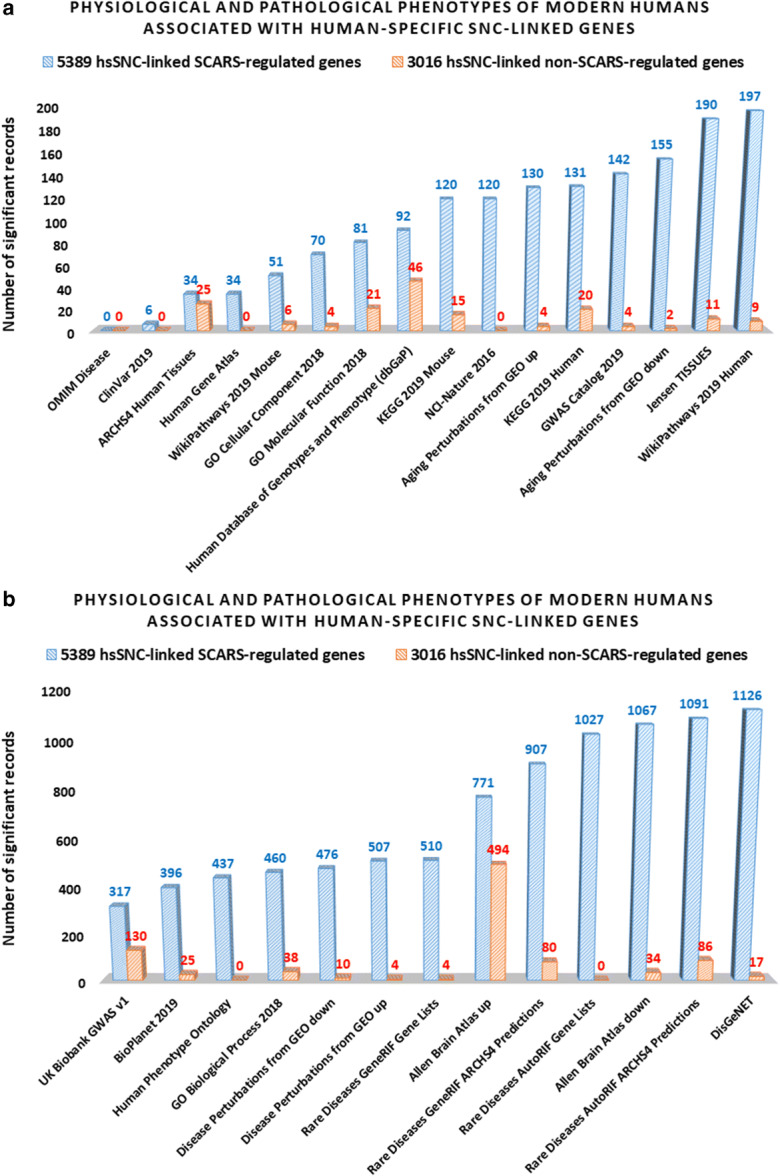
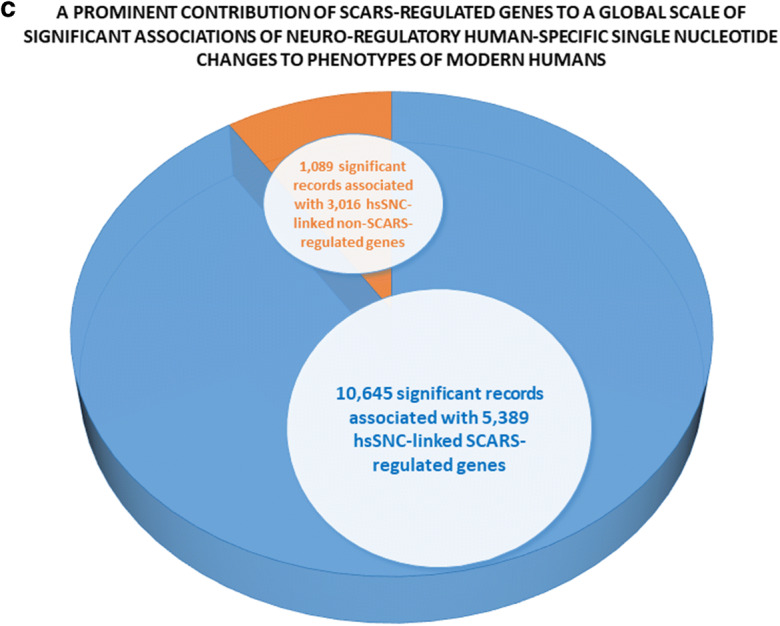
SCARS-regulated genes associated with neuro-regulatory human-specific SNCs manifest prominent patterns of significant associations with physiological and pathological phenotypes of modern humans. Among all neuro-regulatory human-specific SNC-linked genes, 5389 SCARS-regulated genes were identified and segregated from the remaining 3016 non-SCARS-regulated hsSNC-linked genes (non-SCARS-regulated genes). These two categories of hsSNC-linked genes were subjected to gene set enrichment analyses to identify statistically significant associations with morphological features and physiological and pathological phenotypes of modern humans. Results of the analyses were separated into two sets (a and b) using a threshold of 250 significant records per database and plotted for visualization. a The results with less than 250 significant records, while the results with more than 250 significant records are reported in the b. c The summary of the results reflecting prominent contributions of SCARS-regulated genes to a global scale of significant associations of neuro-regulatory human-specific SNC-linked genes to morphological features and physiological and pathological phenotypes of modern humans.
