

Abstract
Numerous studies propose that epigallocatechin-3-gallate (EGCG), an abundant polyphenol in green tea, has anti-cancer properties. However, its mechanism of action in breast cancer remains unclear. This study investigated the capacity of EGCG to suppress breast cancer cell growth in vitro and in vivo, characterizing the underlying mechanisms, focusing on the effect of EGCG on glucose metabolism. EGCG reduced breast cancer 4T1 cell growth in a concentration- (10–320 μM) and time- (12–48 h) dependent manner. EGCG induced breast cancer apoptotic cell death at 24 h, as evidenced by Annexin V/PI, caspase 3, caspase 8 and caspase 9 activation. Furthermore, EGCG affected the expression of 16 apoptosis-related genes, and promoted mitochondrial depolarization. EGCG induced autophagy concentration-dependently in 4T1 cells by modulating the levels of the autophagy-related proteins Beclin1, ATG5 and LC3B. Moreover, EGCG affected glucose, lactate and ATP levels. Mechanistically, EGCG significantly inhibited the activities and mRNA levels of the glycolytic enzymes hexokinase (HK), phosphofructokinase (PFK), and lactic dehydrogenase (LDH), and to a lesser extent the activity of pyruvate kinase (PK). In addition, EGCG decreased the expression of hypoxia-inducible factor 1α (HIF1α) and glucose transporter 1 (GLUT1), critical players in regulating glycolysis. In vivo, EGCG reduced breast tumor weight in a dose-dependent manner, reduced glucose and lactic acid levels and reduced the expression of the vascular endothelial growth factor (VEGF). In conclusion, EGCG exerts anti-tumor effect through the inhibition of key enzymes that participate in the glycolytic pathway and the suppression of glucose metabolism.
Keywords: Breast cancer, Epigallocatechin-3-Gallate, glycolysis, green tea polyphenols
1. Introduction
With over one million new cases diagnosed worldwide every year, breast cancer represents a significant health problem1. Despite advances in early detection, breast cancer remains a major disease burden, due to an increase in its incidence, particularly in the developing world2–4. The dramatic increase in breast cancer incidence mandates for efforts to prevent this disease5, 6, being the evaluation of agents with chemopreventive properties a critical component.
Numerous dietary bioactives have been reported to have chemopreventive effects in a large number of cancer patients7. Among these, Epigallocatechin-3-gallate (EGCG), the most abundant catechin in green tea8, presents multiple health benefits9–11. For example, EGCG has been documented to have beneficial role in various diseases like diabetes12, Parkinson13, stroke14, obesity15, as well as cancer16. Mechanistically, EGCG has been proposed to block and inhibit various signaling pathways that lead to the initiation and progression of various types of cancer. However, the exact mechanisms related to the preventive activity of EGCG in breast cancer are incompletely understood.
Among multiple mechanisms proposed, EGCG has been recently shown to affect pathways related to glucose metabolism. Glucose metabolism allows for energy to be coupled in the form of ATP, through the oxidation of its carbon bonds. This process is essential for sustaining all mammalian life. Glycolysis, a critical metabolic pathway in the metabolism of glucose, is common in anaerobic environments. However, unlike the normal cells, cancer cells can instead rely primarily on glycolysis to generate energy, even in an aerobic environment. This phenomenon, termed as aerobic glycolysis or the Warburg effect17, allows tumor cells to generate a high rate of glycolytic intermediates, which become substrates for several biosynthetic pathways crucial for cell proliferation and cancer progression. Thus, agents, such as EGCG, that can affect glucose metabolism in cancer cells are of particular interest. However, there is a dearth of information on the effect of EGCG on glucose metabolism in breast cancer.
In this study, we investigated the anticancer effect of EGCG in breast cancer in vitro and in vivo, as well as the underlying molecular mechanisms, focusing on the effect of EGCG on tumor glucose metabolism. Our data show that EGCG inhibits the growth of breast cancer cells in vitro and in vivo, by inducing apoptosis and altering autophagy. Furthermore, EGCG strongly suppresses the activity of the enzymes hexokinase (HK), phosphofructokinase (PFK), and lactic dehydrogenase (LDH), enzymes related to the glycolytic pathway, indicating that modulating glucose metabolism could represent a key event in EGCG’s anticancer effect in breast cancer.
2. Materials and Methods
2.1. Chemicals and reagents
EGCG (purity>98%) was purchased from Aladdin Industrial Corporation (Shanghai, China). The CCK8 kit was purchased from Biosharp (Hefei, China). The Annexin V-FITC/PI kit was purchased from Dojindo (Shanghai, China). The kits for caspase 3, caspase 8, caspase 9 activity, JC-1, ATP levels, Ad-mCherry-GFP-LC3B and the Bradford protein assay were purchased from Beyotime (Nantong, China). The kits to determine glucose, lactic acid, HK activity, PFK activity, PK activity and LDH activity were purchased from Jiancheng (Nanjing, China). The PureLink RNA Mini Kit was purchased from Ambion (Shanghai, China). ReverTra Ace qPCR RT Master Mix and the SYBR Green Realtime PCR Master Mix were purchased from TOYOBO (Shanghai, China). Designed oligonucleotide primers were generated by TsingKe (Beijing, China). The RIPA lysis buffer and Halt protease inhibitor cocktail were purchased from Applygen (Beijing, China). The 2X Laemmli sample buffer and the PVDF membranes were purchased from Bio-Rad (Shanghai, China). The HIFα, Beclin1, ATG5, LC3B and β-Actin antibodies were purchased from Cell Signaling Technology Company (Shanghai, China). GLUT1 antibody was purchased from Abcam (Shanghai, China), while the VEGF antibody was from Fisher Scientific (Wuhan, China). Hematoxylin and eosin were purchased from GuGe (Wuhan, China). 5-fluorouracil (5-FU) was purchased from Meilunbio (Dalian, China).
2.2. Cell culture
The 4T1 cells line was purchased from the Cell Bank of Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China), while the HC11 mammary epithelial cells were from the American Type Culture Collection (Manassas, VA).
We have not authenticated these cell lines; however we routinely test for mycoplasma contamination in every cell line every three months. These cells were grown as monolayers in the specific medium and under conditions suggested by the vendor. Both cell lines were characterized by cell morphology and growth rate and passaged in our laboratory less than 6 months after being received. The cells were passaged every 2–3 days at a ratio of 1:3.
2.3. Cell viability
Cell viability was measured by the Cell Counting Kit-8 (CCK8). Briefly, cells seeded in 96-well plates overnight, were treated with escalating concentrations of EGCG for 12, 24 or 48 h. Following the incubation, CCK8 reagent was added, and incubated for 2.5 h. The optical density at 450 nm was measured in an Infinite® microplate reader (Tecan; Männedorf, Switzerland).
2.4. Annexin V-FITC/PI staining
Cells were seeded overnight and then treated with various concentrations of EGCG for 24 h and 48 h. Following the incubation, cells were collected, washed twice with PBS, stained with Annexin V-FITC (Dojindo; Shanghai, China) and propidium iodide (0.5 μg/ml), and then analyzed by FACScaliber (BD Biosciences; San Jose, CA).
2.5. q-PCR
Cells were seeded overnight and then treated with various concentrations of EGCG for 24 h. Following the incubation, total RNA was isolated using the PureLink RNA Mini Kit and transcribed reversely by using ReverTra Ace qPCR RT Master Mix with the Veriti Thermal Cycler (Thermo Fisher Scientific; Waltham, MA). The levels of mRNA were detected by using the SYBR Green Realtime PCR Master Mix and were monitored by the StepOne Realtime PCR system (Thermo Fisher Scientific; Waltham, MA).
2.6. JC-1 staining
The mitochondrial membrane potential (ΔΨm) was determined by fluorescence microscopy using the JC-1 cationic dye.17 Briefly, after treatment with EGCG for 24 h, cells were washed twice with PBS, and incubated with JC-1 reagent for 20 min at 37°C. Subsequently, cells were washed twice with JC-1 staining buffer and the fluorescence of both JC-1 monomer (green) and aggregates (red) were detected by using a fluorescence microscope (Nikon Ti-s; Tokyo, Japan).
2.7. Caspase activity
Caspase 3, 8 and caspase 9 activities were measured using caspase activity assay kits, following the manufacturers’ protocol (Beyotime; Nantong, China). Briefly, caspase activities were measured by the chromogenic products of caspase substrates: Ac-DEVE-pNA (caspase3 substrate), Ac-IETD-pNA (caspase8 substrate) and Ac-LEHD-pNA (caspase9 substrate). After treating with EGCG for 24 h, cells were harvested and centrifuged at 600 g, at 4°C for 5 min. After washing with cold PBS twice, cells were resuspended with 50 μl lysis buffer and incubated on ice for 15 min. Following centrifuged at 18000 × g, at 4 °C for 15 min, the supernatant was transferred into cold fresh tubes. Then, 50 μl cell extract, 40 μl buffer and 10 μl of specific caspase substrate: Ac-DEVE-pNA, Ac-IETD-pNA or Ac-LEHD-pNA were mixed together and incubated at 37°C for 80 min. Following the incubation period, the absorbance of the yellow pNA cleavages products were determined at 405 nm in a microplate reader (Tecan; Männedorf, Switzerland). Concurrently, protein content in the cell lysates was determined to normalize the results by protein content.
2.8. Western blot
Following treatment with EGCG for 24 h, cells were lysed with RIPA lysis buffer and total cell fractions were obtained as previously described.18 Aliquots of total fractions containing 25–40 μg protein were separated by reducing 10–12.5% (w/v) polyacrylamide gel electrophoresis and electroblotted to PVDF membranes. The membranes were probed overnight with primary antibodies (1:1,000 dilution). β-actin was used as the loading control. After incubation, for 90 min at room temperature, in the presence of the secondary antibody (HRP-conjugated; 1:5,000 dilution), the conjugates were visualized by chemiluminescence.
2.9. m-Cherry GFP LC3B transfection
Cell were transfected with the m-Cherry-GFP-LC3B adenovirus following the manufacturer’s protocol (Beyotime; Nantong, China). Briefly, cells were seeded in the 6-well plate overnight, then transfected with the m-Cherry-GFP-LC3B. After 24 h, media was replaced, and treated with EGCG for an additional 24 h. At the end of the incubation period, cells were visualized using a Confocal Laser Scanning Microscope (Nikon; Tokyo, Japan).
2.10. Transmission electron microscopy (TEM)
Cells were seeded in 6-well plates overnight and then treated with various concentrations of EGCG for 24 h. Following treatment, cells were washed twice with PBS, and fixed overnight with 2.5% glutaraldehyde in phosphate buffer (0.1 M, pH 7.0). After washing thrice with PBS, cells were incubated with 1% osmium tetroxide for 2 h, subsequently washed three times with PBS, followed by dehydration with an ascending gradient of ethanol and absolute acetone. Following a transition with ethanol and Spurr resin 1:1, cells were embedded with Spurr resin and incubated at 70°C overnight. Then, samples were sectioned using the ultramicrotome EM UC7 (Leica; Wetzlar, Germany), stained with uranyl acetate and alkaline lead citrate for 10 min respectively, and scanned by using the JEM-1230 transmission electron microscope (Gatan Inc.; Tokyo, Japan).
