

 A general non-binary definition for on- and off-pathway intermediates is developed, enabling comparison of amyloid oligomers' contributions to fibril formation.
A general non-binary definition for on- and off-pathway intermediates is developed, enabling comparison of amyloid oligomers' contributions to fibril formation.
Abstract
The misfolding and aberrant aggregation of proteins into fibrillar structures is a key factor in some of the most prevalent human diseases, including diabetes and dementia. Low molecular weight oligomers are thought to be a central factor in the pathology of these diseases, as well as critical intermediates in the fibril formation process, and as such have received much recent attention. Moreover, on-pathway oligomeric intermediates are potential targets for therapeutic strategies aimed at interrupting the fibril formation process. However, a consistent framework for distinguishing on-pathway from off-pathway oligomers has hitherto been lacking and, in particular, no consensus definition of on- and off-pathway oligomers is available. In this paper, we argue that a non-binary definition of oligomers' contribution to fibril-forming pathways may be more informative and we suggest a quantitative framework, in which each oligomeric species is assigned a value between 0 and 1 describing its relative contribution to the formation of fibrils. First, we clarify the distinction between oligomers and fibrils, and then we use the formalism of reaction networks to develop a general definition for on-pathway oligomers, that yields meaningful classifications in the context of amyloid formation. By applying these concepts to Monte Carlo simulations of a minimal aggregating system, and by revisiting several previous studies of amyloid oligomers in light of our new framework, we demonstrate how to perform these classifications in practice. For each oligomeric species we obtain the degree to which it is on-pathway, highlighting the most effective pharmaceutical targets for the inhibition of amyloid fibril formation.
Oligomers are of paramount importance in protein aggregation diseases. They are believed to be the primary species responsible for the pathology of human disorders associated with amyloid formation, such as Alzheimer's disease and Parkinson's disease.1–6 They have also been identified as kinetic intermediates formed by, for example, amyloid β (Aβ40, Aβ42), tau, and α-synuclein (αS) proteins as precursors to these amyloid fibrils.7–10
Oligomers have variously been defined in the literature as species having smaller size, lower growth rate, less ordered structure, distinct surface properties or higher toxicity compared to fibrils8,11,12,14–19 (Fig. 2). Each of these definitions is associated with, and frequently motivated by, one or more detection techniques. Additionally, some studies use operational definitions based on preparation methods.20 Recent advances in single molecule experimental techniques, as well as isotope-based quantification using radio-assays or mass spectrometry,9,21 have allowed researchers to record the time-dependence of the concentration of oligomeric species present during amyloid fibril formation.8,21–23 This has prompted the development of new theoretical models that leverage the framework of chemical kinetics to determine from such data the mechanisms of oligomer-mediated amyloid fibril formation.10,11,13,21,24–26,64
Fig. 2. Amyloid oligomers have been distinguished from fibrils using several different working definitions. (a) Size differences may be detected using sedimentation properties under centrifugation,11 single-molecule fluorescence microscopy,8 or using cross-linking followed by SDS PAGE.12 (b) Differences in growth rate result in a much lower concentration of fibrillar species; the concentration of small aggregates may therefore often be identified with that of oligomers alone.13 (c) Structural differences may be detected using cryo-EM. The examples shown illustrate oligomers formed on fibril surfaces during the fibril assembly process (left) and end state fibrils (right) (images adapted from ref. 14), or using solid-state NMR, in which case oligomers produce less well-resolved spectra (left, adapted from ref. 15) than fibrils (right, adapted from ref. 16). (d) Differences in surface properties, for example hydrophobicity, may be detected using fluorescence probes such as 8-anilino-1-naphthalenesulfonic acid (ANS), Nile red,17 or by cryo-EM, in which case oligomers may have a higher tendency than fibrils to adsorb to the grid surface (adapted from ref. 14). (e) Toxicity differences can be detected using for example Ca2+ influx assays,18 or measurements of Long Term Potentiation (LTP) in rats – a process related to memory and learning (adapted from ref. 19).

A fundamental goal of such quantitative studies is to elucidate the precise relationship between oligomers and fibrils. Such knowledge would enhance our understanding of the molecular mechanisms underlying Alzheimer's and other protein misfolding diseases, and in so doing could transform current approaches to rational drug design for the treatment of these conditions. Oligomers that ultimately react to form fibrils are prime targets for inhibitory drugs designed to arrest amyloid fibril formation. The targeting of comparatively rare oligomeric species as opposed to native protein or fibrils could in principle be achieved with far lower doses of drug, avoiding potential toxic effects from the drug itself. Moreover, some amyloidogenic proteins have physiological function in their native monomeric state,27–30 and the direct targeting of the monomeric species by aggregation inhibitors may therefore have adverse health effects. Crucially, as the prime toxic species, the oligomers themselves are interesting targets, and understanding the factors that can reduce their concentrations has the potential to open up new therapeutic strategies.
To date, many studies have investigated the kinetic role of oligomers, labelling as “on-pathway” those oligomeric intermediates that react onward to form fibrils, and labelling those that are produced in side-reactions as “off-pathway”.31–39 This terminology originates from early studies of protein folding,40 in which it was hypothesised that proteins fold via a single, well-defined “pathway” through the complex high-dimensional potential energy landscape representing their possible conformations.41 However, the property that “on-pathway” encapsulates is not well defined, even in circumstances where this hypothesis actually holds. For example, although most extant definitions would consider the oligomers in Fig. 1a as on-pathway since they are obligate intermediates in fibril formation, some would not because most of them dissociate to monomers instead of converting to fibrils.42 Furthermore, in situations involving multiple competing reaction paths, that likely arise in oligomer-mediated fibril formation (see e.g.Fig. 1b and c) and in protein folding,42 the terminology itself can be ambiguous due to its binary nature. Consequently, some historical conclusions about the roles of certain experimentally-characterized oligomeric species in fibril formation may remain open to dispute. Despite these challenges, this terminology is already widely adopted and represents a valuable concept. What is needed, therefore, is a fully rigorous and general chemical kinetics-based definition of the terms on- and off-pathway, that successfully indicates the importance of an intermediate to the overall reaction under a wide range of possible scenarios.
Fig. 1. Amyloid oligomers defy easy classification as on- or off-pathway. Ambiguity may arise for several reasons; some of the most important are illustrated here. (a) Most oligomers dissociate into monomers rather than convert to fibrillar species. (b) One oligomer species converts to fibrils but also forms another oligomer species reversibly through a side reaction. (c) Two or more types of oligomer form from monomers, both of which can convert into fibrils with differing rates. (d) Two or more types of oligomer form from monomers, only one of which can convert into fibrils.

In this paper, we seek to resolve these issues and provide consistent definitions motivated by the underlying physics as well as by biochemical relevance. We argue that rather than a binary definition of oligomers as being either on- or off-pathway, it may be more fruitful to use a non-binary and quantitative definition in which an oligomer is assigned a value between 0 and 1 reporting on its relevance to fibril formation. We start by formalizing the definition of amyloid oligomers, and using this to clarify the distinction between oligomers and fibrils. We then explain how the various oligomeric species seen during an aggregation reaction can be represented by a reaction network, and how experimental considerations usually result in this network being coarse-grained into only one or a few distinct species. We provide a rigorous quantitative method for classifying oligomers in terms of their contribution to the overall reactive flux toward fibril formation, and demonstrate its utility by applying it to Monte Carlo simulations of a simple amyloid-forming system. For situations where it is still fruitful to invoke a binary view to discuss on- versus off-pathway oligomers, we propose that a cut-off value be used as a discriminator between these classes of oligomers. We show how our approach may be used in practice by revisiting recent publications discussing oligomers formed by the Parkinson's disease-associated protein α-synuclein,25 and the Alzheimer's disease associated proteins Aβ42 (ref. 21) and tau.11 We finally discuss how this work represents an advance over previous attempts to define on- and off-pathway oligomers.
