

Abstract
Organic solute transporter alpha/beta (OSTα/β) is a heteromeric solute carrier protein that transports bile acids, steroid metabolites and drugs into and out of cells. OSTα/β protein is expressed in various tissues, but its expression is highest in the gastrointestinal tract where it facilitates the recirculation of bile acids from the gut to the liver. Previous studies established that OSTα/β is upregulated in liver tissue of patients with extrahepatic cholestasis, obstructive cholestasis, and primary biliary cholangitis (PBC), conditions that are characterized by elevated bile acid concentrations in the liver and/or systemic circulation. The discovery that OSTα/β is highly upregulated in the liver of patients with nonalcoholic steatohepatitis (NASH) further highlights the clinical relevance of this transporter because the incidence of NASH is increasing at an alarming rate with the obesity epidemic. Since OSTα/β is closely linked to the homeostasis of bile acids, and tightly regulated by the nuclear receptor farnesoid X receptor, OSTα/β is a potential drug target for treatment of cholestatic liver disease, and other bile acid-related metabolic disorders such as obesity and diabetes. Obeticholic acid, a semi-synthetic bile acid used to treat PBC, under review for the treatment of NASH, and in development for the treatment of other metabolic disorders, induces OSTα/β. Some drugs associated with hepatotoxicity inhibit OSTα/β, suggesting a possible role for OSTα/β in drug-induced liver injury (DILI). Furthermore, clinical cases of homozygous genetic defects in both OSTα/β subunits resulting in diarrhea and features of cholestasis have been reported. This review article has been compiled to comprehensively summarize the recent data emerging on OSTα/β, recapitulating the available literature on the structure-function and expression-function relationships of OSTα/β, the regulation of this important transporter, the interaction of drugs and other compounds with OSTα/β, and the comparison of OSTα/β with other solute carrier transporters as well as adenosine triphosphate-binding cassette transporters. Findings from basic to more clinically focused research efforts are described and discussed.
Keywords: bile acids, cholestasis, drug interactions, genetic variation, NASH, SLC51
1. Introduction
Nearly two decades ago, organic solute transporter alpha/beta (OSTα/β/SLC51A/B) was identified in a screen of a hepatic cDNA library from the little skate (Leucoraja erinacea) (Wang, Seward, Li, Boyer, & Ballatori, 2001), but even today this heteromeric transport protein is relatively poorly understood and understudied. OSTα/β has been detected on the basolateral membrane of epithelial cells in tissues ranging from the zona reticularis of the adrenal cortex to the renal tubules and the rectum (Ballatori, et al., 2005; Fang, et al., 2010; Uhlen, et al., 2015), with the highest mRNA and protein levels found in the small intestine (ileum and duodenum). In conjunction with the apical sodium-dependent bile acid transporter (ASBT/SLC10A2) on the luminal membrane of the intestinal epithelial cells, intestinal OSTα/β localized on the basolateral membrane plays a key role in the reabsorption and enterohepatic circulation of bile acids (Ballatori, Fang, Christian, Li, & Hammond, 2008; Frankenberg, et al., 2006; Rao, et al., 2008; Sultan, et al., 2018). Furthermore, OSTα/β-mediated efflux of bile acids protects the ileal epithelium against intracellular bile acid accumulation and intestinal injury in mice (Ferrebee, et al., 2018). While OSTα/β is known primarily for its important role in the transport and homeostasis of bile acids, other steroids and some drugs also have been identified as OSTα/β substrates (Ballatori, et al., 2005; Wang, et al., 2001). In addition, some drugs/xenobiotics inhibit the transport function of OSTα/β (Malinen, Ali, Bezencon, Beaudoin, & Brouwer, 2018; Malinen, Kauttonen, et al., 2019). Although assessing interactions with OSTα/β is not yet a requirement in the drug development pipeline, the International Transporter Consortium has acknowledged the potential of OSTα/β-mediated drug interactions (Kenna, et al., 2018; Zamek-Gliszczynski, et al., 2018). OSTα/β is also expressed in other organs central to drug absorption, distribution, metabolism, excretion and toxicity (ADME-Tox) including the kidneys and liver. Interestingly, hepatic OSTα/β is markedly upregulated in certain liver diseases (Malinen, et al., 2018; Soroka, Ballatori, & Boyer, 2010), and when bile flow is interrupted in humans or rodents (Boyer, et al., 2006; Chai, et al., 2015; Schaap, van der Gaag, Gouma, & Jansen, 2009).
To date, review articles on OSTα/β have focused on the role of OSTα/β as a bile acid transporter (Ballatori, et al., 2009; Dawson, Hubbert, & Rao, 2010; Soroka, Ballatori, et al., 2010), summarizing the expression (Ballatori, 2005, 2011; Ballatori, Christian, Wheeler, & Hammond, 2013), structure (Dawson, et al., 2010) and regulation of OSTα/β (Ballatori, et al., 2013; Ballatori, et al., 2009). Recently, OSTα/β has received renewed attention in relation to its potential roles in nonalcoholic steatohepatitis (NASH), bile acid-related metabolic disorders such as cholestatic liver disease, obesity and diabetes, as well as in drug-induced liver injury (DILI). The present review provides an update on OSTα/β from a pharmaceutical perspective. Recent data emerging on this transporter are highlighted, and available information on the structure-function and expression-function relationships of OSTα/β, the regulation of this important transporter, the interaction of drugs and other compounds with OSTα/β, and the comparison of OSTα/β with other solute carrier (SLC) and adenosine triphosphate (ATP)-binding cassette (ABC) transporters is summarized.
2. Expression of OSTα/β
2.1. Co-expression of SLC51A/OSTα and SLC51B/OSTβ mRNAs and Proteins
Transporters properly expressed on the basolateral or apical plasma membranes are important determinants of the disposition of endogenous and exogenous compounds in the body. Human SLC51A and SLC51B genes are transcribed on chromosomes 3q29 and 15q22.31, respectively. Both OSTα and OSTβ subunits are translated in the endoplasmic reticulum and translocated onto the basolateral plasma membrane of epithelial cells to form a functional transporter (Ballatori, et al., 2005; Boyer, et al., 2006; Dawson, et al., 2005). It seems that OSTα and OSTβ stabilize each other in mammalian cells to form the heteromeric OSTα/β protein complex (Li, Cui, Fang, Lee, & Ballatori, 2007). Protein expression of OSTα and OSTβ in co-expressing human embryonic kidney (HEK) 293 cells decreased only modestly when cells were treated for 24 hr with the protein synthesis inhibitor cycloheximide, suggesting a half-life of OSTα/β beyond 24 hr when both subunits were co-expressed (Li, et al., 2007). Pulse-chase experiments supported a half-life of OSTα beyond 24 hr when co-expressed with OSTβ, and revealed an OSTα half-life of ~2 hr in the absence of OSTβ (Dawson, et al., 2010). Furthermore, both subunits are required to enable the transport function of OSTα/β at the plasma membrane (Wang, et al., 2001). The necessary co-expression for transport function was confirmed with transport studies in transfected African green monkey kidney fibroblast-like COS-7 cells: increased uptake of taurocholate (TCA) and estrone sulfate (ES) was evident in cells expressing both OSTα and OSTβ as compared to cells transfected with only one of the subunits (Sun, et al., 2007). Similarly, HEK 293 and Madin-Darby canine kidney (MDCK) cells only express OSTα and OSTβ protein at the plasma membrane when both genes are transfected simultaneously (Dawson, et al., 2005). In renal and intestinal tissue of OSTα-knockout mice, SLC51B mRNA is present, while OSTβ protein was not detected (Li, et al., 2007). Interestingly, in clawed frog (Xenopus laevis) oocytes, both OSTα and OSTβ subunits were able to separately reach the plasma membrane when singly expressed, although each subunit alone lacked transporter activity (Seward, Koh, Boyer, & Ballatori, 2003). Although both subunits are needed for functional OSTα/β on the plasma membrane, OSTα and OSTβ can be expressed at different protein levels, as evidenced in many tissues (Ballatori, 2005) [Table 1; (Uhlen, et al., 2015)]. These differences may be explained by unknown OSTα/β stoichiometry on the plasma membrane, and/or variable intracellular expression of the subunits.
Table 1.
Protein Levels of Human OSTα and OSTβ in Various Tissues and Cell Types from Healthy Individuals.
| Tissue | Adrenal Gland | Appendix | Cerebellum | Colon | |
|---|---|---|---|---|---|
| Cell Type | Glandular Cells | Glandular Cells | Purk inje Cells | Glandular Cells | Peripheral Nerve/Ganglion |
| OSTα Level | Not detected* | Medium | Not detected* | Low | Low |
| OSTβ Level | Not detected* | Medium | Not detected* | Medium | Not detected |
| Tissue | Epididymis | Hippocampus | Kidney | Liver | |
| Cell Type | Glandular Cells | Neuronal Cells | Cells in Tubules | Bile Duct Cells | Hepatocytes |
| OSTα Level | Low | Not detected* | Low | Not detected* | Low |
| OSTβ Level | Not detected | Not detected* | Low | Not detected | Not detected |
| Tissue | Prostate | Rectum | Seminal Vesicle | Skin | |
| Cell Type | Glandular Cells | Glandular Cells | Glandular Cells | Keratinocytes | Epidermal Cells |
| OSTα Level | Low | Low | Low | Low | Low |
| OSTβ Level | Not detected | Medium | Not detected | Not detected | Not detected |
| Tissue | Small Intestine | Stomach | Testis | ||
| Cell Type | Glandular Cells | Glandular Cells | Cells in Seminiferous Ducts | Leydig Cells | |
| OSTα Level | Medium | Low | Not detected | Medium | |
| OSTβ Level | High | Low/Medium | Low | Not detected | |
Data on protein levels were obtained from The Human Protein Atlas version 19 and Ensembl version 92.38 and were determined using microarray-based immunohistochemistry. A color scale was applied to each column separately, with dark green and red denoting the highest and lowest levels of expression, respectively. When comparing this table to the main text, discrepancies can be noted with other publications, which may be due to differences in sample preparation, antibodies utilized, data analysis, and/or other procedures (e.g., colorimetric versus fluorescence-based detection, with varying sensitivities).
Although The Human Protein Atlas did not report positive staining for OSTα and/or OSTβ in adrenal glandular cells, cerebellar cells, hippocampal cells or bile duct cells, previous studies revealed that OSTα and OSTβ levels were detected in the zona reticularis of the adrenal gland, Purkinje cells of the cerebellum, and cornu ammonis cells of the hippocampus (Fang, et al., 2010), while OSTα levels have been reported in cholangiocytes (Ballatori, et al., 2005). Various other cell types not included in the table also were evaluated by The Human Protein Atlas. OSTα and OSTβ protein were not detectable in adipose tissue (adipocytes), appendix (lymphoid tissue), bone marrow (hematopoietic cells), breast (adipocytes, glandular and myoepithelial cells), bronchus (respiratory epithelial cells), caudate (glial and neuronal cells), cerebellum (granular and molecular layer cells), cerebral cortex (endothelial, glial, neuronal cells and neuropil), colon (endothelial cells), endometrium (endometrial stroma and glandular cells), esophagus (squamous epithelial cells), Fallopian tube (glandular cells), gallbladder (glandular cells), heart muscle (myocytes), hippocampus (glial cells), kidney (glomerular cells), lung (macrophages and pneumocytes), lymph node (non-germinal center cells), nasopharynx (respiratory epithelial cells), oral mucosa (squamous epithelial cells), ovary (ovarian stroma cells), pancreas (exocrine glandular cells and islets of Langerhans), parathyroid gland (glandular cells), placenta (trophoblastic cells), salivary gland (glandular cells), skeletal muscle (myocytes), skin (fibroblasts, Langerhans and melanocytes), smooth muscle (smooth muscle cells), soft tissue (chondrocytes, fibroblasts and peripheral nerve), spleen (red pulp cells), thyroid gland (glandular cells), tonsil [(non)-germinal center and squamous epithelial cells], urinary bladder (urothelial cells), uterine cervix (glandular and squamous epithelial cells), and vagina (squamous epithelial cells). OSTα protein was not detectable in lymph node (germinal center cells) and spleen (white pulp cells). OSTβ protein was not detectable in placenta (decidual cells).
2.2. Tissue expression of SLC51A/OSTα and SLC51B/OSTβ
The expression of OSTα/β mRNA and protein has been reviewed in several species (Ballatori, 2005, 2011; Ballatori, et al., 2013). More recently, the Human Protein Atlas project [www.proteinatlas.org; (Uhlen, et al., 2015)] has shown that OSTα and OSTβ protein (Table 1), as well as SLC51A (14 splice variants) and SLC51B (one transcript) mRNA (Table 2) are expressed in various human tissues and cell types. In tissues, the expression levels of SLC51A/OSTα and SLC51B/OSTβ are usually not equal. Furthermore, when comparing various publicly available resources on OSTα and OSTβ protein levels, some discrepancies are evident, as described below – particularly for those tissues where expression is relatively low, which could be the result of differences in procedures (e.g., tissue handling and storage, manual evaluation of immunohistochemical data) or antibody quality (e.g., selectivity and specificity).
Table 2.
mRNA Isoform Expression of Human SLC51A and SLC51B in Various Tissues and Cell Types from Healthy Individuals.
| SLC51A Level | SLC51B Level | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tissue / Cell Type | ENST 00000 296327 |
ENST 00000 415111 |
ENST 00000 416660 |
ENST 00000 428985 |
ENST 00000 442203 |
ENST 00000 471430 |
ENST 00000 472653 |
ENST 00000 475271 |
ENST 00000 475672 |
ENST 00000 476129 |
ENST 00000 479732 |
ENST 00000 484407 |
ENST 00000 492794 |
ENST 00000 496737 |
Average | Sum | ENST 00000 334287 |
| Adipose Tissue | 0.07 | 0.42 | 0.04 | 0.06 | 0.14 | 0.04 | 0.00 | 0.06 | 0.25 | 0.06 | 1.45 | 0.05 | 0.74 | 1.51 | 0.35 | 4.89 | 0.86 |
| Adrenal Gland | 3.40 | 0.66 | 0.68 | 0.43 | 0.34 | 1.02 | 0.41 | 0.00 | 1.03 | 0.28 | 2.13 | 5.66 | 1.38 | 1.86 | 1.38 | 19.29 | 1.32 |
| Appendix | 1.02 | 0.59 | 0.16 | 0.12 | 0.80 | 0.49 | 0.58 | 0.00 | 0.54 | 0.08 | 1.45 | 0.70 | 0.63 | 2.79 | 0.71 | 9.96 | 4.48 |
| Basophil | 0.00 | 0.00 | 0.00 | 0.00 | 0.59 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.21 | 0.00 | 0.00 | 0.93 | 0.12 | 1.72 | 0.01 |
| Bone Marrow | 0.52 | 1.16 | 0.91 | 1.37 | 3.02 | 1.00 | 1.05 | 0.14 | 0.58 | 3.70 | 1.17 | 5.29 | 1.38 | 4.54 | 1.84 | 25.82 | 0.53 |
| Breast | 0.00 | 0.22 | 0.00 | 0.06 | 0.26 | 0.00 | 0.03 | 0.04 | 0.00 | 0.00 | 0.00 | 0.26 | 0.39 | 0.11 | 0.10 | 1.38 | 0.06 |
| Cerebral Cortex | 0.00 | 0.33 | 0.10 | 0.03 | 0.20 | 0.45 | 0.07 | 0.00 | 0.41 | 0.17 | 1.24 | 0.13 | 0.62 | 1.68 | 0.39 | 5.45 | 5.95 |
| Cervix, Uterine | 0.60 | 0.17 | 0.00 | 0.13 | 0.29 | 0.11 | 0.00 | 0.00 | 0.00 | 0.13 | 0.07 | 0.10 | 1.07 | 0.00 | 0.19 | 2.67 | 59.69 |
| Classical Monocyte | 0.00 | 0.04 | 0.00 | 0.00 | 0.24 | 0.00 | 0.00 | 0.01 | 0.06 | 0.01 | 0.31 | 0.00 | 0.08 | 0.33 | 0.08 | 1.06 | 0.00 |
| Colon | 9.30 | 0.37 | 0.02 | 0.24 | 0.31 | 0.66 | 0.87 | 0.09 | 0.35 | 0.27 | 0.67 | 3.73 | 5.63 | 0.61 | 1.65 | 23.14 | 40.73 |
| Duodenum | 28.07 | 0.60 | 0.11 | 0.58 | 1.65 | 3.51 | 3.11 | 0.00 | 1.22 | 1.82 | 1.78 | 7.61 | 7.44 | 3.38 | 4.35 | 60.87 | 85.37 |