Based on results of the above analyses, it has been concluded that 5389 SCARS-regulated genes linked to fixed neuro-regulatory human-specific SNCs appear to drive the associations with a preponderance of physiological processes, morphological features, and pathological conditions of modern humans. This set of genes and associated human-specific regulatory loci should be regarded as high priority genomic targets for follow-up functional validation studies of phenotypic traits distinguishing the human lineage from non-human primates.
Contributions of 5389 SCARS-regulated genes and 3016 non-SCARS-regulated genes linked with human-specific neuro-regulatory SNCs to a global scale of significant associations with aging and diseases of modern humans
Genes implicated in aging and disease phenotypes were consistently identified among significantly enriched records associated with neuro-regulatory hsSNC-linked genes (Table 1). The above observations suggest that SCARS-regulated genes appear to make a prominent contribution to a global scale of significant associations to phenotypes of modern humans attributed to neuro-regulatory hsSNC-linked genes. It was of interest to evaluate differences between SCARS-regulated and non-SCARS-regulated genes in aging and disease-related categories (Table 5). It has been observed that differences appear particularly notable for SCARS-regulated genes altered expression of which is associated with human diseases (Table 5; 48-fold and 127-fold for down- and upregulated disease-associated genes, respectively) and genes altered expression of which is associated with aging (Table 5; 78-fold and 33-fold for down- and upregulated aging-associated genes, respectively). Collectively, these observations indicate that the dysregulation of SCARS and aberrant expression of SCARS-regulated genes should be considered as mechanistic contributors to pathogenesis of aging and broad spectrum of human disorders.
Table 5.
Comparisons of impacts of 5389 SCARS-regulated genes and 3016 non-SCARS-regulated genes linked with human-specific neuro-regulatory SNCs on a global scale of significant associations to aging and diseases of modern humans
| Aging-associated gene expression changes | |||
| Aging upregulated genes | Number of significantly enriched records (n) | Percent | Enrichment ratio |
| All genes linked with human-specific neuro-regulatory SNCs (n = 8405) | 34 | ||
| SCARS-regulated hsSNC-linked genes (n = 5389) | 130 | 97.0 | 32.5 |
| Non-SCARS-regulated hsSNC-linked genes (n = 3016) | 4 | 3.0 | |
| Aging downregulated genes | Number of significantly enriched records (n) | Fold enrichment | |
| All genes linked with human-specific neuro-regulatory SNCs (n = 8405) | 67 | ||
| SCARS-regulated hsSNC-linked genes (n = 5389) | 155 | 98.7 | 77.5 |
| Non-SCARS-regulated hsSNC-linked genes (n = 3016) | 2 | 1.3 | |
| Disease-associated gene expression changes | |||
| Diseases GEO database (upregulated genes) | Number of significantly enriched records (n) | Fold enrichment | |
| All genes linked with human-specific neuro-regulatory SNCs (n = 8405) | 204 | ||
| SCARS-regulated hsSNC-linked genes (n = 5389) | 507 | 99.2 | 126.8 |
| Non-SCARS-regulated hsSNC-linked genes (n = 3016) | 4 | 0.8 | |
| Diseases GEO database (downregulated genes) | Number of significantly enriched records (n) | Fold enrichment | |
| All genes linked with human-specific neuro-regulatory SNCs (n = 8405) | 240 | ||
| SCARS-regulated hsSNC-linked genes (n = 5389) | 476 | 97.9 | 47.6 |
| Non-SCARS-regulated hsSNC-linked genes (n = 3016) | 10 | 2.1 | |
| DisGeNET database | Number of significantly enriched records (n) | Fold enrichment | |
| All genes linked with human-specific neuro-regulatory SNCs (n = 8405) | 1313 | ||
| SCARS-regulated hsSNC-linked genes (n = 5389) | 1126 | 98.5 | 66.2 |
| Non-SCARS-regulated hsSNC-linked genes (n = 3016) | 17 | 1.5 | |
Differential GSEA were carried out independently on 5389 SCARS-regulated and 3016 non-SCARS-regulated neuro-regulatory hsSNC-linked genes employing 29 genomic databases. Results were recorded and reported for comparisons of the numbers of significantly enriched records. Percent and enrichment ratio columns report metrics reflective of the relative contributions of SCARS-regulated and non-SCAS-regulated hsSNC-linked genes
Discussion
