

Integrating recent findings, Chen and Schmid present a more dynamic, flexible, and nonlinear model for clathrin-coated vesicle formation.
Abstract
Clathrin-mediated endocytosis occurs via the assembly of clathrin-coated pits (CCPs) that invaginate and pinch off to form clathrin-coated vesicles (CCVs). It is well known that adaptor protein 2 (AP2) complexes trigger clathrin assembly on the plasma membrane, and biochemical and structural studies have revealed the nature of these interactions. Numerous endocytic accessory proteins collaborate with clathrin and AP2 to drive CCV formation. However, many questions remain as to the molecular events involved in CCP initiation, stabilization, and curvature generation. Indeed, a plethora of recent evidence derived from cell perturbation, correlative light and EM tomography, live-cell imaging, modeling, and high-resolution structural analyses has revealed more complexity and promiscuity in the protein interactions driving CCP maturation than anticipated. After briefly reviewing the evidence supporting prevailing models, we integrate these new lines of evidence to develop a more dynamic and flexible model for how redundant, dynamic, and competing protein interactions can drive endocytic CCV formation and suggest new approaches to test emerging models.
Introduction
The roles of clathrin-coated vesicles (CCVs) in nutrient uptake (Roth and Porter, 1964) and rapid recycling of synaptic vesicles (Heuser and Reese 1973) were first observed decades ago by EM. Subsequent purification of coated vesicles from pig brains (Pearse, 1975) led to the identification of clathrin triskelia (Fig. 1 A) as the major constituents of these coated vesicles (Pearse, 1976; Ungewickell and Branton, 1981). Clathrin was found to spontaneously self-assemble into closed empty “cages” under low-pH and low-salt conditions (Woodward and Roth, 1978) and form “coats” under physiological conditions in the presence of AP2 (adaptor protein 2) complexes (Fig. 1 C), the second major coat constituent (then named “assembly proteins”; Pearse and Robinson, 1984; Zaremba and Keen, 1983). These findings led naturally to a model in which assembly of the clathrin coat was sufficient to drive deformation of the underlying membrane to create invaginated clathrin-coated pits (CCPs). While subsequent in vitro studies of clathrin and AP2 assembly on lipid monolayers (Ford et al., 2001) or liposomes (Dannhauser and Ungewickell, 2012) have supported this still-prevalent model, in cells, the situation appears to be more complex and certainly far from understood. Thus, while in its late 50s, clathrin-mediated endocytosis (CME) is considered a “mature” field, many questions regarding the mechanisms controlling CCP assembly, maturation, and fission remain un- or incompletely answered. As the American historian Daniel Boorstin said, “The greatest obstacle to discovery is not ignorance, it is the illusion of knowledge.”
Figure 1.

Known AP2–clathrin interactions. (A) Diagram of a clathrin triskelion that is composed of three heavy chains and three light chains. Each heavy chain contains an NTD (dark green), which connects to an ankle region (magenta), a distal leg (light green), and a proximal leg (pink) where light chain (purple) binds. (B) Structure of the NTD, which contains four known binding sites defined by key residues (shown by spheres): the clathrin-box (red), W-box (orange), arrestin-box (blue), and Royle-box (magenta; Lemmon and Traub, 2012; Willox and Royle, 2012; modified from PDB accession no. 1BPO). (C) Diagram of the heterotetrameric AP2 complex composed of α, β2, μ2, and σ2 subunits. The clathrin binding sites are located on the hinge region (i.e., the LLNLD clathrin-box motif) of the β2 subunit (shown in red). (D) Structure of the β2-appendage domain (modified from PDB accession no. 1E42), which contains two additional clathrin binding sites bearing key residues, Y815 and Y888 (shown by spheres). Solid red and magenta lines indicate interactions mediated by the clathrin-box and β-appendage domains, respectively.
In this perspective, we discuss past and recent literature that has spurred new and lively debates regarding the mechanisms underlying clathrin assembly and curvature generation during CCP invagination. We then integrate these new findings and propose a dynamic, flexible, and nonlinear model for CCP assembly and maturation that begs for additional experiments to rigorously test it. We do not discuss curvature generation at the neck of deeply invaginated CCPs or the dynamin-driven fission machinery that mediates the final stages of CCV formation, as these have been extensively reviewed (and debated) elsewhere (Antonny et al., 2016; Kaksonen and Roux, 2018; Mettlen et al., 2018; Schmid and Frolov, 2011). Importantly, CCV formation continues to serve as a paradigm for much of vesicular transport; thus, understanding the mechanisms underlying CME provides a foundation for understanding intracellular membrane trafficking.
AP2-mediated clathrin assembly
Insights from biochemical and structural studies
Clathrin by itself does not interact with cell membranes. Rather, clathrin assembly is triggered on the plasma membrane (PM) by adaptor proteins, in particular AP2, a heterotetrameric complex composed of α, β2, µ2, and σ2 subunits (Fig. 1 C). AP2 binds to clathrin, PM-enriched phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2), and endocytic sorting motifs encoded in cytoplasmic tails of cell surface receptors, for example nutrient receptors for transferrin (Tfn) or low-density lipoproteins, and signaling receptors such as the EGF receptor or G-protein–coupled receptors (i.e., cargo; Cremona and De Camilli, 2001; Owen et al., 2004; Traub and Bonifacino, 2013). Thus, AP2 complexes couple coat assembly to cargo recruitment. Rigorous biochemical, biophysical, and structural studies have mapped clathrin interactions to two regions on the β2 subunit of AP2 (Fig. 1): (1) a now-canonical clathrin-box motif (encoded by amino acids, LLNLD) located on the unstructured hinge region, which binds to the clathrin-box located on the N-terminal β-propeller domain (NTD) of the clathrin heavy chain (Shih et al., 1995; ter Haar et al., 2000); and (2) two less-well-defined binding sites located on the sandwich and platform domains of the β2 appendage, which require Y815 or Y888, respectively, and likely bind to a region within the ankle/distal leg of the clathrin heavy chain (Edeling et al., 2006; Schmid et al., 2006). Although the β2 hinge and appendage domains cooperatively interact with the NTD, in vitro pull-down experiments have shown that the clathrin-box motif plays the strongest role in mediating AP2–clathrin interactions. However, the relative importance of the two binding sites on the β2-appendage domain in clathrin interactions remains unclear, as studies differ in whether mutation of Y888 (Schmid et al., 2006) or Y815 (Edeling et al., 2006) most disrupted AP2–clathrin interactions.
Beyond AP2–clathrin interactions, the appendage domains of the α and β2 subunits, as well as the NTD, are interaction “hubs” that recruit a myriad of endocytic accessory proteins (EAPs) to coated pits in a dynamic and multivalent manner to facilitate clathrin assembly, cargo loading, curvature generation, fission, and coat disassembly (McMahon and Boucrot, 2011; Schmid and McMahon, 2007).
An unexpected complexity in AP2–clathrin interactions detected in living cells
While seemingly well understood biochemically, in-cell experiments are not fully consistent with prevailing models that focus on the clathrin-box–mediated interactions as the drivers of coat assembly. For example, β2 subunits lacking the LLNLD clathrin-box motif are still recruited to CCPs, while those bearing mutations in both appendage domain binding sites (Y815A/Y888A) are not (Edeling et al., 2006). Correspondingly, chemically induced targeting of an FK506-binding protein fusion of the β2 hinge-appendage domain to the PM was much more effective in recruiting clathrin and “hot-wiring” CME (Wood et al., 2017) than similar constructs derived from the β1 or β3 subunits of related clathrin adaptor protein complexes, AP1 and AP3, respectively, even though the clathrin-box motif in their hinge regions is conserved. In addition, knockdown and reconstitution studies have shown that CME cargo uptake can be restored by clathrin bearing point mutations that disrupt clathrin-box interactions in vitro (Collette et al., 2009; Willox and Royle, 2012). Indeed, point mutations disrupting any of the four known binding sites on the NTD, even in combinations of threes, do not impair the ability of clathrin to support Tfn endocytosis; CME is only severely inhibited when all four sites are disrupted or the NTD is deleted in its entirety (Willox and Royle, 2012).
Given the diversity and divergence of clathrin interacting motifs (Lemmon and Traub, 2012), AP2 could bind to clathrin in a promiscuous manner, as has been proposed in a “line-fishing” model for AP180–clathrin interactions (Zhuo et al., 2010). In this model, degenerate binding motifs in the unstructured/intrinsically disordered domain of AP180, another assembly protein/adaptor, are proposed to interact with the clathrin NTD through weak, promiscuous, and transient interactions. In support of this model, a recent x-ray crystallography study revealed that the clathrin-box motif of β2-adaptin can, indeed, bind to multiple sites on the NTD, including the clathrin-box site (Muenzner et al., 2017). This promiscuity, which appears to be encoded both in the degenerative binding motifs (Lemmon and Traub, 2012) and their binding sites, allows for multiple, low-affinity, and transient interactions between AP2 complexes and clathrin triskelia. This would give added flexibility to mechanisms by which AP2 nucleates clathrin assembly and enable structural diversity among CCPs (Sochacki and Taraska, 2019) while maintaining the stability of the clathrin coat. The dynamic, nonspecific interactions could also accommodate interchanges between AP2 and other clathrin-binding EAPs, which are critical for CCP maturation (McMahon and Boucrot, 2011; Mettlen et al., 2018; Schmid and McMahon, 2007).