Theoretical methods
Distinguishing between oligomers and fibrils
The term “oligomers” is frequently used in the context of amyloid fibril formation with different ad hoc definitions. The resultant ambiguity in what species can be considered oligomeric has the potential to lead to confusion. Supramolecular chemistry in fact provides a long-established and appropriate definition, found in the authoritative IUPAC Compendium of Chemical Terminology.43 In abbreviated form this reads: “molecules whose structure comprises a small plurality of units derived from molecules of lower relative molecular mass, and whose properties vary significantly with the removal of one or a few of the units”. The species meeting this formal definition are highly heterogeneous, varying significantly both in conformation and in size; and in propensity to grow into fibrils. However, this definition excludes mesoscopic droplets formed by liquid–liquid phase separation, such as the S-phase of Posey et al.,44 whose properties are expected to be unaffected by modest size fluctuations.45
Perhaps the most obvious oligomers detectable34,46 during amyloid fibril formation reactions are amyloid fibrils that are still short enough to be colloidally-suspended (see Fig. 3). These meet the formal definition of oligomers since addition of a few subunits dramatically increases their sedimentation propensity, and removal of a few subunits causes their disintegration back into monomers. These species have been termed “fibrillar oligomers” by Glabe et al.,34 an appropriate label given that they are both oligomeric and fibrillar in nature. However, whereas Glabe et al. define these species structurally, by their affinity for fibril-specific antibodies, we propose that they can be better defined on a kinetic basis: oligomeric species that are capable of the rapid elongation by monomer addition that is the hallmark of fibrillar aggregates. Fibrillar oligomers are thus not actually intermediates but products of the fibril formation process.
Fig. 3. Schematic illustration of the role of oligomers in amyloid fibril formation. Pre-nucleation clusters and short colloidal fibrils are both oligomeric and both detectable by experiment. They are termed “non-fibrillar oligomers” and “fibrillar oligomers” respectively. Note that fibrillar oligomers are simultaneously fibrils and oligomers. Note also that, once formed, fibrils can catalyse the formation of non-fibrillar oligomers from monomers and/or their conversion to fibrillar species.

Oligomers that are non-fibrillar are also detected in most or all fibril-forming systems studied to date (see Fig. 3). Since elongation of existing fibrils is much faster than formation of new fibrils (a necessary condition for large linear structures to form), fibrillar oligomers are expected to be present at much lower concentrations than any such non-fibrillar species. It is therefore reasonable to question whether fibrillar oligomers bear any practical relevance. However, these species are important even at low concentrations due to their characteristic powerful seeding effect on fibril proliferation. This property allows these species to be easily detected experimentally, even when they cannot be resolved via direct concentration measurements; for instance, the presence of fibrillar oligomers in supernatant taken from tau aggregation reactions after centrifugation to remove macroscopic fibrils is conclusively demonstrated by observation of the powerful seeding effect induced by aliquots of this supernatant.11 By contrast, during an ongoing Aβ42 aggregation reaction, only species retained by 200 nm filters were found to have strong seeding capacity, while no such activity was detected in the flow through of the same filters suggesting a non-significant population of fibrillar oligomers in this case.47
Representing oligomers in a reaction network
To fully understand the role of oligomers in amyloid fibril formation, it is useful to consider the underlying reaction network connecting monomers to fibrillar species (see Fig. 4a). The network, which is essentially a coarse-grained view of the free energy landscape, contains a node for monomers (reactants), nodes for fibrils and fibrillar oligomers (products), and nodes for each distinct oligomeric species. Species are considered distinct if the free energy barrier separating them is significantly greater than kT, the energy of thermal fluctuations (k is the Boltzmann constant; T is the temperature in Kelvin).
Fig. 4. Coarse-graining the oligomer reaction network. (a) A hypothetical full amyloid oligomer reaction network, in which 4 well-separated free energy wells exist in the space spanned by the internal degrees of freedom, corresponding to different generic oligomer conformations. (b) A coarse-grained reaction network seen by experimental methods that can only distinguish large conformational differences, and not oligomer aggregation number. (c) The same reaction network seen by a different experimental technique, the time-scale of which is slow relative to the rate of exchange between oligomer types A and B, which thus cannot be resolved separately by kinetic modelling. A study aiming to identify on- and off-pathway oligomers in this network must then treat them as a single coarse-grained species.

Each reaction process directly connecting distinct species is represented by an arrow. For the purposes of illustration in Fig. 4a, we assume that all reaction processes are either unimolecular or involve addition or loss of monomers. We may then represent all reaction processes by arrows with only one origin and one destination by treating monomer addition or dissociation implicitly in those arrows that connect oligomers of different size.
Note that in principle there may be more than one stable monomer conformation, in which case only native state (i.e. correctly folded) monomer would be designated as reactant. The other monomer conformations would then be intermediates of the aggregation reaction, and their on/off-pathway status determined by our framework. However, we restrict our attention to oligomeric intermediates in the present work. Note also that on/off-pathway terminology refers exclusively to intermediates; by definition all reaction paths start at the reactants and end at the products. As such, where monomers are considered reactants, they do not require an on/off-pathway designation.
Coarse-graining the reaction network
This picture fully describes the system, accounting for all populations that are separated by a significant barrier. However, it is rare that there are sufficient data to fully constrain a kinetic model with this level of detail. Model selection theory48 dictates that our chosen kinetic model should contain no more detail than is necessary to describe the data; we must therefore coarse-grain our network according to experimental considerations, and consider only total fluxes between the resultant combined populations.
Firstly, although available experimental techniques can often obtain separate reaction profiles for different groupings of oligomeric species, these groupings correspond better to distinct conformations, or internal structure, than to distinct sizes. For instance, both in α-synuclein aggregation25 and in yeast prion self-assembly,13 two separate oligomer populations, distinguished by their Förster Resonance Energy Transfer (FRET) efficiency, can be identified; in tau aggregation11 two oligomeric populations can be identified by their differing stabilities under different solution conditions. The experimental distinction of these two populations is primarily due to differences in the nature of the interactions between monomeric subunits, with size playing a minor role. We must therefore typically coarse-grain reaction networks (see Fig. 4a and b) such that oligomer populations are summed over all sizes and over all structures that are detected as one population.
Secondly, species can in practice be considered kinetically distinct only if the timescale of their interconversion is not much shorter than the timescale over which experiments can be accurately carried out (see Fig. 4b and c). This follows because if this condition is not satisfied, these species will be in effective equilibrium over the timescale of the experiment. Finally, reaction steps that proceed too slowly for their rates to be quantified over the experimental time course are ignored.