2.11. Glucose and lactic acid levels
Cells were seeded overnight and then treated with various concentrations of EGCG (20–240 μM) for 24 h. At the end of the treatment, the cell culture medium was collected and the levels of glucose and lactic acid were determined following the manufacturers’ protocol (Jiancheng; Nanjing, China). Glucose and lactic acid levels in tumors were measured following the manufacturers’ protocol (Jiancheng; Nanjing, China).
2.12. Cellular ATP levels
ATP levels were measured using an ATP assay kit. After treatment with EGCG for 24 h, cells were lysed and centrifuged at 12,000 × g, at 4°C for 5 min. Subsequently, 20 μl of the supernatant was mixed together with 100 μl ATP detection working dilution, and Luminance (RLU) was measured using a HRPCS5 system (Photek; East Sussex, UK). A standard curve was generated using ATP and the protein concentration of each treatment group was determined using the Bradford Protein assay. Total ATP levels were expressed as nmol/mg protein.
2.13. Glycolysis-related enzymes activity
Cells were seeded into the 6-well plates overnight and treated with EGCG for 24 h. Following the incubation, cells were collected, washed with cold PBS and homogenized using an ultrasonic tip sonicator. After centrifugation, the supernatant was collected and used to measure the enzyme activity following the manufacturer’s protocol (Jiancheng; Nanjing, China). Protein concentration was determined using the Bradford protein assay.
2.14. Animal study
All animal studies were performed in accordance with the guidelines of the Laboratory Animal Center of Zhejiang University. And all animal experimental protocols were approved by the Laboratory Animal Center of Zhejiang University.
Four week-old Balb/c mice, purchased from the Shanghai Laboratory Animal Center (Shanghai, China), were maintained in pathogen-free conditions and fed irradiated chow. After two weeks of acclimation, mice were injected subcutaneously with 4T1 cancer cells (200 μl, 4×106 cells/ml). Once tumors were palpable, mice (n=5/group) were randomized into groups receiving PBS (control), 5-FU (20 mg/kg) or EGCG at various doses (5mg/kg/d, 10mg/kg/d, 20mg/kg/d) in PBS, given once daily by intraperitoneally injections for 14 d. Body weight was determined every 2 days during the experimental period. At euthanasia, blood was collected, and tumors were carefully excised, weighed and stored for analysis.
2.15. Histological examination
At necropsy, tumors were fixed in 4% paraformaldehyde for 24 h and then processed and embedded in paraffin according to standard protocols. Tissue sections (4 μm) stained with hematoxylin and eosin (H&E) were evaluated histologically under an inverted fluorescence microscope (Nikon Eclipse TI-SR; Tokyo, Japan).
2.16. Immunofluorescence staining
The process of preparing paraffin sections was performed as described above. After antigen retrieval, immunostaining was performed by incubating slides with the primary antibody overnight at 4°C. After PBS washing, sections were incubated with a fluorophore-linked secondary antibody (Alexa Fluor 488–anti-rabbit IgG; Life Technologies). After staining, slides were mounted with DAPI and photographed under an inverted fluorescence microscope (Nikon Eclipse TI-SR; Tokyo, Japan).
2.17. Statistical analysis
The data, obtained from at least three independent experiments, were expressed as the mean ± SD. Statistical evaluation was performed by one-factor analysis of variance (ANOVA) followed by the Tukey test for multiple comparisons. P<0.05 was regarded as being statistically significant.
3. Results
3.1. EGCG inhibits the growth of 4T1 breast cancer cells in culture
Our first goal was to establish the efficacy of EGCG in breast cancer cells in culture. For this purpose, we treated mouse 4T1 breast cancer cells with or without escalating concentrations of EGCG (10–320 μM) for 12, 24 or 48 h. As shown in Fig. 1A, EGCG reduced 4T1 breast cancer cell growth in a concentration- and time-dependent manner. For instance, EGCG 20 μM significantly reduced cell viability at 12 h and 24 h treatment (p<0.05), whereas after 48 h, EGCG at 10 μM significantly reduced cell growth (p<0.05).
Figure 1: EGCG inhibited 4T1 breast cancer cells growth.
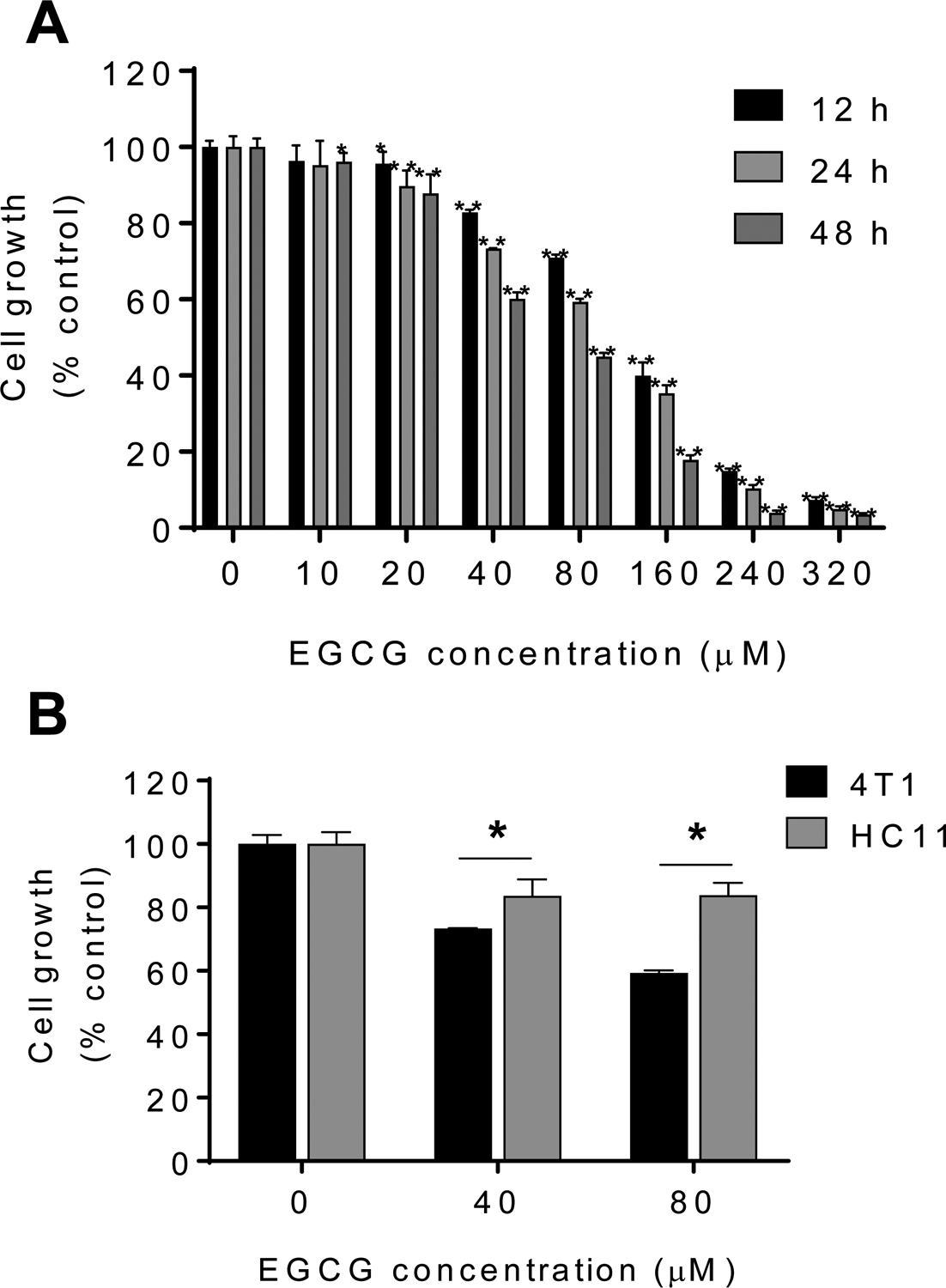
A: EGCG reduced 4T1 cell growth in a time- and concentration-dependent manner. Cell viability was determined in 4T1 cells after 12, 24 and 48 h of incubation with EGCG. Results are expressed as percent control. (*p<0.05, **p<0.01 vs. control). B: Differential cytotoxic effect in 4T1 cells compared with that of mouse breast normal epithelial cells (HC11) after treatment with escalating concentrations of EGCG for 24 h. Results are expressed as percent control. (*p<0.05, **p<0.01 vs. control).
We next compared the effect of EGCG in mouse breast cancer 4T1 cells to that of the mouse mammary epithelial cells (HC11). For this purpose, we treated 4T1 cells and HC11 mammary epithelial cells with or without EGCG (40–80 μM) for 24 h. As shown in Fig. 1B, EGCG reduced cell growth more potently in 4T1 cells compared to HC11 cells. For instance, at 24 h, EGCG 80 μM reduced cell growth in 4T1 cells by 41% (p<0.05), respectively. In contrast, under the same experimental conditions, EGCG 80 μM for 24 h had minimal effect on the HC11 mouse mammary epithelial cells, reducing cell growth by only 17% (Fig. 1B).
3.2. EGCG induces cell death by apoptosis
We next explored whether the reduction in cell growth is due to an induction of apoptosis by EGCG. Staining with propidium iodide (PI) and annexin V revealed that EGCG displayed a concentration- and time- dependent induction of apoptosis (Fig. 2A). For example, compared to the control group, treatment with EGCG 20 μM for 24 h significantly increased the rate of early apoptosis (Annexin V positive but PI negative) from 2.52 ± 0.33% to 6.95 ± 0.63%, and of late apoptosis (Annexin V positive and PI positive cells) from 1.50 ± 0.22% to 8.44 ± 0.67%. This effect was even more pronounced in the EGCG 40 μM and 80 μM groups, whereas EGCG 160 μM and 240 μM, the percent of cells in late apoptosis accounted for 20.82 ± 0.39% and 33.63 ± 0.67%, respectively (Fig. 2B). Moreover, following treatment with EGCG 160 μM and 240 μM for 48 h, >60% of the cells were experiencing late apoptosis (64.11 ± 0.23% and 69.34 ± 0.87%, respectively). To note, EGCG predominantly induced apoptotic cell death, since minimum amount of necrosis (Annexin V negative but PI positive) was observed following EGCG treatment (Fig. 2B).
Figure 2: EGCG induced cell death by apoptosis in 4T1 breast cancer cells.