Consistent with the potential promiscuity of β2–NTD interactions, another recent study (Chen et al., 2020) reported the unexpected finding that a linear peptide encoding key residues that define the W-box site, which was predicted to bind to only SNX9 and amphiphysin (Lemmon and Traub, 2012), potently and rapidly displaced AP2 complexes from CCPs and inhibited NTD–AP2 interactions in vitro. The W-box binding site spans the surface of three blades of the β-propeller and is oriented toward the membrane. That a linear peptide can compete with AP2–NTD interactions at low-micromolar concentrations suggests that binding between these two proteins involves surface residues rather than residues buried within binding pockets. Together, these observations suggest that prevailing models for AP2–clathrin interactions should be revisited.
Recent structural studies have also questioned the role of clathrin-box motifs and their binding site in mediating AP2–clathrin interactions. Two new cryo-EM studies have determined the structures of AP2 and clathrin-coated buds formed on PI(4,5)P2- and cargo-containing membranes (Kovtun et al., 2020) and of native CCVs purified from bovine brains (Paraan et al., 2020). Surprisingly, the β2-hinge could not be detected in either structure, and no evidence for its binding to the NTD binding sites could be found, suggesting that these interactions are weak and transient, variable, or both. Instead of the expected β2-hinge–NTD interactions, both cryo-EM structures revealed that one β2-appendage domain cross-links two adjacent clathrin NTDs, bringing the NTD into contact with its neighboring clathrin ankle region, presumably to promote clathrin cage assembly (Paraan et al., 2020). Consistent with their predominant role in the recruitment of overexpressed β2 adaptins to CCPs in vivo (Edeling et al., 2006), the β2-appendage binding sites (Y815 and Y888) seem to interact more consistently with two sites on the clathrin ankle and the Royle-box site on the NTD (Kovtun et al., 2020; Paraan et al., 2020). Interestingly, these two sites on the β2 appendage are also binding sites for other EAPs and adaptors; Y815 in the sandwich domain is required for binding AP180/CALM (phosphatidylinositol binding clathrin assembly protein), Eps15, and autosomal recessive hypercholesterolemia, whereas Y888 in the platform domain is required for binding β-arrestins and epsin (Edeling et al., 2006; Schmid et al., 2006). This engenders a dynamic competition between EAPs and clathrin for binding sites on AP2.
CCP stabilization and maturation
Once triggered, clathrin assembles in a cooperative manner on the PM (Moskowitz et al., 2005). However, early live-cell studies revealed that a significant portion (30–50%) of nascent CCPs fail to mature and spontaneously abort (Ehrlich et al., 2004; Loerke et al., 2009). Several factors appear to be required to stabilize nascent CCPs, including conformational changes in AP2 (Kadlecova et al., 2017) required to enhance PI(4,5)P2 and cargo binding (Edeling et al., 2006; Jackson et al., 2010; Kelly et al., 2014), cargo loading (Ehrlich et al., 2004; Kadlecova et al., 2017; Loerke et al., 2009), and several “pioneer” EAPs, so named because they act during early stages of CME (Cocucci et al., 2012; Ma et al., 2016; Ritter et al., 2013; Wang et al., 2020).
Among the pioneer EAPs required for early stabilization of nascent CCPs are the scaffold proteins Eps15 and Fcho1 (Cocucci et al., 2012; Ma et al., 2016; Wang et al., 2020), both of which bind AP2 and each other through multiple low-affinity binding motifs and protein interaction domains. Interestingly, a recent study has revealed that Eps15 coassembles with substoichiometric amounts of Fcho1 to form phase-separated liquid-like protein droplets (Day et al., 2019 Preprint), a property shared by its yeast homologue, Ede1 (Kozak and Kaksonen, 2019 Preprint). In an elegant series of experiments, the Stachowiak group used different degrees of light-inducible oligomerization of an Eps15–Cyr2 fusion protein to show that low levels of light-induced protein interactions, compatible with liquid-like assemblies, were essential for efficient endocytosis but that more extensive interactions induced at higher light intensities inhibited CME (Day et al., 2019 Preprint). The authors suggest that rapid assembly and dynamic exchange of protein components, analogous to those involved in liquid–liquid phase transitions, were required for CCP initiation and maturation. Indeed, components of the endocytic machinery bear many of the hallmarks of proteins involved in liquid–liquid phase separation, including (1) the presence of large intrinsic disorder domains (IDDs; Elbaum-Garfinkle et al., 2015; Wei et al., 2017; e.g., CALM, epsin, and AP2), (2) multivalency (Banjade and Rosen, 2014; Li et al., 2012; e.g., AP2, clathrin, Eps15, and intersectin), and (3) low-affinity interaction domains (e.g., EH and SH3 domains, AP2 appendage domains, and the clathrin NTD) that recognize short linear motifs (Brett et al., 2002; Miliaras and Wendland, 2004; Mittag and Parker, 2018). However, these features are also integral to properties previously ascribed to interactions among the components of the CME machinery (McMahon and Boucrot, 2011; Schmid and McMahon, 2007). These include cooperativity, avidity (or coincidence detection when multiple interacting partners are involved) and “matricity,” which was defined by McMahon as stabilized interactions with a geometrically defined scaffold, such as the clathrin lattice (McMahon and Boucrot, 2011; Schmid and McMahon, 2007). Whether the additional, well-defined physical properties of a liquid–liquid phase separation (Banani et al., 2017; Shin and Brangwynne, 2017) are met during the short lifetime and within the restricted volume of a CCP remains to be determined.
Nonetheless, CCP stabilization and growth clearly require a combination of multiple, likely redundant and promiscuous low-affinity interactions characterized by dynamic instability of the early intermediates. What then drives the vectorial process of CCP maturation? Recent evidence suggests that curvature generation at CCPs might be the tipping point (Wang et al., 2020). However, the mechanisms driving curvature generation at CCPs are incompletely understood and remain controversial.
Two historical models for curvature generation during coated pit invagination
From the outset, after Kanaseki and Kadota first observed the polygonal proteinaceous lattice surrounding isolated coated vesicles by negative stain (Kanaseki and Kadota, 1969), they hypothesized that membrane curvature was driven by the coat and would require the conversion of hexagons to the prerequisite 12 pentagons needed to generate a closed structure. This model was supported by John Heuser’s seminal observations made by quick-freeze deep etch of clathrin lattices on the adherent surface of fibroblasts (Heuser, 1980). Heuser observed flat lattices composed almost entirely of hexagonal arrays, curved lattices composed of hexagons and pentagons and completed coated vesicles bearing 12 pentagons. He also frequently observed adjacent pentagons and heptagons, which he suggested were intermediates in the conversion of hexagons to pentagons, structural rearrangements in the coat needed to generate curvature. This structural diversity of CCPs has since been observed in many cell types using multiple modes of light microscopy and EM (Sochacki and Taraska, 2019).
Support for a flat-to-curved transition derived from EM studies of the reassembly of coated pits on the upper surfaces of fibroblasts after recovery from K+ depletion, which arrests CCP formation (Larkin et al., 1983). The authors observed the early (2–5 min after restoring K+) appearance of small flat lattices composed entirely of hexagons, which grew in size. Curved pits were not detected until later time points (5–10 min) after recovery (Larkin et al., 1986). Based on these early studies, conversion of initially flat lattices to curved CCPs was the predominant model for decades. However, starting in the late 1980s, increasingly higher resolution cryo-EM structural studies of clathrin cages (Fotin et al., 2004; Smith et al., 1998; Vigers et al., 1986) revealed the extensive nature of interactions between two pairs of antiparallel proximal and distal legs of assembled triskelia that form the ridged edges of the completed polygon. Therefore, it was argued that disentangling the clathrin lattice once assembled would be energetically unfavorable and that indeed curvature must be built into the coats as they assemble (Kirchhausen, 2009; Kumar and Sain, 2016).
Two extreme models have emerged from these clathrin-centric views for curvature generation at CCPs, referred to as the “constant curvature” model and the “constant area” model (Fig. 2). In their simplest forms, the two models make distinct predictions that, in principle, can be tested by high-resolution microscopy methods. The constant curvature model predicts that curvature generation accompanies coat assembly, whereas the constant area model predicts that complete coat assembly precedes a flat-to-curved transition to generate curvature. Several recent studies applying correlative light-EM (CLEM) tomography (Avinoam et al., 2015), correlative light/quick-freeze deep etch microscopy (Bucher et al., 2018), live-cell polarized total internal refection fluorescence (TIRF) microscopy (Scott et al., 2018), live-cell high-speed atomic force microscopy (Yoshida et al., 2018), and live-cell TIRF structured illumination microscopy (SIM; Willy et al., 2019 Preprint), as well as theoretical modeling of these data (Kumar and Sain, 2016), have attempted to distinguish between these two models by testing these predictions. However, no consensus has emerged, and data are interpreted to support the constant area model (Avinoam et al., 2015), the constant curvature model (Willy et al., 2019 Preprint), a two-stage flat-to-curved process (Bucher et al., 2018; Yoshida et al., 2018), or a versatile process dependent on membrane tension and EAPs (Scott et al., 2018).