In recent years, the elucidation of genetic and molecular mechanisms defining the phenotypic uniqueness of modern humans attained a significant progress in illuminating the potentially broad role of thousands human-specific regulatory sequences (HSRS) in contrast to the relatively modest impact of human-specific changes of a limited number of coding genes (Kronenberg et al. 2018; Glinsky 2016a,2020a; Kanton et al. 2019). Previous reports reflect this progress by focusing on analyses of sub-sets of human-specific structural variations to highlight mechanistically distinct pathways of their evolutionary origins (Glinsky 2016a) or their potential impacts on evolution of defined human-specific traits (reviewed in Levchenko et al. 2018). For example, a recent review of Levchenko et al. (2018) was primarily focused on human accelerated regions (HARs) and their regulatory impacts on specific target genes of potential relevance to development of human brain. Major limitations of the previous reports were a lack of considerations of 35,074 neuro-regulatory human-specific SNCs discovered by Kanton et al. (2019) and the absence of analyses of potential contributions of SCARS as well as SCARS-regulated genes to human-specific genomic regulatory networks.
Observations reported in this contribution are in agreement with the previous reports (Glinsky 2015,2016a, 2020a; Levchenko et al. 2018), emphasizing the significant contributions of human-specific regulatory sequences (HSRS) to development and functions of human brain. However, results of the present analyses revealed unexpectedly broad impacts of HSRS, including neuro-regulatory human-specific SNCs, and downstream target genes on morphological structures, physiological functions, aging, and pathological phenotypes of modern humans. Collectively, reported herein findings argue that a preponderance of anatomical features, physiological functions, and pathological phenotypes of modern humans appear to evolve within genomic regulatory frameworks controlled by HSRS, including neuro-regulatory human-specific SNCs.
Macromolecules comprising the essential building blocks of life at the cellular and organismal levels remain highly conserved during the evolution of humans and other Great Apes. Identification and initial structural-functional characterization of nearly hundred thousand candidate HSRS (Kronenberg et al. 2018; Glinsky 2020a; Kanton et al. 2019; this contribution) validate the idea that unique to human phenotypes may result from human-specific changes to genomic regulatory sequences defined as regulatory mutations (King and Wilson 1975). Technological advances enabled the exquisite degree of accuracy of molecular definition of 35,074 SNCs that are fixed in humans, distinct from other primates, and located in DA chromatin regions during human brain development (Kanton et al. 2019). Notably, 99.8% of candidate regulatory hsSNCs that overlap DA chromatin regions during brain development are shared with the archaic humans while only 64 hsSNCs are unique to modern humans. The conservation on the human lineage of a vast majority of neuro-regulatory hsSNCs associated with early stages of human brain development suggests that coding genes associated with hsSNCs may have a broad effect on human-specific traits beyond embryonic development. This concept has been substantiated by the multiple lines of evidence acquired and reported in the present contribution.
Employing the GREAT algorithm (McLean et al. 2010, 2011), 8405 genes have been identified that are linked to 35,074 hsSNCs via genomic proximity co-localization analysis, indicating that the expression of these hsSNC-linked genes might be affected by hsSNCs located in DA chromatin regions during brain development. Comprehensive gene set enrichment analyses (GSEA) of these 8405 genes revealed the large scale of associations with physiological processes, morphological features, and pathological conditions of H. sapiens. Significantly enriched records include more than 1000 anatomically distinct regions of the adult human brain, many human tissues and cell types, more than 200 common human disorders, and more than 1000 rare diseases. Notably, similar patterns of phenotypic associations have been observed for genes linked to 59,089 previously defined structurally, functionally, and evolutionary distinct classes of HSRS.