It is important to note that an interpretation of coarse-grained models in terms of elementary reaction steps can lead to erroneous conclusions. Rate constants and reaction orders in such models are effective, and will generally change (albeit slowly) in response to large concentration changes. For instance, eliminating oligomers entirely from kinetic models of fibril formation leads to a single coarse-grained nucleation-type reaction step.26 The fitted nucleation reaction order is then often fractional and is not straightforward to interpret using homogeneous classical nucleation theory.49,50
Monte Carlo simulations
To help illustrate the variety of kinetic scenarios for oligomers, we first turn to simulations. We simulate fibril formation from free monomers via globular oligomeric intermediates using a minimal model of aggregation developed in ref. 51. In this model a protein is described as a hard rod decorated with an attractive patch, the interaction of which represents the net interaction between the proteins, i.e. the net sum of electrostatic, van der Waals', and H-bonding interactions and the hydrophobic effect. For the oligomer-forming state of the protein, one weakly-attractive patch is placed on the tip of the rod, driving the formation of globular oligomers. Proteins in the fibril-forming state form interactions via a side-positioned patch, which are strong and drive the formation of stable fibrils. We run Monte Carlo simulations, with small translational and rotational moves, with random swaps between the soluble and fibril-forming state. The swap is penalised with an excess in chemical potential to capture the fact that amyloidogenic proteins are rarely found in a fibrillar conformation on their own, without binding partners. All the simulations use a starting configuration of 600 proteins randomly dispersed in a box of constant volume, the size of which corresponds to the target protein concentration. Oligomer concentration is analysed using an in-house cluster algorithm, while the probability of oligomer conversion is computed as in ref. 50.
Results & discussion
Identification of on- and off-pathway oligomers
A general distinction between oligomers that are on- and off-pathway with respect to the formation of fibrils from monomers must recognize that an oligomer's role is not binary, since in reality every oligomer will have some finite (albeit in some cases very small) probability of ultimately becoming a growth-competent fibril. It must also be able to take into account that more than one reaction path may carry significant flux towards the final fibril state. We note that the on- or off-pathway identity of an oligomer with respect to fibril formation has no direct connection to its toxicity.
As a first step, we require a quantitative indicator of the contribution of an oligomer to the overall fibril formation reaction, which can be more precisely cast as the share of the overall flux from monomers to fibrils that passes through the free energy well corresponding to this oligomer. This cannot typically be precisely measured, since any practical measurement technique will perturb the pattern of fluxes across the reaction network. One relatively non-disruptive approach is to calculate the reduction in overall flux to fibrils caused by the interaction of a particular oligomeric species with an agent that prevents further reaction. A particularly useful property of this metric is that it directly informs on the suitability of the oligomer as a pharmacological target for inhibition of fibril formation; it may be calculated as follows. Having identified the coarse-grained reaction network, we may write down the associated rate equations for oligomer and fibril concentrations. The total flux to fibrils, f, is simply the total rate of formation of new fibrils from all oligomeric precursors. The reduced flux associated with oligomer i, 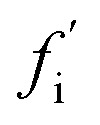 , is the rate of formation of new fibrils when the rate constants for the reaction steps undergone by oligomer i are set to zero. If the rate laws for each species in the reaction network have been identified and solved analytically in terms of the rate constants and time,
, is the rate of formation of new fibrils when the rate constants for the reaction steps undergone by oligomer i are set to zero. If the rate laws for each species in the reaction network have been identified and solved analytically in terms of the rate constants and time, 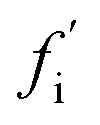 is straightforward to compute. This approach effectively constitutes a discrete sensitivity analysis where the corresponding sensitivity index is:
is straightforward to compute. This approach effectively constitutes a discrete sensitivity analysis where the corresponding sensitivity index is:
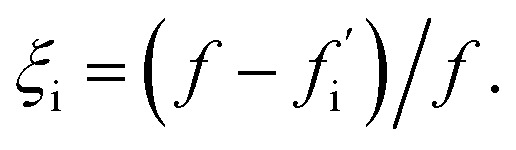 |
1 |
This sensitivity index ξi can be interpreted as the fractional reduction in flux upon suppression of oligomer i. The theoretical maximum value of 1 would be reached if all the flux to fibrils passes through oligomer i. By contrast, a small value of ξi indicates that oligomer i makes only a small contribution to total fibril formation.
Although the sensitivity index ξi quantifies how important a particular intermediate i is to the overall fibril formation reaction, it may not provide a clear indication of the degree to which the intermediate is on-pathway, since when many competing reaction paths are available, those species through which the majority of the flux passes may still all have low values of ξ. Therefore, a suitable way of determining the relative importance of different species is instead to normalize these index ξi by calculating the pathway index pi for each species i, which we define as:
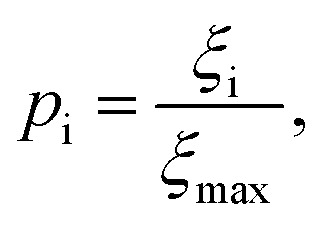 |
2 |
where ξmax is the largest ξ value in the system. Then species with p values near 1 are relatively important to the overall reactive flux, and species with values near 0 are comparatively unimportant. This is a continuous, rather than a binary, metric describing their on- and off-pathway nature, permitting the ranking of intermediates according to their importance to the overall reaction. However, if a binary classification is required, a species i may be considered on-pathway if its value for pi is greater than some investigator-specified threshold value, e.g. 0.1.
The magnitudes of the fluxes between each node in the reaction network will in general change over the course of the aggregation reaction. The p values are thus time-dependent, reflecting the fact that the contribution of a given oligomer to fibril formation can change over time. Although this time-dependence information can be valuable, it is convenient to use a single set of representative p values to rank the importance of intermediates for the entire reaction. The most appropriate values to use in this context are those computed at the time for which the overall flux to fibrils in the unperturbed reaction has reached its maximum. We therefore use these values by default unless we explicitly specify a reaction time. For open systems where monomer concentration remains more or less constant, such as in vivo environments, these are simply the steady-state values. For closed systems such as most in vitro experiments, these are the p values calculated at approximately the time corresponding to the end of the lag phase in the unperturbed reaction.
Here, we are interested in the importance of different oligomeric intermediates to the formation of fibrils (products) from monomers (reactants). However, our framework is fully general and can be applied to intermediates of any chemical reaction, simply by changing the identities of the reactant and product nodes in the reaction network.
Our general definition for on- and off-pathway oligomers (eqn (1) and (2)) allows us to clarify several key questions, including those illustrated in Fig. 1, that have caused particular confusion in the past. Firstly, if an oligomeric intermediate predominantly dissociates back to monomers rather than forming fibrils (Fig. 1a), it does not follow that this species is off-pathway. It is not the fraction of oligomers that form fibrils, but rather the fraction of fibrils that are formed from these oligomers, that determines whether the species is on-pathway. It is therefore perfectly possible for a kinetically unstable oligomer that forms reversibly to be on-pathway, as was recently found for tau,11 Aβ42,21 Ure2 (ref. 13) and PrP.52 To highlight that this is a desired property of the definition, consider the case where all fibrils are formed via a specific oligomeric species, yet the fate of most of those oligomers is to dissociate and not to from fibrils; given that this particular species is a necessary intermediate on the path to fibrils it would be assigned a p value of 1.0, and thus correctly identified as on-pathway in our framework. Any alternative definition by which this oligomeric species would be classified as off-pathway would be misleading.
Secondly, the identification of on- and off-pathway species depends critically on the coarse-graining. For instance, if a species capable of converting to fibrils is in fast equilibrium (on the timescale of the experimental measurements) with one that is not (Fig. 1b), the two species must be represented as a single on-pathway ensemble (such as species AB in Fig. 4c). Equally, where oligomeric species are in rapid equilibrium with monomeric protein, both should be considered as a single reactant ensemble. Further fine-graining of such ensembles requires additional experiments with greater time resolution.
