

Abstract
Conditional gene inactivation and restoration are powerful tools for studying gene functions in the nervous system and for modeling neuropsychiatric diseases. The combination of the two is necessary to interrogate specific cell types within defined developmental stages. However, very few methods and animal models have been developed for such purpose. Here we present a versatile method for conditional gene inactivation and in situ restoration through reversibly inverting a critical part of its endogenous genomic sequence by Cre- and Flp-mediated recombinations. Using this method, we generated a mouse model to manipulate Mecp2, an X-linked dosage-sensitive gene whose mutations cause Rett syndrome. Combined with multiple Cre- and Flp-expressing drivers and viral tools, we achieved efficient and reliable Mecp2 inactivation and restoration in the germline and several neuronal cell types, and demonstrated phenotypic reversal and prevention on cellular and behavioral levels in male mice. This study not only provides valuable tools and critical insights for Mecp2 and Rett syndrome, but also offers a generally applicable strategy to decipher other neurologic disorders.
SIGNIFICANCE STATEMENT Studying neurodevelopment and modeling neurologic disorders rely on genetic tools, such as conditional gene regulation. We developed a new method to combine conditional gene inactivation and restoration on a single allele without disturbing endogenous expression pattern or dosage. We applied it to manipulate Mecp2, a gene residing on X chromosome whose malfunction leads to neurologic disease, including Rett syndrome. Our results demonstrated the efficiency, specificity, and versatility of this new method, provided valuable tools and critical insights for Mecp2 function and Rett syndrome research, and offered a generally applicable strategy to investigate other genes and genetic disorders.
Keywords: cell type, conditional gene inactivation and restoration, Cre and Flp, Mecp2, mouse model, Rett syndrome
Introduction
The nervous system is composed of highly diverse cell types, assembled via intricate developmental processes, and harbors extensive plasticity. In this complex system, many genes function in context-dependent manners, playing distinct roles in different cell types or across different developmental stages. Therefore, it is desirable to conditionally inactivate and then rescue a gene in a specific cell type within a defined temporal window to precisely interrogate its function, access the contribution of its malfunction to diseases, and evaluate the therapeutic potential of genetic rescue. For many neuropsychiatric disease-related genes that are dosage-sensitive or residing on sex chromosomes (Raymond, 2006; Rice and McLysaght, 2017), it is also necessary to establish genetic models that inactivate and restore a gene in situ on the same allele, and avoids disturbing endogenous expression pattern before inactivation and after restoration. However, very few genetic methods have been developed to fulfill all these requirements (Zeng et al., 2008; Chaiyachati et al., 2012; Robles-Oteiza et al., 2015; Andersson-Rolf et al., 2017), and none of them has been applied to establish animal models for genes linked to neurologic diseases to demonstrate cell-type-specific conditional inactivation and in situ restoration within defined time window in the brain.
Here we present a versatile method, which can conditionally inactivate and restore a gene of interest in situ by changing the orientation of a critical part of its genomic sequence via Cre- and Flp-mediated recombinations. We named it SGIRT (i.e., single-allele conditional gene inactivation and restoration via recombinase-based flipping of targeted genomic region). The targeted genomic region is flanked by two pairs of heterologous loxP sites and two pairs of heterologous FRT sites (see Fig. 1A). When only Cre or Flp is present, the targeted genomic region is inverted once, rendering it inaccessible for splicing or translation, and leads to gene inactivation. When both recombinases are present, simultaneously or sequentially, the targeted genomic region is inverted twice to resume the native orientation, restoring gene expression with endogenous pattern and dosage maximally preserved. This method does not require the generation of two separate alleles for KO and restoration (Giacometti et al., 2007; Jugloff et al., 2008; Zeng et al., 2008; Chaiyachati et al., 2012; Kerr et al., 2012). It also differs significantly from previously reported single-allele methods based on reversible gene trapping (Schnutgen et al., 2005; Robles-Oteiza et al., 2015; Andersson-Rolf et al., 2017) in that the gene inactivation is not limited by gene-trapping efficiency.
Figure 1.

A SGIRT mouse model for conditional inactivation and restoration of Mecp2. A, ES targeting strategy. Exon 3 of Mecp2 encoding part of the methyl-CpG-binding domain was flanked by four pairs of recombinase recognition sites: lox2272, LoxN, Frt, and F5. One round of Cre- or Flp-mediated recombination inverts this exon (Mecp2IVC or Mecp2IVF), while two rounds of recombination restore its orientation (Mecp2R). B, Southern blot confirmation of correctly targeted ES clone, which was used to generate Mecp2CF/Y mice. A, Short black bar represents Southern probe. C, Genomic PCR confirmation of successful recombinations and changes of exon 3 orientation in different alleles. The positions of three primer pairs are shown in A. D, RT-PCR with primers encompassing exon 3 confirmed the exclusion of exon 3 in mRNA extracted from Mecp2IVC/Y (shorter band) and the inclusion of exon 3 in mRNAs extracted from Mecp2WT/Y, Mecp2CF/Y, and Mecp2R/Y brains (longer bands). E, Western blot confirmation of normal MeCP2 protein expression in Mecp2CF/Y, loss of its expression in Mecp2IVC/Y, and restoration of expression in Mecp2R/Y. F, Indistinguishable body and brain weights between Mecp2WT/Y, Mecp2CF/Y, and Mecp2R. n = 7. G, Representative photographs of brains taken from 8-week-old adult mice of the four genotypes showing comparable sizes of Mecp2WT/Y, Mecp2CF/Y, and Mecp2R/Y, and reduced size in Mecp2IVC/Y. The weights of these brains are (left to right) as follows: 413, 423, 351, and 417 mg. Scale bar, 500 μm. Data are mean ± SEM. Dots, squares, and triangles represent data from individual mice. n.s., Not significant at p > 0.05. For more statistical details, see Table 2.
As a proof of principle, we applied SGIRT to the X-chromosome-linked gene encoding methyl-CpG-binding protein 2 (MeCP2). Mecp2 functions in a cell-type- and dosage-dependent manner in the developing and mature nervous system (Ip et al., 2018; Qiu, 2018). Its mutations and duplication lead to Rett syndrome (RTT) (Amir et al., 1999; Leonard et al., 2017) and autism spectrum disorders (Van Esch, 2012), respectively. Mouse models of constitutive or conditional Mecp2 KO or reexpression were widely used to model RTT (Qiu, 2018). However, no single-allele in situ reversible conditional inactivation model had been reported. Here we generated such a model and combined it with a collection of Cre and Flp drivers and viral tools for multifaceted and flexible control of Mecp2 in germline and several neuronal cell types. Importantly, we have achieved, to our knowledge for the first time, in situ Mecp2 restoration following conditional inactivation in specific cell types defined by molecular marker or the combination of molecular marker and projection pattern, in temporally and spatially controllable manners, in the mouse brain. We have also assessed phenotypic reversal ranging from cellular to behavioral levels after the genetic rescue. Our study not only demonstrated the efficiency, specificity, and versatility of SGIRT, but also provided valuable tools and critical insights for Mecp2 function and RTT research. The SGIRT strategy we developed offered one of the most compelling and necessary approaches to decipher how mutations may impact cellular and organismic functions and to systematic analyze the complex genetic architecture of brain disorders.