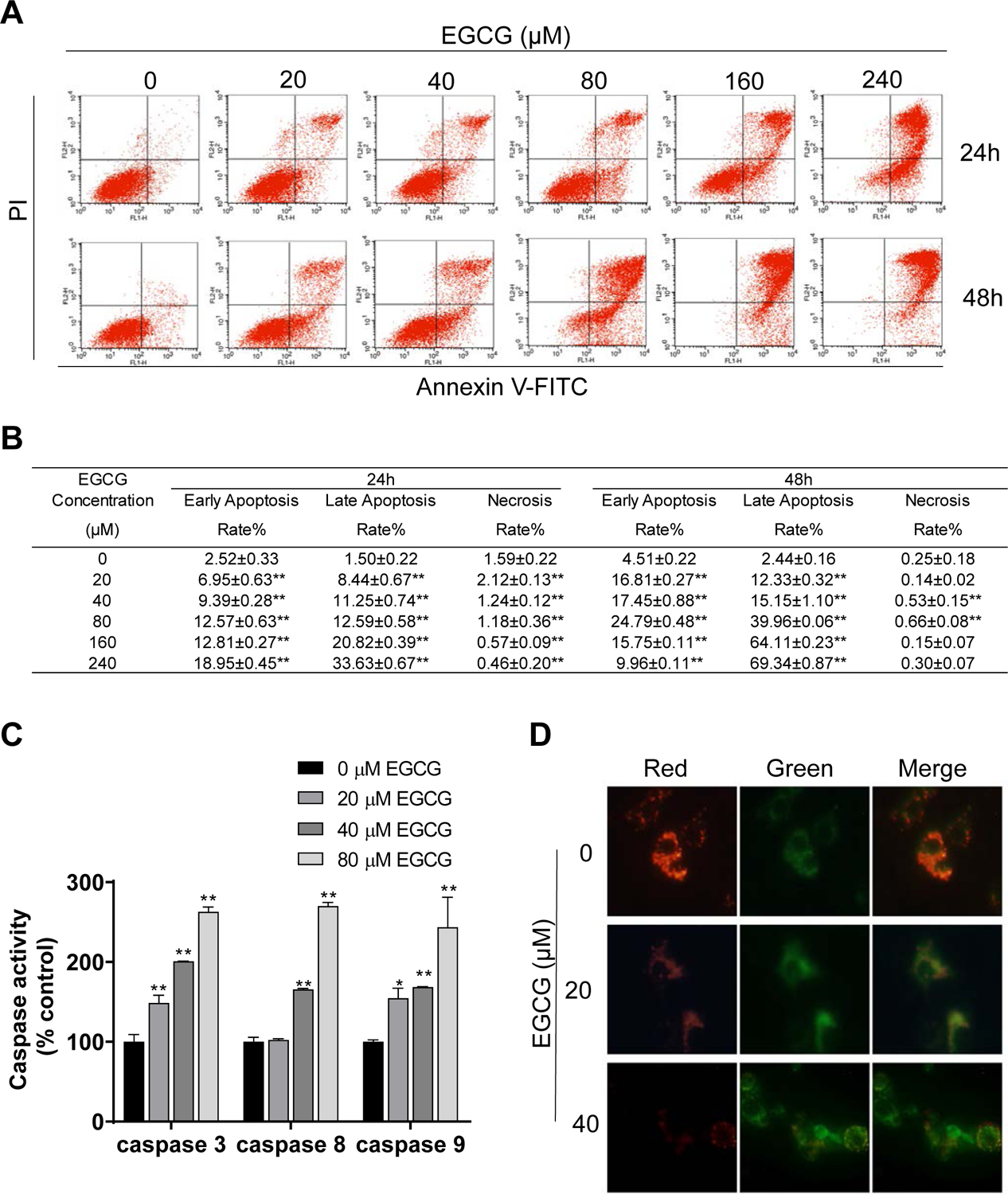
A: 4T1 cells treated with EGCG for 24 h and 48 h were stained with Annexin V/propidium iodide, and the percentage of apoptotic cells was determined by flow cytometry. B: Results are expressed as the mean ± SD. (*p<0.05, **p<0.01 vs. control). C: EGCG activated caspase activity in 4T1 cells. Results are expressed as percent control. (*p<0.05, **p<0.01 vs. control). D: EGCG promoted mitochondrial depolarization after 24 h incubation. Representative images (x400) of 4T1 cells treated with either vehicle (control) or EGCG and stained for JC-1 fluorescence.
Caspases play a vital role in initiating and executing cell apoptosis19. Thus, we determined if EGCG affected the activity of the caspases 9, 8, and 3. As shown in Fig. 2C, EGCG treatment for 24 h increased the activities of all three caspases in a concentration-dependent manner. For instance, treatment with EGCG 80 μM resulted in an increase (p<0.01) of 2.63, 2.70 and 2.44 fold in the activity of caspase 3, caspase 8 and caspase 9, respectively (Fig. 2C).
We next evaluated whether EGCG could affect the mitochondrial membrane potential, by means of the JC-1 probe. In healthy cells, the JC-1 probe aggregates in the normal mitochondrial matrix and emits a red fluorescence. When the mitochondrial depolarization occurs, the mitochondrial membrane becomes permeable and the JC-1 probe disperses to the cytoplasm, disaggregates into monomers, and thus, red fluorescence turns into green fluorescence. As shown in Figure 2D, in control 4T1 cells, JC-1 accumulated in the mitochondria and majority red fluorescence with a little green fluorescence was observed. On the other hand, EGCG 20 μM reduced the red fluorescence intensity, and consequently increased the green fluorescence. This was even more pronounced in the cells treated with EGCG 40 μM (Fig. 2D). These results suggest that EGCG induces the collapse of the mitochondrial membrane potential.
To gain insight into which apoptotic-related proteins were modified by EGCG, we performed a microarray including multiple apoptotic genes. After 24 h treatment, total RNA was extracted and reversely transcribed, and then a hierarchical clustering analysis was performed to represent the expression level of these apoptosis-related genes. Compared to control cells, EGCG increased the expression of the pro-apoptotic genes puma, caspase3, caspase8, caspase9, fas, apaf1, bax, fas-L, cytochrome c, pten, bad and smac in a concentration-dependent manner. In addition, EGCG reduced the expression levels of the anti-apoptotic genes: survivin, bcl-2, bcl-xl and c-myc (Fig. 3).
Figure 3: EGCG affected the expression of apoptosis-related proteins.
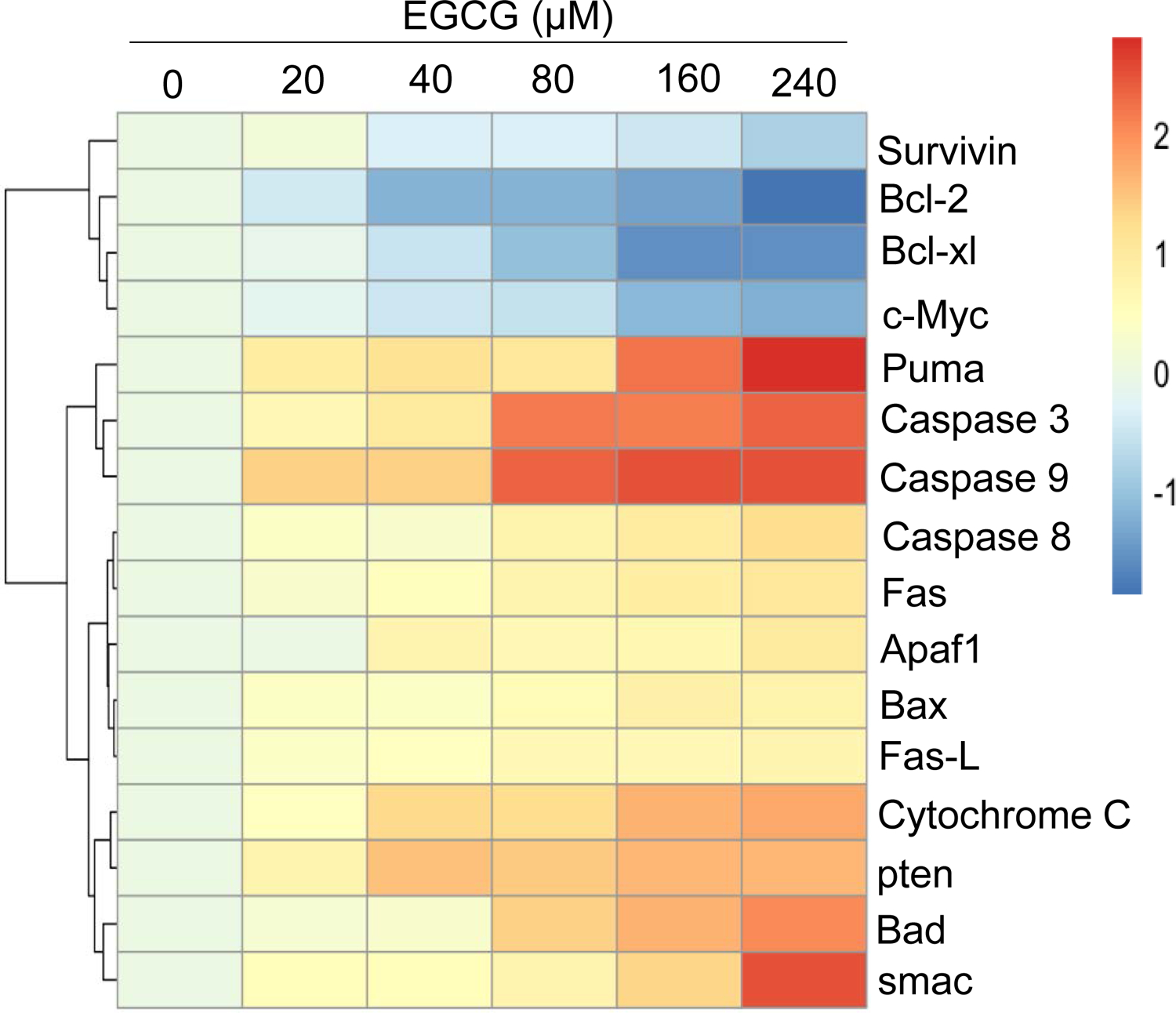
Microarray heat map of differentially expressed genes (as indicated) levels within control cells or cells treated with EGCG at various concentrations (20–240 μM) for 24 h. Heat map results are shown as the mean fold changes (log2) of the relative mRNA expression level for each protein (2−△△t).
3.3. EGCG induces autophagy in breast cancer cells
Autophagy, a lysosomal catabolic pathway of self-degradation and recycling of cellular macromolecules and organelles, is often involved in the response to treatment with anti-cancer agents. Thus, we evaluated whether autophagy played a role in the cytokinetic effect of EGCG, by exploring key characteristic features associated with the autophagy process. These included the determination of autophagy-related proteins by immunoblot and the formation of autophagosomes by electron microscopy.
As shown in Fig. 4A, EGCG 10 and 20 μM for 24 h significantly increased Beclin1, Atg5 and LC3B-II/LC3B-I levels. Interestingly, while EGCG 40 μM increased Beclin1 and Atg5 levels, LC3B-II/LC3B-I levels were reduced compared to control.
Figure 4: EGCG affected autophagy in 4T1 breast cancer cells.