| Endometrium | 0.04 | 0.53 | 0.00 | 0.04 | 0.29 | 0.00 | 0.02 | 0.02 | 0.22 | 0.00 | 1.22 | 0.22 | 0.62 | 1.75 | 0.36 | 4.97 | 5.52 |
| Eosinophil | 0.22 | 0.01 | 0.00 | 0.00 | 0.33 | 0.00 | 0.00 | 0.00 | 0.00 | 0.03 | 0.34 | 0.24 | 0.00 | 0.71 | 0.13 | 1.87 | 0.00 |
| Epididymis | 0.97 | 0.00 | 0.00 | 0.00 | 0.41 | 0.66 | 0.30 | 0.00 | 0.00 | 0.00 | 0.30 | 1.58 | 0.00 | 0.14 | 0.31 | 4.37 | 0.39 |
| Esophagus | 0.00 | 0.61 | 0.00 | 0.00 | 0.13 | 0.27 | 0.03 | 0.10 | 0.49 | 0.00 | 2.44 | 0.00 | 1.01 | 2.16 | 0.52 | 7.23 | 2.71 |
| Fallopian Tube | 2.08 | 0.30 | 0.00 | 0.16 | 0.32 | 0.25 | 0.25 | 0.00 | 0.25 | 0.06 | 0.59 | 0.95 | 0.73 | 0.37 | 0.45 | 6.30 | 6.01 |
| Gallbladder | 0.13 | 0.84 | 0.08 | 0.00 | 0.25 | 0.18 | 0.00 | 0.02 | 0.44 | 0.08 | 1.95 | 0.10 | 0.34 | 3.47 | 0.56 | 7.88 | 4.33 |
| gdTCR | 0.00 | 0.01 | 0.00 | 0.00 | 0.34 | 0.00 | 0.00 | 0.00 | 0.12 | 0.00 | 0.62 | 0.00 | 0.22 | 0.56 | 0.13 | 1.87 | 0.03 |
| Heart Muscle | 0.00 | 0.09 | 0.11 | 0.00 | 0.10 | 0.07 | 0.02 | 0.00 | 0.23 | 0.11 | 1.78 | 0.04 | 0.57 | 0.85 | 0.28 | 3.98 | 1.66 |
| Intermediate Monocyte | 0.00 | 0.02 | 0.06 | 0.00 | 0.16 | 0.00 | 0.00 | 0.00 | 0.04 | 0.00 | 0.27 | 0.00 | 0.07 | 0.28 | 0.06 | 0.91 | 0.00 |
| Kidney | 4.42 | 0.71 | 0.04 | 0.21 | 0.17 | 0.37 | 0.08 | 0.40 | 0.51 | 0.11 | 0.85 | 3.28 | 3.65 | 0.59 | 1.10 | 15.37 | 19.35 |
| Liver (Fetal) | Detected* | Detected* | |||||||||||||||
| Liver | 31.95 | 1.48 | 0.00 | 1.42 | 0.75 | 2.96 | 1.73 | 0.88 | 0.98 | 0.45 | 0.56 | 18.97 | 24.38 | 0.40 | 6.21 | 86.90 | 0.93 |
| Lung | 0.20 | 0.36 | 0.16 | 0.03 | 0.24 | 0.26 | 0.13 | 0.03 | 0.22 | 0.10 | 1.22 | 0.32 | 0.30 | 1.38 | 0.35 | 4.95 | 9.37 |
| Lymph Node | 0.00 | 0.70 | 0.04 | 0.01 | 0.35 | 0.08 | 0.00 | 0.03 | 0.16 | 0.11 | 1.18 | 0.46 | 0.65 | 3.37 | 0.51 | 7.14 | 0.75 |
| MAIT T-cell | 0.00 | 0.01 | 0.00 | 0.00 | 0.26 | 0.00 | 0.00 | 0.00 | 0.01 | 0.00 | 0.43 | 0.00 | 0.02 | 0.20 | 0.07 | 0.93 | 0.03 |
| Memory B-cell | 0.00 | 0.01 | 0.00 | 0.00 | 0.22 | 0.00 | 0.00 | 0.00 | 0.04 | 0.00 | 0.29 | 0.00 | 0.12 | 0.30 | 0.07 | 0.98 | 0.00 |
| Memory CD4 T-cell | 0.00 | 0.01 | 0.02 | 0.00 | 0.20 | 0.00 | 0.00 | 0.00 | 0.13 | 0.00 | 0.48 | 0.00 | 0.16 | 0.39 | 0.10 | 1.39 | 0.02 |
| Memory CD8 T-cell | 0.00 | 0.00 | 0.00 | 0.00 | 0.22 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.25 | 0.00 | 0.00 | 0.20 | 0.05 | 0.68 | 0.02 |
| Myeloid DC | 0.00 | 0.00 | 0.00 | 0.00 | 0.09 | 0.00 | 0.00 | 0.00 | 0.05 | 0.01 | 0.29 | 0.00 | 0.12 | 0.34 | 0.06 | 0.90 | 0.01 |
| Naive B-cell | 0.00 | 0.03 | 0.00 | 0.00 | 0.46 | 0.00 | 0.00 | 0.00 | 0.07 | 0.00 | 0.34 | 0.00 | 0.14 | 0.36 | 0.10 | 1.40 | 0.00 |
| Naive CD4 T-cell | 0.00 | 0.02 | 0.00 | 0.00 | 0.30 | 0.05 | 0.00 | 0.00 | 0.00 | 0.00 | 0.40 | 0.03 | 0.02 | 0.29 | 0.08 | 1.10 | 0.01 |
| Naive CD8 T-cell | 0.00 | 0.08 | 0.01 | 0.00 | 0.31 | 0.00 | 0.00 | 0.00 | 0.24 | 0.01 | 0.53 | 0.00 | 0.28 | 0.45 | 0.14 | 1.91 | 0.02 |
| Neutrophil | 0.00 | 0.03 | 0.14 | 0.00 | 1.24 | 0.00 | 0.01 | 0.00 | 0.07 | 0.00 | 0.73 | 0.00 | 0.07 | 0.64 | 0.21 | 2.93 | 0.03 |
| NK-cell | 0.00 | 0.09 | 0.00 | 0.00 | 0.54 | 0.00 | 0.00 | 0.00 | 0.03 | 0.00 | 0.43 | 0.00 | 0.00 | 0.51 | 0.11 | 1.59 | 0.00 |
| Non-Classical Monocyte | 0.00 | 0.05 | 0.02 | 0.00 | 0.19 | 0.02 | 0.00 | 0.00 | 0.08 | 0.01 | 0.57 | 0.00 | 0.11 | 0.57 | 0.12 | 1.63 | 0.01 |
| Ovary | 0.07 | 0.89 | 0.10 | 0.00 | 0.11 | 0.39 | 0.00 | 0.00 | 0.38 | 0.11 | 1.00 | 6.21 | 0.35 | 0.76 | 0.74 | 10.36 | 1.52 |
| Pancreas | 0.00 | 0.00 | 0.00 | 0.01 | 0.11 | 0.00 | 0.00 | 0.00 | 0.05 | 0.00 | 0.08 | 0.06 | 0.23 | 0.32 | 0.06 | 0.86 | 0.05 |
| Parathyroid Gland | 3.18 | 1.94 | 0.00 | 0.00 | 0.00 | 3.39 | 0.56 | 0.00 | 0.53 | 0.00 | 0.11 | 3.51 | 1.94 | 0.21 | 1.10 | 15.37 | 0.00 |
| Pituitary Gland | Detected* | Detected* | |||||||||||||||
| Placenta | 0.01 | 0.34 | 0.00 | 0.00 | 0.25 | 0.04 | 0.02 | 0.06 | 0.20 | 0.02 | 0.88 | 0.09 | 0.29 | 0.73 | 0.21 | 2.93 | 1.00 |
| Plasmacytoid DC | 0.00 | 0.00 | 0.00 | 0.00 | 0.40 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 | 0.28 | 0.00 | 0.08 | 0.36 | 0.08 | 1.14 | 0.00 |
| Prostate | 0.16 | 0.41 | 0.00 | 0.05 | 0.37 | 0.13 | 0.05 | 0.05 | 0.35 | 0.33 | 0.87 | 0.16 | 0.75 | 0.94 | 0.33 | 4.62 | 2.94 |
| Rectum | 0.53 | 0.07 | 0.00 | 0.20 | 0.11 | 0.05 | 0.09 | 0.00 | 0.00 | 0.12 | 0.09 | 0.60 | 0.66 | 0.10 | 0.19 | 2.61 | 31.41 |
| Salivary Gland | 0.21 | 0.30 | 0.00 | 0.04 | 0.08 | 0.14 | 0.01 | 0.04 | 0.06 | 0.19 | 0.44 | 0.06 | 0.18 | 0.57 | 0.17 | 2.33 | 0.47 |
| Seminal Vesicle | 0.06 | 0.07 | 0.00 | 0.00 | 0.23 | 0.00 | 0.04 | 0.00 | 0.00 | 0.00 | 0.00 | 0.03 | 0.06 | 0.03 | 0.04 | 0.53 | 0.94 |
| Skeletal Muscle | 0.03 | 0.21 | 0.38 | 0.00 | 0.40 | 0.15 | 0.06 | 0.00 | 0.36 | 0.29 | 1.20 | 0.24 | 0.33 | 0.83 | 0.32 | 4.50 | 0.38 |
| Skin | 0.03 | 1.35 | 0.00 | 0.00 | 0.59 | 0.26 | 0.03 | 0.16 | 1.00 | 0.08 | 4.27 | 0.14 | 1.86 | 6.55 | 1.17 | 16.32 | 1.03 |
| Small Intestine | 65.43 | 1.49 | 0.15 | 4.22 | 4.28 | 6.97 | 8.62 | 0.60 | 1.53 | 3.79 | 2.11 | 20.32 | 16.80 | 5.20 | 10.11 | 141.52 | 118.33 |
| Smooth Muscle | 0.31 | 0.04 | 0.00 | 0.12 | 0.21 | 0.10 | 0.21 | 0.00 | 0.00 | 0.02 | 0.00 | 0.58 | 0.35 | 0.00 | 0.14 | 1.94 | 2.56 |
| Spleen | 0.00 | 0.93 | 0.04 | 0.00 | 0.42 | 0.00 | 0.04 | 0.00 | 0.30 | 0.29 | 1.28 | 0.03 | 0.44 | 3.13 | 0.49 | 6.89 | 1.20 |
| Stomach | 0.15 | 0.32 | 0.03 | 0.00 | 0.18 | 0.00 | 0.00 | 0.00 | 0.16 | 0.05 | 0.91 | 0.03 | 0.18 | 1.33 | 0.24 | 3.35 | 0.80 |
| T-reg | 0.00 | 0.02 | 0.00 | 0.00 | 0.34 | 0.00 | 0.00 | 0.00 | 0.04 | 0.01 | 0.14 | 0.00 | 0.07 | 0.24 | 0.06 | 0.85 | 0.00 |
| Testis | 1.23 | 1.02 | 2.67 | 0.75 | 1.18 | 1.00 | 0.44 | 0.48 | 2.66 | 0.24 | 1.63 | 12.32 | 6.06 | 1.75 | 2.39 | 33.44 | 13.12 |
| Thyroid Gland | 0.51 | 1.03 | 0.00 | 0.00 | 0.44 | 0.13 | 0.20 | 0.12 | 0.17 | 0.32 | 1.48 | 0.57 | 0.70 | 1.42 | 0.51 | 7.10 | 1.16 |
| Tonsil | 0.22 | 0.22 | 0.00 | 0.04 | 0.07 | 0.13 | 0.02 | 0.12 | 0.06 | 0.00 | 0.06 | 1.01 | 0.26 | 0.12 | 0.17 | 2.35 | 0.06 |
| Total PBMC | 0.00 | 0.01 | 0.00 | 0.00 | 0.07 | 0.01 | 0.00 | 0.00 | 0.02 | 0.00 | 0.13 | 0.00 | 0.06 | 0.08 | 0.03 | 0.38 | 0.01 |
| Urinary Bladder | 0.04 | 0.37 | 0.00 | 0.00 | 0.21 | 0.00 | 0.00 | 0.00 | 0.26 | 0.00 | 0.78 | 0.14 | 0.00 | 1.43 | 0.23 | 3.23 | 3.19 |
| Average | 2.77 | 0.39 | 0.11 | 0.18 | 0.46 | 0.45 | 0.34 | 0.06 | 0.30 | 0.24 | 0.83 | 1.71 | 1.51 | 1.14 | 0.75 | 10.50 | 7.69 |
Data on transcript levels were obtained from The Human Protein Atlas version 19 and Ensembl version 92.38, and were determined using next-generation sequencing-based RNA-sequencing. Values are presented as transcripts per million. A color scale was applied to each column separately, with green and red denoting the highest and lowest levels of expression, respectively.
Some tissues were not analyzed in The Human Protein Atlas for SLC51A and SLC51B mRNA levels, but positive levels were detected in another study (Seward, et al., 2003). According to ensembl.org, only ENST00000296327 (340 amino acids), ENST00000415111 (23 amino acids), ENST00000416660 (66 amino acids) and ENST00000428985 (212 amino acids) are protein-coding SLC51A transcripts. gdTCR, gamma delta T cell; DC, dendritic cell; MAIT T-cell, mucosal associated invariant T-cell; NK-cell, natural killer cell; T-reg, regulatory T cell; PBMC, peripheral blood mononuclear cell.
2.2.1. SLC51A and SLC51B mRNA expression in tissues
In one of the first OSTα/β studies, human SLC51A and SLC51B mRNA was found in a variety of tissues, primarily the testis, colon, liver, fetal liver, small intestine, kidney, adrenal gland and ovary, and at lower levels in the heart, lung, brain, pituitary gland, uterus, prostate and adipose tissue (Seward, et al., 2003). Some SLC51A mRNA was observed in the human thyroid and mammary glands, but SLC51B mRNA was below the detection limit (Seward, et al., 2003). Differences in SLC51A mRNA expression among the duodenum, terminal ileum and colon were negligible in a study with eight healthy human subjects, and a similar trend among these tissues was observed for SLC51B mRNA expression (Schwarz, 2012). This study also showed higher mRNA expression of SLC51A compared to SLC51B in the human liver; the opposite was observed in the human kidney (Schwarz, 2012), in agreement with a previous study (Ballatori, et al., 2005) and the Human Protein Atlas project (Table 2). The Human Protein Atlas project detected some level (e.g., ≥0.01 transcripts per million RNA molecules per sample) of SLC51A and SLC51B expression in nearly all analyzed cell types and tissues (Table 2). Compared to the average expression of SLC51A mRNA in all analyzed human tissue/cellular samples, SLC51A mRNA expression was higher in the adrenal gland, bone marrow, colon, duodenum (small intestine), kidney, liver, parathyroid gland, skin and testis. SLC51B mRNA expression was higher in the uterine cervix, colon, duodenum (small intestine), kidney, lung, rectum and testis, compared to the average expression of SLC51B mRNA in all analyzed human tissue/cellular samples. The abundance of human SLC51A and SLC51B mRNA compared to other transporters in histologically normal tissues (small intestine, liver and kidney) is depicted in Figure 1.
Figure 1.
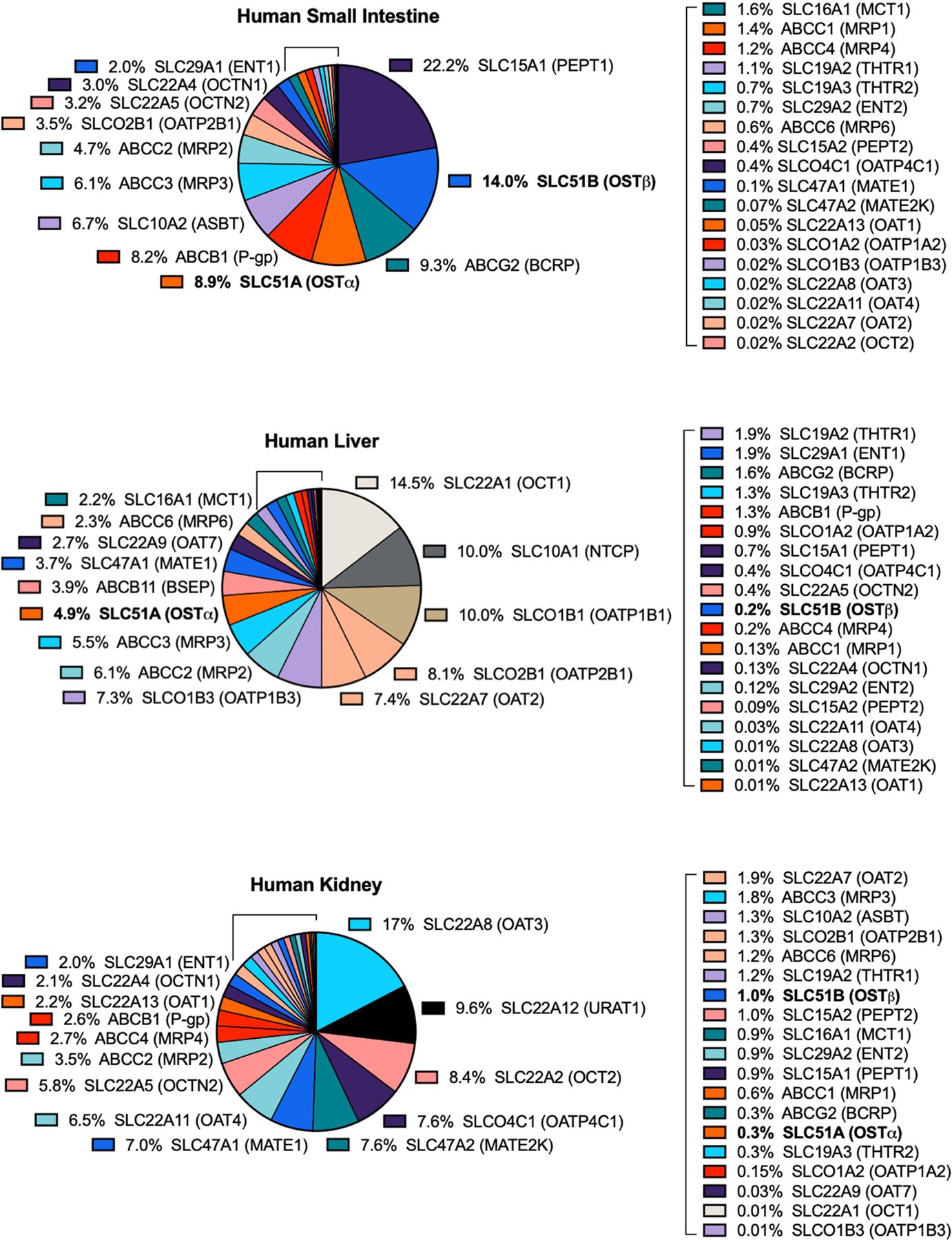
Abundance of Human SLC51A and SLC51B mRNAs Compared to Other Transporters in Histologically Normal Small Intestine, Liver and Kidney. The data were obtained from The Human Protein Atlas version 19 and Ensembl version 92.38, and represent consensus normalized expression levels from three transcriptomics datasets (HPA, GTEx and FANTOM5). Data are expressed as a percentage of total tissue transporter abundance.
In the mouse, the highest expression of SLC51A and SLC51B mRNA was found in the ileum, followed by the jejunum (Dawson, et al., 2005). In pigs, both SLC51A and SLC51B mRNA had the highest intestinal expression in the ileum, followed by the jejunum, duodenum, cecum and colon (Fang, et al., 2018). Interestingly, while SLC51A and SLC51B mRNA was detected in the small intestines, colons and kidneys from mice, rats and humans, the transcripts were nearly undetectable in mouse and rat liver, while these transcripts were detectable in human liver (Ballatori, et al., 2005; Schwarz, 2012; Seward, et al., 2003). Laser capture microdissection-isolated mouse Purkinje and hippocampal cells showed mRNA expression of SLC51A and SLC51B (Fang, et al., 2010).
2.2.2. OSTα and OSTβ protein expression in tissues
Studies on protein expression and localization have been limited due to the unavailability of commercial OSTα/β antibodies until relatively recently. The first OSTα/β protein expression studies were performed with antibodies produced in-house (Ballatori, et al., 2005; Dawson, et al., 2005; Li, et al., 2007). In those early studies, OSTα and/or OSTβ protein expression was reported in ADME-Tox organs, including the human ileum, kidney and liver using indirect immunofluorescence (Ballatori, et al., 2005). In the human liver, OSTα protein was expressed in hepatocytes and cholangiocytes (Ballatori, et al., 2005). Using these same in-house-produced OSTα and OSTβ antibodies, immunolocalization experiments revealed that human OSTα and OSTβ protein levels were detectable in the steroidogenic Purkinje cells and cornu ammonis cells of the cerebellum and hippocampus, respectively, as well as in the zona reticularis of the human adrenal gland (Fang, et al., 2010). In another study with healthy human subjects and using custom-made antibodies, western blot analysis demonstrated that OSTα protein levels were higher in the colon than the terminal ileum and duodenum (Schwarz, 2012). More recently, a quantitative proteomic analysis identified similarly high levels of OSTα and OSTβ protein in four jejunum and twelve ileum samples from macroscopically normal tissue; these tissues were resected from healthy subjects and individuals with underlying inflammatory bowel disease, colon cancer or ischemia (Couto, et al., 2020). Protein levels of OSTα and OSTβ were higher than P-glycoprotein (P-gp/MDR1/ABCB1). New antibodies have been produced as part of the Human Protein Atlas project (Uhlen, et al., 2015), which confirmed, using microarray-based immunohistochemistry, the protein expression of OSTα and OSTβ in several tissues and cell types in which expression was reported previously (Table 1). For example, expression of OSTα and/or OSTβ in hepatocytes, renal tubular cells and intestinal cells was reported before (Ballatori, et al., 2005), and confirmed in the Human Protein Atlas project. The project revealed the highest expression of OSTα protein (medium level) in the duodenum (small intestine), appendix and testis, while the highest expression of OSTβ protein (high level) was found in the duodenum (small intestine), followed by the appendix, colon, rectum and stomach (medium level). Expression of one or both subunits was detected in other tissues (low level), while undetectable levels were reported in the majority of tested cell types. Interestingly, while OSTα protein was detected previously in human cholangiocytes (Ballatori, et al., 2005), the Human Protein Atlas project reported undetectable levels in bile duct cells (Table 1). Furthermore, protein levels of OSTα and OSTβ were reported in cerebellar, hippocampal and adrenal glandular cells (Fang, et al., 2010), but neither of the subunits were detected in these cell types in the Human Protein Atlas project. It is unclear whether these discrepancies are due to antibody quality, or procedural and/or sample differences.
In addition to protein analyses in humans, early studies found OSTα and/or OSTβ protein expression in the rat and/or mouse small intestine, colon, kidney and cholangiocytes by immunoblot analysis and/or tissue immunolocalization (Ballatori, et al., 2005; Dawson, et al., 2005). Furthermore, murine OSTα and OSTβ protein localization was shown in Purkinje cells and cornu ammonis cells (Fang, et al., 2010).
2.3. Expression in ADME-Tox models
Cellular and in silico ADME-Tox models play an important role in drug development and pharmacology, allowing for the evaluation of ADME-Tox properties of drug candidates prior to undertaking time- and resource-intensive animal studies and clinical trials. These systems can be used to assess the impact of transporters (and metabolizing enzymes, among other factors) on the disposition of compounds, and data generated using in vitro and/or in silico models can be extrapolated to humans (Alqahtani, 2017). SLC51A and SLC51B mRNA has been detected in several cell lines widely used in drug development, including Caco-2, HEK 293, HeLa and HepG2, and in cell lines used as models for in vitro pharmacology studies, including MCF7, PC-3 and SH-SY5Y [Table 3; (Uhlen, et al., 2015)]. While SLC51A mRNA was found to be highest in REH cells (a childhood B acute lymphoblastic leukemia cell line), SLC51B transcripts were not detectable in these cells. Conversely, while SLC51B mRNA was highest in Caco-2, HepG2 and EFO-21 cells (epithelial colorectal adenocarcinoma, well-differentiated hepatocellular carcinoma, and metastatic ovarian serous cystadenocarcinoma cell lines, respectively), SLC51A mRNA was relatively low in these cells, albeit detectable. A similar trend between SLC51A and SLC51B gene expression levels in Caco-2 cells was observed in another study, in which SLC51B mRNA levels were ~11-fold higher than SLC51A levels (Li, et al., 2012). Similarly, in the human and rodent colon (Ballatori, et al., 2005), and particularly the human rectum (Table 2), SLC51B mRNA levels were found to be higher than SLC51A mRNA levels.