Materials and Methods
Mice
All mouse studies were conducted in strict accordance with the guidelines of the Institutional Animal Care and Use Committee of Fudan University. All husbandry and experimental procedures were reviewed and approved by the same committee. All applicable institutional and/or national guidelines for the care and use of animals were followed. The following transgenic mouse lines were used in this study: E2a-Cre (Jax 003314), Actin-Flp (Jax 003380), PVFlpe (Jax 021191), PVCreER (Jax 028580), SOMFlp (Jax 028579), VIPCre (Jax 010908), Ai14 (Jax 007914), Ai65 (Jax 021875), and tdT-Frt (Flp-dependent RFP reporter derived from Ai65) (Madisen et al., 2015) by removing LoxP-STOP-LoxP with CMV-Cre (Jax 006054) (M. He et al., 2016). Mice included in the survival curve and body weight analysis were examined weekly.
Transgenic mice generation
The Fezf2CreER mouse line was generated and characterized by Miao He in Z. Josh Huang's laboratory at Cold Spring Harbor Laboratory by knocking in 2A-CreER in frame just before the STOP codon of Fezf2 gene. More details will be revealed in a separate manuscript currently under preparation. The Mecp2CF mouse line was generated by substituting the endogenous exon 3 with a recombinase recognition sites flanked exon 3 using homologous recombination. Gene targeting vectors were generated using PCR-based cloning approach as described previously (M. He et al., 2016). More specifically for Mecp2CF, a 1168 bp NsiI-NcoI genomic piece containing exon 3 and part of intron 2 and 3 of the Mecp2 gene, a 5.2 kb 5′ homology arm and a 2 kb 3′ homology arm were sequentially cloned into a building vector containing the recombination sites, an attP-attB flanking Neo-positive selection cassette and a TK-negative selection cassette to generate the targeting vector. Targeting vectors were linearized and transfected into a C57/black6 ES cell line. ES clones were first screened by PCR, then confirmed by Southern blotting using appropriate DIG-dUTP-labeled probes. For each strain, one positive ES cell clone was used for tetraploid complementation to obtain male mice carrying the modified allele following standard procedures. ES cell transfections, blastocyst injections, and tetraploid complementation were performed by the Rodent Genetic Engineering Laboratory, New York University School of Medicine. Founder males were bred with C57BL/6J females to confirm germline transmission. F1 siblings were bred with one another to establish the colony. The LoxP-Neo-LoxP-positive selection cassette in the Fezf2CreER founder line was removed by breeding with CMV-Cre transgenic mice (JAX 006054). The AttP-Neo-AttB cassette in the Mecp2CF founder line was left intact because of unsuccessful attempts to delete it by breeding with Rosa26-PhiC31 transgenic mice (JAX 007743).
The following primers were used for PCR screening of Mecp2 targeted ES cells: Mecp2-5'KI-exF: 5′-AAGCAGATGTCTATGAGTTCAAGG-3′; Mecp2-5'KI-inR: 5′-GTAGAATTTCGACGACCTGC-3′; Mecp2-3'KI-inF: 5′-TGGGGCTCGACTAGAGCTTGC-3′; and Mecp2-3'KI-exR: 5′-GTGTCACCATTTCAAGCCAG-3′.
The following primers were used for generating southern blot probe of Mecp2: Mecp2-Sprobe-F: 5′- AGGAATGAATGAGTGAGTGAATG-3′; and Mecp2-Sprobe-R: 5′- ATCCTAGAACCAGGGTATGGTAG-3′.
Genomic PCR
Genomic DNA was prepared from mouse tails. Tissues were lysed by incubation in lysis buffer (DirectPCR Lysis Reagent, Viagen Biotech, 102-T) with 0.1 mg/ml proteinase K overnight at 55°C followed by 45 min at 90°C air bath to inactivate proteinase K. The lysate was cleared by centrifugation at maximum speed (21,130 × g) for 15 min in a tabletop centrifuge. Supernatant-containing genomic DNA was use as the PCR template for amplifying DNA products no longer than 2 kb. To amplify longer fragments, the heat-inactivation step was skipped, and genomic DNA was extracted from the supernatant by ethanol precipitation following standard protocol. The following primers were used for genomic PCR: Mecp2-wtF: 5′- CCTGGATTCAGTCCCACCAATC-3′; Mecp2-wtR: 5′- AAACTGGAACACAACCGGGC-3′; Mecp2-5'F: 5′- TCAGGAGGCTGGAACGAAGG-3′; Mecp2-5'R: 5′- ACAGGCTGCAGGTCGAAAGG-3′; Mecp2-3'F: 5′- ACAAAGGAAGTCTGGCCGATCTG-3′; and Mecp2-3'R: 5′- CAAACTGGAACACAACCGGGC-3′.
Tamoxifen (Tam) induction
Tam (Sigma Millipore, T5648-5Q) was prepared by dissolving in corn oil (20 mg/ml) at room temperature with constant rotation overnight. For Mecp2CF/Y::PVFlpe/CreER, Tam (0.1 mg per gram body weight) was administered by intraperitoneal injection for 1, 3, or 5 times, once every other day, starting right after the first set of rotarod test. For Mecp2CF/Y::Fezf2CreER, five injections were administered in the same manner 10 d after virus injection, and mice were perfused 3 weeks after induction.
Immunohistochemistry and microscopy
Mice were anesthetized by an intraperitoneal injection of 1.5% sodium pentobarbital (0.09 mg/g body weight), then intracardially perfused with saline followed by 4% PFA in 0.1 m PB. Following 24 h of postfixation at 4°C, brain slices at 30 or 50 μm were sectioned using 1000s vibratome (Leica Microsystems). Sections were blocked in PBS containing 0.05% Triton and 5% normal goat or donkey serum, depending on the secondary antibodies being used, and then incubated with the following primary antibodies in the blocking solution at room temperature overnight: MeCP2 (rabbit polyclonal antibody, 1:500, Cell Signaling Technology, 3456S), RFP (goat polyclonal antibody, 1:2000, Sicgen, AB0081-200; rabbit polyclonal antibody,1:2000, Rockland, 600-401-379), GFP (chicken polyclonal antibody, 1:1000, Aves Labs, GFP-1020), parvalbumin (PV, mouse monoclonal antibody, 1:1000, Sigma Millipore, P3088; goat polyclonal antibody, 1:1000, Swant PVG-213), somatostatin (SOM, rat monoclonal antibody, 1:300, Millipore, MAB354; goat polyclonal antibody, 1:1000, Santa Cruz Biotechnology, SC-7819), and NeuN (mouse monoclonal antibody, 1:250, Millipore, MAB377). Sections were then incubated with appropriate AlexaFluor dye-conjugated IgG secondary antibodies (1:500, Thermo Fisher Scientific) in blocking solution, counterstained with DAPI, and mounted in Aqua-mount (Lerner Laboratories). Sections were imaged with confocal microscopy (Nikon, AIR), fluorescent microscopy (Nikon Eclipse Ni; Olympus VS120), and fluorescent stereoscope (Nikon, SMZ25). Anatomical regions were identified according to the Paxinos and Franklin's The mouse brain in stereotaxic coordinates (Franklin and Paxinos, 2013). To minimize variability, samples from the same batch of experiments were processed simultaneously at all steps, including perfusions, sectioning, immunostaining, and imaging, whenever possible. To quantify the average fluorescence intensity of MeCP2 immunostaining within a certain brain structure, three FOVs on each hemisphere (i.e., 6 on both hemispheres) per brain structure per sample were identified and averaged; within each FOV, 15 small regions without MeCP2 staining were randomly sampled to calculate the average background fluorescence, which was subtracted from the average fluorescence of the entire FOV. To quantify colocalization of MeCP2 with markers or fluorescent proteins, the structure of interest on both hemispheres from three sections per brain were analyzed. To measure intensity of MeCP2 staining in interneuron subtypes, the ROI was drawn manually along the boundary of each cell, and the average fluorescence of each ROI was measured. Three sections containing the structure of interest from each brain were analyzed, and the average value from all ROIs of the three sections was taken as the value for that brain. Imaging was performed by one experimenter with the knowledge of the genotypes to make sure the anatomic positions were matched between the groups. Another experimenter who was blinded to the genotypes performed the measurement and quantification.