One of main conclusions of this work is that structurally and functionally diverse HSRS appear to display the genomic regulatory alignment with the relatively restricted elite set of genetic targets (Fig. 3; Table 3; Supplemental Figure S9; Supplemental Table Set S3). The estimated timeline of the evolutionary origin of HSRS and neuro-regulatory hsSNCs is likely to span hundred thousand years, and their creation has been driven by stochastic events and mechanistically distinct processes. Therefore, these seemingly apparent functional redundancy in building human-specific GRNs is likely to have biologically meaningful implications contributing to manifestations of human-specific phenotypes. The common patterns of the genomic placement of HSRS appear uniformly associated with sub-sets of genes comprising the putative regulatory targets of neuro-regulatory human-specific SNCs (Fig. 3; Table 3; Supplemental Table Set S3), perhaps, implying that imposing of human-specific elements of genomic regulatory control on this elite set of genes may reflect their importance for the emergence and refinement of human-specific traits. Impacts of HSRS span from multi-lineage markers expressing (MLME) cells of human preimplantation embryos (Glinsky et al. 2018) to cells engaged in the early stages of human brain development (Kanton et al. 2019) and functions of the dorsolateral prefrontal cortex (DLPFC) of the adult human brain (Guffanti et al. 2018). Therefore, combinatorial contributions of distinct families of HSRS may be necessary to facilitate human-specific patterns of expression of the elite set of target genes in different types of human cells at distinct developmental stages and in the adulthood.
Based on the reported above observations, it has been concluded that genes linked to neuro-regulatory hsSNCs appear contributing to development, morphological architecture, and biological functions of the adult human brain, other components of the central nervous system, and many tissues and organs across human body. They were implicated in the extensive range of human physiological and pathological conditions, thus supporting the hypothesis that phenotype-altering effects of neuro-regulatory hsSNCs are not restricted to the early stages of human brain development. Results of the analyses utilizing the Mouse Genome Informatics (MGI) database (http://www.informatics.jax.org/) revealed that neuro-regulatory hsSNC-associated genes affect wide spectra of traits defining both physiology and pathology of modern humans, perhaps reflecting the global scale of human-specific regulatory impacts on thousands essential mammalian phenotypes. Significantly, outlined herein analytical approaches and reported end points provide readily available access to mouse models for precise molecular definitions of unique to humans regulatory effects of neuro-regulatory human-specific SNCs and other types of HSRS on genes causally affecting thousands of defined mammalian phenotypes and hundreds of common and rare human disorders.
Methods
Data source and analytical protocols
Candidate human-specific regulatory sequences and African apes–specific retroviral insertions
A total of 94,806 candidate HSRS, including 35,074 neuro-regulatory human-specific SNCs, detailed descriptions of which, and corresponding references of primary original contributions, are reported elsewhere (Glinsky 2015, 2016a, b, c, 2017, 2018, 2020a; Glinsky and Barakat 2019; Kanton et al. 2019). Solely publicly available datasets and resources were used in this contribution. The significance of the differences in the expected and observed numbers of events was calculated using two-tailed Fisher’s exact test. Additional placement enrichment tests were performed for individual classes of HSRS taking into account the size in base pair of corresponding genomic regions.
Data analysis
Categories of DNA sequence conservation
Identification of highly conserved in primates (pan-primate), primate-specific, and human-specific sequences was performed as previously described (Glinsky 2015, 2016a, b, c, 2017, 2018, 2020a, b). In brief, all categories were defined by direct and reciprocal mapping using LiftOver. Specifically, the following categories of candidate regulatory sequences were distinguished:
Highly conserved in primates’ sequences: DNA sequences that have at least 95% of bases remapped during conversion from/to human (Homo sapiens, hg38), chimp (Pan troglodytes, v5), and bonobo (Pan paniscus, v2; in specifically designated instances, Pan paniscus, v1 was utilized for comparisons). Similarly, highly conserved sequences were defined for hg38 and latest releases of genomes of Gorilla, Orangutan, Gibbon, and Rhesus.
Primate-specific: DNA sequences that failed to map to the mouse genome (mm10).