Thirdly, the identities of the on- and off-pathway species can change over time. An advantage of the present framework is that it captures this time dependence, which can be of critical importance for the identification not just of appropriate inhibition targets, but also of optimal intervention times. For instance, formation of new fibrils sometimes occurs not just by primary nucleation but also by fibril surface-catalyzed secondary nucleation. Where these nucleation processes proceed via separate oligomeric intermediate species, our framework correctly identifies the secondary oligomers as being off-pathway and unimportant to fibril formation when there is a low concentration of fibrillar species, e.g. early on in a reaction from monomers. However, it also recognises that these species become on-pathway, and thus critical inhibition targets, later in the reaction once fibrillar material has built up. In many cases, including Aβ42 aggregation, this threshold is passed early in the lag phase.53
A key conclusion from this treatment is that the identity of the intermediates most responsible for flux to fibrils may change with conditions such as the monomer concentration, over the course of the reaction, or with the level of coarse-graining. Thus the classification of oligomers into on- and off-pathway species is not an absolute, invariant property of a given oligomeric species, but should instead be considered a property of the reaction network. As such, it depends on both the specific conditions and the level of detail with which one can measure the various oligomeric species. Thus, the concept of “being on-pathway” as an invariable property of a given species is not meaningful.
Illustrating the framework using Monte Carlo simulations
Current experimental techniques effectively report on ensembles of oligomers and thus result in a high degree of coarse-graining, with only one or two coarse-grained oligomer populations typically distinguishable. However, as experimental resolution of oligomers continues to improve, a quantitative definition of on-pathway species as presented here will become vital for proper classification of the multitude of recorded intermediates into on- and off-pathway species.
To illustrate how our kinetic framework (eqn (1) and (2)) performs when data on more fine-grained reaction networks are available, and to demonstrate how to compute ξi and pi in practice, we carried out simulations of amyloid nucleation via oligomeric intermediates using the minimal model of aggregation developed in ref. 51 (see Methods). In this system, there is only one type of stable non-fibrillar oligomer of a given size, and it has a micelle-like structure (Fig. 5a). As soon as two proteins in an oligomer convert to the fibril-forming state, the remaining proteins in the same oligomer convert much more rapidly; once all have converted, the fibrillar oligomer grows similarly rapidly by monomer addition. As expected, therefore, the fibrillar oligomer concentration is vanishingly low compared to that of non-fibrillar oligomers. The product of the conversion rate constant of each oligomer (Fig. 5b) with its concentration (Fig. 5c) gives the flux fj of each oligomer directly to fibrillar species (Fig. 5d), which sum to the overall reactive flux to fibrils f.
Fig. 5. Monte Carlo simulations of fibril formation via globular pre-fibrillar oligomers. (a) Reaction network; all fibrillar oligomers are included in the product node. The flux to fibrils is due to conformational conversion of pre-fibrillar to fibrillar oligomers. (b) Conversion rate constant vs. oligomer size j; this peaks at j = 6. (c) Size distribution of oligomers at steady state; this decreases with j apart from a j = 4–14 plateau. (d) The proportion of the total flux to fibrillar species going via each conversion reaction (given by the normalized product of (b) and (c)). (e) Pathway index pi (equal in this case to the sensitivity index ξ) vs. pre-fibrillar oligomer size. An oligomer of size j carries all the flux for all conversion reactions for oligomers ≥j; therefore, ξj is given by summing (d) over all conversion reactions ≥j. Dashed lines illustrate the result of alternative investigator-defined cut-offs (the examples given are 5, 20 and 50%) for a binary on-/off-pathway definition.

An oligomer of size j = N sits on all possible reaction paths to fibrils that pass through oligomers with j ≥ N. Since non-fibrillar oligomers are in near-equilibrium with monomers on the timescale of conversion in these simulations, and since the concentration of oligomers is far lower than that of monomers, the suppression of an oligomer of size j = N has a minimal effect on the populations of oligomers of size j < N. Therefore 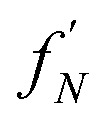 is well-approximated by fj
is well-approximated by fj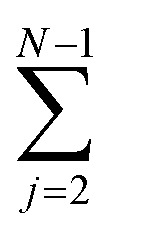 , and the sensitivity index of oligomers of size N is well-approximated by the normalized reverse-cumulative flux at size N, i.e.
, and the sensitivity index of oligomers of size N is well-approximated by the normalized reverse-cumulative flux at size N, i.e.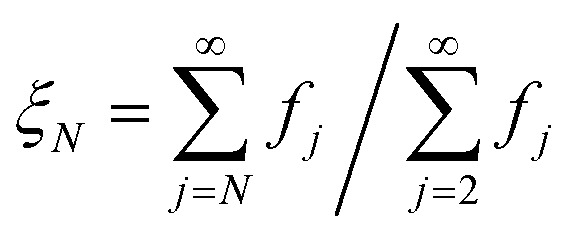 (Fig. 5e). Thus, since all flux to fibrils passes through oligomers of size 2, ξ2 = ξmax = 1 and the pathway index pN is recovered directly as pN = ξN.
(Fig. 5e). Thus, since all flux to fibrils passes through oligomers of size 2, ξ2 = ξmax = 1 and the pathway index pN is recovered directly as pN = ξN.
Both the conversion rate constant (Fig. 5b) and the concentration (Fig. 5c) of such globular micellar oligomers increase with size initially, since the increase in coordination number increases both the total oligomer bonding energy and the chances of forming a fibrillar bonding pair. The conversion rate constant and oligomer concentration both then peak and decline, due to increased steric hindrance within the oligomer destabilizing micellar oligomers,54,55 and disfavouring the conformational changes required for conversion into a fibril. The decline in oligomer concentration with size was also observed in earlier Monte Carlo simulations using a soft Go-type potential;56 the decline in conversion propensity follows since the first step of conversion is the transformation of a single monomer in the oligomer into a fibrillar state, moving its attractive patch from the end to the side of the rod-shaped monomer (see Fig. 5a). In order for this new state to be stabilized, the monomer must move its side into the position previously occupied by its end.
Since they are governed by common factors, we expect that in general for micelle-like oligomers the plots of conversion rate constant and oligomer concentration versus size have similar form. As a result, the species corresponding to the majority of the flux should correspond well to those that account for the majority of the population. Indeed, in our simulations we find that oligomers of size N = 15 and below constitute 95% of the total population of oligomers and contribute 95% of the total flux to fibrils (Fig. 5e). It is therefore in general unlikely that small on-pathway globular, micelle-like oligomers can coexist with a significant population of larger, off-pathway oligomers of the same morphology. Coarse-graining such oligomers by size is thus unlikely to merge on-pathway and off-pathway populations. This finding is highly relevant given that the appearance of globular, micelle-like oligomers is expected on physical grounds to be a common feature of protein aggregation reactions.54
Experimental identification of reaction networks
The experimental methods employed in kinetic studies to date have been able to resolve non-fibrillar oligomers into typically at most two distinct populations. The consequent low complexity of possible reaction networks in these studies makes application of the framework described here straightforward. We illustrate how this may be done using kinetic data on oligomers produced transiently during fibril formation reactions at 37 °C for Aβ42 (ref. 21) under conditions dominated by secondary nucleation, and for α-synuclein,25 at conditions where secondary nucleation is absent,57 and at 22 °C for K18 tau,11i.e. residues 243–274 of the major isoform tau441, under conditions where aggregation is induced by heparin. Both α-synuclein and tau oligomers could be resolved into distinct earlier-forming and later-forming species, referred to as type-A and type-B, respectively. By contrast, only a single population of Aβ42 oligomers could be resolved.