Western blot
Total protein was extracted from brain and other tissues using lysis buffer containing 20 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EGTA, 1 mm Na2EDTA,1% NP-40, 1% sodium deoxycholate, 0.1% SDS with Protease Inhibitor Cocktail (BBI, C600387-0001), and concentration was quantified using BCA Protein Assay Kit (Thermo Fisher Scientific, catalog #23227) per the manufacturer's instructions; 20 μg total protein was loaded per lane and run on 12% SDS-PAGE gel, and electrophoretically transferred onto PVDF membranes (GE Healthcare, NL1160). Membranes were blocked with 5% nonfat milk in 1× PBS, pH 7.4, for 1 h and probed with primary antibodies: MeCP2 (1:1000, rabbit polyclonal antibody, Cell Signaling Technology, 3456S) and GAPDH (1:5000, mouse monoclonal antibody, KangChen Bio-tech, KC-5G4). Then they were washed 3 times with PBST (1× PBS with 0.2% Tween 20), and incubated with HRP-conjugated secondary antibodies against rabbit and mouse (1:10,000, Bioworld, BS13278, BS12478). After three washes of PBST, they were treated with SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific, 34577). Signals were acquired with a luminescent image analyzer (Bio-Rad ChemiDoc Touch Imaging System) and quantified with accompanying Image Lab software.
RT-PCR and qRT-PCR
RNA was extracted using TRIzol (Invitrogen, 15596018), and cDNA was synthesized using SuperScriptIII reverse transcriptase (Invitrogen, 18080093). To examine whether exon 3 was excluded from mRNA, Hotstart Taq Polymerase (Tiandz, 90805) was used for PCR with forward primer landing in exon 2 and the reverse primer landing in exon 4. PCR product was loaded on a 1% agarose gel for electrophoresis. The primer sequences are as follows: Mecp2- PCR-F: 5′- TAGCTGGGATGTTAGGGCTC-3′; and Mecp2- PCR-R: 5′- CACTTCCTTGACCTCGATGC-3′.
SYBR Green qPCR mix (TB green Premix Ex Taq II, Takara, RR820A) was used to perform qPCR (Applied Biosystems, 7300 Real Time PCR System) according to the manufacturer's instructions. Expression of Mecp2 was normalized to that of Actin using the ΔCt method. Relative expression was calculated using the 2-ΔΔCt method. The following primers were used to detect Mecp2 and Actin: Mecp2-qPCR-F: 5′- TGGGAGACACCTCCTTGGAC-3′; Mecp2-qPCR-R: 5′- GATTTGGGCTTCTTAGGTGGTTTC-3′; Actin-qPCR-F: 5′- TTTGCAGCTCCTTCGTTGC-3′; and Actin-qPCR-R: 5′- CCATTCCCACCATCACACC-3′
Virus injection
AAV virus was purchased from Shanghai Tailtool Bioscience and S&E Shanghai Medical Biotech. CAV virus was purchased from IGMM vector core. For adult injection, mice were anesthetized by intraperitoneal injection of 1.5% sodium pentobarbital (0.09 mg/g body weight). Stereotactic injections were performed via rodent stereotax (RWD Life Science, 68025; Narishige, SR-5M-HT). Bregma coordinates were identified for two brain areas: primary visual cortex (A/P −2.8 mm; M/L: 2.5 mm; D/V: 0.5 mm depth from the pial surface); and thalamus (A/P: −2.18 mm; M/L: −1 mm; D/V: 3.25 mm). An incision was made over the scalp, a small burr hole was drilled into the skull using a dental drill (Strong 102Z), and brain surface was exposed. A glass micropipette (tip size ∼10-30 μm) containing the virus was lowered below the pial surface to the specified coordinates. Pulses were delivered using Nanoject 2 (Drummond) or Nanoject 2010 (WPI) at a rate of 30 nl/min. After the desired amount of virus was injected, the pipette was left in the brain for 10-15 min to prevent backflow. After the injection, the pipette was withdrawn, the incision was sutured, and animals recovered. For neonatal injection, P0 pups were ice-anesthetized and taped on to an ice-cold metal mouse adaptor. A glass micropipette (tip size ∼10-30 μm) attached to a Hamilton syringe containing the virus was directly inserted into the lateral ventricle through temporal cortex (anteroposterior: 2 mm anterior to λ; ML: 1-2 mm; DV: 2-4 mm), penetrating the skin and skull; 3 μl virus was injected into each hemisphere by pushing the syringe manually at a constant speed within 10-20 s, and both sides were injected for each pup. The injected pups were allowed to recover on a heating plate until normal breathing and movement were observed, then return to the home cage or a foster mother.
Cortical slice preparation and in vitro electrophysiology
Mice were anesthetized with isoflurane, and transcardially perfused with ice-cold oxygenated (95% O2/5% CO2) cutting solution (pH 7.3-7.4, 300-305 mOsm) containing the following (in mm): 93 N-methyl-D-glucamine, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 Na-ascorbate, 2 thiourea, 3 Na-pyruvate, 10 MgCl2, 0.5 CaCl2, and 12 N-acetyl-cysteine. Coronal slices were sectioned at 300 µm with a vibratome (Leica Microsystems, VT1200). Slices were incubated in an chamber filled with cutting solution at 34°C for 10 min and then transferred to oxygenated HEPES solution containing the following (in mm): 94 NaCl, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 Na-ascorbate, 2 thiourea, 3 Na-pyruvate, 2 MgCl2, 2 CaCl2, and 12 N-acetylcysteine, pH 7.3-7.4, 300-305 mOsm. After 15 min incubation at room temperature, the slices were transferred again to ACSF (pH 7.3-7.4, 300-305 mOsm) containing the following (in mm): 118 NaCl, 2.5 KCl, 26 NaHCO3, 1 NaH2PO4, 22 D-glucose, 2 MgCl2, 2 CaCl2. After 1 h incubation at room temperature, slices were transferred to a recording chamber of an upright microscope (Olympus BW51) and submerged in ACSF. Whole-cell patch-clamp was performed in the primary visual cortex using the morphology of hippocampus and subcortical white matter as primary landmarks according to the Paxinos and Franklin's The mouse brain in stereotaxic coordinates (Franklin and Paxinos, 2013). A Sutter P97 puller was used to pull patch pipettes from borosilicate glass capillaries with filament (1.5 mm outer diameter and 0.86 mm inner diameter, Sutter, BF150-86-10) with a resistance of 3-8 mΩ. The patch pipettes were filled with the following solution (in mm): 120 K-gluconate, 16 KCl, 2 MgCl2, 10 HEPES, 0.2 EGTA, 2.5 MgATP, 0.5 Na3GTP, 10 Na-phosphocreatine, pH 7.3. All voltages reported were not corrected for junction potential. Series resistance was compensated in current-clamp mode and was kept to <30 mΩ. Signals were recorded 10 kHz and filtered at 2 kHz (Digidata 1550A, Molecular Devices). Data were analyzed with Multiclamp 700B (Molecular Devices) using pClamp 10.6. Resting membrane potential was measured in I = 0 mode. Input resistance was calculated from the voltage response to a −40 pA current pulse. Rheobase was defined as the minimal current injection to evoke a spike. Properties of the single action potential (AP) were measured from the first spike fired at rheobase. All recordings were performed at 29°C-31°C with the chamber perfused with oxygenated ACSF.