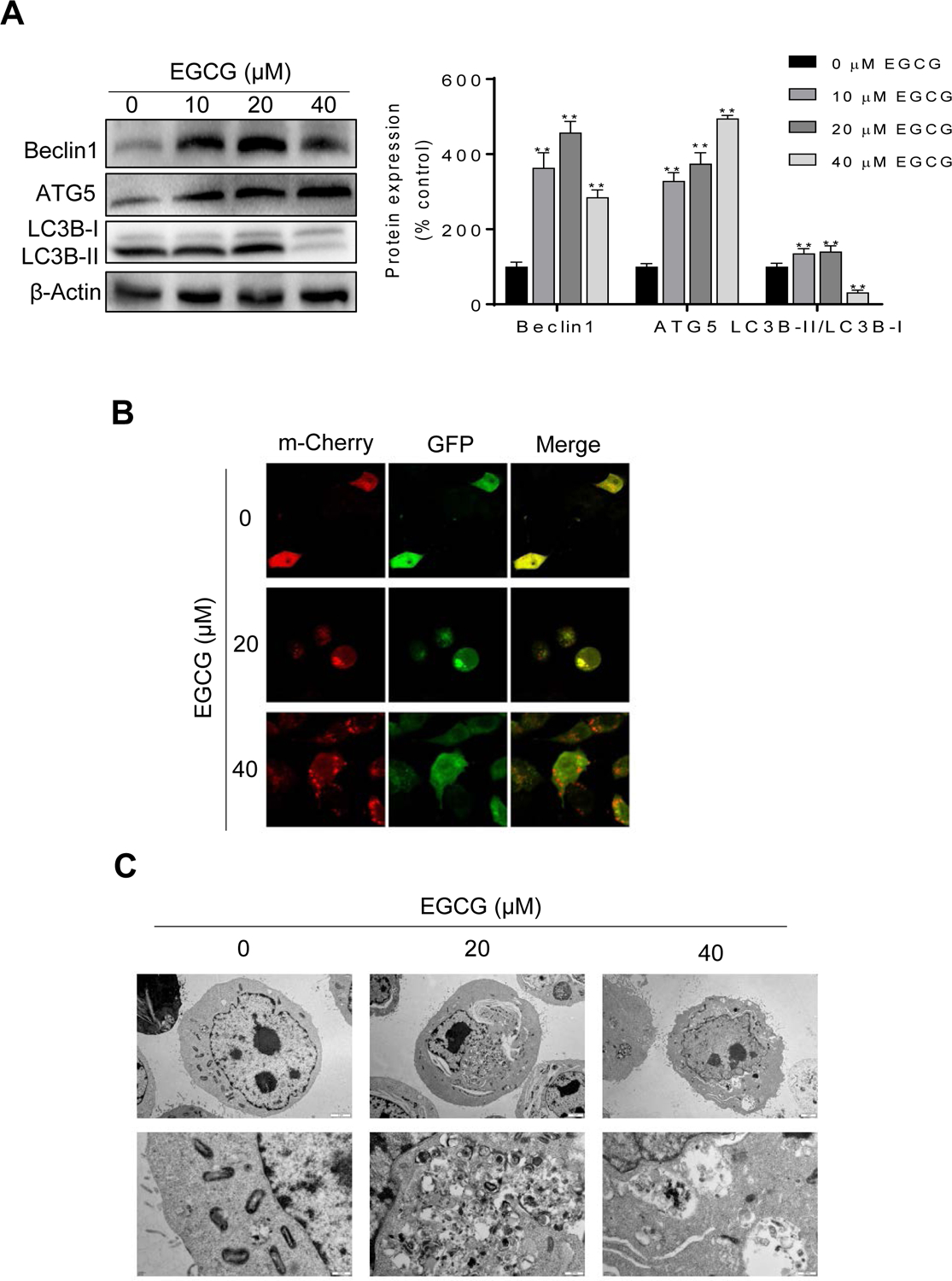
A: 4T1 cells lysates were analyzed for Beclin1, ATG5 and LC3B by immunoblotting. Loading control: β-actin. Bands were quantified and results are expressed as percentage of control. (*p<0.05, **p<0.01 vs. control). B: Representative images (x400) of 4T1 cells treated with either vehicle (control) or EGCG and stained for mCherry-GFP-LC3B. C: Autophagosome formation was depicted using transmission electron microscope in 4T1 cells treated with or without EGCG for 24 h. Representative images (x8000, x30000) are shown.
Next, to monitor the effect of EGCG on autophagy in live cells, we transfected 4T1 cells with an adenovirus expressing m-Cherry-GFP-LC3B fusion protein (Ad-mCherry-GFP-LC3B)20. In normal condition, the mCherry-GFP-LC3B protein disperses in the cytoplasm and displays homogeneous yellow fluorescence. When the autophagic process is triggered, the mCherry-GFP-LC3B will gather around at the membrane of the autophagosome, and yellow dots appear. However, when the autolysosome is formed, the acid environment in the lysosome quenches the fluorescence of the GFP, and as a result, only red dots are present21. Compared to control, EGCG 20 μM increased the formation of autophagosomes, as depicted by the presence of yellow aggregates. Moreover, the formation of autolysosomes was observed following treatment with EGCG 40 μM, as depicted by the red aggregates and by the decrease in green fluorescence due to the acidic environment (Fig. 4B).
Finally, we evaluated whether EGCG induced autophagy in 4T1 cells by transmission electron microscopy, which allows for the direct visualization of the autophagic process. In control cells, no autophagy vacuoles were visibly present and mitochondria were clearly observed. Following treatment with 20 μM EGCG, the presence of autophagosomes with a bilayer was observed, with some organelles wrapped inside. Furthermore, in the cells treated with EGCG 40 μM, autophagosomes fused with the lysosomes were observed, suggesting that autophagy was at a more advanced stage (Fig. 4C). Taken together, these data indicate that EGCG induces autophagy in breast cancer cells.
3.4. EGCG affects the glycolytic pathway in 4T1 breast cancer cells
Given that the glycolytic pathway can modulate the autophagy process22, we next explored the effect of EGCG on multiple parameters of the glycolytic pathway. In 4T1 cells, EGCG treatment displayed lower capacity of glucose uptake and lactic acid production in a concentration-dependent manner (Fig. 5A–B). Glucose consumption was reduced in cells treated with EGCG becoming significant at EGCG 40 μM and reduced by 30% and 35% in the EGCG 160 and 240 μM groups, respectively, compared to control (Fig. 5A). Moreover, the levels of lactic acid were reduced in the EGCG 20 μM and 40 μM groups (p<0.05), and this reduction was even more pronounced (over 70% reduction) in the 160 and 240 μM EGCG groups (p<0.01; Fig. 5B). The decrease in the consumption of glucose and in the production of lactic acid by EGCG resulted in a reduction in ATP levels (Fig. 5C). For example, EGCG 40 μM and 80 μM reduced ATP levels by almost half (p<0.01), compared to the control group, and EGCG 240 μM reduced ATP production by over 80% compared of control (p< 0.01; Fig. 5C).
Figure 5: Effect of EGCG on glucose uptake, lactic acid levels and ATP production.
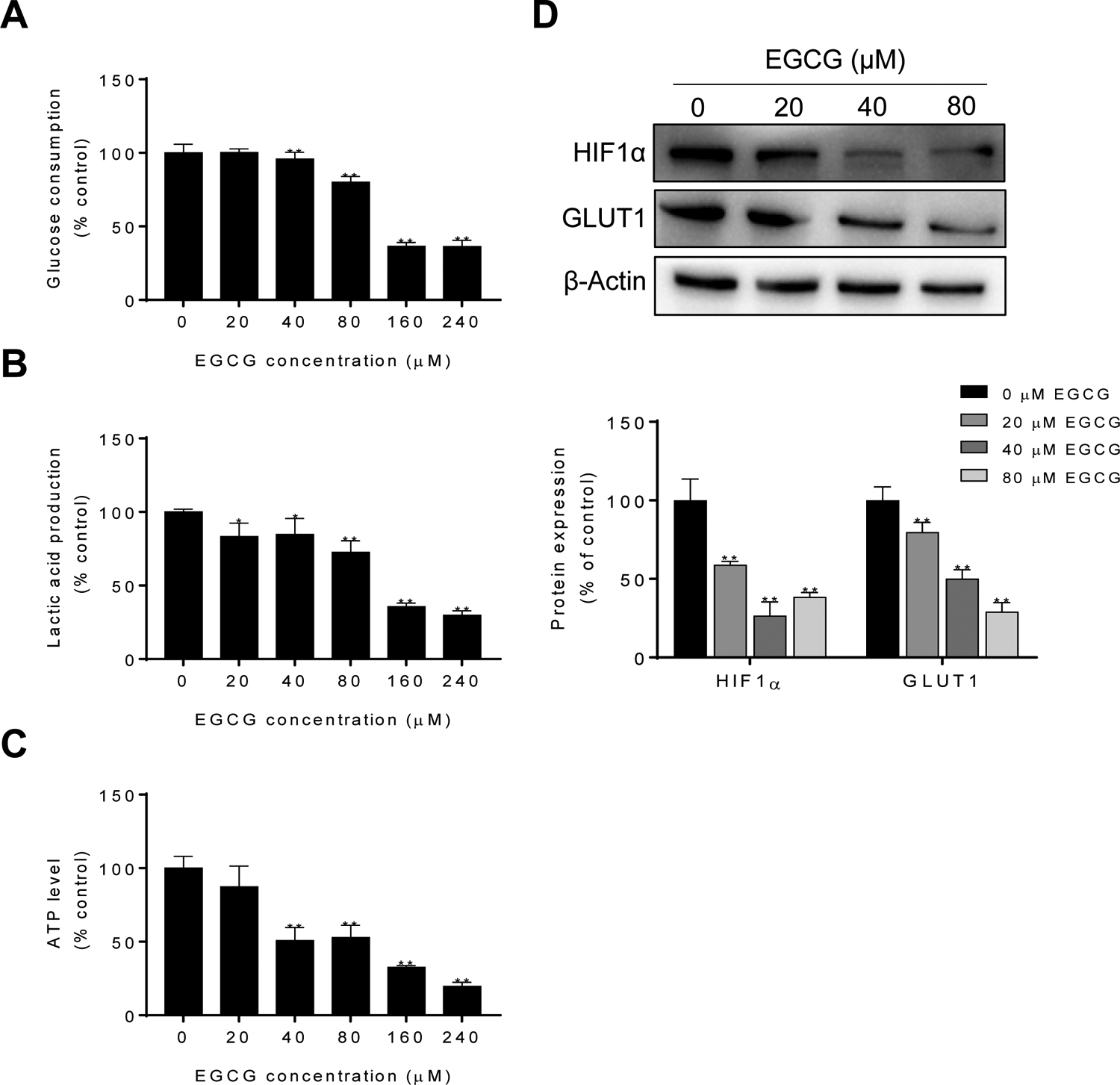
A: EGCG decreased the uptake of glucose. Results are expressed as percent control. (*p<0.05, **p<0.01 vs. control). B: EGCG reduced lactic acid levels. Results are expressed as percent control. (*p<0.05, **p<0.01 vs. control). C: EGCG reduced intracellular ATP levels in 4T1 cells. Results are expressed as percent control. (*p<0.05, **p<0.01 vs. control). D: Immunoblots of HIF1α and GLUT1 in 4T1 cell protein extracts incubated with or without EGCG for 24 h. Loading control: β-actin. Bands were quantified and results are expressed as percent control (*p<0.05, **p<0.01 vs. control).