The first step in establishing whether an oligomer population is on- or off-pathway is to determine its kinetic relationship to monomers. The qualitative features of the data can often allow one to quickly narrow down possible mechanisms; for instance, both types of tau oligomers (Fig. 6a–d) as well as type-A α-synuclein oligomers (Fig. 7a) have initially convex kinetic curves, forming at their maximal rate at t = 0. In an unseeded aggregation reaction such as these, only monomers are present initially; therefore, such oligomers must be generated directly from monomers or species that are in fast equilibrium with monomers and thus part of the reactant ensemble. By contrast, type B α-synuclein oligomers have an initially concave kinetic curve (see Fig. 7b) and an initial formation rate of zero, so must instead originate from a later-appearing species. If, as is the case here, their formation rate reaches a maximum before substantial fibril mass has accumulated, this indicates that they are forming from another species present at early time, in this case type-A oligomers, rather than by disaggregation from fibrils or by secondary nucleation on fibril surfaces. Finally, Aβ42 oligomers are found to only form in appreciable quantities in the presence of both monomers and fibrils (see Fig. 8a and b), suggesting that their formation from monomers is catalyzed by fibrils.21 The validity of each of these observations can subsequently be confirmed by fitting the kinetic rate equations.
Fig. 6. Determining the reaction network for tau aggregation. Oligomer and fibril concentrations measured by single-molecule FRET;8 initial tau monomer concentration is 10 μM. Data from ref. 11. (a and b) Type-A tau oligomers equilibrate with monomers before the first t > 0 time point, i.e. faster than the experiments' time resolution. (c and d) Fits of the type-B tau oligomer concentration data to kinetic models in which they are formed directly from monomers and from type-A oligomers, respectively. As expected, the fits are too similar in quality to favour one model over the other, verifying that in this dataset type-A oligomers and monomers appear as a single reactant ensemble. (e) The maximum rate of tau fibril formation occurs before the first time point at 15 minutes, demonstrating that negligible numbers of new fibrils are produced after this time. However, type-B tau oligomer concentrations continue to rise long after 15 minutes; therefore, fibrils are not generated from type-B tau oligomers. (f) Resultant tau reaction network; type-B oligomers are identified as off-pathway.

Fig. 7. Kinetic model fitting used to establish the reaction network for α-synuclein monomers, oligomers and fibrils. Initial αS monomer concentrations are 35, 70 and 140 μM (increasing darkness); oligomer concentrations measured by single-molecule FRET.8 Data from ref. 25 (a and b), and ref. 8 (c and d). (a) Type-A αS oligomers show initially-convex kinetic profiles, requiring a model in which they form directly from monomeric αS. (b) Type-B αS oligomers show initially-concave kinetic profiles, requiring a model in which they form by conformational conversion from type-A oligomers. (c) Distribution of oligomer FRET efficiencies halfway through an αS aggregation reaction. Two peaks in the distribution can be seen, with the lower FRET peak corresponding to type-A oligomers, and the higher peak corresponding to type-B oligomers. (d) Distribution of oligomer FRET efficiencies near the start of an αS fibril disaggregation reaction performed under similar reaction conditions, showing that only high-FRET type-B oligomers are produced directly from fibrils. Thus to satisfy the requirement of microscopic reversibility, new fibrils must be produced from type-B oligomers and not from type-A oligomers during aggregation. (e) The resultant reaction network; all oligomers are clearly on-pathway. Note the extent to which type-B oligomers convert back to type-A oligomers is not known; this is due to relatively large errors in oligomer concentration data at late times in the aggregation reaction.

Fig. 8. Identifying the Aβ42 reaction network through kinetic model fitting and network perturbation with an inhibitor. Aggregation of 5 μM Aβ42 monomers into fibrils is investigated in the presence of a Brichos chaperone domain at a concentration of 5 μM; data from ref. 21. Fibril mass concentration measured by ThT fluorescence; oligomer concentration measured by radiolabelling. (a) Without Brichos, it can be seen that oligomers form only in the presence of both monomers and fibrils, demonstrating that oligomers are formed through secondary nucleation of monomers at the fibril surface. Addition of Brichos completely prevents formation of these oligomers (by binding the fibril surface nucleation sites). (b) The chaperone simultaneously completely prevents formation of new fibrils via secondary nucleation at the fibril surface; therefore, at least some of the oligomers formed in this way must later form fibrils. (c) Aβ42 reaction network. Only one population of oligomers is detected, which is identified as on-pathway. Note kinetic modelling suggests that Aβ42 oligomers are also formed directly from monomers through primary nucleation at a much slower rate, although the data are not sufficiently accurate to confirm this.

Fitting of the relevant kinetic equations moreover remains vital to determine the reaction rates of the individual processes and thus identify those species which need to be coarse-grained into a single population due to rapid exchange. For instance, kinetic modelling demonstrates that the exchange between tau monomers and type-A tau oligomers is too rapid relative to the experimental time resolution for the rates of formation and dissociation to be determined (see Fig. 6a and b). Type-A oligomers cannot therefore be identified as on- or off-pathway, but instead must be considered as part of the reactant (monomer) ensemble for the purposes of kinetic modelling. Thus we can only conclude that type-B tau oligomers and tau fibrils form either directly from monomers or from type-A oligomers, as it is in fact not possible given the current data to determine whether they form from the monomeric or the type-A oligomeric component of the reactant ensemble (see Fig. 6c and d).
The next step is to determine the oligomers' relationships to fibrils; this can be practically achieved in a number of ways. Ideally, one can measure the rate of new fibril formation over time directly. In the case of tau, this measurement reveals that the rate of maximal new fibril formation occurs in the first 15 minutes of the aggregation reaction (Fig. 6e). This is before appreciable numbers of type-B oligomers have formed, and long before the type-B oligomer population reaches its maximum (Fig. 6c and d). We therefore conclude that type-B oligomers have ξB ≃ 0 and therefore pB ≃ 0 and so should be classified as off-pathway (Fig. 6f).
Another method is to use microscopic reversibility. No reaction is fully unidirectional, so if an oligomeric species reacts to form fibrils, then starting from close to pure fibrils and diluting or transferring them to a solution condition where their concentration is above the equilibrium state, the same species can be generated by disaggregation. Type-B α-synuclein oligomers are formed by disaggregation from resuspended fibrils, followed by type-A oligomers (Fig. 7c and d), and finally monomers.8 This demonstrates that the sole significant reaction path from monomers to new fibrils connects first via type-A and then via type-B oligomers, and that therefore ξA ≃ ξB ≃ 1 (Fig. 7e).
Finally, inhibitors or activators may be employed to probe oligomer sensitivity directly: if the rate of formation of a measurable coarse-grained oligomeric species is decreased or increased but the rate of fibril formation is not affected, then this species is likely to be off-pathway. The converse of this is shown in the case of Aβ42 oligomers, where the Brichos chaperone domain can be used to inhibit fibril formation.58 Addition of 5 μM Brichos is found to fully inhibit the formation of oligomers on fibril surfaces during aggregation of 5 μM Aβ42. Simultaneously, secondary nucleation of new fibrils, ordinarily the predominant source of new Aβ42 fibrils, is completely blocked (Fig. 8a and b). This provides strong evidence that the oligomers detected have ξ ≃ 1 and therefore p = 1, and should thus be classified as on-pathway (Fig. 8c).
Discussion
We have explored how a non-binary classification of oligomers, taking into account the degree to which they contribute to fibril formation, can facilitate mechanistic understanding and inhibitor design in a more straight-forward manner than oligomers being viewed as simply on or off pathway. We have defined the pathway index of a given intermediate as the reduction in flux to fibrils caused by suppressing the reactions undergone by this intermediate. In addition to mimicking the effect of binding the species with e.g. an antibody, this definition has the benefit of minimizing disruption to the overall reaction network, and so comparatively accurately reporting on the share of flux to fibrils passing through this intermediate in an unperturbed reaction. Our definition facilitates the identification of oligomers whose inhibition will lead to a dramatic reduction of fibril formation. What really controls the overall flux to fibrils is the pattern of fluxes across the energy landscape, not the energy wells that correspond to specific intermediates. The most purist approach would be to only use on/off-pathway terminology to describe specific fluxes between chemical species, not the species themselves; however, these cannot be directly measured.