Human-specific: DNA sequences that failed to map at least 10% of bases from human to both chimpanzee and bonobo. All candidate HSRS identified based on the sequence alignments failures to genomes of both chimpanzee and bonobo were subjected to more stringent additional analyses requiring the mapping failures to genomes of Gorilla, Orangutan, Gibbon, and Rhesus. These loci were considered created de novo human-specific regulatory sequences (HSRS).
To infer the putative evolutionary origins, each evolutionary classification was defined independently by running the corresponding analyses on all candidate HSRS representing the specific category. For example, human-rodent conversion identify sequences that are absent in the mouse genome based on the sequence identity threshold of 10%. Additional comparisons were performed using the same methodology and exactly as stated in the manuscript text. Human brain regions’ marker genes were identified among genes linked to hsSNCs by analyzing genes significantly upregulated in specified human brain regions using the Allen Brain Atlas database. Brain region–specific records manifesting significantly increased expression at 1.5-fold cutoff were selected for analyses. Genes differentially expressed in human versus chimpanzee adult brains were identified among hsSNC-linked genes by analyzing genes differentially expressed in eight regions of human versus chimpanzee adult brains (Xu et al. 2018).
Gene set enrichment and genome-wide proximity placement analyses
Gene set enrichment analyses were carried out using the Enrichr bioinformatics platform, which enables the interrogation of nearly 200,000 gene sets from more than 100 gene set libraries. The Enrichr API (January 2018 through January 2020 releases) (Chen et al. 2013; Kuleshov et al. 2016) was used to test genes linked to HSRS of interest for significant enrichment in numerous functional categories. In all tables and plots (unless stated otherwise), in addition to the nominal p values and adjusted p values, the “combined score” calculated by Enrichr is reported, which is a product of the significance estimate and the magnitude of enrichment (combined score c = log(p) × z, where p is the Fisher’s exact test p value and z is the z-score deviation from the expected rank). When technically feasible, larger sets of genes comprising several thousand entries were analyzed. Regulatory connectivity maps between HSRS and coding genes and additional functional enrichment analyses were performed with the GREAT algorithm (McLean et al. 2010, 2011) at default settings. The reproducibility of the results was validated by implementing two releases of the GREAT algorithm: GREAT version 3.0.0 (February 15, 2015 to August 18, 2019) and GREAT version 4.0.4 (August 19, 2019). The GREAT algorithm allows investigators to identify and annotate the genome-wide connectivity networks of user-defined distal regulatory loci and their putative target genes. Concurrently, the GREAT algorithm performs functional annotations and analyses of statistical enrichment of annotations of identified genes, thus enabling the inference of potential biological significance of interrogated genomic regulatory networks. Genome-wide proximity placement analysis (GPPA) of distinct genomic features co-localizing with HSRS was carried out as described previously and originally implemented for human-specific transcription factor binding sites (Glinsky 2015, 2016a, b, c, 2017, 2018, 2020a).
Mammalian phenotype ontology and human disease ontology analyses
To validate and extend findings afforded by the gene set enrichment analyses and to identify all genes linked with human-specific regulatory SNCs that are associated with defined mammalian phenotypes as well as implicated in development of human diseases with one or more mouse models, the additional analyses have been carried out utilizing the Mouse Genome Informatics (MGI) database (http://www.informatics.jax.org/).
Statistical analyses of the publicly available datasets
All statistical analyses of the publicly available genomic datasets, including error rate estimates, background and technical noise measurements and filtering, feature peak calling, feature selection, assignments of genomic coordinates to the corresponding builds of the reference human genome, and data visualization, were performed exactly as reported in the original publications and associated references linked to the corresponding data visualization tracks (http://genome.ucsc.edu/). Any modifications or new elements of statistical analyses are described in the corresponding sections of the Results. Statistical significance of the Pearson correlation coefficients was determined using GraphPad Prism version 6.00 software. Both nominal and Bonferroni adjusted p values were estimated. The significance of the differences in the numbers of events between the groups was calculated using two-sided Fisher’s exact and chi-square test, and the significance of the overlap between the events was determined using the hypergeometric distribution test (Tavazoie et al. 1999).