Table 3.
mRNA Expression of Human SLC51A and SLC51B in Various Cell Lines.
| SLC51A Level | SLC51B Level | |||||
|---|---|---|---|---|---|---|
| Cell Line | TPM | pTPM | NX | TPM | pTPM | NX |
| A-431 | 0.4 | 0.5 | 1.0 | 0.0 | 0.0 | 0.0 |
| A549 | 0.7 | 0.8 | 1.6 | 1.3 | 1.6 | 1.4 |
| AF22 | 0.6 | 0.8 | 1.4 | 0.2 | 0.3 | 0.2 |
| AN3-CA | 0.7 | 0.9 | 1.7 | 0.7 | 0.9 | 0.7 |
| ASC diff | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| ASC TERT1 | 0.3 | 0.3 | 1.0 | 0.1 | 0.1 | 0.1 |
| BEWO | 0.0 | 0.0 | 0.1 | 0.4 | 0.5 | 0.4 |
| BJ | 0.3 | 0.4 | 0.8 | 0.0 | 0.0 | 0.0 |
| BJ hTERT+ | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| BJ hTERT+ SV40 Large T+ | 0.0 | 0.0 | 0.1 | 0.2 | 0.2 | 0.2 |
| BJ hTERT+ SV40 Large T+ RasG12V | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Caco-2 | 0.2 | 0.3 | 0.7 | 14.9 | 18.3 | 16.5 |
| CAPAN-2 | 0.2 | 0.2 | 0.4 | 0.0 | 0.1 | 0.0 |
| Daudi | 0.0 | 0.0 | 0.1 | 0.0 | 0.1 | 0.1 |
| EFO-21 | 0.8 | 1.0 | 1.9 | 11.0 | 13.5 | 11.1 |
| fHDF/TERT166 | 0.4 | 0.4 | 1.1 | 0.0 | 0.0 | 0.0 |
| HaCaT | 0.3 | 0.4 | 0.9 | 0.0 | 0.0 | 0.0 |
| HAP1 | 0.0 | 0.1 | 0.2 | 0.0 | 0.0 | 0.0 |
| HBEC3-KT | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| HBF TERT88 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| HDLM-2 | 0.3 | 0.4 | 0.7 | 0.1 | 0.1 | 0.1 |
| HEK 293 | 1.2 | 1.5 | 2.8 | 0.2 | 0.3 | 0.2 |
| HEL | 0.4 | 0.5 | 1.0 | 0.0 | 0.0 | 0.0 |
| HeLa | 0.2 | 0.3 | 0.8 | 0.1 | 0.1 | 0.1 |
| HepaRG | Detected* | Detected* | ||||
| HepG2 | 0.5 | 0.7 | 1.7 | 10.4 | 12.7 | 13.8 |
| HHSteC | 0.1 | 0.1 | 0.2 | 0.1 | 0.1 | 0.1 |
| HL-60 | 0.5 | 0.6 | 1.4 | 0.1 | 0.2 | 0.2 |
| HMC-1 | 0.5 | 0.7 | 1.4 | 0.0 | 0.0 | 0.0 |
| HSkMC | 0.1 | 0.1 | 0.3 | 0.0 | 0.0 | 0.0 |
| hTCEpi | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| hTEC/SVTERT24-B | 0.0 | 0.1 | 0.2 | 0.1 | 0.1 | 0.1 |
| hTERT-HME1 | 0.4 | 0.5 | 1.3 | 0.0 | 0.0 | 0.0 |
| HuH-7 | Detected* | Detected* | ||||
| HUVEC TERT2 | 0.0 | 0.1 | 0.2 | 0.0 | 0.0 | 0.0 |
| K-562 | 0.1 | 0.1 | 0.2 | 0.2 | 0.2 | 0.2 |
| Karpas-707 | 1.0 | 1.7 | 3.3 | 0.1 | 0.2 | 0.2 |
| LHCN-M2 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| MCF7 | 0.2 | 0.3 | 0.6 | 0.3 | 0.4 | 0.4 |
| MOLT-4 | 0.0 | 0.0 | 0.1 | 0.0 | 0.1 | 0.0 |
| NB-4 | 0.3 | 0.4 | 1.2 | 0.2 | 0.3 | 0.3 |
| NTERA-2 | 0.0 | 0.0 | 0.1 | 0.4 | 0.5 | 0.4 |
| PC-3 | 0.8 | 1.0 | 2.0 | 0.4 | 0.5 | 0.4 |
| REH | 3.4 | 4.4 | 7.9 | 0.0 | 0.0 | 0.0 |
| RH-30 | 0.0 | 0.0 | 0.1 | 0.0 | 0.0 | 0.0 |
| RPMI-8226 | 0.1 | 0.2 | 0.5 | 0.6 | 0.8 | 0.8 |
| RPTEC TERT1 | 0.0 | 0.0 | 0.0 | 0.3 | 0.3 | 0.4 |
| RT4 | 0.3 | 0.4 | 0.9 | 0.0 | 0.0 | 0.0 |
| SCLC-21H | 0.2 | 0.2 | 0.3 | 0.8 | 1.0 | 0.6 |
| SH-SY5Y | 0.2 | 0.3 | 0.5 | 0.5 | 0.7 | 0.5 |
| SiHa | 0.2 | 0.3 | 0.5 | 0.0 | 0.0 | 0.0 |
| SK-BR-3 | 1.1 | 1.3 | 3.2 | 0.0 | 0.0 | 0.0 |
| SK-MEL-30 | 0.1 | 0.1 | 0.3 | 0.0 | 0.0 | 0.0 |
| T-47d | 1.0 | 1.2 | 2.3 | 0.4 | 0.5 | 0.4 |
| THP-1 | 0.1 | 0.2 | 0.5 | 0.0 | 0.0 | 0.0 |
| TIME | 0.2 | 0.2 | 0.5 | 0.0 | 0.0 | 0.0 |
| U-138 MG | 0.7 | 0.9 | 1.6 | 0.3 | 0.3 | 0.3 |
| U-2 OS | 0.3 | 0.4 | 0.7 | 0.0 | 0.0 | 0.0 |
| U-2197 | 0.5 | 0.7 | 1.3 | 0.1 | 0.1 | 0.1 |
| U-251 MG | 0.3 | 0.4 | 0.9 | 0.1 | 0.1 | 0.1 |
| U-266/70 | 1.1 | 1.8 | 3.3 | 0.1 | 0.2 | 0.2 |
| U-266/84 | 1.3 | 2.0 | 4.0 | 0.2 | 0.4 | 0.3 |
| U-698 | 0.2 | 0.2 | 0.6 | 0.1 | 0.2 | 0.2 |
| U-87 MG | 0.5 | 0.6 | 1.1 | 0.0 | 0.1 | 0.0 |
| U-937 | 0.2 | 0.3 | 0.6 | 0.1 | 0.1 | 0.1 |
| WM-115 | 0.2 | 0.3 | 0.6 | 0.1 | 0.2 | 0.1 |
Data on transcript expression levels per gene in 64 cell lines were obtained from The Human Protein Atlas version 19 and Ensembl version 92.38, and were determined using next-generation sequencing-based RNA-sequencing. Values are presented as transcripts per million (TPM) RNA molecules per sample, protein-coding transcripts per million (pTPM) and normalized expression (NX). NX data for every gene in each sample were obtained by normalizing TPM data using trimmed mean of M values (Robinson & Oshlack, 2010), followed by Pareto scaling (van den Berg, Hoefsloot, Westerhuis, Smilde, & van der Werf, 2006). A color scale was applied to each column separately, with green and red denoting the highest and lowest levels of expression, respectively.
HepaRG and HuH-7 cell lines both express SLC51A and SLC51B mRNA (Landrier, et al., 2006; Sissung, et al., 2019), but were not included in the current dataset from the Human Protein Atlas.
Caco-2 cells form tight junctions and resemble enterocytes when appropriately differentiated and polarized. OSTα/β has been suggested to facilitate basolateral uptake of TCA and ES transport in Caco-2 cells (Grandvuinet & Steffansen, 2011), while inhibitors of OSTα/β reduced basolateral uptake of ES in these cells (Grandvuinet, Gustavsson, & Steffansen, 2013). A role for OSTα/β as an uptake transporter in the basolateral-to-apical transport of rosuvastatin was speculated in Caco-2 cells (Li, et al., 2012). Furthermore, a novel physiologically-based pharmacokinetic model of rosuvastatin disposition incorporating contributions of OSTα/β among a few other transporters improved predictions of interactions with rifampin, compared to previous models in which OSTα/β was not included (Wang, Zheng, & Leil, 2017). Studies using P-gp-expressing cells, including Caco-2 cells and human primary proximal tubule cells, have shown that the kinetic models for the P-gp substrate digoxin needed to incorporate one or two basolateral uptake transporters in addition to a passive permeability component in order to adequately describe the observed data (Chaudhry, et al., 2018; Ellens, Meng, Le Marchand, & Bentz, 2018). A role for OSTα/β as an uptake transporter has been proposed for the interpretation of kinetic digoxin data in human primary proximal tubule cells in the presence of verapamil (Ellens, et al., 2018), and OSTα/β may play a role in digoxin disposition in Caco-2 cells; single concentration studies in X. laevis oocytes suggested that digoxin was an OSTα/β substrate (Seward, et al., 2003; Wang, et al., 2001).
The hepatoma cell lines HepG2 as well as HuH-7 and HepaRG cells (two additional well-differentiated hepatocellular carcinoma cell lines) express SLC51A and SLC51B mRNA in standard cell culture conditions. Furthermore, HepG2 and HuH-7 cells express OSTα and OSTβ at the protein level (Boyer, et al., 2006; Landrier, Eloranta, Vavricka, & Kullak-Ublick, 2006; Malinen, Ito, Kang, Honkakoski, & Brouwer, 2019; Schaffner, et al., 2015; Sissung, et al., 2019; Xu, Sun, & Suchy, 2014), and have been used to study this transporter. Particularly the effect of farnesoid X receptor (FXR) ligands on SLC51A/B mRNA and OSTα/β protein expression has been evaluated in HepG2 and HuH-7 cells (Boyer, et al., 2006; Landrier, et al., 2006; Schaffner, et al., 2015; Soroka, Xu, Mennone, Lam, & Boyer, 2008; Xu, et al., 2014). Furthermore, protein expression of OSTα, OSTβ, and the organic anion transporting polypeptide (OATP/SLCO) 1B3, as well as transport of the SLC substrate dehydroepiandrosterone sulfate (DHEAS) increased over time in HuH-7 cell cultures (Malinen, Ito, et al., 2019).
3. Endogenous and exogenous OSTα/β substrates and inhibitors
OSTα/β is an important transporter for bile acids and other steroid substrates. The first OSTα/β-mediated transport studies were performed in X. laevis oocytes injected with synthetic transcripts coding for skate OSTα and OSTβ protein (Wang, et al., 2001); some endogenous and exogenous OSTα/β substrates and inhibitors were identified in these studies. Subsequent studies have discovered several novel OSTα/β substrates and inhibitors.
3.1. OSTα/β substrates
In X. leavis oocytes, the heteromeric OSTα/β transported the endogenous compounds TCA, ES and prostaglandin E2, but p-aminohippurate and S-dinitrophenyl glutathione were not substrates (Seward, et al., 2003; Wang, et al., 2001). Subsequent studies in mice and oocytes confirmed TCA and ES as OSTα/β substrates, and identified DHEAS as a substrate (Ballatori, et al., 2008; Fang, et al., 2010). Pregnenolone sulfate was shown to be an OSTα/β substrate, although pregnenolone and dehydroepiandrosterone were not (Fang, et al., 2010). Furthermore, individual bile acid species have been evaluated as OSTα/β substrates, consistent with the important role this transporter plays in bile acid homeostasis. OSTα/β transported unconjugated, taurine- and glycine-conjugated forms of cholate, chenodeoxycholate (CDCA), deoxycholate (DCA) and lithocholate, as well as the taurine- and glycine-conjugated forms of ursodeoxycholate (Table 4) (Ballatori, et al., 2005; Suga, Yamaguchi, Ogura, & Mano, 2019).
Table 4.
In Vitro Substrates of OSTα/β and Other Transporters.
Reported Km values were higher than the maximum substrate concentration tested. CA, cholate; CDCA, chenodeoxycholate; DHEAS, dehydroepiandrosterone sulfate; ES, estrone sulfate; GCA, glycocholate; GCDCA, glycochenodeoxycholate; GDCA, glycodeoxycholate; GLCA, glycolithocholate; GUDCA, glycoursodeoxycholate; Km, substrate concentration at one-half of the maximum velocity; LCA; lithocholate; NA, data not available in the literature; PGE2, prostaglandin E2; PREGS, pregnenolone sulfate; TCA, taurocholate; TCDCA, taurochenodeoxycholate; TDCA, taurodeoxycholate; TLCA, taurolithocholate; TUDCA, tauroursodeoxycholate
Single concentration studies in X. laevis oocytes suggested that digoxin is an OSTα/β substrate (Seward, et al., 2003; Wang, et al., 2001). In a study with Caco-2 cells and OSTα/β inhibitors causing cis-inhibition (i.e., inhibition from the extracellular side of the cell), a plausible role for OSTα/β in mediating the basolateral-to-apical transport of rosuvastatin was inferred (Li, et al., 2012). This speculation is supported by experiments showing that rosuvastatin uptake was increased in OSTα/β-overexpressing HeLa cells compared to control cells (Schwarz, 2012). However, the role of OSTα/β in rosuvastatin absorption was less clear based on studies in OSTα knockout mice (Schwarz, 2012). Atorvastatin, sulfasalazine and docetaxel, but not the structurally related paclitaxel, were transported by OSTα/β in HeLa cells (Schwarz, 2012).
Compounds that have been identified as OSTα/β substrates are also substrates for other SLC and some ABC transporters (Table 4). In terms of transport of exogenous compounds, OSTα/β is most similar to OATP1B3 and P-gp, which also transport atorvastatin, rosuvastatin, docetaxel and digoxin. With regards to endogenous compounds, OATP1B1 and OATP1B3 typically transport OSTα/β substrates, but Na+-taurocholate co-transporting polypeptide (NTCP/SLC10A1), ASBT, the multidrug resistance-associated protein (MRP/ABCC) 4, breast cancer resistance protein (BCRP/ABCG2) and the bile salt export pump (BSEP/ABCB11) also have affinity for multiple OSTα/β substrates.
OSTα/β is reported to be a low affinity/high capacity transporter for multiple substrates, particularly bile acid species (Malinen, et al., 2018; Suga, et al., 2019). In systems in which both high affinity/low capacity transporters and low affinity/high capacity transporters are present, high affinity transporters typically play a more dominant role in transport at low substrate concentrations (Lin & Smith, 1999). However, when substrate concentrations are substantially elevated, high capacity transporters become more dominant. In situations when high affinity transporters are not able to handle increased substrate concentrations, a high capacity transporter may be induced to become the dominant transporter for substrates in that system. For example, the induction of OSTα/β in hepatocytes under cholestatic conditions (see Section 8) suggests that when hepatocellular bile acids are elevated, OSTα/β is upregulated and becomes the dominant transporter to efflux bile acids across the basolateral plasma membrane.
3.2. OSTα/β inhibitors
More compounds have been evaluated as inhibitors of OSTα/β than as substrates. Thus far, relatively few compounds have been shown to inhibit OSTα/β compared to other SLC transporters. Initial studies in X. laevis oocytes using TCA and ES as the substrates suggested that spironolactone, digoxin, probenecid and indomethacin, in addition to various endogenous bile acids (e.g., the skate bile acid scymnol sulfate) and other steroid molecules, were OSTα/β inhibitors at concentrations ≥200 μM; bromosulfophthalein inhibited OSTα/β at 100 μM (Wang, et al., 2001). However, these compounds were tested only at a single concentration. These findings were largely reproduced in a follow-up study with X. laevis oocytes (Seward, et al., 2003). The inhibitory effect of digoxin, bromosulfophthalein and probenecid on OSTα/β-mediated TCA uptake was not reproduced in a more recent study using OSTα/β-overexpressing Flp-In 293 cells, whereas spironolactone and indomethacin inhibited OSTα/β function (Malinen, et al., 2018). In addition to these compounds, the BSEP and MRP4 inhibitor troglitazone sulfate (Funk, Ponelle, Scheuermann, & Pantze, 2001; Yang, Pfeifer, Köck, & Brouwer, 2015), the MRP3 inhibitor fidaxomicin (Ali, Welch, Lu, Swaan, & Brouwer, 2017) and the BSEP inhibitor ethinyl estradiol (Morgan, et al., 2013) inhibited OSTα/β-mediated DHEAS and/or TCA uptake (Malinen, et al., 2018; Malinen, Kauttonen, et al., 2019). In a fluorescent resonance energy transfer-based, high-throughput screen, 1,280 compounds were tested as OSTα/β inhibitors using taurochenodeoxycholate as the substrate, but only a single compound, clofazimine, was found to be an OSTα/β-specific inhibitor (van de Wiel, de Waart, Oude Elferink, & van de Graaf, 2018). Additional studies revealed that 25 μM sulfasalazine, and both unconjugated and glucuronidated ezetimibe inhibited OSTα/β-mediated transport of TCA (Schwarz, 2012).
Several purported OSTα/β substrates (e.g., digoxin, sulfasalazine, multiple bile acid species) also inhibit transport of other OSTα/β substrates, most likely via competitive inhibition, although most of these studies evaluated inhibitors only at a single concentration. Theoretically, all substrates become (competitive) inhibitors at sufficiently high concentrations. To the knowledge of the authors, the majority of the inhibitors listed in this section have not been studied as substrates. One explanation for this is that substrate quantification requires compound-specific analytical methods or generation of a stable, labeled substrate.
3.3. Current limitations in studying OSTα/β substrates and inhibitors
The published in vitro OSTα/β studies attempting to identify OSTα/β substrates or inhibitors may have been limited by a variety of factors. For instance, the functional evaluation of one transporter may be confounded when the in vitro system is influenced by a second transporter such as co-expression systems involving OSTα/β and ABST in MDCK-II epithelial cells (Ballatori, et al., 2005; van de Wiel, et al., 2018), or OSTα/β and NTCP in U-2 OS (van de Wiel, et al., 2018) or HeLa (Schwarz, 2012) cells, even when cells expressing only ASBT or NTCP are used as controls, respectively. This is especially important to consider because OSTα/β can function as both an uptake and efflux transporter. A novel fluorescent resonance energy transfer-based bile acid sensor (van de Wiel, et al., 2018) elegantly measured bile acid-mediated intracellular activation of FXR, but evaluated the inhibition of OSTα/β-mediated bile acid transport only indirectly using taurochenodeoxycholate as the substrate. Additionally, since there is a relationship between transporter expression and kinetic parameters (Balakrishnan, et al., 2007; Kalvass & Pollack, 2007; Tachibana, et al., 2010), a variable extent of transporter expression at the plasma membrane such as in the OSTα/β-overexpressing Flp-In 293 cells (Malinen, et al., 2018; Malinen, Kauttonen, et al., 2019) can lead to under- or overestimation of substrate parameters such as the maximum transport rate achieved (Vmax), or inhibitor parameters such as the half-maximal inhibitory concentration (IC50). Additionally, poor solubility of a substrate may limit the range of concentrations that can be studied, and hamper accurate determination of kinetic parameters of the transporter in the particular cell system.
4. Transport mechanism of OSTα/β
4.1. Transport direction in vivo and in vitro
OSTα/β belongs to the SLC transporter family, and it is thought to function primarily as an efflux transporter in vivo on the basolateral membrane of enterocytes involved in the enterohepatic recycling of bile acids (Dawson, et al., 2010), and in the cholestatic liver to protect hepatocytes from toxic bile acid accumulation (Boyer, et al., 2006; Chai, et al., 2015; Malinen, et al., 2018). OSTα/β expressed on the basolateral membrane of kidney cells may play a role in salvaging bile acids that escaped hepatic extraction (Dawson, et al., 2010). Interestingly, however, in OSTα−/− mice that underwent bile duct ligation, adaptive responses in the kidney, including reduced apical ASBT and increased apical MRP2 and MRP4 protein levels, resulted in increased urinary excretion of bile acids; no compensatory increase in basolateral MRP3 was observed in the kidneys of these mice (Soroka, Mennone, Hagey, Ballatori, & Boyer, 2010).
Despite the hypothesized, primary role of OSTα/β as an efflux transporter in vivo, the majority of OSTα/β-based transport studies in vitro have evaluated the uptake function of OSTα/β. Some studies utilizing cells expressing both OSTα/β and a different transporter capable of uptake [e.g., ASBT or NTCP (Ballatori, et al., 2005; Schwarz, 2012; van de Wiel, et al., 2018)] have attempted to analyze the efflux direction of OSTα/β by comparing transport in OSTα/β/ASBT- or OSTα/β/NTCP-expressing cells with cells only expressing ASBT or NTCP, respectively. In addition, a study evaluating the hepatobiliary uptake and efflux kinetics of TCA disposition showed that human hepatocytes in which OSTα/β expression was induced exhibited significantly increased basolateral efflux clearance of TCA (Guo, LaCerte, Edwards, Brouwer, & Brouwer, 2018). Although other factors could have contributed to the observed increase in basolateral efflux of TCA, these data suggest a plausible role for OSTα/β as a bile acid efflux transporter in hepatocytes under the conditions used in this study.
Some transporters demonstrate symmetric transport (e.g., the same Km for both uptake and efflux directions); however, this is not necessarily the case for all bidirectional transporters (Baird, et al., 2004; Bosdriesz, et al., 2018; Elbing, et al., 2004; Maier, Volker, Boles, & Fuhrmann, 2002). It is unclear how the uptake kinetics of OSTα/β compares to the efflux kinetics. Examination of differences in OSTα/β-mediated substrate transport in both directions warrants further investigation, since kinetic parameters determined for the uptake direction may not accurately reflect the transporter’s function in the physiologically more relevant efflux direction. Unfortunately, to date it has been inherently more complex to study a bidirectional (ATP-independent) transporter in isolation in the efflux direction, because it involves loading cells with a probe substrate prior to initiation of the efflux phase, which may introduce additional experimental variability. The existing in vitro methodologies for evaluation of efflux kinetics result in high system-to-system and lab-to-lab variability (Heikkinen, Korjamo, Lepikko, & Monkkonen, 2010; Korjamo, Kemilainen, Heikkinen, & Monkkonen, 2007).
4.2. Driving force for transport
Experiments in X. laevis oocytes indicated that OSTα/β transport operated by facilitated diffusion (Ballatori, et al., 2005; Seward, et al., 2003). However, the question remains whether OSTα/β functions as a uniporter, symporter, or antiporter. Early studies suggested that ion (Na+, K+, H+, Cl−) gradients, ATP and pH (Ballatori, et al., 2005; Seward, et al., 2003; Wang, et al., 2001) did not alter OSTα/β-mediated uptake of model substrates, implying that OSTα/β most likely functions as a uniporter that is regulated by the substrate concentration on either side of the plasma membrane. However, recent functional OSTα/β in vitro studies have been performed in K+-rich buffers (Malinen, Kauttonen, et al., 2019; Sultan, et al., 2018), sometimes in the complete absence of Na+. Extracellular replacement of sodium chloride (NaCl) with choline chloride (C5H14NOCl) stimulated OSTα/β-mediated uptake of probe substrates in OSTα/β-overexpressing Flp-In 293 cells (Malinen, et al., 2018), suggesting that the regulation of OSTα/β transport may be ion-dependent. Furthermore, low extracellular pH conditions also stimulated OSTα/β-mediated TCA uptake in OSTα/β-overexpressing Flp-In 293 cells in addition to low Na+ conditions (Malinen, et al., 2018).
Transport mediated by some other members of the SLC transporter family is influenced by extracellular pH, including OATP (SLCO) (Kobayashi, et al., 2003; Leuthold, et al., 2009; Stieger & Hagenbuch, 2014) and monocarboxylate transporters (MCT/SLC16A) (Halestrap & Price, 1999). Mechanisms of transport are fundamental to our understanding of individual transporters and transport protein families, and could be linked to health and disease, but there are still many knowledge gaps that need to be addressed in future investigations, particularly with respect to OSTα/β.