HIF-1α, which a subunit of the transcription factor HIF-1, is involved in regulating energy metabolism under hypoxia conditions23. On the other hand, GLUT1 is a key transporter, responsible for the uptake of glucose from the blood into the cell24. Given that HIF1α and GLUT1 are critical players regulating glucose metabolism, we then explored whether EGCG could affect its expression levels. As shown in Figure 5D, EGCG markedly reduced the expression levels of HIF1α and GLUT1 in a concentration-dependent manner (Fig. 5D).
The glycolytic enzyme hexokinase (HK) facilitates autophagy in response to glucose deprivation (HK substrate deprivation) to protect cells, suggesting that HK may function as a molecular switch from glycolysis to autophagy to ensure cellular energy homeostasis under starvation conditions25. Thus, we next examined the effect of EGCG on the HK enzyme activity and levels. After 24 h, EGCG inhibited HK activity and levels in a concentration-dependent manner (p<0.01; Fig 6A–B). We then explored the effect of EGCG on the activities of PFK, PK, and LDH; all key enzymes that regulate glucose metabolism and lactic acid production. After 24 h, EGCG significantly inhibited the activities of PFK and LDH compared to control group (p<0.05). On the other hand, EGCG showed weaker effect on the activity of PK, only reaching significance at EGCG 80 μM treatment (Fig. 6A). Furthermore, after EGCG treatment for 24 h, reduced the mRNA expression of the HK concentration-dependently (p<0.05). However, the mRNA expression levels of PKM2, PFK1 and LDHA were significantly reduced only at EGCG 80 μM (Fig. 6B).
Figure 6: EGCG inhibited the activity and the mRNA expression of glycolysis-related enzymes.
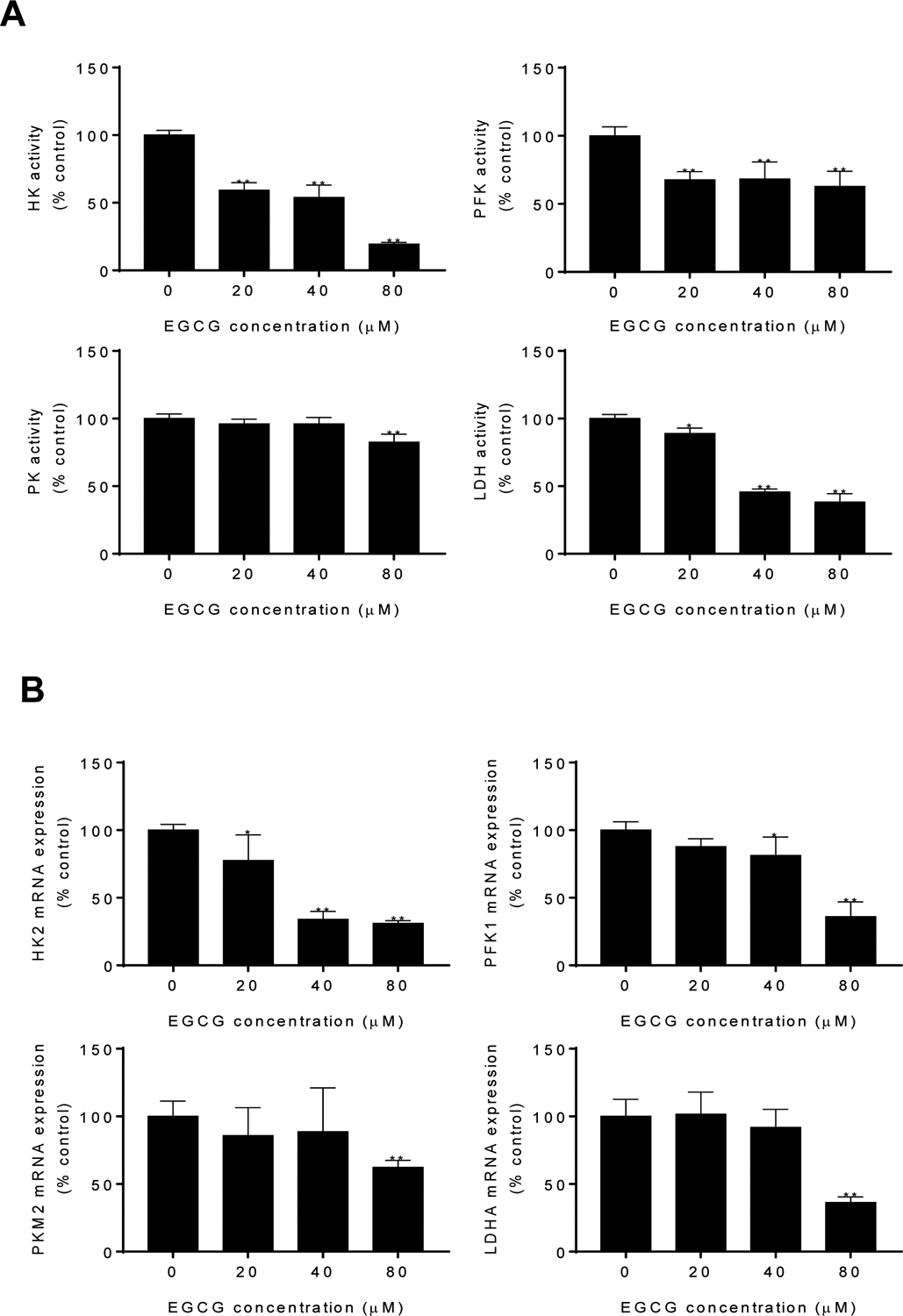
A: Enzymatic activity of HK, PFK, PK and LDH was determined in 4T1 cells treated with EGCG for up to 24 h. Results are expressed as percent control. (*p<0.05, **p<0.01 vs. control). B: mRNA expression of HK2, PFK1, PKM2 and LDHA was measured by qPCR in 4T1 cells treated with EGCG. Results are expressed as percent control. (*p<0.05, **p<0.01 vs. control).
3.5. EGCG reduces breast cancer xenograft growth
Finally, we assessed the in vivo chemotherapeutic potential of EGCG using a breast cancer syngeneic model. 4T1 cells subcutaneously injected into Balb/c mice gave rise to exponentially growing tumors. Once the tumors reached ~300 mm3, the mice were treated with either EGCG 5, 10 or 20 mg/kg, 5-FU 20 mg/kg or with vehicle given once daily intraperitoneally. On day 15 of treatment, mice were euthanized, tumors were carefully resected and weighed. Compared to vehicle control, EGCG 5, 10 and 20 mg/kg significantly reduced tumor weight by 20%, 31% and 34%, respectively. As expected 5-FU strongly inhibited tumor growth, reducing it by 77% (p<0.01; Fig. 7A).
Figure 7: EGCG inhibited the growth of breast cancer in Balb/c mice.
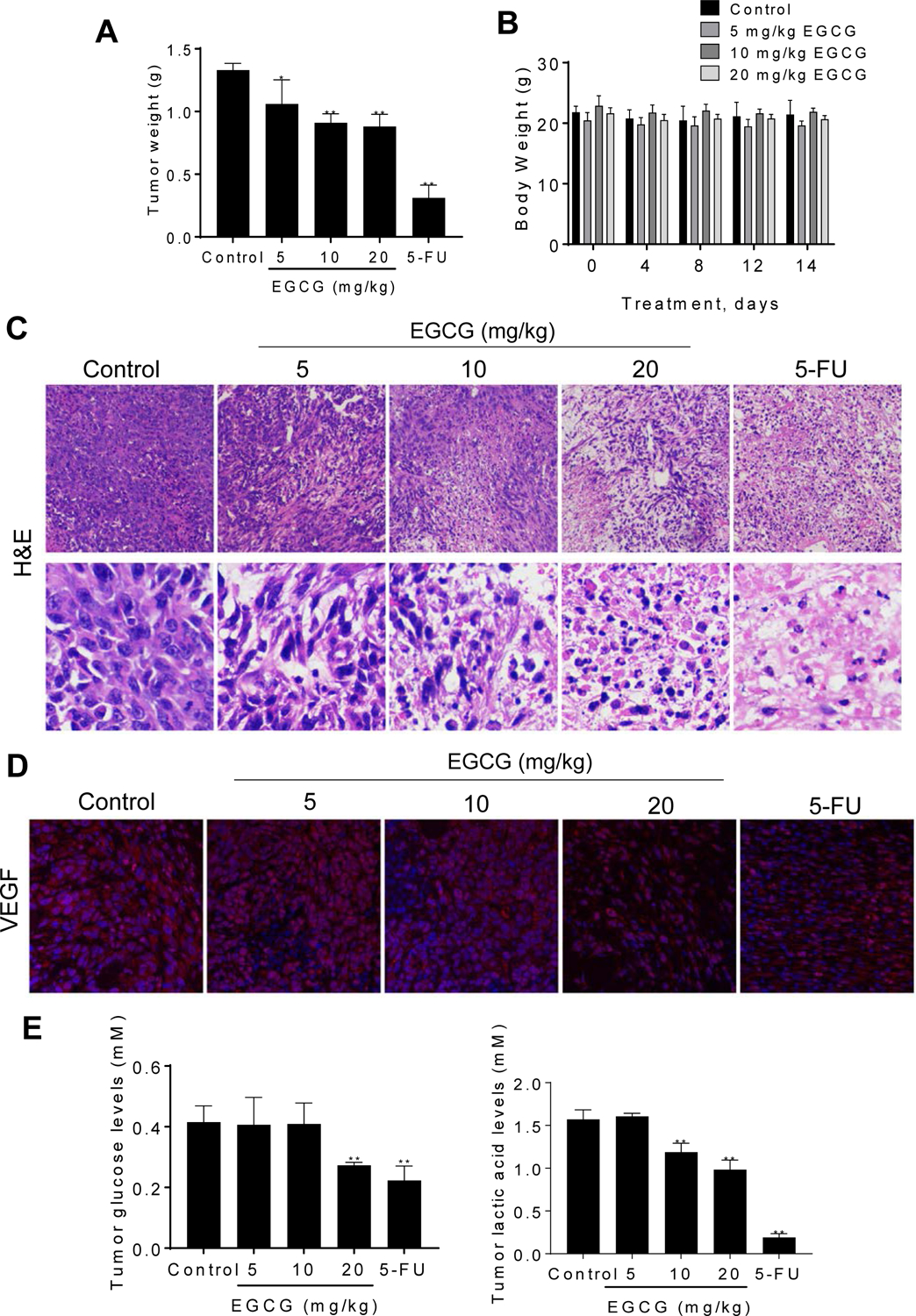
A: Effect of EGCG on tumor weight at sacrifice. Results are expressed as the mean ± SD (*p<0.05, **p<0.01 vs. control). B: Mice body weight over time for control and EGCG treated groups. Results are expressed as the mean ± SD. C: H&E staining of isolated tumors. Representative images (x100, x400) of tumor tissues treated with either vehicle (control) or EGCG and stained for H&E. D: Immunofluorescence staining for VEGF expression of tumor tissues. Representative images (×200) of tumor tissues treated with or without EGCG and stained for VEGF. E: Glucose and lactic acid levels in tumors. Results are expressed as the mean ± SD. (*p<0.05, **p<0.01 vs. control).