Electronic supplementary material
Identification of genes expression of which distinguishes thousands of anatomically distinct areas of the adult human brain, various regions of the central nervous system, and many different cell types and tissues in the human body using the Enrichr bioinformatics platform (see Methods for details). (PDF 422 kb)
Identification and characterization of genes expression of which is altered during aging of humans, rats, and mice using the Enrichr bioinformatics platform (see Methods for details). (PDF 326 kb)
Identification of genes implicated in development and manifestations of hundreds physiological and pathological phenotypes and autosomal inheritance in Modern Humans using the Enrichr bioinformatics platform (see Methods for details). (PDF 422 kb)
Identification of genes expression of which is altered in several hundred common human disorders using the Enrichr bioinformatics platform (see Methods for details). (PDF 436 kb)
Identification of genes implicated in more than 1,000 records classified as human rare diseases using the Enrichr bioinformatics platform (see Methods for details). (PDF 480 kb)
Gene ontology analyses of putative regulatory targets of genetic loci harboring human-specific SNCs using the Enrichr bioinformatics platform (see Methods for details). (PDF 456 kb)
KEGG analyses of putative regulatory targets of genetic loci harboring human-specific SNCs using the Enrichr bioinformatics platform (see Methods for details). (PDF 369 kb)
Interrogation of MGI Mammalian Phenotype databases identifies genes associated with human-specific SNCs and implicated in premature death and embryonic, perinatal, neonatal, and postnatal lethality phenotypes using the Enrichr bioinformatics platform (see Methods for details). (PDF 373 kb)
Structurally, functionally, and evolutionary distinct classes of HSRS share the relatively restricted elite set of common genetic targets. (PDF 456 kb)
Supplemental Note 1. 59K HSRS and target genes at 100Kb and 1Mb distances of single nearest maximum extensions. (PDF 4828 kb)
Supplemental Note 2. 59K HSRS and target genes at 1 Mb distances of single nearest maximum extensions. (PDF 3018 kb)
Supplemental Table Set S1. (XLSX 268 kb)
Supplemental Table Set S2. (XLSX 11757 kb)
Supplemental Table Set S3. (XLSX 846 kb)
Supplemental Table Set S4. (XLSX 686 kb)
Supplemental Table Set S5. (XLSX 9753 kb)
Supplemental Table Set S6. (XLSX 25 kb)
Acknowledgments
This work was made possible by the open public access policies of major grant funding agencies and international genomic databases and the willingness of many investigators worldwide to share their primary research data. This work was supported, in part, by OncoScar, Inc.
Abbreviations
- GSEA
Gene set enrichment analyses
- hsSNCs
Human-specific neuro-regulatory single-nucleotide changes
- HSRS
Human-specific regulatory sequences
- SCARS
Stem cell–associated retroviral sequences
- GRNs
Genomic regulatory networks
- GWAS
Genome-Wide Association Study
- GO
Gene Ontology
- GEO
Genome-Wide Association Study
- GREAT
Genomic Regions Enrichment of Annotations Tool
- DA
Differentially accessible
- ATAC-seq
Assay for Transposase-Accessible Chromatin using sequencing
- ECAs
Extinct common ancestors
- hVIPs
Human virus–interacting proteins
Authors’ contributions
This is a single author contribution. All elements of this work, including the conception of ideas, formulation, and development of concepts, execution of experiments, analysis of data, and writing of the paper, were performed by the author.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Barakat TS, Halbritter F, Zhang M, Rendeiro AF, Perenthaler E, Bock C, Chambers I. Functional dissection of the enhancer repertoire in human embryonic stem cells. Cell Stem Cell. 2018;23:276–288.e8. doi: 10.1016/j.stem.2018.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma'ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinf. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimpanzee Sequencing and Analysis Consortium Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinsky GV. Transposable elements and DNA methylation create in embryonic stem cells human-specific regulatory sequences associated with distal enhancers and non-coding RNAs. Genome Biol Evol. 2015;7:1432–1454. doi: 10.1093/gbe/evv081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinsky GV. Mechanistically distinct pathways of divergent regulatory DNA creation contribute to evolution of human-specific genomic regulatory networks driving phenotypic divergence of Homo sapiens. Genome Biol Evol. 2016;8:2774–2788. doi: 10.1093/gbe/evw185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinsky GV. Activation of endogenous human stem cell-associated retroviruses (SCARs) and therapy-resistant phenotypes of malignant tumors. Cancer Lett. 2016;376:347–359. doi: 10.1016/j.canlet.2016.04.014. [DOI] [PubMed] [Google Scholar]