5. Structure of OSTα/β
In contrast to many well-studied transporters, OSTα/β consists of two different protein subunits, OSTα and OSTβ, encoded by the SLC51A and SLC51B genes, respectively. When OSTα/β was discovered in the little skate, OSTα protein consisting of 352 amino acids with seven putative transmembrane domains (TMDs) and OSTβ protein consisting of 182 amino acids and one or two putative TMDs were identified (Wang, et al., 2001). Human OSTα is a protein of 340 amino acids with seven potential TMDs, whereas OSTβ consists of 128 amino acids with a single predicted TMD (Christian, Li, Hinkle, & Ballatori, 2012). X-ray crystallography and high-resolution cryo-electron microscopy studies of OSTα/β are lacking, but valuable information on OSTα/β structure has been generated using other approaches, including topology-prediction algorithms, protein subunit expression and mutagenesis. Differences in the prediction algorithms of available topology-prediction models estimate five-to-seven TMDs in OSTα. However, the transmembrane hidden Markov model (Krogh, Larsson, von Heijne, & Sonnhammer, 2001), one of the most commonly applied topology-prediction tools used by the Universal Protein Resource (UniProt), among several other models predict that OSTα likely contains seven TMDs, and OSTβ only one (Figure 2). Although OSTα and OSTβ depend on each other for stable expression and function at the plasma membrane, the interaction of the two subunits is unresolved, and the exact stoichiometry of the OSTα/β heteromer is unknown. Protein expression studies suggest that OSTα/β may exist in the form of a heterodimer (i.e., OSTα-OSTβ), or as a heteromultimer consisting of several units of OSTα and/or OSTβ, such as OSTα-OSTα-OSTβ (Li, et al., 2007; Schwarz, 2012). Also, there is some evidence suggesting that OSTα is able to form homodimers (Li, et al., 2007). Whether disproportionate regulation in the expression of the OSTα and OSTβ subunits impacts the type of heteromeric interaction and function of OSTα/β is unknown.
Figure 2.
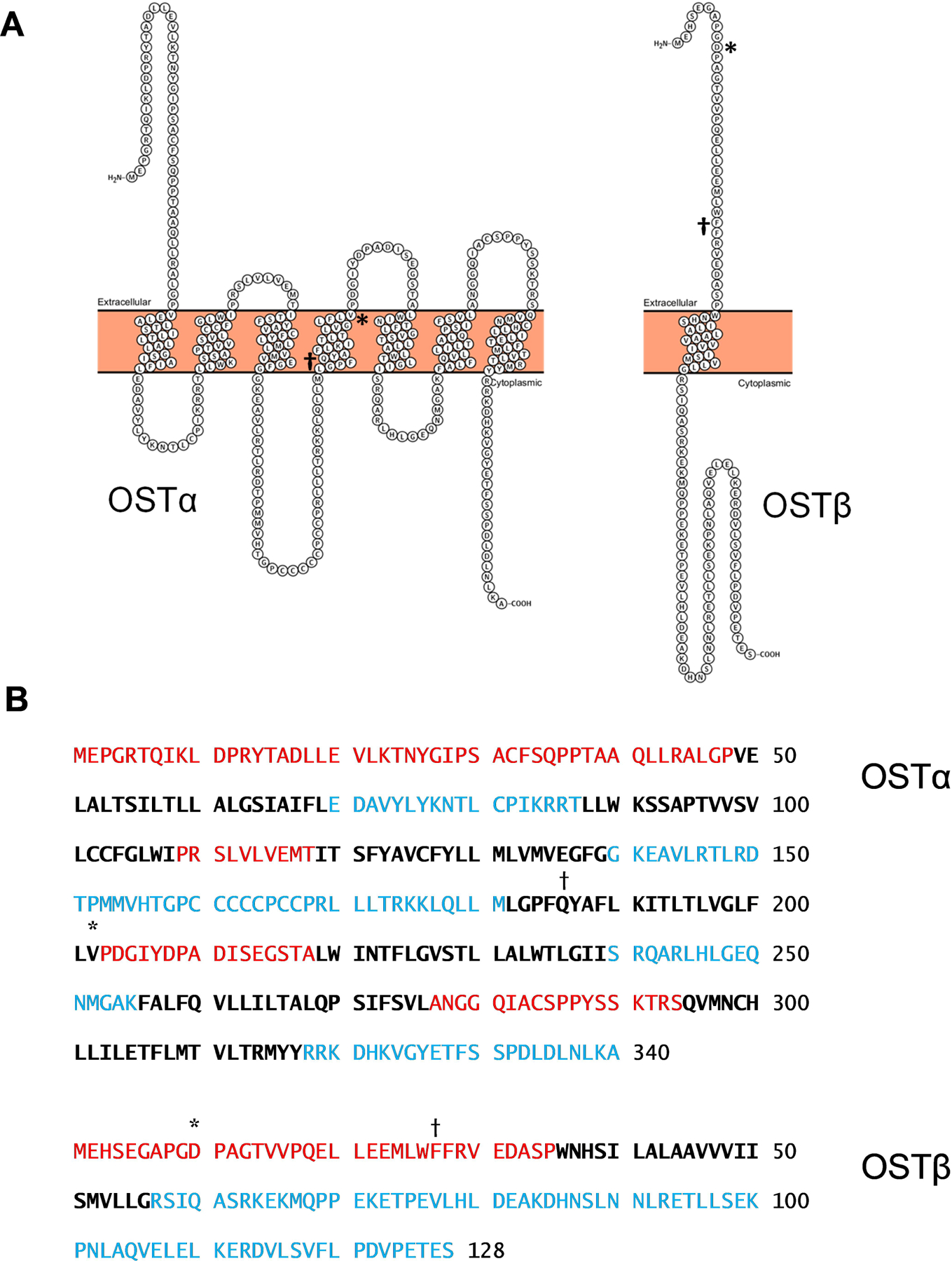
Membrane Topology of OSTα and OSTβ According to the Transmembrane Hidden Markov Model (TMHMM). The figures of the two p otein subunits in (A) were generated using Protter v1.0, an open-source tool for visualization of the extracellular, transmembrane and intracellular domains of membrane proteins. In (B), red text represents extracellular residues; bold black text represents transmembrane residues; blue text represents intracellular residues. *, amino acids with common missense mutations in the general population (see Tables 7 and 8); †, homozygous mutations p.Q186* (premature st p codon) in SLC51A and p.F27fs (frame shift leading to premature stop codon at position 50) in SLC51B were found in the first cases of OSTα (Gao, et al., 2019) and OSTβ (Sultan, et al., 2018) deficiency, respectively.
Several studies have evaluated the role of structural domains essential for the interaction between OSTα and OSTβ. The first of these studies examined the impact of truncating the amino- (N-) and carboxy- (C-)terminal fragments of OSTα on the interaction with OSTβ (Sun, et al., 2007). While the C-terminal tail (23 amino acids) and five amino acids of OSTα’s predicted last TMD were not essential for the interaction with OSTβ, truncation of the N-terminal tail (48 amino acids) and two amino acids of OSTα’s predicted first TMD eliminated the protein interaction, and resulted in intracellular accumulation of both subunits (Sun, et al., 2007).
When mouse OSTα and OSTβ are co-expressed, the OSTα subunit undergoes N-glycosylation-dependent maturation (Dawson, et al., 2005). N-glycosylation is typically associated with the Asn-X-Ser/Thr motif, where X can be any amino acid (Soroka, et al., 2008), except proline. While the mouse, rat and skate have this consensus motif in the N-terminus of the OSTα subunit, the human protein lacks this motif (Soroka, et al., 2008). N-glycosylation also appears to take place in human OSTα (Soroka, et al., 2008), as reported for some other proteins lacking the consensus motif (Valliere-Douglass, et al., 2009). Western blot experiments indicated that mouse and human OSTα have an unglycosylated form (~31 kDa), a core precursor, immature form (~35 kDa) found in the endoplasmic reticulum that is likely glycosylated, and a mature, complex glycoprotein (~40 kDa) (Dawson, et al., 2005; Soroka, et al., 2008). While the mature, glycosylated form of OSTα is generated upon co-expression of OSTβ, immunoprecipitation experiments and studies with the glycosylation inhibitor tunicamycin have shown that OSTα glycosylation is not required for heteromerization with OSTβ, nor for expression at the plasma membrane (Dawson, et al., 2005; Soroka, et al., 2008).
Multiple research groups have examined the structural domains of the relatively small OSTβ subunit. Two studies have indicated that the N-terminal tail of OSTβ is critical for transport activity and membrane localization (Sun, et al., 2012; Xu, et al., 2016). The deletion of 35 amino acids of OSTβ’s N-terminus prevented proper interaction with OSTα and expression at the membrane (Sun, et al., 2012). In a follow-up study, substituting the two leucines in the highly conserved acidic di-leucine motif (-EL20L21EE) at the extracellular N-terminus of OSTβ with two alanines abolished OSTβ’s association with OSTα, and resulted in a lack of expression of both subunits at the plasma membrane (Xu, et al., 2016). However, a more recent study with chimeras of OSTβ and the single TMD receptor activity-modifying protein 1 suggests that OSTβ’s TMD is important in the interaction with OSTα and for maintaining OSTα/β transport function (Christian & Hinkle, 2017). Replacement of OSTβ’s extracellular N-terminal domain by a segment of receptor activity-modifying protein 1 resulted in no loss of function, while the cytoplasmic C-terminal domain demonstrated an involvement with OSTα association (Christian & Hinkle, 2017). Finally, there is clinical evidence from two brothers with OSTβ deficiency (leading to congenital diarrhea and mild cholestasis) that OSTβ’s amino acid sequence from codon position 27 onward, which includes a portion of OSTβ’s extracellular N-terminus as well as OSTβ’s entire predicted TMD and C-terminus, is important for OSTα/β protein expression and transport activity (Sultan, et al., 2018).
More work is needed to elucidate the exact stoichiometry of OSTα/β and the roles and structures of the individual subunits, including the identification of potential sites for, and types of, post-translational modifications besides glycosylation. Molecular modeling approaches may lead to a better understanding of OSTα/β function in health and disease. For example, crystal structures of acetyl CoA synthetase and leucyl-tRNA synthetase have been used to construct a homology model of human OSTβ to clarify the impact of a synthetic mutation (Xu, et al., 2016). Establishing the overall structure of OSTα/β will likely facilitate ongoing and stimulate novel investigations of OSTα/β, and aid in predicting the potential for drug- and/or disease-mediated alterations in function.
6. Genetic variation in SLC51A and SLC51B
Transcript ENST00000296327 is the protein-coding SLC51A transcript with the highest overall expression in the human body (Table 2); this transcript codes for the 340 amino acid form of OSTα. Three other SLC51A transcripts (ENST00000415111, ENST00000416660 and ENST00000428985) also contain an open reading frame, but it is unclear whether these shorter protein-coding sequences (23, 66 and 212 amino acids, respectively) result in functional protein. OSTβ only has one reported transcript (ENST00000334287). Numerous common and rare variants have been detected in SLC51A and SLC51B by the 1000 Genomes Project, the Exome Aggregation Consortium, and others. These variants are located in many types of transcript structure locations, with intron variants being the most common, followed by 239 SLC51A and 92 SLC51B missense (i.e., resulting in an amino acid substitution) variants (Table 5). Some of these are located within the splice site region, within 1–3 bases of the exon boundary. A variety of tools have been employed to predict the functional consequences of these missense variants, including Sorting Intolerant From Tolerant [SIFT; (Kumar, Henikoff, & Ng, 2009)], Polymorphism Phenotyping v2 [PolyPhen-2; (Adzhubei, et al., 2010)], Rare Exome Variant Ensemble Learner [REVEL; (Ioannidis, et al., 2016)] and MutationAssessor (Reva, Antipin, & Sander, 2011) that use tool-specific algorithms to arrive at predictions (Table 6). Only 1 out of 239 SLC51A missense variants (rs939885, V202I) and 1 out of 92 SLC51B missense variants (rs537053592, D10A) are common with a minor allele frequency (MAF) ≥ 0.01 (Tables 7–8). Both of these are predicted to be tolerated by SIFT, benign by PolyPhen-2, likely benign by REVEL, and neutral by MutationAssessor. Only some rare variants found in both genes are predicted to have potentially harmful consequences to the function of the protein. Although these rare variants have been detected in a few individuals (e.g., by the Exome Aggregation Consortium), their health status related to bile acid homeostasis or liver disease is unknown. Further work is needed to obtain more information on the physiological/pathophysiological consequences of the different variants found in both genes.
Table 5.
Distinct Genetic Variants Reported in the General Population in SLC51A and SLC51B Categorized by Variant Type.
| Genetic Variant Type | SLC51A | SLC51B |
|---|---|---|
| 3’ UTR Variant | 44 | 73 |
| 5’ UTR Variant | 83 | 68 |
| Coding Sequence Variant | 68 | 26 |
| Frameshift Variant | 10 | 9 |
| Inframe Deletion | 5 | 2 |
| Inframe Insertion | 2 | 1 |
| Intron Variant | 4,263 | 1,798 |
| Missense Variant | 226 | 89 |
| Missense Variant~Splice Region Variant | 13 | 3 |
| Protein Altering Variant | 1 | 0 |
| Splice Acceptor Variant | 4 | 3 |
| Splice Acceptor Variant~Intron Variant | 2 | 0 |
| Splice Donor Variant | 7 | 2 |
| Splice Region Variant~5’ UTR Variant | 0 | 2 |
| Splice Region Variant~Coding Sequence Variant | 7 | 0 |
| Splice Region Variant~Intron Variant | 46 | 15 |
| Splice Region Variant~Synonymous Variant | 8 | 1 |
| Start Lost | 0 | 2 |
| Start Retained Variant | 1 | 0 |
| Stop Gained | 6 | 1 |
| Stop Gained~Frameshift Variant | 1 | 0 |
| Stop Lost | 1 | 0 |
| Synonymous Variant | 103 | 28 |
| Total | 4,901 | 2,123 |
The data were obtained from Ensembl version 92.38. UTR, untranslated region.
Table 6.
Tools Predicting the Functional Consequence of Distinct Missense Variants Reported in the General Population in SLC51A and SLC51B.
| SLC51A | SLC51B | ||||
|---|---|---|---|---|---|
| Prediction Tool | Classification of Variant Tolerability | Missense Variant | Missense Variant~Splice Region Variant | Missense Variant | Missense Variant~Splice Region Variant |
| SIFT | Tolerated | 104 | 6 | 52 | 0 |
| Tolerated - Low Confidence | 1 | 0 | 0 | 0 | |
| Deleterious - Low Confidence | 1 | 0 | 0 | 0 | |
| Deleterious | 120 | 7 | 37 | 3 | |
| PolyPhen-2 | Benign | 113 | 7 | 55 | 1 |
| Possibly Damaging | 38 | 2 | 19 | 1 | |
| Probably Damaging | 75 | 4 | 15 | 1 | |
| REVEL | NA | 0 | 2 | 0 | 1 |
| Likely Disease Causing | 14 | 2 | 0 | 0 | |
| Neutral | 9 | 0 | 17 | 1 | |
| MutAs | NA | 0 | 2 | 0 | 1 |
| Low | 41 | 0 | 39 | 0 | |
| Medium | 176 | 11 | 33 | 1 | |
Data were obtained from Ensembl version 92.38. Predictions of whether the observed missense variants in SLC51A and SLC51B are harmful to the function of the protein were made based on four tools: SIFT, Sorting Intolerant From Tolerant; PolyPhen-2, Polymorphism Phenotyping v2; REVEL, Rare Exome Variant Ensemble Learner; MutAs, MutationAssessor. NA, not available.
Table 7.
Common Variants in SLC51A.
| Variant ID | Alleles | MAF Allele | MAF | Class | Type | Variant ID | Alleles | MAF Allele | MAF | Class | Type |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs111754451 | A/- | - | 0.01 | Deletion | Intron Variant | rs111996482 | C/T | T | 0.04 | SNP | Intron Variant |
| rs59792018 | C/G/T | G | 0.01 | SNP | Intron Variant | rs78843541 | T/C | C | 0.05 | SNP | Intron Variant |
| rs559074935 | A/G | G | 0.01 | SNP | Intron Variant | rs182482018 | A/G | G | 0.06 | SNP | Intron Variant |
| rs144777514 | T/C | C | 0.01 | SNP | Intron Variant | rs78622644 | T/G | G | 0.06 | SNP | Intron Variant |
| rs12633332 | G/C/T | T | 0.01 | SNP | Intron Variant | rs138047603 | A/C | C | 0.07 | SNP | Intron Variant |
| rs74350809 | G/A | A | 0.01 | SNP | Intron Variant | rs78736929 | T/C/G | C | 0.08 | SNP | Intron Variant |
| rs73083261 | C/T | T | 0.011 | SNP | Intron Variant | rs75096911 | C/T | T | 0.09 | SNP | Intron Variant |
| rs115944258 | G/A | A | 0.011 | SNP | Intron Variant | rs12638322 | C/T | T | 0.1 | SNP | Intron Variant |
| rs78135195 | G/A | A | 0.011 | SNP | Intron Variant | rs35551560 | G/A/T | A | 0.12 | SNP | Intron Variant |
| rs115746134 | A/G | G | 0.011 | SNP | Intron Variant | rs11185518 | C/T | T | 0.17 | SNP | Intron Variant |
| rs143984572 | A/G | G | 0.012 | SNP | Intron Variant | rs78771288 | A/G | G | 0.18 | SNP | Intron Variant |
| rs9835817 | T/C | C | 0.012 | SNP | Intron Variant | rs56030157 | C/A/G/T | T | 0.21 | SNP | 5’ UTR Variant |
| rs142534043 | C/T | T | 0.012 | SNP | Intron Variant | rs78530091 | C/T | T | 0.24 | SNP | Intron Variant |
| rs144713505 | C/T | T | 0.013 | SNP | Intron Variant | rs4916521 | G/A | A | 0.25 | SNP | Intron Variant |
| rs116735912 | C/T | T | 0.013 | SNP | Intron Variant | rs35516030 | G/- | - | 0.26 | Deletion | Intron Variant |
| rs146021046 | G/T | T | 0.013 | SNP | Intron Variant | rs73212130 | G/A/C | C | 0.26 | SNP | Intron Variant |
| rs147722295 | C/T | T | 0.013 | SNP | Intron Variant | rs73083248 | A/G | G | 0.26 | SNP | Intron Variant |
| rs367976475 | C/T | T | 0.013 | SNP | Intron Variant | rs71323710 | A/G/T | G | 0.27 | SNP | Intron Variant |
| rs115878588 | G/A | A | 0.014 | SNP | Intron Variant | rs7429803 | C/G | G | 0.3 | SNP | Intron Variant |
| rs111732200 | C/T | T | 0.014 | SNP | Intron Variant | rs72611184 | C/T | T | 0.31 | SNP | Intron Variant |
| rs116446766 | C/T | T | 0.015 | SNP | Intron Variant | rs13064065 | G/A | A | 0.31 | SNP | Intron Variant |
| rs191597067 | A/G | G | 0.015 | SNP | Intron Variant | rs7625886 | T/C | T | 0.31 | SNP | Intron Variant |
| rs138984296 | G/A | A | 0.015 | SNP | Intron Variant | rs73083250 | G/A | A | 0.32 | SNP | Intron Variant |
| rs73083235 | G/T | T | 0.016 | SNP | Intron Variant | rs1543975 | T/C | C | 0.34 | SNP | Intron Variant |
| rs138755866 | G/A | A | 0.016 | SNP | Intron Variant | rs7641135 | T/C | C | 0.37 | SNP | Intron Variant |
| rs76992230 | C/T | T | 0.017 | SNP | Intron Variant | rs67044678 | G/A | A | 0.37 | SNP | Intron Variant |
| rs111386747 | C/T | T | 0.017 | SNP | Intron Variant | rs56867360 | C/T | T | 0.39 | SNP | Intron Variant |
| rs114844186 | G/A | A | 0.018 | SNP | Intron Variant | rs9840089 | G/A | G | 0.39 | SNP | Intron Variant |
| rs73083243 | G/A | A | 0.019 | SNP | Intron Variant | rs1875088 | G/A | A | 0.39 | SNP | Intron Variant |
| rs9844310 | C/T | T | 0.019 | SNP | Intron Variant | rs7637009 | C/G | G | 0.39 | SNP | Intron Variant |
| rs112960794 | A/G | G | 0.02 | SNP | Intron Variant | rs72611185 | T/A/C | C | 0.39 | SNP | Intron Variant |
| rs79398212 | T/C | C | 0.021 | SNP | Intron Variant | rs67261052 | C/T | T | 0.39 | SNP | Intron Variant |
| rs142969419 | G/A/C | C | 0.021 | SNP | Intron Variant | rs60276076 | C/G/T | G | 0.39 | SNP | Intron Variant |
| rs112174518 | A/G | G | 0.021 | SNP | Intron Variant | rs56653551 | A/G | A | 0.4 | SNP | Intron Variant |
| rs114618254 | G/T | T | 0.022 | SNP | Intron Variant | rs61608982 | G/A | A | 0.4 | SNP | Intron Variant |
| rs113316551 | T/C | C | 0.023 | SNP | Intron Variant | rs11185519 | T/A/C | A | 0.41 | SNP | Intron Variant |
| rs34352044 | C/T | T | 0.023 | SNP | Synonymous Variant (S30) | rs56875789 | C/T | T | 0.41 | SNP | Intron Variant |
| rs76496703 | T/C | C | 0.023 | SNP | Intron Variant | rs7638797 | A/C | C | 0.41 | SNP | Intron Variant |
| rs199580327 | T/- | - | 0.024 | Deletion | Intron Variant | rs9343 | A/G | A | 0.41 | SNP | 3’ UTR Variant |
| rs117874525 | C/T | T | 0.027 | SNP | Intron Variant | rs1476331 | T/C | T | 0.43 | SNP | Intron Variant |
| rs118117782 | T/G | G | 0.027 | SNP | Intron Variant | rs939885 | G/A | A | 0.46 | SNP | Missense Variant (V202I) |
| rs73083237 | G/A/C | A | 0.028 | SNP | Intron Variant | rs68119320 | C/G/T | T | 0.46 | SNP | Intron Variant |
| rs73083239 | G/A | A | 0.028 | SNP | Intron Variant | rs4916441 | A/C/T | T | 0.47 | SNP | Intron Variant |
| rs181512712 | C/T | T | 0.029 | SNP | 5’ UTR Variant | rs58021463 | A/G | G | 0.47 | SNP | Intron Variant |
| rs116216921 | G/A/C/T | C | 0.031 | SNP | Intron Variant | rs4916531 | A/G | A | 0.48 | SNP | Intron Variant |
| rs149309867 | G/A | A | 0.035 | SNP | Intron Variant | rs1476332 | C/G/T | C | 0.48 | SNP | Intron Variant |
| rs113705710 | C/G | G | 0.036 | SNP | Intron Variant | rs4916440 | T/A/C/G | T | 0.48 | SNP | Intron Variant |
| rs73083244 | A/G | G | 0.037 | SNP | Intron Variant | rs6807596 | G/A | G | 0.48 | SNP | Intron Variant |
| rs112216889 | G/A/C | C | 0.039 | SNP | Intron Variant | rs4916519 | C/G/T | C | 0.49 | SNP | Intron Variant |
| rs116434121 | C/T | T | 0.041 | SNP | Intron Variant | rs12491227 | G/A | G | 0.49 | SNP | Intron Variant |
| rs73212134 | G/A | A | 0.041 | SNP | Intron Variant | rs17852687 | T/C | C | 0.5 | SNP | Synonymous Variant (L225) |
Data were obtained from Ensembl version 92.38. MAF, minor allele frequency; SNP, single-nucleotide polymorphism.