Behavioral tests
All behavioral assays were conducted during the light phase with the investigator blinded to the genotype. Mice were handled daily by the experimenter for a week before the day of testing and allowed to habituate for 30 min in the testing room before the test.
Rotarod
Mice were placed on an accelerating rotarod and allowed to move freely as the rotation increased from 4 to 40 rpm over 5 min. Each animal was tested for 4 trials in 1 d, with a 15 min interval between trials. Latency to fall was automatically recorded with an infrared detector in the rotarod apparatus.
Open field
Mice were placed in the center of a gray Plexiglas box (40 cm length × 40 cm width × 40 cm height), and allowed to explore freely for 30 min. The center region was defined as a square (20 cm × 20 cm) in the middle of the open field. The arena was filmed using an overhead digital camera, and the digital tracks for each mouse were analyzed using Ethovision 8.5 tracking software.
Tail suspension
Mice were suspended by their tails to the edge of suspension bar using adhesive tape placed ∼1-2 cm from the end of their tails, and videotaped for 6 min. Immobility was scored manually by an experienced experimenter using stopwatch. Small movements confined to the front legs but without the involvement of the hind legs and oscillations and pendulum-like swings that are the result of the momentum gained during the earlier mobility bouts are not counted as mobility.
Elevated plus maze
Mice were tested in an opaque gray Plexiglas cross-shaped maze with four arms (77 cm length × 7 cm width) elevated 70 cm above the floor. Two opposing arms were protected by walls (19 cm height), and the other two were left open. Each mouse was placed at the center of the maze and allowed to explore freely for 8 min. Only when the whole body (excluding the tail) entered one arm would it be counted as an entry into that arm.
Three-chamber test
A Plexiglas box (62 cm length × 42 cm width × 23 cm height) with clear walls and a gray floor was divided into three equally sized chambers by removable clear plates and filmed using an overhead digital camera. Each chamber on the side contained a cylindrical wire cage. Each subject mouse was placed into the center chamber and habituated for 10 min with both sides closed and then allowed to freely explore all three chambers for 10 min to test for innate direction bias of motion. In the sociability test session, one age- and sex-matched C57BL/6 unfamiliar mouse was placed randomly into one of the two wire cages on either side and considered “stranger.” The other empty wire cage was considered “inanimate.” The subject mouse was allowed to explore all three chambers for 10 min, interacting with the “stranger” and “inanimate.” The digital tracks of each subject mouse were analyzed using the Any-maze tracking system (Stoelting), and the time spent in each chamber was measured.
Fear conditioning
Each mouse was placed into a noise-insulated conditioning cuboid chamber with a metal grid floor for three rounds of training. Each round consisted of 120 s silence followed by a 5 kHz pure tone (90 dB, 30s duration) and a 0.75 mA foot shock for 2 s at the end of the tone. Twenty-four hours after training, contextual memory was tested by placing the mouse back in the same box for 5 min. Five hours after the contextual test, the mouse was placed into a cylindrical chamber with a smooth gray floor and aloe scent and subjected to a cue test consisting of 180 s of silence followed by the same 5 kHz, 90 dB, 180 s tone. Freezing behavior was scored using Freeze Frame software (Actimetics).
Hindlimb clasping (HC)
Mice were suspended by their tails by hand for 30 s. The posture of their hindlimbs was observed and scored manually by an experienced experimenter. No clasping was scored as 0, and clasping was scored as 1.
Statistical analysis
GraphPad Prism version 7.0 was used for statistical calculations. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications. Data collection and analysis were performed blind to the experimental conditions whenever possible. No animals or data points were excluded from the analysis. Equal variances were assessed using the F test or the Bartlett's test. Normalcy was assessed using the Kolmogorov–Smirnov test. Statistical significance was tested using two-tailed paired or unpaired t test, or two-way ANOVA with Bonferroni's post hoc analysis. Data are presented as mean ± SEM. p < 0.05 was considered significant. Statistical values for all experiments, including all post hoc test p values, are shown in Tables 1 and 2.
Table 1.
Intrinsic properties of PV+ neurons in three groups and statistical information of the comparison among groups
| Group | Mecp2CF/Y::PVFlp+ AAV-fDIO-GFP (inactivation) Cell, n = 25; mouse, n = 4 | Mecp2CF/Y::PVFlp+ AAV-fDIO-Cre-GFP (rescue) Cell, n = 30; mouse, n = 4 | Mecp2WT/Y::PVFlp+ AAV-fDIO-GFP (control) Cell, n = 28; mouse, n = 3 | Statistical values of unpaired two-tailed t test |
||
|---|---|---|---|---|---|---|
| p t(53)a | p t(51)b | p t(56)c | ||||
| Input resistance (mΩ) | 146.22 ± 11.19 | 114.15 ± 7.36 | 110.90 ± 7.14 | 0.0170 | 0.0089 | 0.7531 |
| 2.465 | 2.718 | 0.3126 | ||||
| Membrane time constant (ms) | 8.61 ± 0.58 | 6.43 ± 0.50 | 5.84 ± 0.58 | 0.0056 | 0.0019 | 0.4575 |
| 2.886 | 3.373 | 0.0014 | ||||
| Rheobase (pA) | 166.0 ± 19.8 | 275.7 ± 34.8 | 282.2 ± 25.7 | 0.0120 | 0.0009 | 0.8822 |
| 2.601 | 3.541 | 0.1488 | ||||
| AP half-width (ms) | 0.38 ± 0.01 | 0.32 ± 0.01 | 0.34 ± 0.01 | 0.0007 | 0.0046 | 0.4385 |
| 3.619 | 3.003 | 0.8559 | ||||
| Maximum firing rate (Hz) | 259.7 ± 8.9 | 298.7 ± 14.3 | 294.7 ± 8.0 | 0.0206 | 0.0052 | 0.8020 |
| 2.388 | 2.911 | 0.2521 | ||||
| Resting membrane potential (mV) | −70.80 ± 1.08 | −73.91 ± 1.24 | −77.33 ± 0.86 | 0.0704 | < 0.0001 | 0.0302 |
| 1.846 | 4.765 | 2.224 | ||||
| AP amplitude (mV) | 55.08 ± 1.83 | 52.06 ± 1.57 | 50.90 ± 1.01 | 0.2122 | 0.0447 | 0.5440 |
| 1.263 | 2.058 | 0.6105 | ||||
| AP threshold (mV) | −32.41 ± 1.01 | −36.39 ± 1.19 | −34.64 ± 1.25 | 0.0162 | 0.1786 | 0.3162 |
| 2.485 | 1.364 | 1.011 | ||||
| Afterhyperpolarization potential (mV) | −22.95 ± 0.45 | −23.04 ± 0.69 | −20.82 ± 0.76 | 0.9185 | 0.0231 | 0.0348 |
| 0.1028 | 2.342 | 2.163 | ||||
aInactivation versus rescue.
bInactivation versus control.
cRescue versus control.