- Glinsky GV. Single cell genomics reveals activation signatures of endogenous SCARS networks in aneuploid human embryos and clinically intractable malignant tumors. Cancer Lett. 2016;381:176–193. doi: 10.1016/j.canlet.2016.08.001. [DOI] [PubMed] [Google Scholar]
- Glinsky GV (2017) Human-specific features of pluripotency regulatory networks link NANOG with fetal and adult brain development. BioRxiv. https://www.biorxiv.org/content/early/2017/06/19/022913; 10.1101/022913
- Glinsky GV. Contribution of transposable elements and distal enhancers to evolution of human-specific features of interphase chromatin architecture in embryonic stem cells. Chromosom Res. 2018;26:61–84. doi: 10.1007/s10577-018-9571-6. [DOI] [PubMed] [Google Scholar]
- Glinsky GV. A catalogue of 59,732 human-specific regulatory sequences reveals unique to human regulatory patterns associated with virus-interacting proteins, pluripotency and brain development. DNA Cell Biol. 2020;39:126–143. doi: 10.1089/dna.2019.4988. [DOI] [PubMed] [Google Scholar]
- Glinsky GV. Tripartite combination of candidate pandemic mitigation agents: vitamin D, quercetin, and estradiol manifest properties of medicinal agents for targeted mitigation of the COVID-19 pandemic defined by genomics-guided tracing of SARS-CoV-2 targets in human cells. Biomedicines. 2020;8:129. doi: 10.3390/biomedicines8050129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinsky GV, Barakat TS (2019) The evolution of Great Apes has shaped the functional enhancers’ landscape in human embryonic stem cells. 37:101456. 10.1016/j.scr.2019.101456 [DOI] [PubMed]
- Glinsky G, Durruthy-Durruthy J, Wossidlo M, Grow EJ, Weirather JL, Au KF, Wysocka J, Sebastiano V (2018) Single cell expression analysis of primate-specific retroviruses-derived HPAT lincRNAs in viable human blastocysts identifies embryonic cells co-expressing genetic markers of multiple lineages. Heliyon 4: e00667. 10.1016/j.heliyon.2018.e00667. eCollection 2018 Jun [DOI] [PMC free article] [PubMed]
- Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, O’meara MJ, Guo JZ, Swaney D, Tummino TA, Huttenhain R et al (2020) A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature:1–13 [DOI] [PMC free article] [PubMed]
- Guffanti G, Bartlett A, Klengel T, Klengel C, Hunter R, Glinsky G, Macciardi F. Novel bioinformatics approach identifies transcriptional profiles of lineage-specific transposable elements at distinct loci in the human dorsolateral prefrontal cortex. Mol Biol Evol. 2018;35:2435–2453. doi: 10.1093/molbev/msy143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanton S, Boyle MJ, He Z, Santel M, Weigert A, Sanchís-Calleja F, Guijarro P, Sidow L, Fleck JS, Han D, Qian Z, Heide M, Huttner WB, Khaitovich P, Pääbo S, Treutlein B, Camp JG. 2019. Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature. 2019;574:418–422. doi: 10.1038/s41586-019-1654-9. [DOI] [PubMed] [Google Scholar]
- King MC, Wilson AC. Evolution at two levels in humans and chimpanzees. Science. 1975;188:107–116. doi: 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
- Kronenberg ZN, et al. High-resolution comparative analysis of great ape genomes. Science. 2018;360:eaar6343. doi: 10.1126/science.aar6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma'ayan A (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. gkw377 [DOI] [PMC free article] [PubMed]
- Levchenko A, Kanapin A, Samsonova A, Gainetdinov RR. Human accelerated regions and other human specific sequence variations in the context of evolution and their relevance for brain development. Genome Biol Evol. 2018;10:166–188. doi: 10.1093/gbe/evx240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Reno PL, Pollen AA, Bassan AI, Capellini TD, Guenther C, Indjeian VB, Lim X, Menke DB, Schaar BT, Wenger AM, Bejerano G, Kingsley DM. Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature. 2011;471:216–219. doi: 10.1038/nature09774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowakowski TJ, Bhaduri A, Pollen AA, Alvarado B, Mostajo-Radji MA, di Lullo E, Haeussler M, Sandoval-Espinosa C, Liu SJ, Velmeshev D, Ounadjela JR, Shuga J, Wang X, Lim DA, West JA, Leyrat AA, Kent WJ, Kriegstein AR. Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science. 2017;358:1318–1323. doi: 10.1126/science.aap8809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontis J, et al. Hominoid-specific transposable elements and KZFPs facilitate human embryonic genome activation and control transcription in naïve human ESCs. Cell Stem Cell. 2019;24:1–12. doi: 10.1016/j.stem.2019.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie S, Hughes JD, Campbell MJ, Cho RJ, Church GM. Systematic determination of genetic network architecture. Nat Genet. 1999;22:281–285. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- Theunissen TW, Friedli M, He Y, Planet E, O’Neil RC, Markoulaki S, Pontis J, Wang H, Iouranova A, Imbeault M, Duc J, Cohen MA, Wert KJ, Castanon R, Zhang Z, Huang Y, Nery JR, Drotar J, Lungjangwa T, Trono D, Ecker JR, Jaenisch R. Molecular criteria for defining the naive human pluripotent state. Cell Stem Cell. 2016;19:502–515. doi: 10.1016/j.stem.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Leemput J, Boles NC, Kiehl TR, Corneo B, Lederman P, Menon V, Lee C, Martinez RA, Levi BP, Thompson CL, Yao S, Kaykas A, Temple S, Fasano CA. CORTECON: a temporal transcriptome analysis of in vitro human cerebral cortex development from human embryonic stem cells. Neuron. 2014;83:51–68. doi: 10.1016/j.neuron.2014.05.013. [DOI] [PubMed] [Google Scholar]
- Wang J, Xie G, Singh M, Ghanbarian AT, Raskó T, Szvetnik A, Cai H, Besser D, Prigione A, Fuchs NV, Schumann GG, Chen W, Lorincz MC, Ivics Z, Hurst LD, Izsvák Z (2014) Primate-specific endogenous retrovirus-driven transcription defines naive-like stem cells. Nature 516 (7531):405–409 [DOI] [PubMed]
- Xu C, Li Q, Efimova O, He L, Tatsumoto S, Stepanova V, Oishi T, Udono T, Yamaguchi K, Shigenobu S, Kakita A, Nawa H, Khaitovich P, Go Y. Human-specific features of spatial gene expression and regulation in eight brain regions. Genome Res. 2018;28:1097–1110. doi: 10.1101/gr.231357.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Identification of genes expression of which distinguishes thousands of anatomically distinct areas of the adult human brain, various regions of the central nervous system, and many different cell types and tissues in the human body using the Enrichr bioinformatics platform (see Methods for details). (PDF 422 kb)
Identification and characterization of genes expression of which is altered during aging of humans, rats, and mice using the Enrichr bioinformatics platform (see Methods for details). (PDF 326 kb)
Identification of genes implicated in development and manifestations of hundreds physiological and pathological phenotypes and autosomal inheritance in Modern Humans using the Enrichr bioinformatics platform (see Methods for details). (PDF 422 kb)
Identification of genes expression of which is altered in several hundred common human disorders using the Enrichr bioinformatics platform (see Methods for details). (PDF 436 kb)
Identification of genes implicated in more than 1,000 records classified as human rare diseases using the Enrichr bioinformatics platform (see Methods for details). (PDF 480 kb)
Gene ontology analyses of putative regulatory targets of genetic loci harboring human-specific SNCs using the Enrichr bioinformatics platform (see Methods for details). (PDF 456 kb)
KEGG analyses of putative regulatory targets of genetic loci harboring human-specific SNCs using the Enrichr bioinformatics platform (see Methods for details). (PDF 369 kb)
Interrogation of MGI Mammalian Phenotype databases identifies genes associated with human-specific SNCs and implicated in premature death and embryonic, perinatal, neonatal, and postnatal lethality phenotypes using the Enrichr bioinformatics platform (see Methods for details). (PDF 373 kb)
Structurally, functionally, and evolutionary distinct classes of HSRS share the relatively restricted elite set of common genetic targets. (PDF 456 kb)
Supplemental Note 1. 59K HSRS and target genes at 100Kb and 1Mb distances of single nearest maximum extensions. (PDF 4828 kb)
Supplemental Note 2. 59K HSRS and target genes at 1 Mb distances of single nearest maximum extensions. (PDF 3018 kb)
Supplemental Table Set S1. (XLSX 268 kb)
Supplemental Table Set S2. (XLSX 11757 kb)
Supplemental Table Set S3. (XLSX 846 kb)
Supplemental Table Set S4. (XLSX 686 kb)
Supplemental Table Set S5. (XLSX 9753 kb)
Supplemental Table Set S6. (XLSX 25 kb)