Table 8.
Common Variants in SLC51B.
| Variant ID | Alleles | MAF Allele | MAF | Class | Type | Variant ID | Alleles | MAF Allele | MAF | Class | Type |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs151094607 | G/A | A | 0.012 | SNP | Intron Variant | rs2946676 | G/A/C | G | 0.086 | SNP | Intron Variant |
| rs542776151 | T/G | G | 0.014 | SNP | Intron Variant | rs2414870 | C/T | C | 0.086 | SNP | Intron Variant |
| rs2414871 | G/A | A | 0.023 | SNP | Intron Variant | rs2919349 | G/A | G | 0.086 | SNP | Intron Variant |
| rs2919347 | T/C | C | 0.023 | SNP | Intron Variant | rs56390972 | C/T | C | 0.103 | SNP | Intron Variant |
| rs143801232 | AGG/- | - | 0.024 | Deletion | Intron Variant | rs10851744 | C/G/T | C | 0.114 | SNP | Intron Variant |
| rs2919352 | A/G | G | 0.024 | SNP | 3’ UTR Variant | rs537053592 | A/C/T | C | 0.122 | SNP | Missense Variant (D10A) |
| rs2946674 | C/G/T | T | 0.025 | SNP | Intron Variant | rs8026292 | A/G | G | 0.35 | SNP | Intron Variant |
| rs142907492 | C/T | T | 0.025 | SNP | Intron Variant | rs6494510 | T/C | C | 0.352 | SNP | Intron Variant |
| rs74981303 | C/A/T | T | 0.025 | SNP | Intron Variant | rs4238399 | C/T | T | 0.412 | SNP | Intron Variant |
| rs2946675 | T/C | C | 0.028 | SNP | Intron Variant | rs12906276 | G/T | T | 0.423 | SNP | Intron Variant |
| rs2919351 | T/C/G | C | 0.028 | SNP | Intron Variant | rs28688080 | T/A/C | C | 0.457 | SNP | Intron Variant |
| rs2919348 | A/G | G | 0.031 | SNP | Intron Variant | rs11071825 | T/A/C | T | 0.471 | SNP | Intron Variant |
| rs1670 | T/C | C | 0.068 | SNP | 3’ UTR Variant | ||||||
Data were obtained from Ensembl version 92.38. MAF, minor allele frequency; SNP, single-nucleotide polymorphism.
A recent description of two brothers with a homozygous mutation in SLC51B leading to a frameshift at codon 27 (in the extracellular N-terminus of OSTβ) and a premature stop at codon 50 (in OSTβ’s predicted TMD) constitutes the first two clinical cases of a genetic defect in one of the OSTα/β subunits (Sultan, et al., 2018). These brothers were diagnosed with chronic diarrhea, features of cholestatic liver disease (e.g., elevated serum gamma-glutamyltransferase activity) and severe fat-soluble vitamin deficiency. Since previous studies have shown the importance of OSTβ’s TMD for proper interaction with OSTα, and the importance of several amino acids in OSTβ’s intracellular C-terminus for proper orientation in the plasma membrane (Christian & Hinkle, 2017; Christian, et al., 2012), it is not surprising that this frameshift mutation (p.F27fs) would lead to defective OSTα/β transport. In vitro studies with OSTα/β-overexpressing COS cells containing this mutation showed a lack of protein expression of both OSTα and OSTβ subunits, and a strong reduction in TCA uptake compared to OSTα/β-overexpressing cells without this mutation. The observation that these first two clinical cases of OSTα/β dysfunction have a homozygous deletion in a SLC51B codon coupled with the fact that both parents reported no gastrointestinal symptoms, and had normal serum liver chemistries, suggests that only one functional copy of SLC51B may be needed for normal health. Another case report of a male Pakistani toddler (2.5 years old) with cholestasis, elevated liver enzymes and congenital diarrhea, revealed that this patient had a homozygous mutation in SLC51A leading to a premature termination at codon 186 (Gao, et al., 2019). Based on the finding that his unaffected parents and at least one of his siblings were heterozygous for this mutation, it appears that only one functional SLC51A allele is required for normal OSTα/β function.
7. Mechanisms of OSTα/β induction
Transporter expression is tightly regulated at the transcriptional level by nuclear receptors (NRs), including the aryl hydrocarbon receptor, constitutive androstane receptor (CAR), nuclear factor erythroid-2-related factor 2, pregnane X receptor (PXR), peroxisome proliferator-activated receptors, and hepatocyte nuclear factors among others (Ferslew, Köck, & Brouwer, 2014) (Table 9). Transcriptional regulation of OSTα/β has been described previously, including the involvement of various NRs and information on promoter binding sites (Ballatori, et al., 2013; Ballatori, et al., 2009). In the present review, the most recent reports on this subtopic are highlighted. The bile acid-activated FXR is a primary NR that is responsible for the induction of OSTα/β (Soroka, Ballatori, et al., 2010). FXR is involved in the regulation of several other transporters playing important roles in bile acid disposition such as NTCP, OATP1B3, BSEP, and MRP2 (Ferslew, et al., 2014); thus, in this manner bile acids are involved in the regulation of their own levels. Activation of hepatic FXR leads to decreased protein levels of bile acid uptake transporters while efflux transporters, including OSTα/β, are induced. In addition to the regulation of bile acid transporters, hepatic FXR regulates bile acid biosynthesis by inducing expression of small heterodimer partner (SHP), a repressor of gene transcription, and indirectly downregulating the key enzyme cytochrome P450 (CYP) 7A1 that metabolizes cholesterol into the bile acid precursor 7α-hydroxycholesterol (Goodwin, et al., 2000). Furthermore, bile acids in the gastrointestinal tract can activate intestinal FXR leading to induction of the hormone fibroblast growth factor (FGF) 19 in humans (or FGF15 in mice). Secreted FGF15/19 activates the receptor tyrosine kinase FGF receptor 4 present on hepatocytes, resulting in subsequent hepatic CYP7A1 downregulation that appears to be SHP-independent; in addition, cholestatic bile acids in the human liver may activate hepatic FXR and lead to FGF19 and FGF receptor 4 signaling in an autocrine or paracrine fashion (Chiang, 2009). In vitro and mouse studies suggest that OSTα/β is strictly dependent on FXR (Boyer, et al., 2006; Liu, et al., 2014; Soroka, et al., 2008; Zollner, et al., 2006).
Table 9.
Comparison of OSTα/β to Other Transporters Sharing the Same Substrate(s).
| Protein | Gene(s) | Cellular Localizationa | Transport Direction | Driving Force | Transcriptional Regulation | References |
|---|---|---|---|---|---|---|
| OSTα/β | SLC51A, SLC51B | Basolateral (intestine, liver, kidney) | Uptake and Efflux | Substrate gradient | FXR, CAR, PXR, HNF1α, HNF4α, LXRα, RARα, RXRs | (Ballatori, et al., 2005; Okuwaki, et al., 2007; Petrov, et al., 2020; Schaffner, et al., 2015; Seward, et al., 2003; Teng & Piquette-Miller, 2007; Wang, et al., 2001; Xu, et al., 2014) |
| NTCP | SLC10A1 | Basolateral (liver) | Uptake | Na+-dependent (symporter) | FXR, PXR, HNF4α, RXR | (Denson, et al., 2001; Ho, et al., 2004) |
| ASBT | SLC10A2 | Apical (intestine, kidney) | Uptake | Na+-dependent (symporter) | FXR | (Craddock, et al., 1998; Hallen, et al., 2002; Kramer, et al., 1999; Love, et al., 2001) |
| OCT1 | SLC22A1 | Basolateral (intestine, kidney, liver) | Uptake and Efflux | Substrate gradient, membrane potential-influenced | PXR, PPAR, HNF1α, HNF4α | (O’Brien, et al., 2013; Saborowski, Kullak-Ublick, & Eloranta, 2006) |
| OCT2 | SLC22A2 | Basolateral (kidney) | Uptake and Efflux | Substrate gradient | HNF1, HNF4α | (Popowski, et al., 2005) |
| OAT1 | SLC22A6 | Basolateral (liver) | Uptake | Anion/Dicarboxylate exchanger | LXR, HNF1α/β | (Jin, Kikuchi, Saji, Kusuhara, & Sugiyama, 2012; Kittayaruksakul, Soodvilai, Asavapanumas, Muanprasat, & Chatsudthipong, 2012) |
| OAT2 | SLC22A7 | Basolateral (kidney, liver), apical (kidney) | Uptake and Efflux | Anion/Dicarboxylate exchanger | FXR, RARα, RXRα, HNF4α | (Fork, et al., 2011; Kobayashi, et al., 2005; Le Vee, Jouan, Stieger, & Fardel, 2013; Shen, et al., 2015) |
| OAT3 | SLC22A8 | Basolateral (kidney) | Uptake | Anion/Dicarboxylate exchanger | HNF1α/β | (Burckhardt & Burckhardt, 2011; Jin, et al., 2012; Kikuchi, et al., 2006) |
| OAT4 | SLC22A11 | Apical (kidney) | Uptake and Efflux | Anion/Dicarboxylate exchanger | HNF1α/β | (Burckhardt, 2012; Jin, et al., 2012; Zhou, et al., 2004) |
| OAT7 | SLC22A9 | Basolateral (liver) | Uptake | Fatty acid exchanger | HNF1α, HNF4α | (Klein, et al., 2010; Emami Riedmaier, et al., 2016) |
| MATE1 | SLC47A1 | Apical (kidney, liver) | Efflux (Uptake at ≥pH 7.4) | pH-dependent, H+ antiporter | HNF4α | (Lu, et al., 2010; Tanihara, et al., 2007; Tsuda, et al., 2007) |
| MATE2-K | SLC47A2 | Apical (kidney) | Efflux | pH-dependent, H+ antiporter | NA | (Tanihara, et al., 2007; Terada & Inui, 2008) |
| OATP1A2 | SLCO1A2 | Apical (intestine, liver, kidney), Basolateral (liver) | Uptake (and Efflux) | Unclear | SXR, PXR, VDR | (Eloranta, Hiller, Juttner, & Kullak-Ublick, 2012; Lee, et al., 2005; Miki, et al., 2006) |
| OATP1B1 | SLCO1B1 | Basolateral (liver) | Uptake | Substrate gradient, pH-influenced | FXR, PXR, LXRα, HNF1α, HNF3β, HNF4α | (Gui, et al., 2008; Kamiyama, et al., 2007; Ohtsuka, et al., 2006; Rodrigues, et al., 2019) |
| OATP1B3 | SLCO1B3 | Basolateral (liver) | Uptake (and Efflux) | Substrate gradient, pH-influenced | FXR, HNF1α, HNF3β, HNF4α | (Gui, et al., 2008; Kamiyama, et al., 2007; Ohtsuka, et al., 2006; Rodrigues, et al., 2019) |
| OATP2B1 | SLCO2B1 | Apical (intestine), Basolateral (liver, intestine) | Uptake | Substrate gradient, pH-influenced | FXR, PXR, HNF1α, HNF3β, HNF4α | (Hagenbuch & Stieger, 2013; Ohtsuka, et al., 2006; Rodrigues, et al., 2019; Yamada, et al., 2007) |
| OATP4C1 | SLCO4C1 | Basolateral (kidney) | Uptake | Substrate gradient | AhR | (Mikkaichi, et al., 2004; Suzuki, et al., 2011) |
| P-gp | ABCB1 | Apical (intestine, kidney, liver) | Efflux | ATP-dependent | CAR, PXR, HNF1α, VDR | (Callaghan, Crowley, Potter, & Kerr, 2008; Chan, Hoque, Cummins, & Bendayan, 2011; Chow, Durk, Cummins, & Pang, 2011; Kimura, Kioka, Kato, Matsuo, & Ueda, 2007; Rodrigues, et al., 2019) |
| MDR3 | ABCB4 | Apical (liver) | Efflux | ATP-dependent | FXR, LXR, PPAR | (Smith, et al., 2000) |
| BSEP | ABCB11 | Apical (liver) | Efflux | ATP-dependent | FXR, RXR | (Hayashi, et al., 2005; Kis, et al., 2009) |
| MRP1 | ABCC1 | Basolateral (intestine, kidney) | Efflux | ATP-dependent, GSH-influenced | PXR, CAR | (Bakos & Homolya, 2007) |
| MRP2 | ABCC2 | Apical (intestine, kidney, liver) | Efflux | ATP-dependent | FXR, CAR, PXR, HNF4α, VDR, NRF2 | (Arana, Tocchetti, Rigalli, Mottino, & Villanueva, 2016; Rodrigues, et al., 2019) |
| MRP3 | ABCC3 | Basolateral (liver, intestine, kidney) | Efflux | ATP-dependent | PXR, CAR, VDR | (Rodrigues, et al., 2019; Zeng, et al., 2000) |
| MRP4 | ABCC4 | Apical (intestine, kidney), Basolateral (liver) | Efflux | ATP-dependent, GSH-influenced | FXR, CAR, PPAR | (Lai & Tan, 2002) |
| BCRP | ABCG2 | Apical (intestine, liver) | Efflux | ATP-dependent | CAR, PXR, PPAR | (Rodrigues, et al., 2019; Lin, et al., 2017) |
Tissue localization: intestine, kidney and/or liver
ABC, ATP-binding cassette transporter; AhR, aryl hydrocarbon receptor; ATP, adenosine triphosphate; CAR, constitutive androstane receptor; FXR, farnesoid X receptor; GSH, reduced glutathione; HNF, hepatocyte nuclear factor; Km, substrate concentration at one-half of the maximum velocity; LXR, liver X receptor; NA, data not available in the literature; NRF2, nuclear factor erythroid-2-related factor 2; PPAR, peroxisome proliferator-activated receptor; PXR; pregnane X receptor; RAR, retinoid acid receptor; RXR, retinoid X receptor; SLC, solute carrier transporter; SXR, steroid and xenobiotic receptor; VDR, vitamin D receptor
Treatment of primary human hepatocytes with the potent FXR agonist CDCA, a prevalent unconjugated primary bile acid produced in the liver, and obeticholic acid (OCA), a semi-synthetic bile acid used to treat primary biliary cholangitis (PBC), under review for the treatment of NASH, and in development for the treatment of other metabolic disorders, induced SLC51A/OSTα and SLC51B/OSTβ at the mRNA and protein level (Guo, et al., 2018; Jackson, Freeman, & Brouwer, 2016; Liu, et al., 2014). Overall, the secondary bile acid DCA, produced by gut bacteria via dehydroxylation of primary bile acids, and the primary bile acid cholic acid, showed an intermediate ability to regulate FXR target genes (Liu, et al., 2014). While CDCA-mediated induction of SLC51A and SLC51B mRNA was observed in human ileal biopsies (Landrier, et al., 2006), OCA-mediated induction of the SLC51A and SLC51B genes was reported in human liver slices (Ijssennagger, et al., 2016). CDCA also induced SLC51A and SLC51B mRNA in HepG2 and HuH-7 cell lines (Landrier, et al., 2006; Schaffner, et al., 2015; Xu, et al., 2014).
Apart from bile acids, additional compounds, NRs or conditions have been tested as potential mediators of OSTα/β induction. For example, in pigs fed with corn-soybean meal diets containing 5% apple pectin compared to pigs fed cornstarch as the control, ileal, cecal and colonic SLC51A and SLC51B mRNA expression trended higher (Fang, et al., 2018). In primary human hepatocytes cultured under hypoxic conditions, a characteristic of cholestasis, OSTα/β expression was increased. Under low oxygen conditions, hypoxia-inducible factor-1α binds to hypoxia responsive elements in the gene promoters of both OSTα/β subunits (Schaffner, et al., 2015). In a humanized liver mouse model, bile acids were elevated compared to the non-humanized control model; furthermore, hepatic human SLC51A and SLC51B mRNA was increased in the humanized liver mouse model compared to human liver (Chow, et al., 2017). Intestinal murine SLC51A mRNA was elevated in the humanized liver mouse model compared to the non-humanized control model. Some of these alterations in expression may be due to miscommunication between human hepatocytes and the murine intestinal cells. PXR may be involved in negatively regulating SLC51A and SLC51B, among other FXR-regulated genes (Ballatori, et al., 2009). For instance, in cholic acid-fed wild-type mice, the PXR activator 5-pregnen-3β-ol-20-one-16α-carbonitrile downregulated Slc51a and Slc51b gene expression (Teng & Piquette-Miller, 2007). Furthermore, gene expression of Slc51a and Slc51b was elevated in PXR−/− mice compared to wild-type control mice. Antibiotics can result in bile acid elevations by enhancing bile acid synthesis; however, in mice treated with ampicillin or bacitracin/neomycin/streptomycin, ileal OSTα protein was not altered even though ASBT protein levels were elevated compared to control mice (Miyata, Hayashi, Yamakawa, Yamazoe, & Yoshinari, 2015). Experiments with Caco-2 cells demonstrated that liver X receptor-α plays a role in the transcriptional regulation of mouse OSTα/β (Okuwaki, et al., 2007). In the LEE-1 cell line derived from a little skate embryo, the eicosanoid precursor arachidonic acid induced OSTβ, while a lipid mixture containing arachidonic acid induced OSTα (Hwang, Parton, Czechanski, Ballatori, & Barnes, 2008). Studies in HuH-7 and HepG2 cells have suggested that OSTβ is regulated by retinoic acid receptor-α and CAR heteromerized with retinoid X receptor-α (Xu, et al., 2014). Furthermore, OSTα/β can be regulated by ligands of the vitamin D receptor and the glucocorticoid receptor in a species-dependent manner (Khan, Chow, Porte, Pang, & Groothuis, 2009).
Aside from extensive research on transcriptional regulation, the post-translational regulation of OSTα/β has been evaluated. For instance, it is evident that OSTα undergoes N-glycosylation-dependent maturation (see Section 5), although identification of other post-translational modifications warrants further investigation. Finally, whether microRNA-mediated post-transcriptional control of gene expression plays a role in regulating OSTα and OSTβ levels is unknown, but could be involved in organ-specific differences in OSTα/β protein expression.
8. Altered hepatic OSTα/β expression in cholestatic liver conditions, NASH and other disorders
Studies in humans and non-clinical models have demonstrated that cholestatic conditions lead to adaptive responses in bile acid synthesis and transporters (Boyer, et al., 2006; Schaap, et al., 2009; Trauner, Wagner, Fickert, & Zollner, 2005). For instance, treatment of the HepG2 human hepatoma cell line with CDCA (50 μM, 24 hr) increased OSTα, OSTβ, SHP and BSEP mRNA, and OSTα and OSTβ proteins (Boyer, et al., 2006). Furthermore, hepatic transcripts of FGF19, the bile acid synthesis enzyme CYP7A1, and several transporters including OSTα, OSTβ, MRP3 and OATP1B1 were significantly altered in patients with extrahepatic cholestasis (i.e., obstruction of bile flow in the ducts from the liver to the duodenum) compared to control subjects (Schaap, et al., 2009). In several other human cholestatic disorders, OSTα and/or OSTβ are substantially induced, suggesting an important detoxifying role for hepatic OSTα/β-mediated efflux when bile constituents (e.g., bile acids) accumulate in the liver. While OSTα protein usually has low hepatic expression and OSTβ protein tends to be nearly undetectable in healthy livers, hepatic OSTα and particularly OSTβ protein were highly expressed in patients with PBC (Boyer, et al., 2006; Malinen, et al., 2018); elevations of SLC51A mRNA and especially SLC51B mRNA have been reported in PBC. In obstructive cholestatic human livers, both SLC51B mRNA and OSTβ protein were found to be significantly elevated compared to control livers, while SLC51A mRNA was unchanged, and OSTα protein was significantly decreased (Chai, et al., 2015). Most recently, SLC51B mRNA and OSTβ protein were found to be significantly increased in livers from patients with NASH, with OSTα protein also trending higher in NASH livers (Malinen, et al., 2018). NASH, the most severe form of non-alcoholic fatty liver disease, is increasing at an alarming rate with the obesity epidemic. Unlike obstructive/extrahepatic cholestasis and PBC, NASH is not typically characterized by clinical symptoms of cholestasis; nevertheless, some patients with NASH exhibit cholestasis (Sorrentino, et al., 2005). The reduction of bile flow in cholestasis can lead to elevated serum bile acid concentrations, potentially attributable, in part, to overexpression of hepatic OSTα/β and other basolateral efflux transporters serving to protect hepatocytes from toxicity. In addition to overexpression of hepatic OSTα/β in NASH, serum bile acid concentrations also are elevated in NASH (Ferslew, et al., 2015). The increase in hepatic OSTα/β expression in individuals with cholestatic disorders could mean that drug interactions with OSTα/β could affect hepatic OSTα/β-mediated transport, which may increase susceptibility to toxicity.