Table 2.
Statistical summary of the data, except recording data showing in Table 1
| Fig | Statistical Test | Values | Bonferroni's post hoc |
|---|---|---|---|
| 1F | One-way ANOVA | Body weight: F(2,18) = 0.0323, p = 0.9683; brain weight: F(2,18) = 0.3663, p = 0.6984 | Between any pair of genotypes: p > 0.9999 |
| 2B | One-way ANOVA | Cor: F(2,6) = 0.0274, p = 0.9731; Cb: F(2,6) = 0.0604, p = 0.9420; liver: F(2,6) = 0.2189, p = 0.8095; lung: F(2,6) = 0.1772, p = 0.8419 | Between any pair of genotypes: p > 0.9999 |
| 2C | One-way ANOVA | Cor: F(2,6) = 0.0753, p = 0.9283; Cb: F(2,6) = 0.0211, p = 0.9792; liver: F(2,6) = 0.1034, p = 0.9033; lung: F(2,6) = 0.3229, p = 0.7359 | Between any pair of genotypes: p > 0.9999 |
| 2D | One-way ANOVA | Cor: F(2,6) = 0.1098, p = 0.8978; hip: F(2,6) = 1.236, p = 0.3552; Str: F(2,6) = 0.1431, p = 0.8696 | Hip: WT vs CF: p = 0.5420; WT vs R: p = 0.9126; any other pairs: p > 0.9999 |
| 3C | Two-way ANOVA | Genotype: F(1,20) = 133.9, p < 0.0001; week: F(8,160) = 302.7, p < 0.0001; genotype and week: F(8,160) = 7.123, p < 0.0001 | Week 1 and week 2: p > 0.9999; week 3: p = 0.0805; week 4: p = 0.0002; week 5 and week 9: p < 0.0001 |
| 3E | Two-way ANOVA | Genotype: F(1,20) = 51.88, p < 0.0001; trial: F(3,60) = 29.23, p < 0.0001; genotype + trial: F(3,60) = 22.08, p < 0.0001 | Trial 1: p = 0.5956; Trial 2: p = 0.0015; Trials 3 and 4: p < 0.0001 |
| 3F | Unpaired t | t(19) = 8.444, p < 0.0001 | NA |
| 3G | Unpaired t | t(16) = 3.779, p = 0.0016 | NA |
| 4C | Two-way ANOVA | Genotype: F(1,45) = 62.8, p < 0.0001; trial: F(3,135) = 40.49, p < 0.0001; genotype + trial: F(3,135) = 6.924, p = 0.0002 | Trial 1: p = 0.0823; Trial 2: p = 0.0005; Trials 3 and 4: p < 0.0001 |
| 4F | Unpaired t | Total distance: t(39) = 0.1740, p = 0.8628; center distance: t(39) = 0.1259, p = 0.9005 | NA |
| 4G | Unpaired t | Contextual memory: t(41) = 1.017, p = 0.3153; cued memory: t(41) = 0.1066, p = 0.9156 |
NA |
| 4H | Two-way ANOVA | Genotype: F(1,44) = 3.007, p = 0.0899; subject: F(1,44) = 15.65, p = 0.0003; genotype + subject: F(1,44) = 1.321, p = 0.2566 | Inanimate: p = 0.2188; stranger: p > 0.9999 |
| 5D | Paired t Unpaired t |
8 week vs 12 week Veh: t(9) = 2.710, p = 0.0240; Tam: t(8) = 4.261, p = 0.0028 Veh vs Tam 8 week: t(17) = 4.725, p = 0.0002; 12 week t(17) = 3.093, p = 0.0066 |
NA |
| 5E | Paired t Unpaired t |
16 week vs 20 week Veh: t(9) = 3.693, p = 0.0050; Tam: t(16) = 6.114, p < 0.0001 Veh vs Tam 16 week: t(10.28) = 4.812, p = 0.0007; 20 week: t(19.82) = 3.361, p = 0.0031 |
NA |
| 10D | Two-way ANOVA | Genotype: F(2,15) = 1.595, p = 0.2354; trial: F(3,45) = 34.27, p < 0.0001; genotype + trial: F(6,45) = 0.874, p = 0.5215 | Trial 2: Mecp2WT/Y::VIPCre vs Mecp2CF/Y::VIPCre: p = 0.5659; Mecp2WT/Y::VIPCre vs Mecp2CF/Y::VIPCre::SOMFlp: p = 0.2844; Trial 3: Mecp2WT/Y::VIPCre vs Mecp2CF/Y::VIPCre::SOMFlp: p = 0.9009; Mecp2CF/Y::VIPCre vs Mecp2CF/Y::VIPCre::SOMFlp: p = 0.8658; Trial 4: Mecp2WT/Y::VIPCre vs Mecp2CF/Y::VIPCre::SOMFlp: p = 0.3164; Mecp2CF/Y::VIPCre vs Mecp2CF/Y::VIPCre::SOMFlp: p = 0.1728; any other pairs: p > 0.9999 |
| 10E | One-way ANOVA | Total distance: F(2,15) = 1.236, p = 0.3186; center distance: F(2,15) = 0.4681, p = 0.6351 |
Total distance: Mecp2WT/Y::VIPCre vs Mecp2CF/Y::VIPCre::SOMFlp: p = 0.4601; Mecp2CF/Y::VIPCre vs Mecp2CF/Y::VIPCre::SOMFlp: p = 0.8065; any other pairs: p > 0.9999 |
| 10F | One-way ANOVA | F(2,15) = 0.1182, p = 0.8893 | Between any pair of genotypes: p > 0.9999 |
Results
Design of SGIRT
SGIRT achieves conditional gene regulation by changing the orientation of a genomic region essential for gene expression or function via site-specific recombinations. Inverting a genomic region renders it inaccessible for normal expression. Exons containing translational start codon, encoding critical functional domain, or leading to frameshift are good candidates. To achieve orientation control, we adopted the FLEx (also known as DIO) design, which enables stable inversion of DNA sequence flanked by two pairs of interleaved, heterotypic, antiparallel recombinase recognition sites via Cre-mediated recombination (Schnutgen et al., 2003). We incorporated two sets of such switches: a Cre-dependent one composed of loxN and Lox2272 and a Flp-dependent one composed of Frt and F5 (fDIO). The fDIO switch was placed inside the DIO switch, which flanks the targeted genomic sequence. One round of Cre- or Flp-mediated recombination inverts the DIO-fDIO flanked genomic region to inactivate a gene of interest, while two rounds restore the original orientation to restore gene expression and function (Fig. 1A). To avoid disrupting endogenous gene expression pattern and dosage, the recombinase recognition sites should be inserted into introns.