There are many examples in the literature of different strategies for classifying a particular observed oligomeric species as either on- or off-pathway, occasionally accompanied by an explicit definition of on- and off-pathway species.31–39 These strategies are often effective in the studies in which they are employed, but are usually not widely applicable, and can be viewed as special cases of the more general approach proposed here.
A notable example of an effective definition of on-pathway oligomers that nonetheless lacks generality is given in ref. 32, and reads: “a misfolded protein monomer or higher-order aggregate that is an obligate intermediate in the formation of amyloid”. This definition is only practical when the reaction networks being considered are sufficiently simple that they contain just one significant reaction path, and when the timescales of interconversion of the identified oligomeric species are slow compared to that of the measurements. These conditions are indeed satisfied by many reaction networks studied to date, including those of αS and Aβ42 considered in the present study. Under such conditions this definition yields identical results to our definition. However, it is unable to classify the type-A tau oligomers of Fig. 6b. Moreover, it is unable to correctly classify the oligomers in our Monte Carlo simulations (Fig. 5), and due to the presence of multiple competing paths mislabels all oligomers containing more than two monomers as off-pathway, despite the fact that their inhibition would almost completely halt fibril formation.
Another classification approach is given in ref. 39, where oligomers are isolated and added to fresh monomeric solution. If fibril formation in this seeded solution is not accelerated compared to unseeded monomeric solution, the oligomers are considered off-pathway. However, on-pathway oligomers are only expected to seed fibril formation effectively when the generation of these oligomers is a relatively slow step in the reaction path for new fibril formation. In all amyloid oligomer systems hitherto studied quantitatively with kinetic models, this is not the case and it is the conformational conversion of pre-fibrillar oligomers to fibrillar species that is the slow step in new fibril formation, not the generation of the pre-fibrillar oligomers themselves.
In the same work it was suggested that the lack of fibril mass produced under conditions promoting the formation of the oligomer species studied indicates that this species is off-pathway under all reaction conditions. However, if an oligomer that is normally on-pathway is sufficiently stabilized by addition of a binding substance, most monomer will end up kinetically trapped in this oligomeric state, leaving no monomer for elongation of existing fibrils. Appreciable fibril mass will then not form on a reasonable timescale, even though the oligomers may continue to slowly convert to fibrillar species. A high affinity binding substance may even cause the oligomer to become thermodynamically more stable than fibrils. This scenario again highlights the importance of taking an approach that considers the whole reaction network.
An alternative operational definition would be to suppress all reaction steps that lead directly to formation of the intermediate, such that it is effectively removed from the reaction network. Under most circumstances this would yield largely similar results to the present definition in terms of the pathway index. However, under certain circumstances, a species described as off-pathway by this alternative definition could still be a useful target for sequestration in order to reduce the overall flux to product; we therefore do not pursue this alternative definition further.
Although we focus on oligomer-mediated fibril formation in this paper, the framework we have developed is actually more broadly applicable to self assembly reactions. Intermediates need not be oligomeric, and end states need not be fibrillar, for on-/off-pathway concepts to have value. An obvious application is protein folding, the study of which is what originally gave rise to the on-/off-pathway terminology. Although the inhibition angle is less relevant in that case, our rigorous definition could help shed light on folding mechanisms by informing appropriate usage of this rather popular terminology.59
An application more closely related to fibril formation is the generation of amorphous aggregates in amyloidogenic systems. In ref. 60, transthyretin (M-TTR) is found to bind to Aβ42 oligomers, in so doing causing the production of amorphous aggregates instead of fibrils. By considering an alternative reaction network in which amorphous aggregates are the product, and by noting that these aggregates contain M-TTR, we can determine that these oligomers must be on-pathway to both fibril and amorphous aggregate formation. Moreover, it has been shown that amyloidogenic proteins are susceptible to both liquid–liquid phase separation and protein aggregation;61 one can thus expect phase-separated protein to sometimes be a competing reaction end-point alongside fibrils for amyloidogenic protein aggregation in vivo. Our framework permits us in principle to determine the on/off-pathway status of different oligomeric and non-oligomeric intermediates with respect to each of these end points, affording greater insight into the overall chemistry.
Liquid–liquid phase-separated protein droplets can also act as non-oligomeric intermediates of either protein aggregation or macroscopic phase separation,44,62 and it is of interest to apply our on/off-pathway concepts to such species. In ref. 62 it is demonstrated that the clusters that are large enough to be unstable with respect to growth are selectively cleared in vivo by chaperones to prevent macroscopic phase separation. Considering this system in the context of our framework, the smaller, growth-stable, clusters can only be considered on-pathway to macroscopic phase separation (and thus reasonable clearance targets) in the absence of larger clusters at the reaction starting point. Once larger clusters have formed, these smaller clusters become off-pathway, since their suppression no longer significantly affects the phase separation reaction. Chaperones have thus likely evolved to selectively target larger clusters since attempting to clear solely small clusters to prevent phase separation is not a useful control strategy in the long run: as soon as some smaller clusters evade clearance long enough to grow into larger clusters, it becomes ineffective. Moreover, attempting to clear both small and large clusters is an inefficient use of cell resources. Note there is a parallel here to fibril formation via distinct primary and secondary oligomers: once such a reaction has got underway, primary oligomers switch from being on-pathway to off-pathway, and cease to represent good target species for the inhibition of protein aggregation.63 These examples highlight the importance of considering carefully the reaction starting point when studying reaction mechanisms.
Conclusions
In this work we have described a general conceptual framework for identifying amyloid oligomers and for classifying them according to their role in fibril formation. Our quantitative, non-binary approach for classifying on- and off-pathway species is applicable to systems featuring multiple distinct reaction paths from monomers to fibrils, and is furthermore able to appropriately classify oligomeric species in fast equilibrium with other species. With continued improvements in experimental techniques, oligomer reaction networks will be resolved in ever greater detail in the future, and a generally applicable definition will become increasingly important.
For the three amyloid systems included in the current analysis we can conclude the following. For Aβ42 in buffer under conditions where secondary nucleation dominates, all oligomers detected experimentally are fully on pathway with ξ ≃ 1. The Aβ42 oligomers dissociate rapidly to monomers, but are constantly reformed thus contribute very productively to the formation of fibrils. For α-synuclein, under conditions where secondary nucleation is suppressed, two kinds of oligomers are detected, both are fully on pathway with ξ ≃ 1 and one type forms from monomers and then converts the other type, which in turn converts to fibrils. For K18 tau, in reactions using heparin to trigger heterogeneous primary nucleation, two kinds of oligomers are detected, with one contributing to fibril formation with a sensitivity index of ξ ≃ 1 and the other one being much less productive with a sensitivity index of ξ ≃ 0.
A key therapeutic goal in neurodegenerative diseases is to interrupt or halt the oligomer formation process; charting the reaction network connecting native protein to fibrillar aggregates is a vital component of this endeavour. On-pathway oligomers, properly identified, present attractive targets for rational drug design; not only due to their inherent and well-documented toxicity, but also due to the critical dependence of the overall fibrillation process on their formation.
Thus, our definition of on-pathway species is advantageous not only for its generality and consistency, but also because it retains its usefulness in the context of inhibition strategies: the only oligomers whose inhibition will lead to a clear reduction in new fibril formation are those identified as on-pathway using our framework. We therefore believe that the present work forms the necessary theoretical basis for future studies of targeted suppression of amyloid formation via oligomer inhibition.