While SLC51A mRNA was largely unchanged in the livers from common bile duct-ligated mice or rats, SLC51B mRNA increased compared to sham-treated rodents (Boyer, et al., 2006). However, despite the lack of alterations in SLC51A mRNA, hepatic OSTα protein was induced in the common bile duct-ligated rodents. These studies suggest that biliary obstruction induces hepatic SLC51B/OSTβ in both humans and rodents, but that obstructive cholestatic human livers and livers from common bile duct-ligated rodents may regulate SLC51A/OSTα differently, likely due to important species differences. Apart from hepatic conditions, SLC51A and SLC51B mRNA was elevated in the livers from rats with chronic renal failure, a condition associated with increased serum bile acid concentrations and changes in bile acid homeostasis (Gai, et al., 2014).
9. Potential role of OSTα/β in drug development
Although OSTα/β has not been a direct target for new therapeutics, the FXR agonist OCA, which is used for the treatment of PBC, significantly induced OSTα/β and increased the intrinsic basolateral efflux clearance of TCA in sandwich-cultured human hepatocytes (Guo, et al., 2018). However, it is unclear to what extent the enhanced efflux mediated by OSTα/β plays a role in OCA-based PBC treatment. It has been hypothesized that therapeutically inducing OSTα/β expression in cholestasis can protect the liver from hepatotoxicity (Zollner, et al., 2006), and this still appears to be a plausible hepatoprotective mechanism against bile acid-mediated liver injury. Apart from inducing OSTα/β expression, in vitro uptake studies have shown that human OSTα/β transport activity can be stimulated by various compounds (Malinen, et al., 2018); whether stimulation of OSTα/β transport also occurs in vivo remains to be elucidated.
Studies in mice have indicated that inhibition of OSTα/β may have therapeutic purposes [e.g., by activating intestinal FXR and enhancing bile acid signaling (van de Wiel, et al., 2018)], but it is important to emphasize that rodents have a distinct bile acid profile and different bile acid regulation compared to humans (Chiang, 2009; Garcia-Canaveras, Donato, Castell, & Lahoz, 2012; Hofmann, 2004, 2009). Therefore, rodent models may not accurately predict human cholestatic conditions.
Aside from potentially targeting OSTα/β for pharmacotherapeutic purposes, OSTα/β may play an important role in predicting xenobiotic-induced hepatotoxicity in drug development. DILI is a major public health concern for patients and healthcare providers, a primary safety issue in drug development, and a frequent reason for drug withdrawal from the market (Mosedale & Watkins, 2017; Onakpoya, Heneghan, & Aronson, 2016; Perez & Briz, 2009; Temple & Himmel, 2002). Among the various mechanisms underlying DILI, inhibition of bile acid transport and alterations in bile acid homeostasis account for almost half of the DILI cases (Weaver, et al., 2019). Since OSTα/β is important in bile acid homeostasis, it is plausible that proper functioning of this transporter protects patients from DILI. As discussed, the transport function of OSTα/β is inhibited by some drugs associated with DILI (Malinen, Kauttonen, et al., 2019), but interactions with OSTα/β are not evaluated routinely due to lack of clinical evidence. The potential role of OSTα/β in bile acid-mediated DILI remains to be elucidated.
10. Conclusion/outlook
The observed expression of OSTα/β in the intestine, liver and kidneys, the major organs determining the absorption, metabolism and elimination of drugs, the upregulation of OSTα/β in hepatic diseases, the interaction of OSTα/β with drugs, and clinical case reports of diarrhea and cholestasis in patients with homozygous genetic defects in OSTα/β have increased awareness among clinicians and scientists about this understudied transporter. Prior reviews on OSTα/β focused on the role of this protein in bile acid and steroid metabolite transport. The present review provides current knowledge on the pharmacological role of OSTα/β, as well as the expression, structure and function of OSTα/β.
OSTα/β is expressed on the basolateral membrane of various types of epithelial cells and is composed of two subunits, OSTα and OSTβ. The need for the expression of two separate proteins for transporter function on the basolateral membrane separates OSTα/β from the well-known drug transporters. Interestingly, in intestinal cells where the physiological function of OSTα/β is evident under basal conditions, or in hepatocytes under cholestatic conditions, the expression of OSTβ protein is higher than OSTα protein, while the opposite tends to be true in the tissues where the role of OSTα/β is still unclear. Although OSTα/β-mediated transport appears to be largely dependent on substrate concentration, a role for pH and some ions has been noted.
Studies during the last two decades have shown that OSTα/β is an important transporter for the disposition of endogenous compounds, particularly relating to the enterohepatic circulation of bile acids, but the involvement of OSTα/β in drug-drug/drug-bile acid interactions has not been thoroughly characterized. It is now evident that OSTα/β can transport probe substrates that are shared by other clinically relevant drug transporters (Table 4). OSTα/β appears to be a low affinity/high capacity transporter for several substrates (see Section 3.1). This is a complicating factor when trying to determine the extent of involvement of OSTα/β in the transport of, or interaction with, a specific compound of interest. To compare substrate affinities of OSTα/β to other transporters of interest, it is ideal to evaluate these transporters under identical experimental conditions, which is not always possible.
The inhibition of OSTα/β by drugs associated with hepatotoxicity suggests a possible role of OSTα/β in DILI; however, the number of compounds interrupting OSTα/β transport is relatively low even though more than a thousand compounds have been evaluated as inhibitors. This low identification rate may correspond to actual physiological phenomena or it may reflect the lack of sensitivity of in vitro systems used thus far to study OSTα/β. The presence of OSTα/β is often ignored in models predicting disposition or toxicity of test compounds, potentially generating a misleading interpretation of the pharmacokinetics, pharmacodynamics and safety of compounds. Future investigations designed to determine the three-dimensional structure of OSTα/β and to identify novel OSTα/β substrates and inhibitors will aid in the development and use of structure-activity relationship models, and advance knowledge regarding the role of OSTα/β in drug disposition and drug interactions.
12. Acknowledgements
This work was supported, in part, by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under award number F31DK120196 (James J. Beaudoin), and the National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM122576 (Dr. Kim L.R. Brouwer). Dr. Melina M. Malinen received funding from the European Union’s Horizon 2020 Research and Innovation program under the Marie Skłodowska-Curie grant agreement number 799510. We thank Drs. Paavo Honkakoski and William A. Murphy Jr. for providing invaluable feedback on the manuscript.
13. Role of the funding source
Any opinions, findings, conclusions, or recommendations expressed in this publication are those of the authors and do not necessarily reflect the views of the National Institutes of Health or the European Union’s Horizon 2020 Research and Innovation program.
List of Abbreviations*
- ASBT/SLC10A2
apical sodium-dependent bile acid transporter
- ATP
adenosine triphosphate
- ADME-Tox
absorption, distribution, metabolism, excretion and toxicity
- BCRP/ABCG2
breast cancer resistance protein
- BSEP/ABCB11
bile salt export pump
- C-
carboxy-
- CAR
constitutive androstane receptor
- CDCA
chenodeoxycholate
- CYP
cytochrome P450
- DCA
deoxycholate
- DHEAS
dehydroepiandrosterone sulfate
- DILI
drug-induced liver injury
- ES
estrone sulfate
- FGF
fibroblast growth factor
- FXR
farnesoid X receptor
- HEK
human embryonic kidney
- MAF
minor allele frequency
- MCT/SLC16A
monocarboxylate transporters
- MDCK
Madin-Darby canine kidney
- MRP/ABCC
multidrug resistance-associated protein
- N-
amino-
- NASH
nonalcoholic steatohepatitis
- NTCP/SLC10A1
Na+-taurocholate cotransporting polypeptide
- NR
nuclear receptor
- OATP/SLCO
organic anion transporting polypeptide
- OCA
obeticholic acid
- OSTα/SLC51A
organic solute transporter alpha
- OSTβ/SLC51B
organic solute transporter beta
- P-gp/MDR1/ABCB1
P-glycoprotein
- PBC
primary biliary cholangitis
- PolyPhen-2
Polymorphism Phenotyping v2
- PXR
pregnane X receptor
- REVEL
Rare Exome Variant Ensemble Learner
- SIFT
Sorting Intolerant From Tolerant
- SLC
solute carrier
- TCA
taurocholate
- TMD
transmembrane domain
*In this review, all letters of mRNA and protein names for all species are in uppercase. Gene names are italicized, with only the first letter in uppercase for mouse gene names.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest statement
Kim L.R. Brouwer is a co-inventor of the sandwich-cultured hepatocyte technology for quantification of biliary excretion (B-CLEAR®) and related technologies, which have been licensed exclusively to Qualyst Transporter Solutions, acquired by BioIVT. James J. Beaudoin and Melina M. Malinen declare no conflicts of interest.
15. References
- Abe T, Unno M, Onogawa T, Tokui T, Kondo TN, Nakagomi R, et al. (2001). LST-2, a human liver-specific organic anion transporter, determines methotrexate sensitivity in gastrointestinal cancers. Gastroenterology, 120, 1689–1699. [DOI] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. (2010). A method and server for predicting damaging missense mutations. Nat Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali I, Welch MA, Lu Y, Swaan PW, & Brouwer KLR (2017). Identification of novel MRP3 inhibitors based on computational models and validation using an in vitro membrane vesicle assay. Eur J Pharm Sci, 103, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alqahtani S (2017). In silico ADME-Tox modeling: progress and prospects. Expert Opin Drug Metab Toxicol, 13, 1147–1158. [DOI] [PubMed] [Google Scholar]
- Arana MR, Tocchetti GN, Rigalli JP, Mottino AD, & Villanueva SS (2016). Physiological and pathophysiological factors affecting the expression and activity of the drug transporter MRP2 in intestine. Impact on its function as membrane barrier. Pharmacol Res, 109, 32–44. [DOI] [PubMed] [Google Scholar]
- Baird FE, Beattie KJ, Hyde AR, Ganapathy V, Rennie MJ, & Taylor PM (2004). Bidirectional substrate fluxes through the system N (SNAT5) glutamine transporter may determine net glutamine flux in rat liver. J Physiol, 559, 367–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakos E, & Homolya L (2007). Portrait of multifaceted transporter, the multidrug resistance-associated protein 1 (MRP1/ABCC1). Pflugers Arch, 453, 621–641. [DOI] [PubMed] [Google Scholar]
- Balakrishnan A, Hussainzada N, Gonzalez P, Bermejo M, Swaan PW, & Polli JE (2007). Bias in estimation of transporter kinetic parameters from overexpression systems: Interplay of transporter expression level and substrate affinity. J Pharmacol Exp Ther, 320, 133–144. [DOI] [PubMed] [Google Scholar]
- Balakrishnan A, Wring SA, & Polli JE (2006). Interaction of native bile acids with human apical sodium-dependent bile acid transporter (hASBT): influence of steroidal hydroxylation pattern and C-24 conjugation. Pharm Res, 23, 1451–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatori N (2005). Biology of a novel organic solute and steroid transporter, OSTalpha-OSTbeta. Exp Biol Med (Maywood), 230, 689–698. [DOI] [PubMed] [Google Scholar]
- Ballatori N (2011). Pleiotropic functions of the organic solute transporter Ostalpha-Ostbeta. Dig Dis, 29, 13–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatori N, Christian WV, Lee JY, Dawson PA, Soroka CJ, Boyer JL, et al. (2005). OSTalpha-OSTbeta: a major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology, 42, 1270–1279. [DOI] [PubMed] [Google Scholar]
- Ballatori N, Christian WV, Wheeler SG, & Hammond CL (2013). The heteromeric organic solute transporter, OSTalpha-OSTbeta/SLC51: a transporter for steroid-derived molecules. Mol Aspects Med, 34, 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatori N, Fang F, Christian WV, Li N, & Hammond CL (2008). Ostalpha-Ostbeta is required for bile acid and conjugated steroid disposition in the intestine, kidney, and liver. Am J Physiol Gastrointest Liver Physiol, 295, G179–G186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatori N, Li N, Fang F, Boyer JL, Christian WV, & Hammond CL (2009). OST alpha-OST beta: a key membrane transporter of bile acids and conjugated steroids. Front Biosci (Landmark Ed), 14, 2829–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazquez AG, Briz O, Romero MR, Rosales R, Monte MJ, Vaquero J, et al. (2012). Characterization of the role of ABCG2 as a bile acid transporter in liver and placenta. Mol Pharmacol, 81, 273–283. [DOI] [PubMed] [Google Scholar]
- Bosdriesz E, Wortel MT, Haanstra JR, Wagner MJ, de la Torre Cortes P, & Teusink B (2018). Low affinity uniporter carrier proteins can increase net substrate uptake rate by reducing efflux. Sci Rep, 8, 5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer JL, Trauner M, Mennone A, Soroka CJ, Cai SY, Moustafa T, et al. (2006). Upregulation of a basolateral FXR-dependent bile acid efflux transporter OSTalpha-OSTbeta in cholestasis in humans and rodents. Am J Physiol Gastrointest Liver Physiol, 290, G1124–1130. [DOI] [PubMed] [Google Scholar]
- Burckhardt G (2012). Drug transport by Organic Anion Transporters (OATs). Pharmacol Ther, 136, 106–130. [DOI] [PubMed] [Google Scholar]
- Burckhardt G, & Burckhardt BC (2011). In vitro and in vivo evidence of the importance of organic anion transporters (OATs) in drug therapy. Handb Exp Pharmacol, 29–104. [DOI] [PubMed]
- Callaghan R, Crowley E, Potter S, & Kerr ID (2008). P-glycoprotein: so many ways to turn it on. J Clin Pharmacol, 48, 365–378. [DOI] [PubMed] [Google Scholar]
- Cha SH, Sekine T, Fukushima JI, Kanai Y, Kobayashi Y, Goya T, et al. (2001). Identification and characterization of human organic anion transporter 3 expressing predominantly in the kidney. Mol Pharmacol, 59, 1277–1286. [DOI] [PubMed] [Google Scholar]
- Cha SH, Sekine T, Kusuhara H, Yu E, Kim JY, Kim DK, et al. (2000). Molecular cloning and characterization of multispecific organic anion transporter 4 expressed in the placenta. J Biol Chem, 275, 4507–4512. [DOI] [PubMed] [Google Scholar]
- Chai J, Feng X, Zhang L, Chen S, Cheng Y, He X, et al. (2015). Hepatic expression of detoxification enzymes is decreased in human obstructive cholestasis due to gallstone biliary obstruction. PLoS One, 10, e0120055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan GN, Hoque MT, Cummins CL, & Bendayan R (2011). Regulation of P-glycoprotein by orphan nuclear receptors in human brain microvessel endothelial cells. J Neurochem, 118, 163–175. [DOI] [PubMed] [Google Scholar]
- Chaudhry A, Chung G, Lynn A, Yalvigi A, Brown C, Ellens H, et al. (2018). Derivation of a System-Independent Ki for P-glycoprotein Mediated Digoxin Transport from System-Dependent IC50 Data. Drug Metab Dispos, 46, 279–290. [DOI] [PubMed] [Google Scholar]
- Chiang JY (2009). Bile acids: regulation of synthesis. J Lipid Res, 50, 1955–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi MK, Shin HJ, Choi YL, Deng JW, Shin JG, & Song IS (2011). Differential effect of genetic variants of Na(+)-taurocholate co-transporting polypeptide (NTCP) and organic anion-transporting polypeptide 1B1 (OATP1B1) on the uptake of HMG-CoA reductase inhibitors. Xenobiotica, 41, 24–34. [DOI] [PubMed] [Google Scholar]
- Chow EC, Durk MR, Cummins CL, & Pang KS (2011). 1Alpha,25-dihydroxyvitamin D3 up-regulates P-glycoprotein via the vitamin D receptor and not farnesoid X receptor in both fxr(−/−) and fxr(+/+) mice and increased renal and brain efflux of digoxin in mice in vivo. J Pharmacol Exp Ther, 337, 846–859. [DOI] [PubMed] [Google Scholar]
- Chow EC, Quach HP, Zhang Y, Wang JZ, Evans DC, Li AP, et al. (2017). Disrupted Murine Gut-to-Human Liver Signaling Alters Bile Acid Homeostasis in Humanized Mouse Liver Models. J Pharmacol Exp Ther, 360, 174–191. [DOI] [PubMed] [Google Scholar]
- Christian WV, & Hinkle PM (2017). Global functions of extracellular, transmembrane and cytoplasmic domains of organic solute transporter beta-subunit. Biochem J, 474, 1981–1992. [DOI] [PubMed] [Google Scholar]
- Christian WV, Li N, Hinkle PM, & Ballatori N (2012). beta-Subunit of the Ostalpha-Ostbeta organic solute transporter is required not only for heterodimerization and trafficking but also for function. J Biol Chem, 287, 21233–21243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couto N, Al-Majdoub Z, Gibson S, Davies P, Achour B, Harwood MD, et al. (2020). Quantitative Proteomics of Clinically Relevant Drug-Metabolizing Enzymes and Drug Transporters and Their Inter-correlations in the Human Small Intestine. Drug Metab Dispos [DOI] [PMC free article] [PubMed]
- Craddock AL, Love MW, Daniel RW, Kirby LC, Walters HC, Wong MH, et al. (1998). Expression and transport properties of the human ileal and renal sodium - dependent bile acid transporter. Am J Physiol, 274, G157–169. [DOI] [PubMed] [Google Scholar]
- Cui Y, Konig J, Leier I, Buchholz U, & Keppler D (2001). Hepatic uptake of bilirubin and its conjugates by the human organic anion transporter SLC21A6. J Biol Chem, 276, 9626–9630. [DOI] [PubMed] [Google Scholar]
- Dahan A, & Amidon GL (2009). Small intestinal efflux mediated by MRP2 and BCRP shifts sulfasalazine intestinal permeability from high to low, enabling its colonic targeting. The American Journal of Physiology: Gastrointestinal and Liver Physiology 297, G371–G377. [DOI] [PubMed] [Google Scholar]
- Dawson PA, Hubbert M, Haywood J, Craddock AL, Zerangue N, Christian WV, et al. (2005). The heteromeric organic solute transporter alpha-beta, Ostalpha-Ostbeta, is an ileal basolateral bile acid transporter. J Biol Chem, 280, 6960–6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson PA, Hubbert ML, & Rao A (2010). Getting the mOST from OST: Role of organic solute transporter, OSTalpha-OSTbeta, in bile acid and steroid metabolism. Biochim Biophys Acta, 1801, 994–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graan AJ, Lancaster CS, Obaidat A, Hagenbuch B, Elens L, Friberg LE, et al. (2012). Influence of polymorphic OATP1B-type carriers on the disposition of docetaxel. Clin Cancer Res, 18, 4433–4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denson LA, Sturm E, Echevarria W, Zimmerman TL, Makishima M, Mangelsdorf DJ, et al. (2001). The orphan nuclear receptor, shp, mediates bile acid-induced inhibition of the rat bile acid transporter, ntcp. Gastroenterology, 121, 140–147. [DOI] [PubMed] [Google Scholar]
- Dong Z, Ekins S, & Polli JE (2015). A substrate pharmacophore for the human sodium taurocholate co-transporting polypeptide. Int J Pharm, 478, 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durmus S, Naik J, Buil L, Wagenaar E, van Tellingen O, & Schinkel AH (2014). In vivo disposition of doxorubicin is affected by mouse Oatp1a/1b and human OATP1A/1B transporters. Int J Cancer, 135, 1700–1710. [DOI] [PubMed] [Google Scholar]
- Elbing K, Larsson C, Bill RM, Albers E, Snoep JL, Boles E, et al. (2004). Role of hexose transport in control of glycolytic flux in Saccharomyces cerevisiae. Appl Environ Microbiol, 70, 5323–5330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellens H, Meng Z, Le Marchand SJ, & Bentz J (2018). Mechanistic kinetic modeling generates system-independent P-glycoprotein mediated transport elementary rate constants for inhibition and, in combination with 3D SIM microscopy, elucidates the importance of microvilli morphology on P-glycoprotein mediated efflux activity. Expert Opin Drug Metab Toxicol, 14, 571–584. [DOI] [PubMed] [Google Scholar]
- Ellis LC, Hawksworth GM, & Weaver RJ (2013). ATP-dependent transport of statins by human and rat MRP2/Mrp2. Toxicol Appl Pharmacol, 269, 187–194. [DOI] [PubMed] [Google Scholar]