Figure 2.
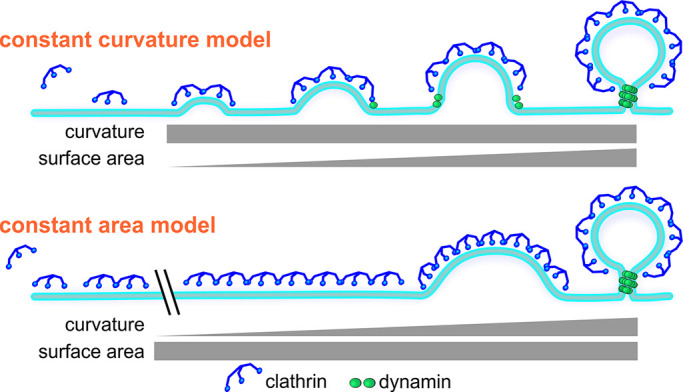
Two extreme models for curvature generation at CCPs: Constant curvature versus constant area. The constant curvature model predicts that curvature is acquired along with clathrin assembly, whereas the constant area model predicts that curvature is acquired by rearrangements of the clathrin lattice after its complete assembly.
The discrepancy regarding the constant curvature versus constant area model may lie in the limitations of current technologies. EM and CLEM studies look at single static snapshots, and whether the different structures are indeed intermediates along a single, productive pathway is ambiguous, especially given the prevalence of abortive CCPs (Ehrlich et al., 2004; Loerke et al., 2009). Live-cell imaging derives an average over many structures that may not reflect the behavior of individuals. Moreover, CLEM, live-cell TIRF-SIM, and high-speed atomic force microscopy all involve analysis of a selected subset of (typically larger and longer-lived) CCPs that may not represent the whole, especially given the structural heterogeneity of CCPs, even within single cells (Heuser, 1980; Sochacki and Taraska, 2019). Thus, there exists a need for technical advances that can ensure unbiased, live-cell tracking and analysis of individual coated pits with higher resolution and more accurate determination of curvature development. Curvature detection by polarized TIRF in live-cell CCP represents a promising technical advancement. Polarized TIRF studies show that approximately half of the clathrin coats acquired curvature at the very beginning of clathrin assembly, whereas the other half showed a variable delay before the onset of curvature acquisition (Scott et al., 2018). Based on these observations, the two extreme models may not be, and indeed are unlikely to be, mutually exclusive, as has been suggested (Sochacki and Taraska, 2019).
Potential mechanisms and players involved in curvature generation and stabilization during coated pit invagination
While the clathrin lattice undoubtedly contributes to curvature generation and/or curvature stabilization at CCPs (Fig. 3), it is now clear that numerous EAPs are also required. Indeed, when AP2 is outcompeted by overexpression of Dab2 and its cargo, the low-density lipoprotein receptor, clathrin assembles into large flat lattices (Mettlen et al., 2010). Similarly, when EAP recruitment is perturbed by replacing endogenous α-adaptins with a C-terminally truncated construct lacking the EAP-recruiting appendage domain, the result is the assembly of diffraction-limited (∼100–200 nm in diameter) flat clathrin lattices that rapidly turn over as abortive CCPs (Aguet et al., 2013).
Figure 3.

Potential mechanisms of curvature generation/stabilization at CCPs. Left to right: Scaffolding effect via imprinting the curved clathrin coats; scaffolding effect via imprinting the intrinsic shape of dimeric BAR domains (e.g., in Fcho1/2, endophilin, amphiphysin, and SNX9); wedging effect via hydrophobic insertion of amphipathic helices (e.g., in epsin1, AP180/CALM, endophilin, and amphiphysin); and crowding effect via steric force generated by the accumulation of proteins encoding large intrinsically disorderd domains (e.g., in epsin1, Fcho1/2, AP180/CALM, and amphiphysin).
EAPs can assist in membrane curvature generation and/or stabilization at CCPs by any of the following general mechanisms, which have been extensively reviewed elsewhere (Bassereau et al., 2018; Haucke and Kozlov, 2018): (1) scaffolding of membranes on intrinsically curved BAR (Bin1/Amphiphysin/Rvs) domains, (2) asymmetric expansion of the inner membrane leaflet by insertion of hydrophobic residues or loops, and/or (3) asymmetric molecular crowding on the inner leaflet (Fig. 3). Epsins (Ford et al., 2002; Snead et al., 2017) and CALM (Miller et al., 2015) have been implicated in curvature generation at CCPs, although the mechanism of action remains in dispute. Both proteins encode ANTH/ENTH (AP180/Epsin N-terminal homology) domains that bear N-terminal amphipathic H0 helices that can insert into the lipid bilayer, as well as long IDDs, which occupy large volumes that can also function as potent membrane curvature generators (Busch et al., 2015; Snead et al., 2019; Zeno et al., 2018). Recent in vitro studies involving careful quantitative measurement of protein density on the membrane and mutagenesis to disrupt the H0 helix have revealed that curvature generation by epsin1 is induced by a crowding effect rather than by hydrophobic insertion (Snead et al., 2017). In contrast, in vivo knockdown and reconstitution studies of CALM (Miller et al., 2015) and overexpression studies of epsin mutants (Ford et al., 2002) have suggested a requirement for a functional H0 helix to support CME. Additional quantitative knockdown and reconstitution studies that more directly measure curvature generation at CCPs are needed to test the relative contributions of these two curvature-generating modes in CME. Nonetheless, given the confined space within a growing CCP and the fact that AP2 complexes, CALM, and epsins, which are among the most abundant components of CCVs after clathrin (Paraan et al., 2020), all encode IDDs, molecular crowding is likely to have a significant effect in developing curvature during CCP maturation. Consistent with this, polarized TIRF (Scott et al., 2018) and superresolution CLEM (Sochacki et al., 2017) revealed that CALM became more concentrated at CCPs during membrane bending. Importantly, depending on the temporal hierarchy of their activities, these EAPs could contribute to either constant curvature and/or constant area modes of curvature generation at CCPs.
Other EAPs could contribute to clathrin-dependent curvature formation at CCPs by catalyzing rearrangements of the clathrin lattice. Thus, the uncoating EAPs (Hsc70, auxilin, and GAK) might be recruited during CCP assembly to allow the dynamic exchange of clathrin that is required for a flat-to-curved transition (Kaksonen and Roux, 2018). Indeed, high turnover rates of clathrin at CCPs were observed in fluorescence recovery after photobleaching experiments, with recovery halftimes varying from ∼2 to ∼10 s depending on experimental differences in cell line, temperature, and bleaching area (Avinoam et al., 2015; Loerke et al., 2005; Wu et al., 2001). This function of uncoating EAPs is supported by a recent study in a cell-free reconstitution system suggesting that Hsc70 regulated the dynamic instability of clathrin assembly (Chen et al., 2019). However, another study was unable to detect either endogenously tagged auxilin or GAK at CCPs during the early stages of maturation (He et al., 2020). Further functional studies in living cells, which will be complicated by functional redundancies described below, will be required to resolve this discrepancy.
The recent cryo-EM–based structural studies of native CCVs and membrane-bound AP2/clathrin buds have also provided some insight into curvature generation. Interestingly, AP2 complexes are not uniformly distributed on the clathrin coat but are depleted from curvature-essential pentagons (Kovtun et al., 2020; Paraan et al., 2020). Consistent with this, the flat-to-curved transition of clathrin coats has been reported to be marked by a decrease in the AP2/clathrin ratio (Bucher et al., 2018). AP2 levels in CCPs begin to decrease 15–20 s before the point when CCV scission occurs, while CALM levels continue to increase until CCV scission (Loerke et al., 2011; Miller et al., 2015). These combined observations suggest two potential mechanisms for the formation of pentagons: (1) clustered AP2 complexes nucleate clathrin assembly as hexagons, and then AP2 complexes in some hexagons are replaced by other adaptors (i.e., CALM or epsin1, which have intrinsic membrane curvature–sensing/generating ability) triggering the formation of pentagons as curvature is acquired; and (2) colocalized AP2 complexes and other adaptors (i.e., CALM or epsin1) simultaneously recruit and assemble clathrin to favor pentagon assembly. While further studies are needed to test whether and how AP2 and EAPs might affect clathrin assembly into hexagons versus pentagons, mechanism 1 fits a flat-to-curved transition model, while mechanism 2 fits the constant curvature model. The two mechanisms may coexist and be determined by the environmental factors that exist within subdomains of the PM, including local membrane tension (Boulant et al., 2011; Sheetz and Dai, 1996), stochastic availability of curvature-stabilizing/generating proteins, and the concentration and bulk of cargo molecules (Busch et al., 2015).