Conflicts of interest
S. Linse and T. P. J. Knowles are founders of Wren Therapeutics Ltd, UK, a drug discovery company focussed on protein misfolding diseases.
Acknowledgments
We are grateful to the Schiff Foundation (AJD), Peterhouse, Cambridge (TCTM), the Swiss National Science foundation (TCTM), Ramon Jenkins Fellowship, Sidney Sussex, Cambridge (GM), the Royal Society (AŠ), the Academy of Medical Sciences and Wellcome Trust (AŠ), the Danish Research Council (MK), the Lundbeck Foundation (MK), the Swedish Research Council (SL), the Wellcome Trust (TPJK), the Cambridge Centre for Misfolding Diseases (TPJK), the BBSRC (TPJK), the Frances and Augustus Newman Foundation (TPJK) for financial support. The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007-2013) through the ERC grants PhysProt (agreement no. 337969), MAMBA (agreement no. 340890) and NovoNordiskFonden (SL).
References
- Winner B., Jappelli R., Maji S. K., Desplats P. A., Boyer L., Aigner S., Hetzer C., Loher T., Vilar M., Campioni S., Tzitzilonis C., Soragni A., Jessberger S., Mira H., Consiglio A., Pham E., Masliah E., Gage F. H., Riek R. Proc. Natl. Acad. Sci. U. S. A. 2011;108:4194–4199. doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Muñoz M. J., Castillo-Carranza D. L., Krishnamurthy S., Paulucci-Holthauzen A. A., Sengupta U., Lasagna-Reeves C. A., Ahmad Y., Jackson G. R., Kayed R. Neurobiol. Dis. 2014;71:14–23. doi: 10.1016/j.nbd.2014.08.008. [DOI] [PubMed] [Google Scholar]
- Conway K. A., Lee S. J., Rochet J. C., Ding T. T., Williamson R. E., Lansbury P. T. Proc. Natl. Acad. Sci. U. S. A. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary J. P., Walsh D. M., Hofmeister J. J., Shankar G. M., Kuskowski M. A., Selkoe D. J., Ashe K. H. Nat. Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Mukherjee A., Morales-Scheihing D., Butler P. C., Soto C. Trends Mol. Med. 2015;21:439–449. doi: 10.1016/j.molmed.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.-Y., Gurlo T., Kayed R., Butler A. E., Haataja L., Glabe C. G., Butler P. C. Diabetes. 2007;56:1324–1332. doi: 10.2337/db06-1579. [DOI] [PubMed] [Google Scholar]
- Narayan P., Orte A., Clarke R. W., Bolognesi B., Hook S., Ganzinger K. A., Meehan S., Wilson M. R., Dobson C. M., Klenerman D. Nat. Struct. Mol. Biol. 2011;19:79. doi: 10.1038/nsmb.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremades N., Cohen S. I. A., Deas E., Abramov A. Y., Chen A. Y., Orte A., Sandal M., Clarke R. W., Dunne P., Aprile F. A., Bertoncini C. W., Wood N. W., Knowles T. P. J., Dobson C. M., Klenerman D. Cell. 2012;149:1048–1059. doi: 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S. I. A., Linse S., Luheshi L. M., Hellstrand E., White D. A., Rajah L., Otzen D. E., Vendruscolo M., Dobson C. M., Knowles T. P. J. Proc. Natl. Acad. Sci. U. S. A. 2013;110:9758–9763. doi: 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shammas S. L., Garcia G. A., Kumar S., Kjaergaard M., Horrocks M. H., Shivji N., Mandelkow E., Knowles T. P. J., Mandelkow E., Klenerman D. Nat. Commun. 2015;6:7025. doi: 10.1038/ncomms8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjaergaard M., Dear A. J., Kundel F., Qamar S., Meisl G., Knowles T. P. J., Klenerman D. ACS Chem. Neurosci. 2018;9:3060–3071. doi: 10.1021/acschemneuro.8b00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitan G., Lomakin A., Teplow D. B. J. Biol. Chem. 2001;276:35176–35184. doi: 10.1074/jbc.M102223200. [DOI] [PubMed] [Google Scholar]
- Yang J., Dear A. J., Michaels T. C. T., Dobson C. M., Knowles T. P. J., Wu S., Perrett S. J. Am. Chem. Soc. 2018;140:2493–2503. doi: 10.1021/jacs.7b10439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Törnquist M. Proc. Natl. Acad. Sci. U. S. A. 2020;117(21):11265–11273. doi: 10.1073/pnas.1918481117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendel C., Bjerring M., Dubnovitsky A., Kelly R. T., Filippov A., Antzutkin O. N., Nielsen N. C., Härd T. Angew. Chem., Int. Ed. 2014;53:12756–12760. doi: 10.1002/anie.201406357. [DOI] [PubMed] [Google Scholar]
- Colvin M. T., Silvers R., Ni Q. Z., Can T. V., Sergeyev I., Rosay M., Donovan K. J., Michael B., Wall J., Linse S., Griffin R. G. J. Am. Chem. Soc. 2016;138:9663–9674. doi: 10.1021/jacs.6b05129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.-E., Sang J. C., Rodrigues M., Carr A. R., Horrocks M. H., De S., Bongiovanni M. N., Flagmeier P., Dobson C. M., Wales D. J., Lee S. F., Klenerman D. Nano Lett. 2018;18:7494–7501. doi: 10.1021/acs.nanolett.8b02916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagmeier P., De S., Wirthensohn D. C., Lee S. F., Vincke C., Muyldermans S., Knowles T. P. J., Gandhi S., Dobson C. M., Klenerman D. Angew. Chem., Int. Ed. 2017;56:7750–7754. doi: 10.1002/anie.201700966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D. M., Klyubin I., Fadeeva J. V., Cullen W. K., Anwyl R., Wolfe M. S., Rowan M. J., Selkoe D. J. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Klein W. L. Neurochem. Int. 2002;41:345–352. doi: 10.1016/s0197-0186(02)00050-5. [DOI] [PubMed] [Google Scholar]
- Michaels T. C. T., Saric A., Curk S., Bernfur K., Arosio P., Meisl G., Dear A. J., Cohen S. I. A., Dobson C. M., Vendruscolo M., Linse S., Knowles T. P. J. Nat. Chem. 2020;12:445–451. doi: 10.1038/s41557-020-0452-1. [DOI] [PubMed] [Google Scholar]
- Horrocks M. H., Tosatto L., Dear A. J., Garcia G. A., Iljina M., Cremades N., Dalla Serra M., Knowles T. P. J., Dobson C. M., Klenerman D. Anal. Chem. 2015;87:8818–8826. doi: 10.1021/acs.analchem.5b01811. [DOI] [PubMed] [Google Scholar]
- Tosatto L., Horrocks M. H., Dear A. J., Knowles T. P. J., Dalla Serra M., Cremades N., Dobson C. M., Klenerman D. Sci. Rep. 2015;5:16696. doi: 10.1038/srep16696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia G. A., Cohen S. I. A., Dobson C. M., Knowles T. P. J. Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys. 2014;89:032712. doi: 10.1103/PhysRevE.89.032712. [DOI] [PubMed] [Google Scholar]
- Iljina M., Garcia G. A., Horrocks M. H., Tosatto L., Choi M. L., Ganzinger K. A., Abramov A. Y., Gandhi S., Wood N. W., Cremades N., Dobson C. M., Knowles T. P. J., Klenerman D. Proc. Natl. Acad. Sci. U. S. A. 2016;113:E1206–E1215. doi: 10.1073/pnas.1524128113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dear A. J., Michaels T. C. T., Meisl G., Dobson C. M., Knowles T. P. J. Proc. Natl. Acad. Sci. U. S. A. 2020 doi: 10.1073/pnas.1922267117. [DOI] [Google Scholar]