Regarding its safety, EGCG was well tolerated, with the mice showing no weight loss or other signs of toxicity during treatment. For instance, on the day before sacrifice, body weight in the control and three EGCG groups of mice was as follows: control = 21.4±2.3 g; EGCG 5 mg/kg = 19.7.7±0.7 g; EGCG 10 mg/kg = 21.9 ± 0.6 and EGCG 20 mg/kg = 20.7±0.6 g (Fig. 7B).
To investigate the tumor morphology, we performed Hematoxylin and Eosin (H&E) staining of the excised tumors. H&E staining showed nuclear condensation, shrinkage of nuclear membrane and less mitotic tumors in tumors treated with EGCG 20 mg/kg as well as in the 5-FU group, compared to control (Fig. 7C), suggesting that the EGCG 20 mg/kg and 5-FU groups damaged the inner structure of the tumor.
We then examined by immunfluorescence the expression of VEGF, which stimulates the growth of new blood vessels. As shown in Figure 7D, there is a progressive decrease in the intensity of VEGF expression from vehicle-treated to 20mg/kg EGCG-treated xenografts. In particular, the expression of VEGF in EGCG 20 mg/kg was similar to that one observed following treatment with 5-FU group; both groups reducing significantly the expression of VEGF, as compared to the vehicle control group.
Finally, we determined the effect of EGCG on glucose and lactic acid levels in vivo. Compared to vehicle-treated controls, the levels of glucose in the tumors were similar in the EGCG 5 and 10 mg/kg, whereas EGCG 20 mg/kg reduced tumor glucose levels by 34% (p<0.05; Fig. 7E). Furthermore, EGCG reduced in a concentration-dependent manner the level of lactic acid in tumor. As expected, 5-FU group had a strong effect on lactic acid levels. These results, together with the in vitro results described above, indicate that EGCG strongly affects glucose metabolism.
4. Discussion
Breast cancer remains a significant health problem in need of new therapeutic options. In the present study, we show that EGCG is effective against breast cancer in mice by suppressing glucose metabolism and inhibiting key enzymes in the glycolytic pathway. The anti-cancer efficacy of EGCG, accompanied by its selectivity toward tumor cells and its lack of toxicity, makes it a promising adjuvant drug candidate for the treatment of breast cancer.
We used in vitro and in vivo preclinical models to evaluate the anticancer effect of EGCG in breast cancer. EGCG reduced breast cancer cell growth in a time- and concentration-dependent manner. A critical characteristic of EGCG was its selectivity toward cancer cells. Ideally, in order to minimize toxicity, anticancer drugs should target specifically the tumor and not the normal surrounding tissue. EGCG displayed such selectivity. Compared with 4T1 breast cancer cell line, the normal breast epithelial cell line HC11 was more resistant to EGCG-induced suppression of its growth. Such selectivity, if broadly confirmed, will be a significant advantage of EGCG. In addition, based on our findings from syngeneic breast cancer mouse model, which allowed us to evaluate the therapeutic potential of EGCG in vivo, the inhibition of breast cancer growth by EGCG was strong, significantly reducing the rate of growth of 4T1 tumors by up to 34% in mice. All in all, these in vitro and in vivo models indicate that EGCG may be efficacious in breast cancer.
The strong growth inhibitory effect of EGCG was due, in part, to its strong apoptotic effect. Both early apoptosis and late apoptosis increased after treatment with EGCG. As shown by the microarray heat map of differentially expressed genes level, the expression of 16 apoptotic-related genes were differentially expressed following treatment with EGCG. For instance, EGCG significantly affected the expression of bax, bad, bcl-2, and bcl-xl, members from Bcl-2 family, which are key players in apoptosis26. EGCG upregulated the expression of pro-apoptotic genes, like bad, bax, smac27, apaf28, puma29, pten; and, on the other hand, EGCG inhibited the expression of anti-apoptosis genes bcl-2, bcl-xl, survivin30, c-myc31. The modulation of these key apoptotic players provides insight into the mechanism by which EGCG induces apoptosis.
Mitochondria dysfunction is a key mediator of apoptosis, playing a vital role in the caspase cascade activation32, 33. After treatment with EGCG, the mitochondrial membrane potential collapsed, and this led to the activation of downstream caspases, including caspase 9 and caspase 3. Consistent with our findings, Chen et al. showed that EGCG enhanced the activity of caspase 3 and caspase 8 and the loss of mitochondrial potential in TSGH-8301 human urinary bladder carcinoma cells34. Moreover, EGCG was shown to induce mitochondrial membrane depolarization and promoted caspase activation in pancreatic cancer cells35. Even though the exact mechanisms of the effects of EGCG on mitochondria-dependent apoptosis mechanism still remains elusive, these findings strongly suggest that EGCG reduces cancer cell growth by inducing intrinsic apoptosis.
Autophagy, an evolutionarily conserved catabolic process, is important in maintenance of cellular homeostasis36. However, autophagy dysfunction is related to many diseases37–39, including cancer40. High levels of autophagy have been observed during cell death, and has been shown to be induced by various chemotherapeutic agents41. We used various complementary assays to evaluate the effect of EGCG on the process of autophagy. These included: 1) the determination of the key autophagy formation proteins; 2) the determination of autophagosome formation by confocal microscopy; and 3) the direct evaluation of the formation of the autophagosomes and the fusion with the lysosomes by TEM. Our findings indicate that EGCG strongly induced autophagy in breast cancer cells. For instance, EGCG affected the levels of the key autophagy proteins ATG5, Beclin1 and LC3B. ATG5 is a member of autophagy related protein family, binds with ATG12 and targets to autophagosome vesicles, and plays essential role in membrane formation of the autophagosome42, 43. Beclin1 is a critical protein in the process of autophagosome formation, and plays an important role in localizing autophagic proteins to form the pre-autophagosomal structure (PAS)44. During the formation of the autophagosome, LC3B is cleaved into LC3B-I (16kD) and a carboxyl terminal glycine. LC3B-I in the cytoplasm is then modified into the membrane-conjunct form, LC3B-II (14kD), which positions at both inner and outer of the autophagosome membrane. When the autophagosome integrates with the lysosome, organelles and the LC3B-II are degraded by lysosomal hydrolases45, 46. The expression of these three proteins was enhanced at 10 and 20 μM EGCG groups, while the expression of Beclin1 and LC3B-II/ LC3B-I decreased after treated with 40 μM EGCG. These findings, together with direct evaluation of the formation of the autophagosomes and its fusion with the lysosomes by TEM, indicate that EGCG induces autophagy in 4T1 breast cancer cells. Consistent with our findings, Satoh et al.47 observed that EGCG induced human mesothelioma cell death by affecting autophagy, and Calgarotto et al.48 reported that green tea as well as quercetin induced autophagy in xenografts of human leukemia HL60 cells.
Mechanistically, the anti-growth effect of EGCG appears to be related to the alteration in glucose metabolism49, 50. Generally, tumors have an increased metabolic rate, with high energy requirements. Glycolysis offers a selective advantage to rapidly growing tumors because it can generate ATP more rapidly than oxidative phosphorylation. Furthermore, the activities of glycolytic pathway enzymes are increased in malignant cells. These increased enzymatic activities lead to reduced levels of pyruvate available to mitochondria, resulting in a low oxidation rate through the citric acid cycle. These observations known as aerobic glycolysis, are characterized by increased lactate production and glucose uptake, and decreased oxidative capacity. Given that glycolytic enzymes are differentially activated in tumor cells compared to normal cells, targeting glycolysis appears promising for the development of new antitumor drugs. EGCG reduced glucose uptake and lactate levels in breast cancer cells and in tumors. As a main byproduct of glycolysis, ATP is a critical marker of energy metabolism. ATP levels were reduced after treated with EGCG, being significant starting at a 40 μM, which is in agreement with the concentrations required to inhibit cell growth or induce apoptosis.
HK, PFK, PK, and LDH are four enzymes that regulate glycolysis and lactic acid formation. As the first step in glycolysis, HK phosphorylates glucose to glucose-6-phosphate (G-6-P). HK has four isoforms encoded by the separated genes: HK1, HK2, HK3 and HK4. High expression levels of HK2 has been observed in the cancer cells51, 52. HK2 also plays a vital role in ErbB2-driven breast cancer and Kras-driven lung cancer in mouse model53. PFK is the key-rate limiting enzyme in glycolysis, being regulated by many factors, e.g. ATP, ADP, citric acid, fatty acid. Interestingly, a positive association between increased rate of glycolysis in tumor cells and increased activity of PFK has been shown. On the other hand, PK catalyzes the phosphoenolpyruvic acid into pyruvic acid, is another “gatekeeper” of glycolysis. Lastly, LDH facilitates the transformation of pyruvic acid into lactic acid. In breast cancer cells, EGCG decreased the activity and levels of HK, PFK and LDH significantly, being HK and PFK the enzymes most strongly affected by EGCG. In agreement with our findings, EGCG inhibited PFK activity in hepatic cancer cells and tumors54.
HIF1α, one of the subunits of HIF1 transcription factor, is highly expressed in the tumor tissue55. Intracellular hypoxia occurring during fast growth of cancer cells, leads to increased levels of HIF-1α. A critical function of HIF-1α is the regulation of genes encoding glucose transporters and improving the activities of glycolysis enzymes56. GLUT is a family of glucose transporters, and has been extensively studied in cancer cells. As a glucose family, GLUT1 is the predominant transporter usually overexpressed in tumors, including hepatic57, lung58, colorectal59, and breast cancer60. The reduction of the expression levels of HIF1α and GLUT1 by EGCG, may represent a mechanism to explain the decrease in glucose consumption following EGCG treatment.
Inhibition of angiogenesis is an attractive therapy for cancer treatment. The growth and expansion of a tumor is mainly dependent on the formation of new blood vessels. Among multiple chemical signals that regulate angiogenesis, VEGF is a critical regulator in the development of the vascular system, being commonly overexpressed in various human solid tumors, including breast cancer, and thus represents an attractive target61, 62. The present study revealed that EGCG inhibits VEGF expression in breast tumors. In agreement, EGCG has been shown to directly target both tumor cells and tumor vasculature, thereby inhibiting tumor growth, and angiogenesis of breast cancer, by the inhibition of HIF-1α and as well as VEGF expression63. Moreover, because EGCG has been shown to also protect endothelial cells through various mechanisms64–66, we cannot rule out that the inhibitory effect of EGCG on VEGF is, in part, the result of its effect on endothelial cells.