The plasticity and resilience of CME
Further complicating the development and testing of models for CCV formation are observations establishing the plasticity and resilience of CME. For example, several whole-genome screens based on receptor internalization have failed to identify EAPs (Collinet et al., 2010; Gulbranson et al., 2019; Kozik et al., 2013), suggesting that they are functionally redundant and/or that compensatory mechanisms can be induced to restore efficient CME. Indeed, Tfn receptor uptake was not significantly impaired even when the ability of AP2 to interact with multiple EAPs was disrupted by deleting the α-appendage domain (Aguet et al., 2013; Motley et al., 2006), and a compensatory mechanism and signaling pathway accounting for this resilience has been identified (Reis et al., 2015). Even AP2 complexes are not required for all forms of CME (Motley et al., 2003; Pascolutti et al., 2019). These observations likely reflect three key features of the protein interaction networks that drive CME: (1) the many EAPs have overlapping and/or redundant functions, i.e., as AP2 activators, assembly proteins, scaffolds, and/or curvature generators (McMahon and Boucrot, 2011; Mettlen and Danuser, 2014); (2) most EAPs function at low, substoichiometric concentrations and mediate multivalent but low-affinity and potentially promiscuous interactions (Perkins et al., 2010); and (3) the interaction hubs, AP2, and clathrin bind to and compete for overlapping partners (Edeling et al., 2006; Schmid and McMahon, 2007). These features ensure the dynamic instability and flexibility of this pathway. They further suggest that current, linear models of CME progression might not adequately capture its dynamic, stochastic, and plastic nature.
A dynamic model for CCP initiation, stabilization, and maturation
Prevalent models of CME suggest that the endocytic machinery is organized in functional “modules,” including nucleators, cargo selectors, coat components, curvature generators, and the scission machinery that act in a prescribed sequential manner. This concept emerged from influential studies on the temporal (Kaksonen et al., 2005) and spatial (Mund et al., 2018) relationships of EAP recruitment to sites of endocytosis in yeast and to CCPs in mammalian cells (Taylor et al., 2011), as well as from analysis of interactions among components of the endocytic machinery (Brett et al., 2002; Olesen et al., 2008; Praefcke et al., 2004). However, the new data described above suggest the need for a more dynamic model for AP2–clathrin interactions and EAP involvement during early stages of CCP initiation and stabilization, followed by a more stochastic phase of curvature generation and maturation, potentially along multiple paths (Fig. 4). This nonlinear model is supported by the structural diversity of CCPs detected in many cell types (Sochacki and Taraska, 2019), the variable effects of EAP depletion on CME and CCP dynamics, and the coexistence of curved, flat, and nonterminal CCPs (Perrais and Merrifield, 2005). It is also supported by the broad, Rayleigh-like distribution of CCP lifetimes, which range from ∼20 to >120 s as measured by quantitative TIRF microscopy (Aguet et al., 2013). A peak of the CCP lifetime distribution curve at ∼30 s identifies the average timing of key regulatory events, which are proposed to reflect a fidelity-monitoring, potentially kinetic, checkpoint governing progression to productive CCPs (Chen et al., 2019; Henry et al., 2012; Loerke et al., 2009; Mettlen et al., 2018). The peak in lifetime distribution is followed by an exponentially declining phase that suggests a more stochastic determination of overall CCP lifetimes, which end upon further recruitment and activation of the dynamin fission machinery during the last ∼10–20 s of a CCP’s lifetime. The lifetimes of CCPs are not limited by the extent of recruitment of AP2, clathrin (Loerke et al., 2011; Mettlen et al., 2010), or cargo (Liu et al., 2010). Thus, what determines the timing of recruitment and activation of the fission machinery remains unknown. Similarly, what properties (molecular, physical, or kinetic) of CCP progression determine whether a pit is productive or aborted remains unknown.
Figure 4.

A more dynamic and flexible model for CCP initiation, stabilization, and maturation. Top: Multivalent interactions between variable subsets of AP2-interacting EAPs (e.g., Eps15, Fcho1, ITSN, and/or NECAP) regulate the nucleation and stabilization of nascent CCPs. Cooperative clathrin lattice assembly further reinforces opportunities for avidity and matricity to stabilize growing CCPs. During CCP assembly and maturation, clathrin, AP2, and EAPs engage in multivalent, transient, and competitive interactions, as detected by rapid exchange of both clathrin and EAPs (blue arrows). Middle: Depending on the local environment, the nature of cargo, or even the state or type of the cell, the effects of the clathrin lattice, BAR-domain–containing proteins, hydrophobic insertions, and molecular crowding could be integrated, to variable and in part interchangeable extents, to facilitate CCP invagination by one of several pathways. Path 1 allows for curvature generation concomitant with CCP growth. Path 2 allows curvature to develop (potentially through a heptagonal intermediate (Heuser, 1980), as illustrated) at the edges of flat lattices, which can serve as nucleating platforms for formation of multiple CCVs (i.e., nonterminal events). Path 3 involves the conversion of flat lattices to curved pits requiring rapid clathrin exchange and rearrangements of the clathrin lattice. Each of these paths is subject to an “endocytic checkpoint” that results in the abortive turnover of nonproductive CCPs. The exact molecular or physical nature of factors sensed by the endocytic checkpoint and the mechanisms for turning over abortive CCPs remain to be determined. Inset shows an image from Steer and Hauser (1991) generously provided by John Heuser) showing evidence for the coexistence of each of these pathways, from quick-freeze, deep-etched, and rotary-shadowed micrographs that show the structural diversity of CCPs on the PM of cultured epithelial cells. Pentagons (yellow) and heptagons (cyan) were observed, frequently adjacent to each other, in curved coats and/or on flat lattices potentially starting to gain curvature.
The importance of cargo and PI(4,5)P2 interactions in the recruitment and allosteric activation of AP2 complexes at the membrane is well established (Edeling et al., 2006; Jackson et al., 2010; Kadlecova et al., 2017; Kelly et al., 2014). In addition, we propose that the regulated nucleation and stabilization of CCPs requires conformational activation and potentially local clustering of AP2 complexes mediated by multivalent interactions between some variable subset of AP2-interacting EAPs (e.g., Eps15, Fcho1, ITSN, and/or NECAP; see also Cocucci et al., 2012; Ma et al., 2016; Wang et al., 2020). Cooperative clathrin lattice assembly would further reinforce opportunities for avidity and matricity (analogous to phase separation?) and CCP stabilization. Early curvature generation would also increase coat stability as the ratio of unstable edges to a more stable interior of a clathrin lattice would be decreased as curvature increases (Fig. 4, path 1). Consistent with this model, a recent analysis of live-cell TIRF microscopy movies of CCP dynamics using a new algorithm based on the fluctuation of clathrin intensities during CCP assembly and growth indeed found that the rate of AP2 and clathrin recruitment and early curvature generation were factors that distinguished abortive coats from stabilized CCPs (Wang et al., 2020).
During CCP maturation, we propose that clathrin, AP2, and EAPs engage in multivalent, transient, and competitive interactions to accommodate fast clathrin assembly/disassembly, perhaps during intermediate stages of lattice assembly. Depending on the local environment, the nature of cargo, or even the state of the cell, the clathrin lattice, BAR-domain–containing proteins, hydrophobic insertions, and molecular crowding could combine, to variable and in part interchangeable extents, to facilitate CCP invagination at divergent stages during coat assembly (Fig. 4, paths 2 and 3).
Dynamin-2 is a weak curvature generator but an effective curvature sensor (Liu et al., 2011) and is recruited to CCPs via interactions between its Pro/Arg-rich domain and SH3-domain–containing EAPs (Bhave et al., 2020; Liu et al., 2011; McMahon et al., 1997; Posor et al., 2013; Rosendale et al., 2019; Takei et al., 1999). As several SH3-domain–containing binding partners of dynamin also encode BAR domains (e.g., endophilin, amphiphysin, and SNX9), the stochastic timing of dynamin-2 recruitment to CCPs could reflect coincidence detection of induced curvature at the necks of deeply invaginated CCPs (Daste et al., 2017; Liu et al., 2011) and SH3-domain interactions.
Testing models for CCP assembly and maturation in living cells
The resilience and plasticity of CME have hindered the functional analysis of AP2–clathrin interactions and the roles played by individual EAPs during CCP assembly, stabilization, and maturation. Previous efforts were primarily focused on using protein knockdown/knockout or mutant reconstitution techniques and usually using Tfn uptake as a readout. An obvious drawback for these traditional approaches is the potential induction of compensatory mechanisms due to the days or even weeks of cell preparation. A much more sensitive approach to detect effects on CME attributed to depletion of individual EAPs is to measure changes in CCP dynamics, which can also be diagnostic of compensatory mechanisms, by quantitative TIRF microscopy (Aguet et al., 2013; Wang et al., 2020). Indeed, a recent study showed that measurements of Tfn-receptor uptake in EAP-depleted cells frequently showed little or no correlation to changes in CCP dynamics (Wang et al., 2020).
An innovative approach to mitigate induction of compensatory mechanisms is to develop acute inhibitors that block specific and/or subsets of protein interactions thought essential for CME. For example, the chemical inhibitor Pitstop2 was shown to block clathrin-box interactions (von Kleist et al., 2011) and hence recruitment of a subset of EAPs to CCPs. However, subsequent studies have questioned its specificity and mechanism of action (Lemmon and Traub, 2012; Liashkovich et al., 2015; Smith et al., 2013; Willox et al., 2014). More recently, a small membrane-permeant, TAT-tagged peptide inhibitor named Wbox2 was designed based on the critical residues that define the W-box binding site on the clathrin NTD (Chen et al., 2020). Wbox2 strongly and acutely inhibited Tfn uptake and perturbed CCP dynamics by interfering with NTD–AP2 and NTD–SNX9 interactions (Chen et al., 2020). This approach might be especially applicable to interfering with selected subsets of low-affinity interactions between short linear motifs and their binding modules (e.g., SH3, EH, NTD, and AP2 appendage domains) abundant among components of the endocytic machinery. Systematic approaches, such as peptide phage display techniques (Sidhu et al., 2003), could be used to identify peptides that can acutely and specifically inhibit subsets of these protein interactions. By combining acute inhibition with quantitative TIRF microscopy, the effects of disrupting these interactions on discrete stages of CME could be measured and model predictions tested.