- Eloranta JJ, Hiller C, Juttner M, & Kullak-Ublick GA (2012). The SLCO1A2 gene, encoding human organic anion-transporting polypeptide 1A2, is transactivated by the vitamin D receptor. Mol Pharmacol, 82, 37–46. [DOI] [PubMed] [Google Scholar]
- Emami Riedmaier A, Burk O, Eijck BA, Schaeffeler E, Klein K, Fehr S, et al. (2016). Variability in hepatic expression of organic anion transporter 7/SLC22A9, a novel pravastatin uptake transporter: impact of genetic and regulatory factors. Pharmacogenomics J, 16, 341–351. [DOI] [PubMed] [Google Scholar]
- Fang F, Christian WV, Gorman SG, Cui M, Huang J, Tieu K, et al. (2010). Neurosteroid transport by the organic solute transporter OSTalpha-OSTbeta. J Neurochem, 115, 220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang W, Zhang L, Meng Q, Wu W, Lee YK, Xie J, et al. (2018). Effects of dietary pectin on the profile and transport of intestinal bile acids in young pigs. J Anim Sci, 96, 4743–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrebee CB, Li J, Haywood J, Pachura K, Robinson BS, Hinrichs BH, et al. (2018). Organic Solute Transporter alpha-beta Protects Ileal Enterocytes From Bile Acid-Induced Injury. Cell Mol Gastroenterol Hepatol, 5, 499–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferslew BC, Köck K, & Brouwer KLR (2014). Drug Transport in the Liver You G & Morris ME (Eds.), In Drug Transporters: Molecular Characterization and Role in Drug Disposition (2nd ed., pp. 245–271). Hoboken, New Jersey: John Wiley & Sons, Inc. [Google Scholar]
- Ferslew BC, Xie G, Johnston CK, Su M, Stewart PW, Jia W, et al. (2015). Altered Bile Acid Metabolome in Patients with Nonalcoholic Steatohepatitis. Dig Dis Sci, 60, 3318–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fork C, Bauer T, Golz S, Geerts A, Weiland J, Del Turco D, et al. (2011). OAT2 catalyses efflux of glutamate and uptake of orotic acid. Biochem J, 436, 305–312. [DOI] [PubMed] [Google Scholar]
- Frankenberg T, Rao A, Chen F, Haywood J, Shneider BL, & Dawson PA (2006). Regulation of the mouse organic solute transporter alpha-beta, Ostalpha-Ostbeta, by bile acids. Am J Physiol Gastrointest Liver Physiol, 290, G912–922. [DOI] [PubMed] [Google Scholar]
- Funk C, Ponelle C, Scheuermann G, & Pantze M (2001). Cholestatic potential of troglitazone as a possible factor contributing to troglitazone-induced hepatotoxicity: in vivo and in vitro interaction at the canalicular bile salt export pump (Bsep) in the rat. Mol Pharmacol, 59, 627–635. [PubMed] [Google Scholar]
- Gai Z, Chu L, Hiller C, Arsenijevic D, Penno CA, Montani JP, et al. (2014). Effect of chronic renal failure on the hepatic, intestinal, and renal expression of bile acid transporters. Am J Physiol Renal Physiol, 306, F130–137. [DOI] [PubMed] [Google Scholar]
- Gao E, Cheema H, Waheed N, Mushtaq I, Erden N, Nelson-Williams C, et al. (2019). OSTalpha deficiency: A disorder with cholestasis, liver fibrosis and congenital diarrhea. Hepatology. [DOI] [PMC free article] [PubMed]
- Garcia-Canaveras JC, Donato MT, Castell JV, & Lahoz A (2012). Targeted profiling of circulating and hepatic bile acids in human, mouse, and rat using a UPLC-MRM-MS-validated method. J Lipid Res, 53, 2231–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, et al. (2000). A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell, 6, 517–526. [DOI] [PubMed] [Google Scholar]
- Grandvuinet AS, Gustavsson L, & Steffansen B (2013). New insights into the carrier-mediated transport of estrone-3-sulfate in the Caco-2 cell model. Mol Pharm, 10, 3285–3295. [DOI] [PubMed] [Google Scholar]
- Grandvuinet AS, & Steffansen B (2011). Interactions between organic anions on multiple transporters in Caco-2 cells. J Pharm Sci, 100, 3817–3830. [DOI] [PubMed] [Google Scholar]
- Grube M, Köck K, Karner S, Reuther S, Ritter CA, Jedlitschky G, et al. (2006). Modification of OATP2B1-mediated transport by steroid hormones. Mol Pharmacol, 70, 1735–1741. [DOI] [PubMed] [Google Scholar]
- Grube M, Köck K, Oswald S, Draber K, Meissner K, Eckel L, et al. (2006). Organic anion transporting polypeptide 2B1 is a high-affinity transporter for atorvastatin and is expressed in the human heart. Clin Pharmacol Ther, 80, 607–620. [DOI] [PubMed] [Google Scholar]
- Grube M, Reuther S, Meyer Zu Schwabedissen H, Köck K, Draber K, Ritter CA, et al. (2007). Organic anion transporting polypeptide 2B1 and breast cancer resistance protein interact in the transepithelial transport of steroid sulfates in human placenta. Drug Metab Dispos, 35, 30–35. [DOI] [PubMed] [Google Scholar]
- Gui C, Miao Y, Thompson L, Wahlgren B, Mock M, Stieger B, et al. (2008). Effect of pregnane X receptor ligands on transport mediated by human OATP1B1 and OATP1B3. Eur J Pharmacol, 584, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, LaCerte C, Edwards JE, Brouwer KR, & Brouwer KLR (2018). Farnesoid X Receptor Agonists Obeticholic Acid and Chenodeoxycholic Acid Increase Bile Acid Efflux in Sandwich-Cultured Human Hepatocytes: Functional Evidence and Mechanisms. J Pharmacol Exp Ther, 365, 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenbuch B, & Stieger B (2013). The SLCO (former SLC21) superfamily of transporters. Mol Aspects Med, 34, 396–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, & Price NT (1999). The proton-linked monocarboxylate transporter (MCT) family: structure, function and regulation. Biochem J, 343 Pt 2, 281–299. [PMC free article] [PubMed] [Google Scholar]
- Hallen S, Bjorquist A, Ostlund-Lindqvist AM, & Sachs G (2002). Identification of a region of the ileal-type sodium/bile acid cotransporter interacting with a competitive bile acid transport inhibitor. Biochemistry, 41, 14916–14924. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Takada T, Suzuki H, Onuki R, Hofmann AF, & Sugiyama Y (2005). Transport by vesicles of glycine- and taurine-conjugated bile salts and taurolithocholate 3-sulfate: a comparison of human BSEP with rat Bsep. Biochim Biophys Acta, 1738, 54–62. [DOI] [PubMed] [Google Scholar]
- Heikkinen AT, Korjamo T, Lepikko V, & Monkkonen J (2010). Effects of experimental setup on the apparent concentration dependency of active efflux transport in in vitro cell permeation experiments. Mol Pharm, 7, 605–617. [DOI] [PubMed] [Google Scholar]
- Hirano M, Maeda K, Shitara Y, & Sugiyama Y (2006). Drug-drug interaction between pitavastatin and various drugs via OATP1B1. Drug Metab Dispos, 34, 1229–1236. [DOI] [PubMed] [Google Scholar]
- Ho RH, Leake BF, Roberts RL, Lee W, & Kim RB (2004). Ethnicity-dependent polymorphism in Na+-taurocholate cotransporting polypeptide (SLC10A1) reveals a domain critical for bile acid substrate recognition. J Biol Chem, 279, 7213–7222. [DOI] [PubMed] [Google Scholar]
- Ho RH, Tirona RG, Leake BF, Glaeser H, Lee W, Lemke CJ, et al. (2006). Drug and bile acid transporters in rosuvastatin hepatic uptake: function, expression, and pharmacogenetics. Gastroenterology, 130, 1793–1806. [DOI] [PubMed] [Google Scholar]
- Hochman JH, Pudvah N, Qiu J, Yamazaki M, Tang C, Lin JH, et al. (2004). Interactions of human P-glycoprotein with simvastatin, simvastatin acid, and atorvastatin. Pharm Res, 21, 1686–1691. [DOI] [PubMed] [Google Scholar]
- Hofmann AF (2004). Detoxification of lithocholic acid, a toxic bile acid: relevance to drug hepatotoxicity. Drug Metab Rev, 36, 703–722. [DOI] [PubMed] [Google Scholar]
- Hofmann AF (2009). The enterohepatic circulation of bile acids in mammals: form and functions. Front Biosci (Landmark Ed), 14, 2584–2598. [DOI] [PubMed] [Google Scholar]
- Hwang JH, Parton A, Czechanski A, Ballatori N, & Barnes D (2008). Arachidonic acid-induced expression of the organic solute and steroid transporter-beta (Ost-beta) in a cartilaginous fish cell line. Comp Biochem Physiol C Toxicol Pharmacol, 148, 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ijssennagger N, Janssen AWF, Milona A, Ramos Pittol JM, Hollman DAA, Mokry M, et al. (2016). Gene expression profiling in human precision cut liver slices in response to the FXR agonist obeticholic acid. J Hepatol, 64, 1158–1166. [DOI] [PubMed] [Google Scholar]
- Imai Y, Asada S, Tsukahara S, Ishikawa E, Tsuruo T, & Sugimoto Y (2003). Breast cancer resistance protein exports sulfated estrogens but not free estrogens. Mol Pharmacol, 64, 610–618. [DOI] [PubMed] [Google Scholar]
- Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. (2016). REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet, 99, 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iusuf D, Hendrikx JJ, van Esch A, van de Steeg E, Wagenaar E, Rosing H, et al. (2015). Human OATP1B1, OATP1B3 and OATP1A2 can mediate the in vivo uptake and clearance of docetaxel. Int J Cancer, 136, 225–233. [DOI] [PubMed] [Google Scholar]
- Jackson JP, Freeman K, & Brouwer KR (2016). Basolateral Efflux Transporters: A Potentially Important Pathway for the Prevention of Cholestatic Hepatotoxicity. Applied In Vitro Toxicology, 2, 207–216. [Google Scholar]
- Jin L, Kikuchi R, Saji T, Kusuhara H, & Sugiyama Y (2012). Regulation of tissue-specific expression of renal organic anion transporters by hepatocyte nuclear factor 1 alpha/beta and DNA methylation. J Pharmacol Exp Ther, 340, 648–655. [DOI] [PubMed] [Google Scholar]
- Kalvass JC, & Pollack GM (2007). Kinetic considerations for the quantitative assessment of efflux activity and inhibition: implications for understanding and predicting the effects of efflux inhibition. Pharm Res, 24, 265–276. [DOI] [PubMed] [Google Scholar]
- Kameyama Y, Yamashita K, Kobayashi K, Hosokawa M, & Chiba K (2005). Functional characterization of SLCO1B1 (OATP-C) variants, SLCO1B1*5, SLCO1B1*15 and SLCO1B1*15+C1007G, by using transient expression systems of HeLa and HEK293 cells. Pharmacogenet Genomics, 15, 513–522. [DOI] [PubMed] [Google Scholar]
- Kamiyama Y, Matsubara T, Yoshinari K, Nagata K, Kamimura H, & Yamazoe Y (2007). Role of human hepatocyte nuclear factor 4alpha in the expression of drug-metabolizing enzymes and transporters in human hepatocytes assessed by use of small interfering RNA. Drug Metab Pharmacokinet, 22, 287–298. [DOI] [PubMed] [Google Scholar]
- Karlgren M, Vildhede A, Norinder U, Wisniewski JR, Kimoto E, Lai Y, et al. (2012). Classification of inhibitors of hepatic organic anion transporting polypeptides (OATPs): influence of protein expression on drug-drug interactions. J Med Chem, 55, 4740–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenna JG, Taskar KS, Battista C, Bourdet DL, Brouwer KLR, Brouwer KR, et al. (2018). Can Bile Salt Export Pump Inhibition Testing in Drug Discovery and Development Reduce Liver Injury Risk? An International Transporter Consortium Perspective. Clin Pharmacol Ther, 104, 916–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keskitalo JE, Zolk O, Fromm MF, Kurkinen KJ, Neuvonen PJ, & Niemi M (2009). ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clin Pharmacol Ther, 86, 197–203. [DOI] [PubMed] [Google Scholar]
- Khan AA, Chow EC, Porte RJ, Pang KS, & Groothuis GM (2009). Expression and regulation of the bile acid transporter, OSTalpha-OSTbeta in rat and human intestine and liver. Biopharm Drug Dispos, 30, 241–258. [DOI] [PubMed] [Google Scholar]
- Kikuchi R, Kusuhara H, Hattori N, Shiota K, Kim I, Gonzalez FJ, et al. (2006). Regulation of the expression of human organic anion transporter 3 by hepatocyte nuclear factor 1alpha/beta and DNA methylation. Mol Pharmacol, 70, 887–896. [DOI] [PubMed] [Google Scholar]
- Kim RB, Leake B, Cvetkovic M, Roden MM, Nadeau J, Walubo A, et al. (1999). Modulation by drugs of human hepatic sodium-dependent bile acid transporter (sodium taurocholate cotransporting polypeptide) activity. J Pharmacol Exp Ther, 291, 1204–1209. [PubMed] [Google Scholar]
- Kimura H, Takeda M, Narikawa S, Enomoto A, Ichida K, & Endou H (2002). Human organic anion transporters and human organic cation transporters mediate renal transport of prostaglandins. J Pharmacol Exp Ther, 301, 293–298. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Kioka N, Kato H, Matsuo M, & Ueda K (2007). Modulation of drug-stimulated ATPase activity of human MDR1/P-glycoprotein by cholesterol. Biochem J, 401, 597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kis E, Ioja E, Nagy T, Szente L, Heredi-Szabo K, & Krajcsi P (2009). Effect of membrane cholesterol on BSEP/Bsep activity: species specificity studies for substrates and inhibitors. Drug Metab Dispos, 37, 1878–1886. [DOI] [PubMed] [Google Scholar]
- Kitamura S, Maeda K, Wang Y, & Sugiyama Y (2008). Involvement of multiple transporters in the hepatobiliary transport of rosuvastatin. Drug Metab Dispos, 36, 2014–2023. [DOI] [PubMed] [Google Scholar]
- Kittayaruksakul S, Soodvilai S, Asavapanumas N, Muanprasat C, & Chatsudthipong V (2012). Liver X receptor activation downregulates organic anion transporter 1 (OAT1) in the renal proximal tubule. Am J Physiol Renal Physiol, 302, F552–560. [DOI] [PubMed] [Google Scholar]
- Klein K, Jüngst C, Mwinyi J, Stieger B, Krempler F, Patsch W, et al. (2010). The human organic anion transporter genes OAT5 and OAT7 are transactivated by hepatocyte nuclear factor-1α (HNF-1α). Mol Pharmacol, 78, 1079–1087. [DOI] [PubMed] [Google Scholar]
- Kobayashi D, Nozawa T, Imai K, Nezu J, Tsuji A, & Tamai I (2003). Involvement of human organic anion transporting polypeptide OATP-B (SLC21A9) in pH-dependent transport across intestinal apical membrane. J Pharmacol Exp Ther, 306, 703–708. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Ohshiro N, Sakai R, Ohbayashi M, Kohyama N, & Yamamoto T (2005). Transport mechanism and substrate specificity of human organic anion transporter 2 (hOat2 [SLC22A7]). J Pharm Pharmacol, 57, 573–578. [DOI] [PubMed] [Google Scholar]
- Korjamo T, Kemilainen H, Heikkinen AT, & Monkkonen J (2007). Decrease in intracellular concentration causes the shift in Km value of efflux pump substrates. Drug Metab Dispos, 35, 1574–1579. [DOI] [PubMed] [Google Scholar]
- Kramer W, Stengelin S, Baringhaus KH, Enhsen A, Heuer H, Becker W, et al. (1999). Substrate specificity of the ileal and the hepatic Na(+)/bile acid cotransporters of the rabbit. I. Transport studies with membrane vesicles and cell lines expressing the cloned transporters. J Lipid Res, 40, 1604–1617. [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, & Sonnhammer EL (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol, 305, 567–580. [DOI] [PubMed] [Google Scholar]
- Kullak-Ublick GA, Fisch T, Oswald M, Hagenbuch B, Meier PJ, Beuers U, et al. (1998). Dehydroepiandrosterone sulfate (DHEAS): identification of a carrier protein in human liver and brain. FEBS Lett, 424, 173–176. [DOI] [PubMed] [Google Scholar]
- Kullak-Ublick GA, Ismair MG, Stieger B, Landmann L, Huber R, Pizzagalli F, et al. (2001). Organic anion-transporting polypeptide B (OATP-B) and its functional comparison with three other OATPs of human liver. Gastroenterology, 120, 525–533. [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, & Ng PC (2009). Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc, 4, 1073–1081. [DOI] [PubMed] [Google Scholar]
- Kusuhara H, Furuie H, Inano A, Sunagawa A, Yamada S, Wu C, et al. (2012). Pharmacokinetic interaction study of sulphasalazine in healthy subjects and the impact of curcumin as an in vivo inhibitor of BCRP. British Journal of Pharmacology, 166, 1793–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai L, & Tan TM (2002). Role of glutathione in the multidrug resistance protein 4 (MRP4/ABCC4)-mediated efflux of cAMP and resistance to purine analogues. Biochem J, 361, 497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrier JF, Eloranta JJ, Vavricka SR, & Kullak-Ublick GA (2006). The nuclear receptor for bile acids, FXR, transactivates human organic solute transporter-alpha and - beta genes. Am J Physiol Gastrointest Liver Physiol, 290, G476–485. [DOI] [PubMed] [Google Scholar]
- Lau YY, Huang Y, Frassetto L, & Benet LZ (2007). effect of OATP1B transporter inhibition on the pharmacokinetics of atorvastatin in healthy volunteers. Clin Pharmacol Ther, 81, 194–204. [DOI] [PubMed] [Google Scholar]
- Le Vee M, Jouan E, Stieger B, & Fardel O (2013). Differential regulation of drug transporter expression by all-trans retinoic acid in hepatoma HepaRG cells and human hepatocytes. Eur J Pharm Sci, 48, 767–774. [DOI] [PubMed] [Google Scholar]
- Lee HH, Leake BF, Kim RB, & Ho RH (2017). Contribution of Organic Anion-Transporting Polypeptides 1A/1B to Doxorubicin Uptake and Clearance. Mol Pharmacol, 91, 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Glaeser H, Smith LH, Roberts RL, Moeckel GW, Gervasini G, et al. (2005). Polymorphisms in human organic anion-transporting polypeptide 1A2 (OATP1A2): implications for altered drug disposition and central nervous system drug entry. J Biol Chem, 280, 9610–9617. [DOI] [PubMed] [Google Scholar]
- Leslie EM, Watkins PB, Kim RB, & Brouwer KLR (2007). Differential inhibition of rat and human Na+-dependent taurocholate cotransporting polypeptide (NTCP/SLC10A1)by bosentan: a mechanism for species differences in hepatotoxicity. J Pharmacol Exp Ther, 321, 1170–1178. [DOI] [PubMed] [Google Scholar]
- Leuthold S, Hagenbuch B, Mohebbi N, Wagner CA, Meier PJ, & Stieger B (2009). Mechanisms of pH-gradient driven transport mediated by organic anion polypeptide transporters. Am J Physiol Cell Physiol, 296, C570–582. [DOI] [PubMed] [Google Scholar]
- Li J, Wang Y, Zhang W, Huang Y, Hein K, & Hidalgo IJ (2012). The role of a basolateral transporter in rosuvastatin transport and its interplay with apical breast cancer resistance protein in polarized cell monolayer systems. Drug Metab Dispos, 40, 2102–2108. [DOI] [PubMed] [Google Scholar]
- Li N, Cui Z, Fang F, Lee JY, & Ballatori N (2007). Heterodimerization, trafficking and membrane topology of the two proteins, Ost alpha and Ost beta, that constitute the organic solute and steroid transporter. Biochem J, 407, 363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CJ, & Smith DE (1999). Glycylsarcosine uptake in rabbit renal brush border membrane vesicles isolated from outer cortex or outer medulla: evidence for heterogeneous distribution of oligopeptide transporters. AAPS PharmSci, 1, E1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Bircsak KM, Gorczyca L, Wen X, & Aleksunes LM (2017). Regulation of the placental BCRP transporter by PPAR gamma. J Biochem Mol Toxicol, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lu H, Lu YF, Lei X, Cui JY, Ellis E, et al. (2014). Potency of individual bile acids to regulate bile acid synthesis and transport genes in primary human hepatocyte cultures. Toxicol Sci, 141, 538–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MW, Craddock AL, Angelin B, Brunzell JD, Duane WC, & Dawson PA (2001). Analysis of the ileal bile acid transporter gene, SLC10A2, in subjects with familial hypertriglyceridemia. Arterioscler Thromb Vasc Biol, 21, 2039–2045. [DOI] [PubMed] [Google Scholar]
- Lu H, Gonzalez FJ, & Klaassen C (2010). Alterations in hepatic mRNA expression of phase II enzymes and xenobiotic transporters after targeted disruption of hepatocyte nuclear factor 4 alpha. Toxicol Sci, 118, 380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Michaud V, Moya LG, Gaudette F, Leung YH, & Turgeon J (2015). Effects of beta-blockers and tricyclic antidepressants on the activity of human organic anion transporting polypeptide 1A2 (OATP1A2). J Pharmacol Exp Ther, 352, 552–558. [DOI] [PubMed] [Google Scholar]
- Maeda K, Kambara M, Tian Y, Hofmann AF, & Sugiyama Y (2006). Uptake of ursodeoxycholate and its conjugates by human hepatocytes: role of Na(+)-taurocholate cotransporting polypeptide (NTCP), organic anion transporting polypeptide (OATP) 1B1 (OATP-C), and oatp1B3 (OATP8). Mol Pharm, 3, 70–77. [DOI] [PubMed] [Google Scholar]
- Maier A, Volker B, Boles E, & Fuhrmann GF (2002). Characterisation of glucose transport in Saccharomyces cerevisiae with plasma membrane vesicles (countertransport) and intact cells (initial uptake) with single Hxt1, Hxt2, Hxt3, Hxt4, Hxt6, Hxt7 or Gal2 transporters. FEMS Yeast Res, 2, 539–550. [DOI] [PubMed] [Google Scholar]