A SGIRT model for conditional Mecp2 regulation
We generated an SGIRT model for conditional Mecp2 regulation by targeting the exon 3 of this gene through homologous recombination in mouse embryonic stem cells (Fig. 1A–C). This exon encodes a large part of the methyl-CpG binding domain, which is indispensable for Mecp2 function. Many disease-causing mutations were discovered in this exon in RTT patients. Its small size ensures high recombination efficiency. The same region has been targeted before in a conditional KO model (Chen et al., 2001). Therefore, targeting this exon promises efficient gene regulation for RTT modeling and allows us to cross-check our observations with published reports. The modified allele is named Mecp2CF (CF: Cre and Flp) (Fig. 1A).
In the absence of Cre or Flp, Mecp2CF functions as a WT allele, expressing full-length transcript and protein (Fig. 1D,E). Mice carrying Mecp2CF were viable, fertile, and phenotypically indistinguishable from WT mice with comparable body and brain weights (Fig. 1F,G). Importantly, the expressing pattern and dosage of Mecp2 were not disturbed by the modifications made in Mecp2CF, as shown by Western blots (Fig. 2A,B), qRT-PCR (Fig. 2C), and immunostaining (Fig. 2D,E).
Figure 2.

Mecp2CF and Mecp2R are functionally equivalent to Mecp2WT. Comparable Mecp2 expression pattern and dosage on protein (A,B,D,E) and mRNA levels (C) in Mecp2WT/Y, Mecp2CF/Y, and Mecp2R/Y mice. A, Representative Western blot images. B–D, Quantifications. n = 3. GAPDH and actin were used as internal control for Western blot and qRT-PCR, respectively. Relative values were calculated by normalizing value from individual sample to the average values of the three Mecp2CF/Y samples. Data are mean ± SEM. Dots, squares, and triangles represent data from individual mice. n.s., Not significant at p > 0.05. For more statistical details, see Table 2. E, Representative immunostaining images. Cor, Cortex; Cb, cerebellum; Hip, hippocampus; Str, striatum. Scale bar, 200 μm.
In order to test Mecp2 inactivation, we crossed Mecp2CF with germline-expressing E2a-cre. Cre-mediated recombination successfully inverted exon 3, rendering it inaccessible for splicing and translation. The resulting Mecp2IVC (IVC: inverted by Cre) allele is functionally null (Fig. 1). We confirmed the exclusion of exon 3 in the mRNA by RT-PCR (Fig. 1D) and the absence of full-length protein by Western blot (Figs. 1E, 3A) in Mecp2IVC/Y male mice. These mice recapitulated previously reported null phenotypes, including reduced life span (Fig. 3B), body weight (Figs. 1F, 3C) and brain size (Fig. 1F,G), and various behavioral deficits (Fig. 3D–G).
Figure 3.

Efficient inactivation of Mecp2 by Cre- or Flp-mediated recombination. A, Western blots proving absence of full-length MeCP2 in all examined tissues in Mecp2IVC/Y. B, C, Reduced lifespan (B) and body weight (C) of Mecp2IVC/Y mice (survival curve, n = 27; body weight, n = 14) compared with Mecp2WT/Y mice (survival curve, n = 12; body weight, n = 8). D–G, Behavior deficits. D, Representative photographs of hindlimb clasping phenotype manifested in Mecp2IVC/Y. Arrowhead indicates clasping hindlimbs. E–G, Reduced motor coordination in rotarod test (E) and locomotor activity in open field (F) and increased immobility time in tail suspension (G) in Mecp2IVC/Y (n = 13, 13, 10) compared with Mecp2WT/Y (n = 9, 8, 8) littermates. H, Representative photograph of a brain from P0 AAV-Flp-injected, adult-perfused Mecp2CF/Y::tdT-Frt mouse. Purple, in bright-field image, represents higher-level RFP expression in a large number of infected cells in the brain. Scale bar, 500 μm. I, J, Efficient Mecp2 inactivation in AAV-Flp-infected cells in olfactory bulb (Ob), cortex (Cor), hippocampus (Hip), striatum (Str), and cerebellum (Cb). Scale bar, 50 μm. I, Representative images. J, Quantification. n = 3. Data are mean ± SEM. Dots and squares represent data from individual mice. n.s., Not significant at p > 0.05, **p < 0.01, ***p < 0.001. For more statistical details, see Table 2.
In order to test Mecp2 restoration, we bred Mecp2IVC with germline-expressing Actin-Flp. Flp-mediated recombination successfully reverted exon 3 back to the native orientation and restored Mecp2 at genomic (Fig. 1C), mRNA (Figs. 1D, 2C), and protein levels (Figs. 1E, 2A,B,D,E). Mice carrying the resulting Mecp2R (R: restoration) allele were viable, fertile, and phenotypically indistinguishable from WT mice and mice carrying Mecp2CF. Importantly, the expressing pattern and dosage of Mecp2 were fully restored in Mecp2IVF, as shown by Western blots (Fig. 2A,B), qRT-PCR (Fig. 2C), and immunostaining (Fig. 2D,E).
We further tested Flp-mediated Mecp2 inactivation by injecting AAV-Flp into lateral ventricles of P0 Mecp2CF/Y::tdT-Frt male pups and examined the brains in adulthood (Fig. 3H,I). tdT-Frt is a Flp-dependent RFP reporter derived from Ai65 (Madisen et al., 2015) by germline removal of its LoxP-STOP-LoxP cassette using CMV-Cre (M. He et al., 2016). High level RFP expression in large number of infected cells made the tissue appear purple under room light (Fig. 3H). Efficient Mecp2 inactivation by Flp-mediated exon 3 inversion was demonstrated by the absence of MeCP2 immunostaining in RFP+ cells distributed from the most rostral end (olfactory bulb) to the most caudal end (cerebellum) of the mouse brain (Fig. 3I,J).
Together, these data demonstrated efficient and reversible Mecp2 inactivation by transgenic and viral Cre- and Flp-mediated recombinations.
Mecp2 inactivation and restoration in PV+ cells
Previous studies have proved a crucial role Mecp2 plays in GABAergic neurons (Chao et al., 2010; Ure et al., 2016; Ip et al., 2018). Deleting Mecp2 in GABAergic neurons recapitulates most of the phenotypes displayed by Mecp2-null mice (Chao et al., 2010), and restoring Mecp2 in GABAergic neurons is sufficient to rescue multiple RTT-like features (Ure et al., 2016). GABAergic interneurons consist of many subtypes with different marker expression, physiological properties, connectivity, and circuit functions (Kepecs and Fishell, 2014). PV, SOM, and vasoactive intestinal peptide (VIP) are the most commonly used markers delineating three major GABAergic neuron subtypes, which play distinct roles in the cortical circuits (Ascoli et al., 2008; Kepecs and Fishell, 2014). Among them, PV+ interneurons are the most extensively studied. Their dysfunction has been reported in constitutive and conditional Mecp2 KO models, and PV-specific Mecp2 deletion affects circuit function and mouse behavior (L. J. He et al., 2014; Ito-Ishida et al., 2015; Krishnan et al., 2015; Patrizi et al., 2020). However, no PV-specific rescue had been achieved before, and its outcome remained elusive. Therefore, we set to perform PV-specific Mecp2 inactivation followed by in situ restoration to test the cellular and behavior effects.