- Pearson H. A., Peers C. J. Physiol. 2006;575:5–10. doi: 10.1113/jphysiol.2006.111203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendor J. T., Logan T. P., Edwards R. H. Neuron. 2013;79:1044–1066. doi: 10.1016/j.neuron.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emamzadeh F. N. J. Res. Med. Sci. 2016;21:29. doi: 10.4103/1735-1995.181989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittner R. A., Albrandt K., Beaumont K., Gaeta L. S. L., Koda J. E., Moore C. X., Rittenhouse J., Rink T. J. J. Cell. Biochem. 1994;55:19–28. doi: 10.1002/jcb.240550004. [DOI] [PubMed] [Google Scholar]
- Baskakov I. V., Legname G., Baldwin M. A., Prusiner S. B., Cohen F. E. J. Biol. Chem. 2002;277:21140–21148. doi: 10.1074/jbc.M111402200. [DOI] [PubMed] [Google Scholar]
- Muchowski P. J., Wacker J. L. Nat. Rev. Neurosci. 2005;6:11. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- Ehrnhoefer D. E., Bieschke J., Boeddrich A., Herbst M., Masino L., Lurz R., Engemann S., Pastore A., Wanker E. E. Nat. Struct. Mol. Biol. 2008;15:558. doi: 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- Glabe C. G. J. Biol. Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemporad F., Chiti F. Chem. Biol. 2012;19:315–327. doi: 10.1016/j.chembiol.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Paslawski W., Mysling S., Thomsen K., Jørgensen T. J. D., Otzen D. E. Angew. Chem., Int. Ed. 2014;53:7560–7563. doi: 10.1002/anie.201400491. [DOI] [PubMed] [Google Scholar]
- Andreasen M., Lorenzen N., Otzen D. Biochim. Biophys. Acta, Biomembr. 2015;1848:1897–1907. doi: 10.1016/j.bbamem.2015.01.018. [DOI] [PubMed] [Google Scholar]
- Breydo L., Uversky V. N. FEBS Lett. 2015;589:2640–2648. doi: 10.1016/j.febslet.2015.07.013. [DOI] [PubMed] [Google Scholar]
- Lee M.-C., Yu W.-C., Shih Y.-H., Chen C.-Y., Guo Z.-H., Huang S.-J., Chan J. C. C., Chen Y.-R. Sci. Rep. 2018;8:4772. doi: 10.1038/s41598-018-23122-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman P. J. Biopolymers. 1977;16:731–747. doi: 10.1002/bip.1977.360160404. [DOI] [PubMed] [Google Scholar]
- Levinthal C. J. Chim. Phys. 1968;65:44. [Google Scholar]
- Dill K. A., Chan H. S. Nat. Struct. Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- Mcnaught A. D. and Wilkinson A., IUPAC. Compendium of Chemical Terminology (the “Gold Book”), Blackwell Scientific Publications, 2nd edn, 1997. [Google Scholar]
- Posey A. E., Ruff K. M., Harmon T. S., Crick S. L., Li A., Diamond M. I., Pappu R. V. J. Biol. Chem. 2018;293:3734–3746. doi: 10.1074/jbc.RA117.000357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klosin A., Oltsch F., Harmon T., Honigmann A., Jülicher F., Hyman A. A., Zechner C. Science. 2020;367:464–468. doi: 10.1126/science.aav6691. [DOI] [PubMed] [Google Scholar]
- Horrocks M. H., Lee S. F., Gandhi S., Magdalinou N. K., Chen S. W., Devine M. J., Tosatto L., Kjaergaard M., Beckwith J. S., Zetterberg H., Iljina M., Cremades N., Dobson C. M., Wood N. W., Klenerman D. ACS Chem. Neurosci. 2016;7:399–406. doi: 10.1021/acschemneuro.5b00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arosio P., Cukalevski R., Frohm B., Knowles T. P. J., Linse S. J. Am. Chem. Soc. 2014;136:219–225. doi: 10.1021/ja408765u. [DOI] [PubMed] [Google Scholar]
- Burnham K. P. and Anderson D. R., Model Selection and Multimodel Inference, Springer-Verlag, New York, 2002. [Google Scholar]
- Vitalis A., Pappu R. V. Biophys. Chem. 2011;159(1):14–23. doi: 10.1016/j.bpc.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šarić A., Michaels T. C. T., Zaccone A., Knowles T. P. J., Frenkel D. J. Chem. Phys. 2016;145:211926. doi: 10.1063/1.4965040. [DOI] [PubMed] [Google Scholar]
- Šarić A., Chebaro Y. C., Knowles T. P. J., Frenkel D. Proc. Natl. Acad. Sci. U. S. A. 2014;111:17869–17874. doi: 10.1073/pnas.1410159111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang J. C., Lee J.-E., Dear A. J., De S., Meisl G., Thackray A. M., Bujdoso R., Knowles T. P. J., Klenerman D. Chem. Sci. 2019;10:4588–4597. doi: 10.1039/c8sc05627g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arosio P., Knowles T. P. J., Linse S. Phys. Chem. Chem. Phys. 2015;17:7606–7618. doi: 10.1039/c4cp05563b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dear A. J., Šarić A., Michaels T. C. T., Dobson C. M., Knowles T. P. J. J. Phys. Chem. B. 2018;122:11721–11730. doi: 10.1021/acs.jpcb.8b07805. [DOI] [PubMed] [Google Scholar]
- Lee C.-T., Terentjev E. M. J. Chem. Phys. 2017;147:105103. doi: 10.1063/1.4995255. [DOI] [PubMed] [Google Scholar]
- Linse B., Linse S. Mol. BioSyst. 2011;7:2296–2303. doi: 10.1039/c0mb00321b. [DOI] [PubMed] [Google Scholar]
- Buell A. K., Galvagnion C., Gaspar R., Sparr E., Vendruscolo M., Knowles T. P. J., Linse S., Dobson C. M. Proc. Natl. Acad. Sci. U. S. A. 2014;111:7671–7676. doi: 10.1073/pnas.1315346111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S. I. A., Arosio P., Presto J., Kurudenkandy F. R., Biverstal H., Dolfe L., Dunning C., Yang X., Frohm B., Vendruscolo M., Johansson J., Dobson C. M., Fisahn A., Knowles T. P. J., Linse S. Nat. Struct. Mol. Biol. 2015;22:207–213. doi: 10.1038/nsmb.2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin R. L. Folding Des. 1996;1:R1–R8. doi: 10.1016/S1359-0278(96)00003-X. [DOI] [PubMed] [Google Scholar]
- Garai K., Posey A. E., Li X., Buxbaum J. N., Pappu R. V. Protein Sci. 2018;27:1252–1261. doi: 10.1002/pro.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E. W., Holehouse A. S., Peran I., Farag M., Incicco J. J., Bremer A., Grace C. R., Soranno A., Pappu R. V., Mittag T. Science. 2020;367:694–699. doi: 10.1126/science.aaw8653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan A., Meriin A., Andrews J. O., Spille J.-H., Sherman M. Y., Cisse I. I. eLife. 2019;8:e39695. doi: 10.7554/eLife.39695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaels T. C. T., Weber C. A., Mahadevan L. Proc. Natl. Acad. Sci. U. S. A. 2019;116:14593–14598. doi: 10.1073/pnas.1904090116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellomo G. Chem. Commun. 2018;54:7601–7604. doi: 10.1039/c8cc01710g. [DOI] [PubMed] [Google Scholar]