- Malinen MM, Ali I, Bezencon J, Beaudoin JJ, & Brouwer KLR (2018). Organic solute transporter OSTalpha/beta is overexpressed in nonalcoholic steatohepatitis and modulated by drugs associated with liver injury. Am J Physiol Gastrointest Liver Physiol, 314, G597–G609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinen MM, Ito K, Kang HE, Honkakoski P, & Brouwer KLR (2019). Protein expression and function of organic anion transporters in short-term and long-term cultures of Huh7 human hepatoma cells. Eur J Pharm Sci, 130, 186–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinen MM, Kauttonen A, Beaudoin JJ, Sjostedt N, Honkakoski P, & Brouwer KLR (2019). Novel in Vitro Method Reveals Drugs That Inhibit Organic Solute Transporter Alpha/Beta (OSTalpha/beta). Mol Pharm, 16, 238–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier PJ, Eckhardt U, Schroeder A, Hagenbuch B, & Stieger B (1997). Substrate specificity of sinusoidal bile acid and organic anion uptake systems in rat and human liver. Hepatology, 26, 1667–1677. [DOI] [PubMed] [Google Scholar]
- Miki Y, Suzuki T, Kitada K, Yabuki N, Shibuya R, Moriya T, et al. (2006). Expression of the steroid and xenobiotic receptor and its possible target gene, organic anion transporting polypeptide-A, in human breast carcinoma. Cancer Res, 66, 535–542. [DOI] [PubMed] [Google Scholar]
- Mikkaichi T, Suzuki T, Onogawa T, Tanemoto M, Mizutamari H, Okada M, et al. (2004). Isolation and characterization of a digoxin transporter and its rat homologue expressed in the kidney. Proc Natl Acad Sci U S A, 101, 3569–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyajima M, Kusuhara H, Fujishima M, Adachi Y, & Sugiyama Y (2011). Organic anion transporter 3 mediates the efflux transport of an amphipathic organic anion, dehydroepiandrosterone sulfate, across the blood-brain barrier in mice. Drug Metab Dispos, 39, 814–819. [DOI] [PubMed] [Google Scholar]
- Miyata M, Hayashi K, Yamakawa H, Yamazoe Y, & Yoshinari K (2015). Antibacterial drug treatment increases intestinal bile acid absorption via elevated levels of ileal apical sodium-dependent bile acid transporter but not organic solute transporter alpha protein. Biol Pharm Bull, 38, 493–496. [DOI] [PubMed] [Google Scholar]
- Miyazaki H, Anzai N, Ekaratanawong S, Sakata T, Shin HJ, Jutabha P, et al. (2005). Modulation of renal apical organic anion transporter 4 function by two PDZ domain-containing proteins. J Am Soc Nephrol, 16, 3498–3506. [DOI] [PubMed] [Google Scholar]
- Morgan RE, van Staden CJ, Chen Y, Kalyanaraman N, Kalanzi J, Dunn RT 2nd, et al. (2013). A multifactorial approach to hepatobiliary transporter assessment enables improved therapeutic compound development. Toxicol Sci, 136, 216–241. [DOI] [PubMed] [Google Scholar]
- Mosedale M, & Watkins PB (2017). Drug-induced liver injury: Advances in mechanistic understanding that will inform risk management. Clin Pharmacol Ther, 101, 469–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio T, Adachi H, Nakagomi R, Tokui T, Sato E, Tanemoto M, et al. (2000). Molecular identification of a rat novel organic anion transporter moat1, which transports prostaglandin D(2), leukotriene C(4), and taurocholate. Biochem Biophys Res Commun, 275, 831–838. [DOI] [PubMed] [Google Scholar]
- Noe J, Portmann R, Brun ME, & Funk C (2007). Substrate-dependent drug-drug interactions between gemfibrozil, fluvastatin and other organic anion-transporting peptide (OATP) substrates on OATP1B1, OATP2B1, and OATP1B3. Drug Metab Dispos, 35, 1308–1314. [DOI] [PubMed] [Google Scholar]
- Nozawa T, Imai K, Nezu J, Tsuji A, & Tamai I (2004). Functional characterization of pH-sensitive organic anion transporting polypeptide OATP-B in human. J Pharmacol Exp Ther, 308, 438–445. [DOI] [PubMed] [Google Scholar]
- O’Brien VP, Bokelmann K, Ramirez J, Jobst K, Ratain MJ, Brockmoller J, et al. (2013). Hepatocyte nuclear factor 1 regulates the expression of the organic cation transporter 1 via binding to an evolutionary conserved region in intron 1 of the OCT1 gene. J Pharmacol Exp Ther, 347, 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuka H, Abe T, Onogawa T, Kondo N, Sato T, Oshio H, et al. (2006). Farnesoid X receptor, hepatocyte nuclear factors 1alpha and 3beta are essential for transcriptional activation of the liver-specific organic anion transporter-2 gene. J Gastroenterol, 41, 369–377. [DOI] [PubMed] [Google Scholar]
- Okuwaki M, Takada T, Iwayanagi Y, Koh S, Kariya Y, Fujii H, et al. (2007). LXR alpha transactivates mouse organic solute transporter alpha and beta via IR-1 elements shared with FXR. Pharm Res, 24, 390–398. [DOI] [PubMed] [Google Scholar]
- Onakpoya IJ, Heneghan CJ, & Aronson JK (2016). Worldwide withdrawal of medicinal products because of adverse drug reactions: a systematic review and analysis. Crit Rev Toxicol, 46, 477–489. [DOI] [PubMed] [Google Scholar]
- Perez MJ, & Briz O (2009). Bile-acid-induced cell injury and protection. World J Gastroenterol, 15, 1677–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov PD, Fernandez-Murga L, Conde I, Martinez-Sena T, Guzman C, Castell JV, et al. (2020). Epistane, an anabolic steroid used for recreational purposes, causes cholestasis with elevated levels of cholic acid conjugates, by upregulating bile acid synthesis (CYP8B1) and cross-talking with nuclear receptors in human hepatocytes. Arch Toxicol [DOI] [PubMed]
- Pfeifer ND, Bridges AS, Ferslew BC, Hardwick RN, & Brouwer KLR (2013). Hepatic basolateral efflux contributes significantly to rosuvastatin disposition II: characterization of hepatic elimination by basolateral, biliary, and metabolic clearance pathways in rat isolated perfused liver. J Pharmacol Exp Ther, 347, 737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzagalli F, Varga Z, Huber RD, Folkers G, Meier PJ, & St-Pierre MV (2003). Identification of steroid sulfate transport processes in the human mammary gland. J Clin Endocrinol Metab, 88, 3902–3912. [DOI] [PubMed] [Google Scholar]
- Popowski K, Eloranta JJ, Saborowski M, Fried M, Meier PJ, & Kullak-Ublick GA (2005). The human organic anion transporter 2 gene is transactivated by hepatocyte nuclear factor-4 alpha and suppressed by bile acids. Mol Pharmacol, 67, 1629–1638. [DOI] [PubMed] [Google Scholar]
- Qian YM, Song WC, Cui H, Cole SP, & Deeley RG (2001). Glutathione stimulates sulfated estrogen transport by multidrug resistance protein 1. J Biol Chem, 276, 6404–6411. [DOI] [PubMed] [Google Scholar]
- Rao A, Haywood J, Craddock AL, Belinsky MG, Kruh GD, & Dawson PA (2008). The organic solute transporter alpha-beta, Ostalpha-Ostbeta, is essential for intestinal bile acid transport and homeostasis. Proc Natl Acad Sci U S A, 105, 3891–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid G, Wielinga P, Zelcer N, van der Heijden I, Kuil A, de Haas M, et al. (2003). The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal antiinflammatory drugs. Proc Natl Acad Sci U S A, 100, 9244–9249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reva B, Antipin Y, & Sander C (2011). Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res, 39, e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rius M, Hummel-Eisenbeiss J, Hofmann AF, & Keppler D (2006). Substrate specificity of human ABCC4 (MRP4)-mediated cotransport of bile acids and reduced glutathione. Am J Physiol Gastrointest Liver Physiol, 290, G640–649. [DOI] [PubMed] [Google Scholar]
- Robinson MD, & Oshlack A (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol, 11, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues AD, Lai Y, Shen H, Varma MVS, Rowland A, & Oswald S (2019). Induction of Human Intestinal and Hepatic Organic Anion Transporting Polypeptides; Where is the Evidence for its Relevance in Drug-Drug Interactions? Drug Metab Dispos. [DOI] [PubMed]
- Saborowski M, Kullak-Ublick GA, & Eloranta JJ (2006). The human organic cation transporter-1 gene is transactivated by hepatocyte nuclear factor-4alpha. J Pharmacol Exp Ther, 317, 778–785. [DOI] [PubMed] [Google Scholar]
- Schaap FG, van der Gaag NA, Gouma DJ, & Jansen PL (2009). High expression of the bile salt-homeostatic hormone fibroblast growth factor 19 in the liver of patients with extrahepatic cholestasis. Hepatology, 49, 1228–1235. [DOI] [PubMed] [Google Scholar]
- Schaffner CA, Mwinyi J, Gai Z, Thasler WE, Eloranta JJ, & Kullak-Ublick GA (2015). The organic solute transporters alpha and beta are induced by hypoxia in human hepatocytes. Liver Int, 35, 1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz UI (2012). The bile acid transporter organic solute transporter (OST) alpha-beta is also an intestinal drug transporter In Intestinal and hepatic drug transporters and their role in the disposition of lipid-lowering drugs. (pp. 81–112). Electronic Thesis and Dissertation Repository: The University of Western Ontario. [Google Scholar]
- Seward DJ, Koh AS, Boyer JL, & Ballatori N (2003). Functional complementation between a novel mammalian polygenic transport complex and an evolutionarily ancient organic solute transporter, OSTalpha-OSTbeta. J Biol Chem, 278, 27473–27482. [DOI] [PubMed] [Google Scholar]
- Shen H, Liu T, Morse BL, Zhao Y, Zhang Y, Qiu X, et al. (2015). Characterization of Organic Anion Transporter 2 (SLC22A7): A Highly Efficient Transporter for Creatinine and Species-Dependent Renal Tubular Expression. Drug Metab Dispos, 43, 984–993. [DOI] [PubMed] [Google Scholar]
- Shirakawa K, Takara K, Tanigawara Y, Aoyama N, Kasuga M, Komada F, et al. (1999). Interaction of docetaxel (“Taxotere”) with human P-glycoprotein. Jpn J Cancer Res, 90, 1380–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sissung TM, Huang PA, Hauke RJ, McCrea EM, Peer CJ, Barbier RH, et al. (2019). Severe Hepatotoxicity of Mithramycin Therapy Caused by Altered Expression of Hepatocellular Bile Transporters. Mol Pharmacol, 96, 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AJ, van Helvoort A, van Meer G, Szabo K, Welker E, Szakacs G, et al. (2000). MDR3 P-glycoprotein, a phosphatidylcholine translocase, transports several cytotoxic drugs and directly interacts with drugs as judged by interference with nucleotide trapping. J Biol Chem, 275, 23530–23539. [DOI] [PubMed] [Google Scholar]
- Soroka CJ, Ballatori N, & Boyer JL (2010). Organic solute transporter, OSTalpha-OSTbeta: its role in bile acid transport and cholestasis. Semin Liver Dis, 30, 178–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soroka CJ, Mennone A, Hagey LR, Ballatori N, & Boyer JL (2010). Mouse organic solute transporter alpha deficiency enhances renal excretion of bile acids and attenuates cholestasis. Hepatology, 51, 181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soroka CJ, Xu S, Mennone A, Lam P, & Boyer JL (2008). N-Glycosylation of the alpha subunit does not influence trafficking or functional activity of the human organic solute transporter alpha/beta. BMC Cell Biol, 9, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino P, Tarantino G, Perrella A, Micheli P, Perrella O, & Conca P (2005). A clinical-morphological study on cholestatic presentation of nonalcoholic fatty liver disease. Dig Dis Sci, 50, 1130–1135. [DOI] [PubMed] [Google Scholar]
- Stieger B, & Hagenbuch B (2014). Organic anion-transporting polypeptides. Curr Top Membr, 73, 205–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suga T, Yamaguchi H, Ogura J, & Mano N (2019). Characterization of conjugated and unconjugated bile acid transport via human organic solute transporter alpha/beta. Biochim Biophys Acta Biomembr, 1861, 1023–1029. [DOI] [PubMed] [Google Scholar]
- Suga T, Yamaguchi H, Sato T, Maekawa M, Goto J, & Mano N (2017). Preference of Conjugated Bile Acids over Unconjugated Bile Acids as Substrates for OATP1B1 and OATP1B3. PLoS One, 12, e0169719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan M, Rao A, Elpeleg O, Vaz FM, Abu-Libdeh B, Karpen SJ, et al. (2018). Organic solute transporter-beta (SLC51B) deficiency in two brothers with congenital diarrhea and features of cholestasis. Hepatology, 68, 590–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun AQ, Balasubramaniyan N, Xu K, Liu CJ, Ponamgi VM, Liu H, et al. (2007). Protein-protein interactions and membrane localization of the human organic solute transporter. Am J Physiol Gastrointest Liver Physiol, 292, G1586–1593. [DOI] [PubMed] [Google Scholar]
- Sun AQ, Zhu L, Luo Y, Xu S, Lin J, & Suchy FJ (2012). Human Organic Solute Transporter (hOST): protein interaction and membrane sorting process. Int J Biochem Mol Biol, 3, 290–301. [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Toyohara T, Akiyama Y, Takeuchi Y, Mishima E, Suzuki C, et al. (2011). Transcriptional regulation of organic anion transporting polypeptide SLCO4C1 as a new therapeutic modality to prevent chronic kidney disease. J Pharm Sci, 100, 3696–3707. [DOI] [PubMed] [Google Scholar]
- Tachibana T, Kitamura S, Kato M, Mitsui T, Shirasaka Y, Yamashita S, et al. (2010). Model analysis of the concentration-dependent permeability of P-gp substrates. Pharm Res, 27, 442–446. [DOI] [PubMed] [Google Scholar]
- Tanihara Y, Masuda S, Sato T, Katsura T, Ogawa O, & Inui K (2007). Substrate specificity of MATE1 and MATE2-K, human multidrug and toxin extrusions/H(+)-organic cation antiporters. Biochem Pharmacol, 74, 359–371. [DOI] [PubMed] [Google Scholar]
- Temple RJ, & Himmel MH (2002). Safety of newly approved drugs: implications for prescribing. JAMA, 287, 2273–2275. [DOI] [PubMed] [Google Scholar]
- Teng S, & Piquette-Miller M (2007). Hepatoprotective role of PXR activation and MRP3 in cholic acid-induced cholestasis. Br J Pharmacol, 151, 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada T, & Inui K (2008). Physiological and pharmacokinetic roles of H+/organic cation antiporters (MATE/SLC47A). Biochem Pharmacol, 75, 1689–1696. [DOI] [PubMed] [Google Scholar]
- Trauner M, Wagner M, Fickert P, & Zollner G (2005). Molecular regulation of hepatobiliary transport systems: clinical implications for understanding and treating cholestasis. J Clin Gastroenterol, 39, S111–124. [DOI] [PubMed] [Google Scholar]
- Troutman MD, & Thakker DR (2003). Efflux ratio cannot assess P-glycoprotein-mediated attenuation of absorptive transport: asymmetric effect of P-glycoprotein on absorptive and secretory transport across Caco-2 cell monolayers. Pharm Res, 20, 1200–1209. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Terada T, Asaka J, Ueba M, Katsura T, & Inui K (2007). Oppositely directed H+ gradient functions as a driving force of rat H+/organic cation antiporter MATE1. Am J Physiol Renal Physiol, 292, F593–598. [DOI] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science, 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Urquhart BL, Ware JA, Tirona RG, Ho RH, Leake BF, Schwarz UI, et al. (2008). Breast cancer resistance protein (ABCG2) and drug disposition: intestinal expression, polymorphisms and sulfasalazine as an in vivo probe. Pharmacogenet Genomics, 18, 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valliere-Douglass JF, Kodama P, Mujacic M, Brady LJ, Wang W, Wallace A, et al. (2009). Asparagine-linked oligosaccharides present on a non-consensus amino acid sequence in the CH1 domain of human antibodies. J Biol Chem, 284, 32493–32506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wiel SMW, de Waart DR, Oude Elferink RPJ, & van de Graaf SFJ (2018). Intestinal Farnesoid X Receptor Activation by Pharmacologic Inhibition of the Organic Solute Transporter α-β. Cell Mol Gastroenterol Hepatol, 5, 223–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg RA, Hoefsloot HC, Westerhuis JA, Smilde AK, & van der Werf MJ (2006). Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genomics, 7, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser WE, Wong WS, van Mullem AA, Friesema EC, Geyer J, & Visser TJ (2010). Study of the transport of thyroid hormone by transporters of the SLC10 family. Mol Cell Endocrinol, 315, 138–145. [DOI] [PubMed] [Google Scholar]
- Wang Q, Zheng M, & Leil T (2017). Investigating Transporter-Mediated Drug-Drug Interactions Using a Physiologically Based Pharmacokinetic Model of Rosuvastatin. CPT Pharmacometrics Syst Pharmacol, 6, 228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Seward DJ, Li L, Boyer JL, & Ballatori N (2001). Expression cloning of two genes that together mediate organic solute and steroid transport in the liver of a marine vertebrate. Proc Natl Acad Sci U S A, 98, 9431–9436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver RJ, Blomme EA, Chadwick AE, Copple IM, Gerets HHJ, Goldring CE, et al. (2019). Managing the challenge of drug-induced liver injury: a roadmap for the development and deployment of preclinical predictive models. Nat Rev Drug Discov [DOI] [PubMed]
- Windass AS, Lowes S, Wang Y, & Brown CD (2007). The contribution of organic anion transporters OAT1 and OAT3 to the renal uptake of rosuvastatin. J Pharmacol Exp Ther, 322, 1221–1227. [DOI] [PubMed] [Google Scholar]
- Xu S, Soroka CJ, Sun AQ, Backos DS, Mennone A, Suchy FJ, et al. (2016). A Novel Di-Leucine Motif at the N-Terminus of Human Organic Solute Transporter Beta Is Essential for Protein Association and Membrane Localization. PLoS One, 11, e0158269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Sun AQ, & Suchy FJ (2014). A novel RARalpha/CAR-mediated mechanism for regulation of human organic solute transporter-beta gene expression. Am J Physiol Gastrointest Liver Physiol, 306, G154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada A, Maeda K, Kamiyama E, Sugiyama D, Kondo T, Shiroyanagi Y, et al. (2007). Multiple human isoforms of drug transporters contribute to the hepatic and renal transport of olmesartan, a selective antagonist of the angiotensin II AT1-receptor. Drug Metab Dispos, 35, 2166–2176. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Kobayashi M, Okada M, Takeuchi T, Unno M, Abe T, et al. (2008). Rapid screening of antineoplastic candidates for the human organic anion transporter OATP1B3 substrates using fluorescent probes. Cancer Lett, 260, 163–169. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Sugie M, Okada M, Mikkaichi T, Toyohara T, Abe T, et al. (2010). Transport of estrone 3-sulfate mediated by organic anion transporter OATP4C1: estrone 3-sulfate binds to the different recognition site for digoxin in OATP4C1. Drug Metab Pharmacokinet, 25, 314–317. [DOI] [PubMed] [Google Scholar]
- Yamashita F, Ohtani H, Koyabu N, Ushigome F, Satoh H, Murakami H, et al. (2006). Inhibitory effects of angiotensin II receptor antagonists and leukotriene receptor antagonists on the transport of human organic anion transporter 4. J Pharm Pharmacol, 58, 1499–1505. [DOI] [PubMed] [Google Scholar]
- Yang K, Pfeifer ND, Köck K, & Brouwer KLR (2015). Species differences in hepatobiliary disposition of taurocholic acid in human and rat sandwich-cultured hepatocytes: implications for drug-induced liver injury. J Pharmacol Exp Ther, 353, 415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamek-Gliszczynski MJ, Taub ME, Chothe PP, Chu X, Giacomini KM, Kim RB, et al. (2018). Transporters in Drug Development: 2018 ITC Recommendations for Transporters of Emerging Clinical Importance. Clin Pharmacol Ther, 104, 890–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelcer N, Reid G, Wielinga P, Kuil A, van der Heijden I, Schuetz JD, et al. (2003). Steroid and bile acid conjugates are substrates of human multidrug-resistance protein (MRP) 4 (ATP-binding cassette C4). Biochem J, 371, 361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Liu G, Rea PA, & Kruh GD (2000). Transport of amphipathic anions by human multidrug resistance protein 3. Cancer Res, 60, 4779–4784. [PubMed] [Google Scholar]
- Zhang DW, Gu HM, Vasa M, Muredda M, Cole SP, & Deeley RG (2003). Characterization of the role of polar amino acid residues within predicted transmembrane helix 17 in determining the substrate specificity of multidrug resistance protein 3. Biochemistry, 42, 9989–10000. [DOI] [PubMed] [Google Scholar]
- Zhou F, Tanaka K, Pan Z, Ma J, & You G (2004). The role of glycine residues in the function of human organic anion transporter 4. Mol Pharmacol, 65, 1141–1147. [DOI] [PubMed] [Google Scholar]
- Zollner G, Wagner M, Moustafa T, Fickert P, Silbert D, Gumhold J, et al. (2006). Coordinated induction of bile acid detoxification and alternative elimination in mice: role of FXR-regulated organic solute transporter-alpha/beta in the adaptive response to bile acids. Am J Physiol Gastrointest Liver Physiol, 290, G923–932. [DOI] [PubMed] [Google Scholar]