Combining high spatial– and temporal–resolution techniques such as SIM-TIRF, polarized TIRF, or epifluorescence/TIRF microscopy with measurements of the temporal hierarchy of appearance of individual EAPs to CCPs could also be used to test model predictions. However, given the overlap in protein interactions and hence competition among EAPs for recruitment to CCPs, these studies need to be performed with genome-edited cells so as not to change relative concentrations. Sensitivity then becomes an issue, as even when endogenously tagged dynamin-1 (Srinivasan et al., 2018) or GAK/auxilin (He et al., 2020) cannot be detected at CCPs, their effects on CCP dynamics can still be measured (Mettlen et al., 2010; Srinivasan et al., 2018). Another problem with this approach is that the kinetics of appearance of EAPs at CCPs is measured as an average of all events; hence, stochastic, transient interactions or the existence of multiple pathways (Fig. 4) can become blurred. Comparing the dynamic behaviors of EAP-positive versus negative subpopulations of CCPs, which could be reflective of different pathways, could be instructive, especially if combined with quantitative measurements of the numbers/stoichiometries of EAP molecules recruited to productive CCPs. The above approaches, coupled to acute interference of specific interactions (i.e., with chemical and peptide inhibitors), would confirm which protein interactions are being disrupted and reveal the function of these players in CCP stabilization and curvature development during CME.
Summary and implications
50 yr of biochemical and structural studies have culminated in fully reconstituting the CCV cycle in vitro, thereby establishing a strong foundation for the mechanisms underlying CME. However, in-cell experiments to probe the nature of clathrin–AP2 interactions, as well as the mechanism underlying curvature development during CCP budding, have frequently yielded unexpected results, highlighting the complexity of CME in living cells. They have also revealed an unexpected degree of flexibility and resilience in CME that is not captured in current linear models of CME as depicted in textbooks and reviews. Here, we have attempted to integrate the current literature, considering not only the experimental findings themselves but also their limitations and assumptions. We present a more dynamic, flexible, and nonlinear model for how functionally redundant EAPs and competing protein interactions can drive endocytic CCV formation and compensate for the loss of individual components of the endocytic machinery. This plasticity could play out in tuning CME to the differing physiology of the multiple cell types in the human body (e.g., neurons vs. hepatocytes vs. myeloid cells) that have different requirements for CME and exist in different physical environments. We know, for example, that autosomal dominant alleles of dynamin-2, thought to be essential for CME, result in muscle-specific and even age-dependent disease phenotypes (Durieux et al., 2010). This result alone brings into question our assumptions regarding mechanisms of endocytosis derived from studies of a small subset of (typically cancer) cells in culture. This plasticity also allows for adaptation of CME under pathological conditions, such as in cancer cells (Schmid, 2017). CME might be a mature field, but technological advancements have opened new horizons that rejuvenate it. As the British statistician George E.P. Box said, “All models are wrong; some are useful.” Thus, future studies using diverse cells not only in culture but also ultimately in whole organisms are needed to test and then evolve these models for understanding CME.
Acknowledgments
We thank all members of the Schmid laboratory for helpful discussions, particularly Dr. Marcel Mettlen for his contributions in conceptualizing and illustrating the nonlinear models for CCV formation.
Work in the Schmid laboratory was gratefully enabled by long-term support from the National Institute of General Medical Sciences (grants R01 GM42455 and R01 GM73165, also to G. Danuser) and the National Institute of Mental Health (grant MH61345).
The authors declare no competing financial interests.
Author contributions: Z. Chen and S.L. Schmid co-conceived and co-wrote this perspective.
References
- Aguet F., Antonescu C.N., Mettlen M., Schmid S.L., and Danuser G.. 2013. Advances in analysis of low signal-to-noise images link dynamin and AP2 to the functions of an endocytic checkpoint. Dev. Cell. 26:279–291. 10.1016/j.devcel.2013.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonny B., Burd C., De Camilli P., Chen E., Daumke O., Faelber K., Ford M., Frolov V.A., Frost A., Hinshaw J.E., et al. . 2016. Membrane fission by dynamin: what we know and what we need to know. EMBO J. 35:2270–2284. 10.15252/embj.201694613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avinoam O., Schorb M., Beese C.J., Briggs J.A.G., and Kaksonen M.. 2015. ENDOCYTOSIS. Endocytic sites mature by continuous bending and remodeling of the clathrin coat. Science. 348:1369–1372. 10.1126/science.aaa9555 [DOI] [PubMed] [Google Scholar]
- Banani S.F., Lee H.O., Hyman A.A., and Rosen M.K.. 2017. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18:285–298. 10.1038/nrm.2017.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banjade S., and Rosen M.K.. 2014. Phase transitions of multivalent proteins can promote clustering of membrane receptors. eLife. 3 e04123 10.7554/eLife.04123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassereau P., Jin R., Baumgart T., Deserno M., Dimova R., Frolov V.A., Bashkirov P.V., Grubmüller H., Jahn R., Risselada H.J., et al. . 2018. The 2018 biomembrane curvature and remodeling roadmap. J. Phys. D Appl. Phys. 51 343001 10.1088/1361-6463/aacb98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhave M., Mettlen M., Wang X., and Schmid S.L.. 2020. Early and non-redundatn functions of dynamin isoforms in clathrin-mediated endocytosis. Mol. Cell. Biol. mbc.E20-06-0363 10.1091/mbc.E20-06-0363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulant S., Kural C., Zeeh J.-C., Ubelmann F., and Kirchhausen T.. 2011. Actin dynamics counteract membrane tension during clathrin-mediated endocytosis. Nat. Cell Biol. 13:1124–1131. 10.1038/ncb2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brett T.J., Traub L.M., and Fremont D.H.. 2002. Accessory protein recruitment motifs in clathrin-mediated endocytosis. Structure. 10:797–809. 10.1016/S0969-2126(02)00784-0 [DOI] [PubMed] [Google Scholar]
- Bucher D., Frey F., Sochacki K.A., Kummer S., Bergeest J.-P., Godinez W.J., Kräusslich H.-G., Rohr K., Taraska J.W., Schwarz U.S., et al. . 2018. Clathrin-adaptor ratio and membrane tension regulate the flat-to-curved transition of the clathrin coat during endocytosis. Nat. Commun. 9:1109 10.1038/s41467-018-03533-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch D.J., Houser J.R., Hayden C.C., Sherman M.B., Lafer E.M., and Stachowiak J.C.. 2015. Intrinsically disordered proteins drive membrane curvature. Nat. Commun. 6:7875 10.1038/ncomms8875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Yong J., Martínez-Sánchez A., Yang Y., Wu Y., De Camilli P., Fernández-Busnadiego R., and Wu M.. 2019. Dynamic instability of clathrin assembly provides proofreading control for endocytosis. J. Cell Biol. 218:3200–3211. 10.1083/jcb.201804136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Mino R.E., Mettlen M., Michaely P., Bhave M., Reed D.K., and Schmid S.L.. 2020. Wbox2: A clathrin terminal domain-derived peptide inhibitor of clathrin-mediated endocytosis. J. Cell Biol. 219 e201908189 10.1083/jcb.201908189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocucci E., Aguet F., Boulant S., and Kirchhausen T.. 2012. The first five seconds in the life of a clathrin-coated pit. Cell. 150:495–507. 10.1016/j.cell.2012.05.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collette J.R., Chi R.J., Boettner D.R., Fernandez-Golbano I.M., Plemel R., Merz A.J., Geli M.I., Traub L.M., and Lemmon S.K.. 2009. Clathrin functions in the absence of the terminal domain binding site for adaptor-associated clathrin-box motifs. Mol. Biol. Cell. 20:3401–3413. 10.1091/mbc.e08-10-1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinet C., Stöter M., Bradshaw C.R., Samusik N., Rink J.C., Kenski D., Habermann B., Buchholz F., Henschel R., Mueller M.S., et al. . 2010. Systems survey of endocytosis by multiparametric image analysis. Nature. 464:243–249. 10.1038/nature08779 [DOI] [PubMed] [Google Scholar]