To note, the beneficial action of EGCG still faces many challenges for clinical application, attributed, in part, to its poor bioavailability. Thus, the xenograft growth inhibition effect observed by EGCG, which was administered by intraperitoneal injections, may represent an underestimation of EGCG’s potential inhibitory capacity. In this regard, extensive efforts have been undertaken to enhance EGCG’s bioavailability and stability, including EGCG’s encapsulation into nanoparticles67–69. For example, formulating EGCG in solid lipid nanoparticles has been shown to enhance its stability and increase cytotoxicity in both breast and prostate cancer cells67. In addition, a lecithin formulation of a green tea catechin extract, which included EGCG, increased bioavailability of EGCG in human breast cancer patients who consumed this lecithin formulation daily for 4 weeks69. Interestingly, EGCG was detectable in breast tumor tissue and was associated with anti-proliferative effects on breast cancer tissue69. Therefore, although not explored in our study, selecting a proper formulation approach, directed at enhancing EGCG’s bioavailability, may represent a critical strategy to enhance EGCG’s anticancer effect.
In summary, EGCG, a naturally occurring compound with glycolysis inhibitory activity, has antitumor activity through the induction of apoptosis and autophagy in breast cancer cells in culture and in tumor-bearing mice. Taken together, our results point to the potential of EGCG as an adjuvant agent against breast cancer. Future studies on its potential efficacy in combination with current chemotherapeutic drugs used to treat breast cancer patients deserves further evaluation.
Acknowledgements
This study was supported by the Agriculture Practical Technology Research and Promotion Project (No. 3A6000-544904) from Zhejiang University; as well as by NIFA-USDA (CA-D-XXX-2397-H) and funds from the University of California, Davis to GGM. Ran Wei is also supported by China Scholarship Council.
Footnotes
Conflicts of interest
There are no conflicts to declare.
5. References
- 1.Siegel RL, Miller KD and Jemal A, Cancer Statistics, 2017, CA: a cancer journal for clinicians, 2017, 67, 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Forouzanfar MH, Foreman KJ, Delossantos AM, Lozano R, Lopez AD, Murray CJ and Naghavi M, Breast and cervical cancer in 187 countries between 1980 and 2010: a systematic analysis, Lancet, 2011, 378, 1461–1484. [DOI] [PubMed] [Google Scholar]
- 3.Jung KW, Won YJ, Oh CM, Kong HJ, Lee DH and Lee KH, Prediction of Cancer Incidence and Mortality in Korea, 2017, Cancer Res Treat, 2017, 49, 306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shin HR, Joubert C, Boniol M, Hery C, Ahn SH, Won YJ, Nishino Y, Sobue T, Chen CJ, You SL, Mirasol-Lumague MR, Law SC, Mang O, Xiang YB, Chia KS, Rattanamongkolgul S, Chen JG, Curado MP and Autier P, Recent trends and patterns in breast cancer incidence among Eastern and Southeastern Asian women, Cancer Causes Control, 2010, 21, 1777–1785. [DOI] [PubMed] [Google Scholar]
- 5.Meyskens FL Jr., Mukhtar H, Rock CL, Cuzick J, Kensler TW, Yang CS, Ramsey SD, Lippman SM and Alberts DS, Cancer Prevention: Obstacles, Challenges and the Road Ahead, Journal of the National Cancer Institute, 2016, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eccles SA, Aboagye EO, Ali S, Anderson AS, Armes J, Berditchevski F, Blaydes JP, Brennan K, Brown NJ, Bryant HE, Bundred NJ, Burchell JM, Campbell AM, Carroll JS, Clarke RB, Coles CE, Cook GJ, Cox A, Curtin NJ, Dekker LV, Silva Idos S, Duffy SW, Easton DF, Eccles DM, Edwards DR, Edwards J, Evans D, Fenlon DF, Flanagan JM, Foster C, Gallagher WM, Garcia-Closas M, Gee JM, Gescher AJ, Goh V, Groves AM, Harvey AJ, Harvie M, Hennessy BT, Hiscox S, Holen I, Howell SJ, Howell A, Hubbard G, Hulbert-Williams N, Hunter MS, Jasani B, Jones LJ, Key TJ, Kirwan CC, Kong A, Kunkler IH, Langdon SP, Leach MO, Mann DJ, Marshall JF, Martin L, Martin SG, Macdougall JE, Miles DW, Miller WR, Morris JR, Moss SM, Mullan P, Natrajan R, O’Connor JP, O’Connor R, Palmieri C, Pharoah PD, Rakha EA, Reed E, Robinson SP, Sahai E, Saxton JM, Schmid P, Smalley MJ, Speirs V, Stein R, Stingl J, Streuli CH, Tutt AN, Velikova G, Walker RA, Watson CJ, Williams KJ, Young LS and Thompson AM, Critical research gaps and translational priorities for the successful prevention and treatment of breast cancer, Breast cancer research : BCR, 2013, 15, R92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Key TJ, Schatzkin A, Willett WC, Allen NE, Spencer EA and Travis RC, Diet, nutrition and the prevention of cancer, Public Health Nutr, 2004, 7, 187–200. [DOI] [PubMed] [Google Scholar]
- 8.Taguchi C, Fukushima Y, Kishimoto Y, Suzuki-Sugihara N, Saita E, Takahashi Y and Kondo K, Estimated Dietary Polyphenol Intake and Major Food and Beverage Sources among Elderly Japanese, Nutrients, 2015, 7, 10269–10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang CS, Wang X, Lu G and Picinich SC, Cancer prevention by tea: animal studies, molecular mechanisms and human relevance, Nature reviews. Cancer, 2009, 9, 429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connors SK, Chornokur G and Kumar NB, New insights into the mechanisms of green tea catechins in the chemoprevention of prostate cancer, Nutr Cancer, 2012, 64, 4–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramshankar V and Krishnamurthy A, Chemoprevention of oral cancer: Green tea experience, J Nat Sci Biol Med, 2014, 5, 3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jang H-J, Ridgeway SD and Kim J.-a., Effects of the green tea polyphenol epigallocatechin-3-gallate on high-fat diet-induced insulin resistance and endothelial dysfunction, American Journal of Physiology-Endocrinology and Metabolism, 2013, 305, E1444–E1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu Y, Zhang Y, Quan Z, Wong W, Guo J, Zhang R, Yang Q, Dai R, McGeer PL and Qing H, Epigallocatechin Gallate (EGCG) Inhibits Alpha-Synuclein Aggregation: A Potential Agent for Parkinson’s Disease, Neurochem Res, 2016, 41, 2788–2796. [DOI] [PubMed] [Google Scholar]
- 14.Yao CY, Zhang JC, Liu GP, Chen F and Lin Y, Neuroprotection by (−)-epigallocatechin-3-gallate in a rat model of stroke is mediated through inhibition of endoplasmic reticulum stress, Molecular Medicine Reports, 2014, 9, 69–76. [DOI] [PubMed] [Google Scholar]
- 15.Byun JK, Yoon BY, Jhun JY, Oh HJ, Kim EK, Min JK and Cho ML, Epigallocatechin-3-gallate ameliorates both obesity and autoinflammatory arthritis aggravated by obesity by altering the balance among CD4(+) T-cell subsets, Immunology Letters, 2014, 157, 51–59. [DOI] [PubMed] [Google Scholar]
- 16.Zhu B-H, (−)-Epigallocatechin-3-gallate inhibits VEGF expression induced by IL-6 бvia Stat3 in gastric cancer, World Journal of Gastroenterology, 2011, 17, 2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao W, Mackenzie GG, Murray OT, Zhang Z and Rigas B, Phosphoaspirin (MDC-43), a novel benzyl ester of aspirin, inhibits the growth of human cancer cell lines more potently than aspirin: a redox-dependent effect, Carcinogenesis, 2009, 30, 512–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mackenzie GG, Queisser N, Wolfson ML, Fraga CG, Adamo AM and Oteiza PI, Curcumin induces cell-arrest and apoptosis in association with the inhibition of constitutively active NF-kappaB and STAT3 pathways in Hodgkin’s lymphoma cells, Int J Cancer, 2008, 123, 56–65. [DOI] [PubMed] [Google Scholar]
- 19.Shi YG, Mechanisms of caspase activation and inhibition during apoptosis, MOLECULAR CELL, 2002, 9, 459–470. [DOI] [PubMed] [Google Scholar]
- 20.Zhang C, Yan J, Xiao Y, Shen Y, Wang J, Ge W and Chen Y, Inhibition of Autophagic Degradation Process Contributes to Claudin-2 Expression Increase and Epithelial Tight Junction Dysfunction in TNF-α Treated Cell Monolayers, International Journal of Molecular Sciences, 2017, 18, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kimura S, Noda T and Yoshimori T, Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3, Autophagy, 2007, 3, 452–460. [DOI] [PubMed] [Google Scholar]
- 22.Lozy F and Karantza V, Autophagy and cancer cell metabolism, Seminars in Cell & Developmental Biology, 2012, 23, 395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pouysségur J, Dayan F and Mazure NM, Hypoxia signalling in cancer and approaches to enforce tumour regression, Nature, 2006, 441, 437–443. [DOI] [PubMed] [Google Scholar]
- 24.Deng D, Xu C, Sun P, Wu J, Yan C, Hu M and Yan N, Crystal structure of the human glucose transporter GLUT1, Nature, 2014, 510, 121–125. [DOI] [PubMed] [Google Scholar]
- 25.Tan VP and Miyamoto S, HK2/hexokinase-II integrates glycolysis and autophagy to confer cellular protection, Autophagy, 2015, 11, 963–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kale J, Osterlund EJ and Andrews DW, BCL-2 family proteins: changing partners in the dance towards death, Cell Death Differ, 2018, 25, 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanafi M, Afzan A, Yaakob H, Aziz R, Sarmidi MR, Wolfender JL and Prieto JM, In Vitro Pro-apoptotic and Anti-migratory Effects of Ficus deltoidea L. Plant Extracts on the Human Prostate Cancer Cell Lines PC3, Front Pharmacol, 2017, 8, 895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei ZZ, Qin QP, Meng T, Deng CX, Liang H and Chen ZF, 5-Bromo-oxoisoaporphine platinum(II) complexes exhibit tumor cell cytotoxcicity via inhibition of telomerase activity and disruption of c-myc G-quadruplex DNA and mitochondrial functions, Eur J Med Chem, 2018, 145, 360–369. [DOI] [PubMed] [Google Scholar]
- 29.Xiong H and Zhang J, Expression and clinical significance of ATM and PUMA gene in patients with colorectal cancer, Oncol Lett, 2017, 14, 7825–7828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Motawi TM, Bustanji Y, El-Maraghy S, Taha MO and Al-Ghussein MA, Evaluation of naproxen and cromolyn activities against cancer cells viability, proliferation, apoptosis, p53 and gene expression of survivin and caspase-3, J Enzyme Inhib Med Chem, 2014, 29, 153–161. [DOI] [PubMed] [Google Scholar]