To inactivate Mecp2 in PV+ cells, we bred Mecp2CF with PVFlp (Madisen et al., 2015). Cortical PV expression starts around postnatal days 10-14. In Mecp2CF/Y::PVFlp, most cortical PV+ cells lost MeCP2 by P28, and close-to-complete inactivation was achieved by P34 (Fig. 4A,B). Efficient Mecp2 inactivation was also achieved in PV+ cells in hippocampus, striatum, and cerebellum (Fig. 4A,B). Consistent with a previous report (Ito-Ishida et al., 2015), these mice developed motor deficits, including worse rotarod performance (Fig. 4C) and HC (Fig. 4D,E), but no seizure. HC is a late-onset phenotype first observed at postnatal week 14 in a few mice and fully manifested in all mice by week 25 (Fig. 4E). However, we did not observe significant deficits in open filed test (Fig. 4F), contextual and auditory fear conditioning test (Fig. 4G), and three-chamber test (Fig. 4H). Moreover HC phenotype (Fig. 4I) and efficient Mecp2 inactivation (Fig. 4J,K) were also observed in Mecp2CF/CF::PVFlp females, indicating highly efficient recombination on both alleles.
Figure 4.

PV-specific Mecp2 inactivation mediated by PVFlp. A, B, Gradual loss of MeCP2 immunostaining in PV+ cells in cortex (Cor), striatum (Str), hippocampus (Hip), and cerebellum (Cb) in Mecp2CF/Y::PVFlp. A, Representative images. Scale bar, 50 μm. B, Quantification. n = 3. C–H, Behavioral performance of Mecp2WT/Y::PVFlp and Mecp2CF/Y::PVFlp littermates in rotarod test (C), HC (D,E), open field test (F), fear conditioning (G), and 3-chamber social test (H). Mecp2WT/Y::PVFlp, n = 26, 26, 24, 23, and 25; Mecp2CF/Y::PVFlp, n = 21, 21, 17, 20, and 21. D, I, Representative photographs HC phenotype in male (D) and female (I) when Mecp2 is inactivated in PV+ cells. White arrowhead indicates clasping hindlimb. J, K, Loss of MeCP2 expression in PV+ cells in the Cor, Str, Hip, and Cb of Mecp2CF/CF::PVFlp females. J, Representative images. Open arrowheads indicate PV+ cells without MeCP2. Filled arrowheads indicate PV+ cells with MeCP2. K, Quantification. n = 3. Data are mean ± SEM. Dots and squares represent data from individual mice. n.s., Not significant at p > 0.05, ***p < 0.001. For more statistical details, see Table 2.
We then designed two paradigms to restore Mecp2 in PV+ cells in temporal and spatially controllable manners. Paradigm 1 used a highly efficient PVCreER (M. He et al., 2016) (Fig. 5), in which the onset and efficiency of rescue were controlled by the time and dosage of Tam induction. Paradigm 2 used Flp-dependent Cre virus (AAV-fDIO-Cre-GFP) (Fig. 6), in which the onset and brain region of rescue were controlled by the time and location of virus injection.
Figure 5.
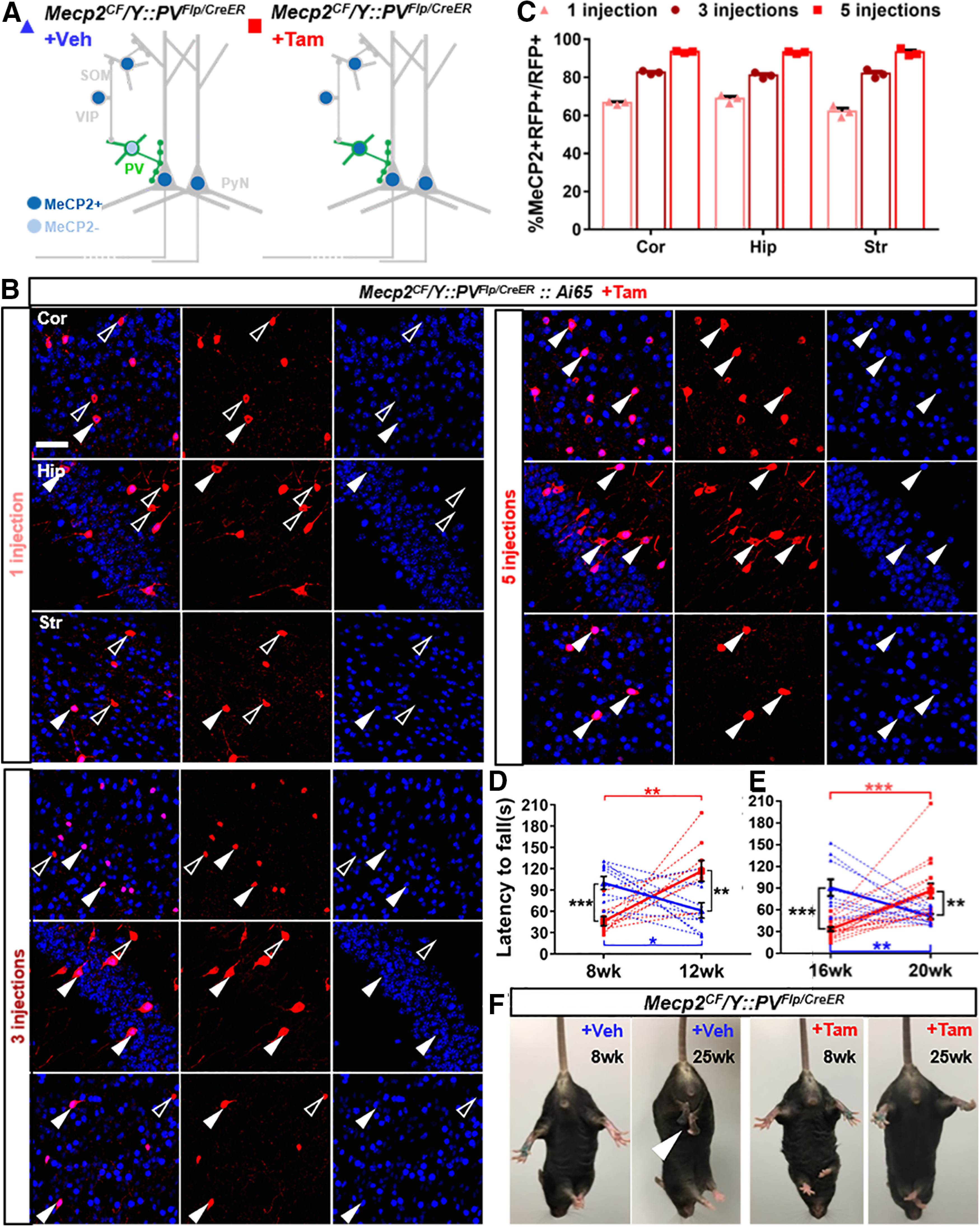
PV-specific Mecp2 restoration mediated by PVCreER. A, Schematic view of experiment design. In the corn oil administration (+Veh) group, Mecp2 remained inactivated in PV+ cells in Mecp2CF/Y::PVFlp/CreER; whereas in the Tam induction group (+Tam), Mecp2 expression was restored by active CreER. B, C, Better restoration of MeCP2 immunostaining in PV+ cells in Cor, Hip, and Str in Mecp2CF/Y::PVFlp/CreER::Ai65 with increasing number of Tam injections. B, Representative images. Open arrowheads indicate PV+ cells without MeCP2. Filled arrowheads indicate PV+ cells with MeCP2. Scale bar, 50 μm. C, Quantifications. D, E, Rescue of rotarod performance in the +Tam group and deterioration of performance in the +Veh group. Tam was administrated earlier in D at 8 weeks (n = 10 for +Veh; n = 9 for +Tam) and later in E at 16 weeks (n = 10 for +Veh; n = 17 for +Tam). F, Prevention of HC by Mecp2 restoration. White arrowhead indicates clasping hindlimb. Data are mean ± SEM. Triangles and squares represent data from individual mice. n.s., Not significant at p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001. For more statistical details, see Table 2.