- Cremona O., and De Camilli P.. 2001. Phosphoinositides in membrane traffic at the synapse. J. Cell Sci. 114:1041–1052. [DOI] [PubMed] [Google Scholar]
- Dannhauser P.N., and Ungewickell E.J.. 2012. Reconstitution of clathrin-coated bud and vesicle formation with minimal components. Nat. Cell Biol. 14:634–639. 10.1038/ncb2478 [DOI] [PubMed] [Google Scholar]
- Daste F., Walrant A., Holst M.R., Gadsby J.R., Mason J., Lee J.E., Brook D., Mettlen M., Larsson E., Lee S.F., et al. . 2017. Control of actin polymerization via the coincidence of phosphoinositides and high membrane curvature. J. Cell Biol. 216:3745–3765. 10.1083/jcb.201704061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day K.J., Kago G., Wang L., Richter J.B., Hayden C.C., Lafer E.M., and Stachowiak J.C.. 2019. Liquid-like protein interactions catalyze assembly of endocytic vesicles. bioRxiv. doi: (Preprint posted December 2, 2019) 10.1101/860684 [DOI] [PMC free article] [PubMed]
- Durieux A.C., Prudhon B., Guicheney P., and Bitoun M.. 2010. Dynamin 2 and human diseases. J. Mol. Med. (Berl.). 88:339–350. 10.1007/s00109-009-0587-4 [DOI] [PubMed] [Google Scholar]
- Edeling M.A., Mishra S.K., Keyel P.A., Steinhauser A.L., Collins B.M., Roth R., Heuser J.E., Owen D.J., and Traub L.M.. 2006. Molecular switches involving the AP-2 β2 appendage regulate endocytic cargo selection and clathrin coat assembly. Dev. Cell. 10:329–342. 10.1016/j.devcel.2006.01.016 [DOI] [PubMed] [Google Scholar]
- Ehrlich M., Boll W., Van Oijen A., Hariharan R., Chandran K., Nibert M.L., and Kirchhausen T.. 2004. Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell. 118:591–605. 10.1016/j.cell.2004.08.017 [DOI] [PubMed] [Google Scholar]
- Elbaum-Garfinkle S., Kim Y., Szczepaniak K., Chen C.C.-H., Eckmann C.R., Myong S., and Brangwynne C.P.. 2015. The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc. Natl. Acad. Sci. USA. 112:7189–7194. 10.1073/pnas.1504822112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford M.G.J., Pearse B.M.F., Higgins M.K., Vallis Y., Owen D.J., Gibson A., Hopkins C.R., Evans P.R., and McMahon H.T.. 2001. Simultaneous binding of PtdIns(4,5)P2 and clathrin by AP180 in the nucleation of clathrin lattices on membranes. Science. 291:1051–1055. 10.1126/science.291.5506.1051 [DOI] [PubMed] [Google Scholar]
- Ford M.G., Mills I.G., Peter B.J., Vallis Y., Praefcke G.J., Evans P.R., and McMahon H.T.. 2002. Curvature of clathrin-coated pits driven by epsin. Nature. 419:361–366. 10.1038/nature01020 [DOI] [PubMed] [Google Scholar]
- Fotin A., Cheng Y., Grigorieff N., Walz T., Harrison S.C., and Kirchhausen T.. 2004. Structure of an auxilin-bound clathrin coat and its implications for the mechanism of uncoating. Nature. 432:649–653. 10.1038/nature03078 [DOI] [PubMed] [Google Scholar]
- Gulbranson D.R., Crisman L., Lee M., Ouyang Y., Menasche B.L., Demmitt B.A., Wan C., Nomura T., Ye Y., Yu H., et al. . 2019. AAGAB Controls AP2 Adaptor Assembly in Clathrin-Mediated Endocytosis. Dev. Cell. 50:436–446.e5. 10.1016/j.devcel.2019.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haucke V., and Kozlov M.M.. 2018. Membrane remodeling in clathrin-mediated endocytosis. J. Cell Sci. 131 jcs216812 10.1242/jcs.216812 [DOI] [PubMed] [Google Scholar]
- He K., Song E., Upadhyayula S., Dang S., Gaudin R., Skillern W., Bu K., Capraro B.R., Rapoport I., Kusters I., et al. . 2020. Dynamics of Auxilin 1 and GAK in clathrin-mediated traffic. J. Cell Biol. 219 e201908142 10.1083/jcb.201908142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry A.G., Hislop J.N., Grove J., Thorn K., Marsh M., and von Zastrow M.. 2012. Regulation of endocytic clathrin dynamics by cargo ubiquitination. Dev. Cell. 23:519–532. 10.1016/j.devcel.2012.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser J. 1980. Three-dimensional visualization of coated vesicle formation in fibroblasts. J. Cell Biol. 84:560–583. 10.1083/jcb.84.3.560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser J.E., and Reese T.S.. 1973. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J. Cell Biol. 57:315–344. 10.1083/jcb.57.2.315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson L.P., Kelly B.T., McCoy A.J., Gaffry T., James L.C., Collins B.M., Höning S., Evans P.R., and Owen D.J.. 2010. A large-scale conformational change couples membrane recruitment to cargo binding in the AP2 clathrin adaptor complex. Cell. 141:1220–1229. 10.1016/j.cell.2010.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadlecova Z., Spielman S.J., Loerke D., Mohanakrishnan A., Reed D.K., and Schmid S.L.. 2017. Regulation of clathrin-mediated endocytosis by hierarchical allosteric activation of AP2. J. Cell Biol. 216:167–179. 10.1083/jcb.201608071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaksonen M., and Roux A.. 2018. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 19:313–326. 10.1038/nrm.2017.132 [DOI] [PubMed] [Google Scholar]
- Kaksonen M., Toret C.P., and Drubin D.G.. 2005. A modular design for the clathrin- and actin-mediated endocytosis machinery. Cell. 123:305–320. 10.1016/j.cell.2005.09.024 [DOI] [PubMed] [Google Scholar]
- Kanaseki T., and Kadota K.. 1969. The “vesicle in a basket”. A morphological study of the coated vesicle isolated from the nerve endings of the guinea pig brain, with special reference to the mechanism of membrane movements. J. Cell Biol. 42:202–220. 10.1083/jcb.42.1.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly B.T., Graham S.C., Liska N., Dannhauser P.N., Höning S., Ungewickell E.J., and Owen D.J.. 2014. Clathrin adaptors. AP2 controls clathrin polymerization with a membrane-activated switch. Science. 345:459–463. 10.1126/science.1254836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhausen T. 2009. Imaging endocytic clathrin structures in living cells. Trends Cell Biol. 19:596–605. 10.1016/j.tcb.2009.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun O., Kane Dickson V., Kelly B.T., Owen D.J., and Briggs J.A.G.. 2020. Architecture of the AP2:clathrin coat on the membranes of clathrin-coated vesicles. bioRxiv:2020.2001.2028.922591. 10.1126/sciadv.aba8381 [DOI] [PMC free article] [PubMed]
- Kozak M., and Kaksonen M.. 2019. Phase separation of Ede1 promotes the initiation of endocytic events. bioRxiv. doi: (Preprint posted December 2, 2019) 10.1101/861203 [DOI]
- Kozik P., Hodson N.A., Sahlender D.A., Simecek N., Soromani C., Wu J., Collinson L.M., and Robinson M.S.. 2013. A human genome-wide screen for regulators of clathrin-coated vesicle formation reveals an unexpected role for the V-ATPase. Nat. Cell Biol. 15:50–60. 10.1038/ncb2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar G., and Sain A.. 2016. Shape transitions during clathrin-induced endocytosis. Phys. Rev. E. 94 062404 10.1103/PhysRevE.94.062404 [DOI] [PubMed] [Google Scholar]
- Larkin J.M., Brown M.S., Goldstein J.L., and Anderson R.G.W.. 1983. Depletion of intracellular potassium arrests coated pit formation and receptor-mediated endocytosis in fibroblasts. Cell. 33:273–285. 10.1016/0092-8674(83)90356-2 [DOI] [PubMed] [Google Scholar]
- Larkin J.M., Donzell W.C., and Anderson R.G.. 1986. Potassium-dependent assembly of coated pits: new coated pits form as planar clathrin lattices. J. Cell Biol. 103:2619–2627. 10.1083/jcb.103.6.2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon S.K., and Traub L.M.. 2012. Getting in touch with the clathrin terminal domain. Traffic. 13:511–519. 10.1111/j.1600-0854.2011.01321.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P., Banjade S., Cheng H.-C., Kim S., Chen B., Guo L., Llaguno M., Hollingsworth J.V., King D.S., Banani S.F., et al. . 2012. Phase transitions in the assembly of multivalent signalling proteins. Nature. 483:336–340. 10.1038/nature10879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liashkovich I., Pasrednik D., Prystopiuk V., Rosso G., Oberleithner H., and Shahin V.. 2015. Clathrin inhibitor Pitstop-2 disrupts the nuclear pore complex permeability barrier. Sci. Rep. 5:9994 10.1038/srep09994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A.P., Aguet F., Danuser G., and Schmid S.L.. 2010. Local clustering of transferrin receptors promotes clathrin-coated pit initiation. J. Cell Biol. 191:1381–1393. 10.1083/jcb.201008117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.W., Lukiyanchuk V., and Schmid S.L.. 2011. Common membrane trafficking defects of disease-associated dynamin 2 mutations. Traffic. 12:1620–1633. 10.1111/j.1600-0854.2011.01250.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loerke D., Wienisch M., Kochubey O., and Klingauf J.. 2005. Differential control of clathrin subunit dynamics measured with EW-FRAP microscopy. Traffic. 6:918–929. 10.1111/j.1600-0854.2005.00329.x [DOI] [PubMed] [Google Scholar]