- 31.Lian Y, Niu X, Cai H, Yang X, Ma H, Ma S, Zhang Y and Chen Y, Clinicopathological significance of c-MYC in esophageal squamous cell carcinoma, Tumour Biol, 2017, 39, 1010428317715804. [DOI] [PubMed] [Google Scholar]
- 32.Schultz DR and Harringto WJ, Apoptosis: Programmed cell death at a molecular level, Seminars in Arthritis and Rheumatism, 2003, 32, 345–369. [DOI] [PubMed] [Google Scholar]
- 33.Jourdain A and Martinou J-C, Mitochondrial outer-membrane permeabilization and remodelling in apoptosis, The International Journal of Biochemistry & Cell Biology, 2009, 41, 1884–1889. [DOI] [PubMed] [Google Scholar]
- 34.Chen NG, Lu CC, Lin YH, Shen WC, Lai CH, Ho YJ, Chung JG, Lin TH, Lin YC and Yang JS, Proteomic approaches to study epigallocatechin gallate-provoked apoptosis of TSGH-8301 human urinary bladder carcinoma cells: roles of AKT and heat shock protein 27-modulated intrinsic apoptotic pathways, Oncol Rep, 2011, 26, 939–947. [DOI] [PubMed] [Google Scholar]
- 35.Qanungo S, Das M, Haldar S and Basu A, Epigallocatechin-3-gallate induces mitochondrial membrane depolarization and caspase-dependent apoptosis in pancreatic cancer cells, Carcinogenesis, 2005, 26, 958–967. [DOI] [PubMed] [Google Scholar]
- 36.Puleston DJ and Simon AK, Autophagy in the immune system, Immunology, 2014, 141, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, Fullgrabe J, Jackson A, Jimenez SM, Karabiyik C, Licitra F, Lopez RA, Pavel M, Puri C, Renna M, Ricketts T, Schlotawa L, Vicinanza M, Won H, Zhu Y, Skidmore J and Rubinsztein DC, Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities, Neuron, 2017, 93, 1015–1034. [DOI] [PubMed] [Google Scholar]
- 38.Ji L, Chen Z, Xu Y, Xiong G, Liu R, Wu C, Hu H and Wang L, Systematic Characterization of Autophagy in Gestational Diabetes Mellitus, Endocrinology, 2017, 158, 2522–2532. [DOI] [PubMed] [Google Scholar]
- 39.Li H, Zhang Y, Min J, Gao L, Zhang R and Yang Y, Astragaloside IV attenuates orbital inflammation in Graves’ orbitopathy through suppression of autophagy, Inflamm Res, 2018, 67, 117–127. [DOI] [PubMed] [Google Scholar]
- 40.Zhao W, Shi F, Guo Z, Zhao J, Song X and Yang H, Metabolite of ellagitannins, urolithin A induces autophagy and inhibits metastasis in human sw620 colorectal cancer cells, Mol Carcinog, 2018, 57, 193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Denton D, Xu T and Kumar S, Autophagy as a pro-death pathway, Immunology and Cell Biology, 2015, 93, 35–42. [DOI] [PubMed] [Google Scholar]
- 42.Mizushima N, Sugita H, Yoshimori T and Ohsumi Y, A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy, J Biol Chem, 1998, 273, 33889–33892. [DOI] [PubMed] [Google Scholar]
- 43.Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y and Yoshimori T, Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells, J Cell Biol, 2001, 152, 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wirth M, Joachim J and Tooze SA, Autophagosome formation--the role of ULK1 and Beclin1-PI3KC3 complexes in setting the stage, Semin Cancer Biol, 2013, 23, 301–309. [DOI] [PubMed] [Google Scholar]
- 45.Mizushima N and Yoshimori T, How to interpret LC3 immunoblotting, Autophagy, 2007, 3, 542–545. [DOI] [PubMed] [Google Scholar]
- 46.Tanida I, Minematsu-Ikeguchi N, Ueno T and Kominami E, Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy, Autophagy, 2005, 1, 84. [DOI] [PubMed] [Google Scholar]
- 47.Satoh M, Takemura Y, Hamada H, Sekido Y and Kubota S, EGCG induces human mesothelioma cell death by inducing reactive oxygen species and autophagy, Cancer Cell Int, 2013, 13, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Calgarotto AK, Maso V, Junior GCF, Nowill AE, Filho PL, Vassallo J and Saad STO, Antitumor activities of Quercetin and Green Tea in xenografts of human leukemia HL60 cells, Scientific Reports, 2018, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao F, Li M, Liu W-B, Zhou Z-S, Zhang R, Li J-L and Zhou K-C, Epigallocatechin gallate inhibits human tongue carcinoma cells via HK2-mediated glycolysis, ONCOLOGY REPORTS, 2015, 33, 1533–1539. [DOI] [PubMed] [Google Scholar]
- 50.Lu Q-Y, Zhang L, Yee JK, Go V-LW and Lee W-N, Metabolic consequences of LDHA inhibition by epigallocatechin gallate and oxamate in MIA PaCa-2 pancreatic cancer cells, Metabolomics, 2015, 11, 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anderson M, Marayati R, Moffitt R and Yeh JJ, Hexokinase 2 promotes tumor growth and metastasis by regulating lactate production in pancreatic cancer, oncotarget, 2017, 8, 56081–56094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu H, Liu N, Cheng Y, Jin W, Zhang P, Wang X, Yang H, Xu X, Wang Z and Tu Y, Hexokinase 2 (HK2), the tumor promoter in glioma, is downregulated by miR-218/Bmi1 pathway, PLOS ONE, 2017, 12, e0189353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patra KC, Wang Q, Bhaskar PT, Miller L, Wang Z, Wheaton W, Chandel N, Laakso M, Muller WJ, Allen EL, Jha AK, Smolen GA, Clasquin MF, Robey RB and Hay N, Hexokinase 2 Is Required for Tumor Initiation and Maintenance and Its Systemic Deletion Is Therapeutic in Mouse Models of Cancer, Cancer Cell, 2013, 24, 213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li S, Wu L, Feng J, Li J, Liu T, Zhang R, Xu S, Cheng K, Zhou Y, Zhou S, Kong R, Chen K, Wang F, Xia Y, Lu J, Zhou Y, Dai W and Guo C, In vitro and in vivo study of epigallocatechin-3-gallate-induced apoptosis in aerobic glycolytic hepatocellular carcinoma cells involving inhibition of phosphofructokinase activity, Scientific Reports, 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patel SA and Simon MC, Biology of hypoxia-inducible factor-2alpha in development and disease, Cell Death Differ, 2008, 15, 628–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu W, Shen SM, Zhao XY and Chen GQ, Targeted genes and interacting proteins of hypoxia inducible factor-1, Int J Biochem Mol Biol, 2012, 3, 165–178. [PMC free article] [PubMed] [Google Scholar]
- 57.Amann T, Maegdefrau U, Hartmann A, Agaimy A, Marienhagen J, Weiss TS, Stoeltzing O, Warnecke C, Schölmerich J, Oefner PJ, Kreutz M, Bosserhoff AK and Hellerbrand C, GLUT1 Expression Is Increased in Hepatocellular Carcinoma and Promotes Tumorigenesis, The American Journal of Pathology, 2009, 174, 1544–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tan Z, Yang C, Zhang X, Zheng P and Shen W, Expression of glucose transporter 1 and prognosis in non-small cell lung cancer: a pooled analysis of 1665 patients, Oncotarget, 2017, 8, 60954–60961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feng W, Cui G, Tang CW, Zhang XL, Dai C, Xu YQ, Gong H, Xue T, Guo HH and Bao Y, Role of glucose metabolism related gene GLUT1 in the occurrence and prognosis of colorectal cancer, Oncotarget, 2017, 8, 56850–56857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oh S, Kim H, Nam K and Shin I, Glut1 promotes cell proliferation, migration and invasion by regulating epidermal growth factor receptor and integrin signaling in triple-negative breast cancer cells, BMB Rep, 2017, 50, 132–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Klukovits A, Schally AV, Szalontay L, Vidaurre I, Papadia A, Zarandi M, Varga JL, Block NL and Halmos G, Novel antagonists of growth hormone-releasing hormone inhibit growth and vascularization of human experimental ovarian cancers, Cancer, 2012, 118, 670–680. [DOI] [PubMed] [Google Scholar]
- 62.Barr MP, Gray SG, Gately K, Hams E, Fallon PG, Davies AM, Richard DJ, Pidgeon GP and O Byrne KJ, Vascular endothelial growth factor is an autocrine growth factor, signaling through neuropilin-1 in non-small cell lung cancer, Molecular Cancer, 2015, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gu JW, Makey KL, Tucker KB, Chinchar E, Mao X, Pei I, Thomas EY and Miele L, EGCG, a major green tea catechin suppresses breast tumor angiogenesis and growth via inhibiting the activation of HIF-1alpha and NFkappaB, and VEGF expression, Vasc Cell, 2013, 5, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang J, Han Y, Chen C, Sun H, He D, Guo J, Jiang B, Zhou L and Zeng C, EGCG attenuates high glucose-induced endothelial cell inflammation by suppression of PKC and NF-kappaB signaling in human umbilical vein endothelial cells, Life Sci, 2013, 92, 589–597. [DOI] [PubMed] [Google Scholar]
- 65.Zhou X, Liang L, Zhao Y and Zhang H, Epigallocatechin-3-Gallate Ameliorates Angiotensin II-Induced Oxidative Stress and Apoptosis in Human Umbilical Vein Endothelial Cells through the Activation of Nrf2/Caspase-3 Signaling, J Vasc Res, 2017, 54, 299–308. [DOI] [PubMed] [Google Scholar]
- 66.Li J, Zhang Z, Lv L, Qiao H, Chen X and Zou C, (−)-Epigallocatechin Gallate Inhibits Asymmetric Dimethylarginine-Induced Injury in Human Brain Microvascular Endothelial Cells, Neurochem Res, 2016, 41, 1868–1876. [DOI] [PubMed] [Google Scholar]
- 67.Radhakrishnan R, Kulhari H, Pooja D, Gudem S, Bhargava S, Shukla R and Sistla R, Encapsulation of biophenolic phytochemical EGCG within lipid nanoparticles enhances its stability and cytotoxicity against cancer, Chem Phys Lipids, 2016, 198, 51–60. [DOI] [PubMed] [Google Scholar]
- 68.Yuan X, He Y, Zhou G, Li X, Feng A and Zheng W, Target challenging-cancer drug delivery to gastric cancer tissues with a fucose graft epigallocatechin-3-gallate-gold particles nanocomposite approach, J Photochem Photobiol B, 2018, 183, 147–153. [DOI] [PubMed] [Google Scholar]
- 69.Lazzeroni M, Guerrieri-Gonzaga A, Gandini S, Johansson H, Serrano D, Cazzaniga M, Aristarco V, Macis D, Mora S, Caldarella P, Pagani G, Pruneri G, Riva A, Petrangolini G, Morazzoni P, DeCensi A and Bonanni B, A Presurgical Study of Lecithin Formulation of Green Tea Extract in Women with Early Breast Cancer, Cancer Prev Res (Phila), 2017, 10, 363–370. [DOI] [PubMed] [Google Scholar]