- Loerke D., Mettlen M., Yarar D., Jaqaman K., Jaqaman H., Danuser G., and Schmid S.L.. 2009. Cargo and dynamin regulate clathrin-coated pit maturation. PLoS Biol. 7 e57 10.1371/journal.pbio.1000057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loerke D., Mettlen M., Schmid S.L., and Danuser G.. 2011. Measuring the hierarchy of molecular events during clathrin-mediated endocytosis. Traffic. 12:815–825. 10.1111/j.1600-0854.2011.01197.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L., Umasankar P.K., Wrobel A.G., Lymar A., McCoy A.J., Holkar S.S., Jha A., Pradhan-Sundd T., Watkins S.C., Owen D.J., et al. . 2016. Transient Fcho1/2⋅Eps15/R⋅AP-2 Nanoclusters Prime the AP-2 Clathrin Adaptor for Cargo Binding. Dev. Cell. 37:428–443. 10.1016/j.devcel.2016.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon H.T., and Boucrot E.. 2011. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 12:517–533. 10.1038/nrm3151 [DOI] [PubMed] [Google Scholar]
- McMahon H.T., Wigge P., and Smith C.. 1997. Clathrin interacts specifically with amphiphysin and is displaced by dynamin. FEBS Lett. 413:319–322. 10.1016/S0014-5793(97)00928-9 [DOI] [PubMed] [Google Scholar]
- Mettlen M., and Danuser G.. 2014. Imaging and modeling the dynamics of clathrin-mediated endocytosis. Cold Spring Harb. Perspect. Biol. 6 a017038 10.1101/cshperspect.a017038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mettlen M., Loerke D., Yarar D., Danuser G., and Schmid S.L.. 2010. Cargo- and adaptor-specific mechanisms regulate clathrin-mediated endocytosis. J. Cell Biol. 188:919–933. 10.1083/jcb.200908078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mettlen M., Chen P.-H., Srinivasan S., Danuser G., and Schmid S.L.. 2018. Regulation of Clathrin-Mediated Endocytosis. Annu. Rev. Biochem. 87:871–896. 10.1146/annurev-biochem-062917-012644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miliaras N.B., and Wendland B.. 2004. EH proteins: multivalent regulators of endocytosis (and other pathways). Cell Biochem. Biophys. 41:295–318. 10.1385/CBB:41:2:295 [DOI] [PubMed] [Google Scholar]
- Miller S.E., Mathiasen S., Bright N.A., Pierre F., Kelly B.T., Kladt N., Schauss A., Merrifield C.J., Stamou D., Höning S., et al. . 2015. CALM regulates clathrin-coated vesicle size and maturation by directly sensing and driving membrane curvature. Dev. Cell. 33:163–175. 10.1016/j.devcel.2015.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittag T., and Parker R.. 2018. Multiple Modes of Protein-Protein Interactions Promote RNP Granule Assembly. J. Mol. Biol. 430:4636–4649. 10.1016/j.jmb.2018.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz H.S., Yokoyama C.T., and Ryan T.A.. 2005. Highly cooperative control of endocytosis by clathrin. Mol. Biol. Cell. 16:1769–1776. 10.1091/mbc.e04-08-0739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motley A., Bright N.A., Seaman M.N.J., and Robinson M.S.. 2003. Clathrin-mediated endocytosis in AP-2-depleted cells. J. Cell Biol. 162:909–918. 10.1083/jcb.200305145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motley A.M., Berg N., Taylor M.J., Sahlender D.A., Hirst J., Owen D.J., and Robinson M.S.. 2006. Functional analysis of AP-2 alpha and mu2 subunits. Mol. Biol. Cell. 17:5298–5308. 10.1091/mbc.e06-05-0452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muenzner J., Traub L.M., Kelly B.T., and Graham S.C.. 2017. Cellular and viral peptides bind multiple sites on the N-terminal domain of clathrin. Traffic. 18:44–57. 10.1111/tra.12457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mund M., van der Beek J.A., Deschamps J., Dmitrieff S., Hoess P., Monster J.L., Picco A., Nédélec F., Kaksonen M., and Ries J.. 2018. Systematic Nanoscale Analysis of Endocytosis Links Efficient Vesicle Formation to Patterned Actin Nucleation. Cell. 174:884–896.e17. 10.1016/j.cell.2018.06.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen L.E., Ford M.G., Schmid E.M., Vallis Y., Babu M.M., Li P.H., Mills I.G., McMahon H.T., and Praefcke G.J.. 2008. Solitary and repetitive binding motifs for the AP2 complex alpha-appendage in amphiphysin and other accessory proteins. J. Biol. Chem. 283:5099–5109. 10.1074/jbc.M708621200 [DOI] [PubMed] [Google Scholar]
- Owen D.J., Collins B.M., and Evans P.R.. 2004. Adaptors for clathrin coats: structure and function. Annu. Rev. Cell Dev. Biol. 20:153–191. 10.1146/annurev.cellbio.20.010403.104543 [DOI] [PubMed] [Google Scholar]
- Paraan M., Mendez J., Sharum S., Kurtin D., He H., and Stagg S.M.. 2020. The Structures of Natively Assembled Clathrin-Coated Vesicles. Science Advances. 6:eaba8397 10.1126/sciadv.aba8397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascolutti R., Algisi V., Conte A., Raimondi A., Pasham M., Upadhyayula S., Gaudin R., Maritzen T., Barbieri E., Caldieri G., et al. . 2019. Molecularly Distinct Clathrin-Coated Pits Differentially Impact EGFR Fate and Signaling. Cell Rep. 27:3049–3061.e6. 10.1016/j.celrep.2019.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearse B.M.F. 1975. Coated vesicles from pig brain: purification and biochemical characterization. J. Mol. Biol. 97:93–98. 10.1016/S0022-2836(75)80024-6 [DOI] [PubMed] [Google Scholar]
- Pearse B.M. 1976. Clathrin: a unique protein associated with intracellular transfer of membrane by coated vesicles. Proc. Natl. Acad. Sci. USA. 73:1255–1259. 10.1073/pnas.73.4.1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearse B.M., and Robinson M.S.. 1984. Purification and properties of 100-kd proteins from coated vesicles and their reconstitution with clathrin. EMBO J. 3:1951–1957. 10.1002/j.1460-2075.1984.tb02075.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins J.R., Diboun I., Dessailly B.H., Lees J.G., and Orengo C.. 2010. Transient protein-protein interactions: structural, functional, and network properties. Structure. 18:1233–1243. 10.1016/j.str.2010.08.007 [DOI] [PubMed] [Google Scholar]
- Perrais D., and Merrifield C.J.. 2005. Dynamics of endocytic vesicle creation. Dev. Cell. 9:581–592. 10.1016/j.devcel.2005.10.002 [DOI] [PubMed] [Google Scholar]
- Posor Y., Eichhorn-Gruenig M., Puchkov D., Schöneberg J., Ullrich A., Lampe A., Müller R., Zarbakhsh S., Gulluni F., Hirsch E., et al. . 2013. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature. 499:233–237. 10.1038/nature12360 [DOI] [PubMed] [Google Scholar]
- Praefcke G.J., Ford M.G., Schmid E.M., Olesen L.E., Gallop J.L., Peak-Chew S.Y., Vallis Y., Babu M.M., Mills I.G., and McMahon H.T.. 2004. Evolving nature of the AP2 alpha-appendage hub during clathrin-coated vesicle endocytosis. EMBO J. 23:4371–4383. 10.1038/sj.emboj.7600445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis C.R., Chen P.H., Srinivasan S., Aguet F., Mettlen M., and Schmid S.L.. 2015. Crosstalk between Akt/GSK3β signaling and dynamin-1 regulates clathrin-mediated endocytosis. EMBO J. 34:2132–2146. 10.15252/embj.201591518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter B., Murphy S., Dokainish H., Girard M., Gudheti M.V., Kozlov G., Halin M., Philie J., Jorgensen E.M., Gehring K., et al. . 2013. NECAP 1 regulates AP-2 interactions to control vesicle size, number, and cargo during clathrin-mediated endocytosis. PLoS Biol. 11 e1001670 10.1371/journal.pbio.1001670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosendale M., Van T.N.N., Grillo-Bosch D., Sposini S., Claverie L., Gauthereau I., Claverol S., Choquet D., Sainlos M., and Perrais D.. 2019. Functional recruitment of dynamin requires multimeric interactions for efficient endocytosis. Nat. Commun. 10:4462 10.1038/s41467-019-12434-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth T.F., and Porter K.R.. 1964. Yolk Protein Uptake in the Oocyte of the Mosquito Aedes Aegypti. L. J. Cell Biol. 20:313–332. 10.1083/jcb.20.2.313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid S.L. 2017. Reciprocal regulation of signaling and endocytosis: Implications for the evolving cancer cell. J. Cell Biol. 216:2623–2632. 10.1083/jcb.201705017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid S.L., and Frolov V.A.. 2011. Dynamin: functional design of a membrane fission catalyst. Annu. Rev. Cell Dev. Biol. 27:79–105. 10.1146/annurev-cellbio-100109-104016 [DOI] [PubMed] [Google Scholar]
- Schmid E.M., and McMahon H.T.. 2007. Integrating molecular and network biology to decode endocytosis. Nature. 448:883–888. 10.1038/nature06031 [DOI] [PubMed] [Google Scholar]
- Schmid E.M., Ford M.G.J., Burtey A., Praefcke G.J.K., Peak-Chew S.-Y., Mills I.G., Benmerah A., and McMahon H.T.. 2006. Role of the AP2 β-appendage hub in recruiting partners for clathrin-coated vesicle assembly. PLoS Biol. 4 e262 10.1371/journal.pbio.0040262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott B.L., Sochacki K.A., Low-Nam S.T., Bailey E.M., Luu Q., Hor A., Dickey A.M., Smith S., Kerkvliet J.G., Taraska J.W., et al. . 2018. Membrane bending occurs at all stages of clathrin-coat assembly and defines endocytic dynamics. Nat. Commun. 9:419 10.1038/s41467-018-02818-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheetz M.P., and Dai J.. 1996. Modulation of membrane dynamics and cell motility by membrane tension. Trends Cell Biol. 6:85–89. 10.1016/0962-8924(96)80993-7 [DOI] [PubMed] [Google Scholar]
- Shih W., Gallusser A., and Kirchhausen T.. 1995. A clathrin-binding site in the hinge of the beta 2 chain of mammalian AP-2 complexes. J. Biol. Chem. 270:31083–31090. 10.1074/jbc.270.52.31083 [DOI] [PubMed] [Google Scholar]
- Shin Y., and Brangwynne C.P.. 2017. Liquid phase condensation in cell physiology and disease. Science. 357:eaaf4382. [DOI] [PubMed]
- Sidhu S.S., Fairbrother W.J., and Deshayes K.. 2003. Exploring protein-protein interactions with phage display. ChemBioChem. 4:14–25. 10.1002/cbic.200390008 [DOI] [PubMed] [Google Scholar]
- Smith C.J., Grigorieff N., and Pearse B.M.F.. 1998. Clathrin coats at 21 A resolution: a cellular assembly designed to recycle multiple membrane receptors. EMBO J. 17:4943–4953. 10.1093/emboj/17.17.4943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C.M., Haucke V., McCluskey A., Robinson P.J., and Chircop M.. 2013. Inhibition of clathrin by pitstop 2 activates the spindle assembly checkpoint and induces cell death in dividing HeLa cancer cells. Mol. Cancer. 12:4 10.1186/1476-4598-12-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snead W.T., Hayden C.C., Gadok A.K., Zhao C., Lafer E.M., Rangamani P., and Stachowiak J.C.. 2017. Membrane fission by protein crowding. Proc. Natl. Acad. Sci. USA. 114:E3258–E3267. 10.1073/pnas.1616199114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snead W.T., Zeno W.F., Kago G., Perkins R.W., Richter J.B., Zhao C., Lafer E.M., and Stachowiak J.C.. 2019. BAR scaffolds drive membrane fission by crowding disordered domains. J. Cell Biol. 218:664–682. 10.1083/jcb.201807119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sochacki K.A., and Taraska J.W.. 2019. From Flat to Curved Clathrin: Controlling a Plastic Ratchet. Trends Cell Biol. 29:241–256. 10.1016/j.tcb.2018.12.002 [DOI] [PubMed] [Google Scholar]
- Sochacki K.A., Dickey A.M., Strub M.-P., and Taraska J.W.. 2017. Endocytic proteins are partitioned at the edge of the clathrin lattice in mammalian cells. Nat. Cell Biol. 19:352–361. 10.1038/ncb3498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S., Burckhardt C.J., Bhave M., Chen Z., Chen P.H., Wang X., Danuser G., and Schmid S.L.. 2018. A noncanonical role for dynamin-1 in regulating early stages of clathrin-mediated endocytosis in non-neuronal cells. PLoS Biol. 16 e2005377 10.1371/journal.pbio.2005377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steer C.J., and Hauser J.E.. 1991. Clathrin and coated vesicles: critical determinants of intracellular trafficking. In Intracellular Trafficking of Proteins. Hanover J.A., and Steer C.J., editors. Cambridge, UK: Cambridge University Press; 47–102. [Google Scholar]
- Takei K., Slepnev V.I., Haucke V., and De Camilli P.. 1999. Functional partnership between amphiphysin and dynamin in clathrin-mediated endocytosis. Nat. Cell Biol. 1:33–39. 10.1038/9004 [DOI] [PubMed] [Google Scholar]
- Taylor M.J., Perrais D., and Merrifield C.J.. 2011. A high precision survey of the molecular dynamics of mammalian clathrin-mediated endocytosis. PLoS Biol. 9 e1000604 10.1371/journal.pbio.1000604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Haar E., Harrison S.C., and Kirchhausen T.. 2000. Peptide-in-groove interactions link target proteins to the β-propeller of clathrin. Proc. Natl. Acad. Sci. USA. 97:1096–1100. 10.1073/pnas.97.3.1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub L.M., and Bonifacino J.S.. 2013. Cargo recognition in clathrin-mediated endocytosis. Cold Spring Harb. Perspect. Biol. 5 a016790 10.1101/cshperspect.a016790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungewickell E., and Branton D.. 1981. Assembly units of clathrin coats. Nature. 289:420–422. 10.1038/289420a0 [DOI] [PubMed] [Google Scholar]
- Vigers G.P., Crowther R.A., and Pearse B.M.. 1986. Three-dimensional structure of clathrin cages in ice. EMBO J. 5:529–534. 10.1002/j.1460-2075.1986.tb04242.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kleist L., Stahlschmidt W., Bulut H., Gromova K., Puchkov D., Robertson M.J., MacGregor K.A., Tomilin N., Pechstein A., Chau N., et al. . 2011. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell. 146:471–484. 10.1016/j.cell.2011.06.025 [DOI] [PubMed] [Google Scholar]
- Wang X., Chen Z., Mettlen M., Noh J., Schmid S.L., and Danuser G.. 2020. DASC, a sensitive classifier for measuring discrete early stages in clathrin-mediated endocytosis. eLife. 9 e53686 10.7554/eLife.53686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M.-T., Elbaum-Garfinkle S., Holehouse A.S., Chen C.C.-H., Feric M., Arnold C.B., Priestley R.D., Pappu R.V., and Brangwynne C.P.. 2017. Phase behaviour of disordered proteins underlying low density and high permeability of liquid organelles. Nat. Chem. 9:1118–1125. 10.1038/nchem.2803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willox A.K., and Royle S.J.. 2012. Functional analysis of interaction sites on the N-terminal domain of clathrin heavy chain. Traffic. 13:70–81. 10.1111/j.1600-0854.2011.01289.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willox A.K., Sahraoui Y.M.E., and Royle S.J.. 2014. Non-specificity of Pitstop 2 in clathrin-mediated endocytosis. Biol. Open. 3:326–331. 10.1242/bio.20147955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willy N.M., Ferguson J.P., Silahli S., Cakez C., Hasan F., Chang H.C., Travesset A., Li S., Zandi R., Li D., Betzig E., Cocucci E., and Kural C.. 2019. Endocytic Clathrin Coats Develop Curvature at Early Stages of Their Formation. bioRxiv. doi: (Preprint posted December 23,2019) 10.1101/715219 [DOI] [PMC free article] [PubMed]
- Wood L.A., Larocque G., Clarke N.I., Sarkar S., and Royle S.J.. 2017. New tools for “hot-wiring” clathrin-mediated endocytosis with temporal and spatial precision. J. Cell Biol. 216:4351–4365. 10.1083/jcb.201702188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward M.P., and Roth T.F.. 1978. Coated vesicles: characterization, selective dissociation, and reassembly. Proc. Natl. Acad. Sci. USA. 75:4394–4398. 10.1073/pnas.75.9.4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Zhao X., Baylor L., Kaushal S., Eisenberg E., and Greene L.E.. 2001. Clathrin exchange during clathrin-mediated endocytosis. J. Cell Biol. 155:291–300. 10.1083/jcb.200104085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida A., Sakai N., Uekusa Y., Imaoka Y., Itagaki Y., Suzuki Y., and Yoshimura S.H.. 2018. Morphological changes of plasma membrane and protein assembly during clathrin-mediated endocytosis. PLoS Biol. 16 e2004786 10.1371/journal.pbio.2004786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaremba S., and Keen J.H.. 1983. Assembly polypeptides from coated vesicles mediate reassembly of unique clathrin coats. J. Cell Biol. 97:1339–1347. 10.1083/jcb.97.5.1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeno W.F., Baul U., Snead W.T., DeGroot A.C.M., Wang L., Lafer E.M., Thirumalai D., and Stachowiak J.C.. 2018. Synergy between intrinsically disordered domains and structured proteins amplifies membrane curvature sensing. Nat. Commun. 9:4152 10.1038/s41467-018-06532-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo Y., Ilangovan U., Schirf V., Demeler B., Sousa R., Hinck A.P., and Lafer E.M.. 2010. Dynamic interactions between clathrin and locally structured elements in a disordered protein mediate clathrin lattice assembly. J. Mol. Biol. 404:274–290. 10.1016/j.jmb.2010.09.044 [DOI] [PMC free article] [PubMed] [Google Scholar]