Figure 6.
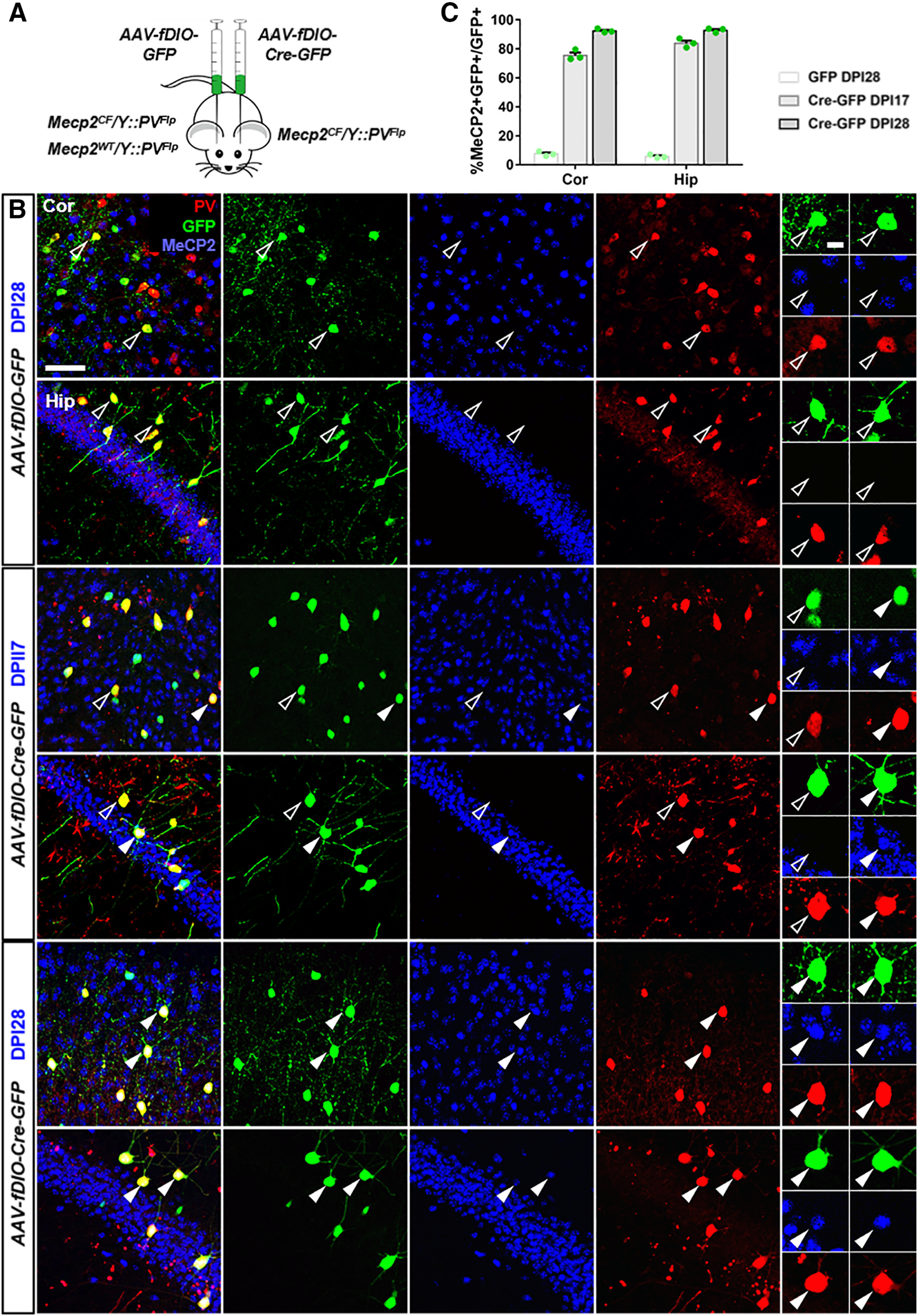
Virus-mediated Mecp2 restoration in PV+ neurons. A, Experiment scheme. AAV-fDIO-GFP was injected into primary visual cortex or hippocampus of Mecp2CF/Y::PVFlp on one hemisphere, and AAV-fDIO-GFP-Cre on the other hemisphere. As a control, AAV-fDIO-GFP was injected Mecp2WT/Y::PVFlp to label WT PV+ neurons. B, C, Immunostaining confirmation and quantification of gradual Mecp2 restoration in AAV-fDIO-Cre-GFP-infected PV+ neurons. B, Top, AAV-fDIO-GFP-infected PV+ neurons in Mecp2CF/Y::PVFlp. Middle, Bottom, AAV-fDIO-Cre-GFP-infected PV+ neurons at 17 or 28 d post injection (DPI) in Mecp2CF/Y::PVFlp. Open arrowheads indicate GFP+ cells without MeCP2. Filled arrowheads indicate GFP+ cells with MeCP2. Panels on the right, High magnification views of representative cells. Scale bars: low magnification, 50 μm; high magnification, 10 μm. Data are mean ± SEM. Dots represent data from individual mice.
In Paradigm 1, Mecp2CF/Y::PVFlp/CreER were divided into two groups. Corn oil as vehicle was administered to the +Veh group, in which CreER was inactive and Mecp2 remained silent in the PV+ cells (Fig. 5A, left). Therefore, these mice were phenotypically equivalent to Mecp2CF/Y::PVFlp. Tam dissolved in corn oil was administered to the +Tam group, in which CreER became active to revert and restore Mecp2 (Fig. 5A, right). The +Veh group serves as a control for the +Tam group to examine cellular and behavioral rescue.
We first tested the dose-responses to Tam inductions. Five Tam injections (5× Tam) performed once every other day fully restored MeCP2 immunostaining in PV+ cells in cortex, hippocampus, and striatum 4 weeks after the last injection, while three (3×) or one injection were less efficient (Fig. 5B,C). Therefore, we decided to use 5× Tam for behavior tests.
We performed rotarod test and assigned Mecp2CF/Y::PVFlp/CreER mice into two groups according to their performance: mice with below-average performance were assigned to the +Tam group and mice with above-average performance were assigned to the +Veh group. Tam or corn oil was administered right after the test to the two groups, respectively. To check whether a critical window of intervention exists, we performed two cohorts of experiments independently: one cohort with 5× Tam or Veh treatments starting at 8 weeks (early treatment, Fig. 5D) and the other starting at 16 weeks (late treatment; Fig. 5E). For each cohort, two batches of mice were treated and tested, and the results were combined. In both cohorts, the performance of mice in the +Veh group deteriorated while mice in the +Tam group improved, on average and individually, 4 weeks after treatment (Fig. 5D,E). Consequently, there was a flip of rotarod performance between the two groups: the +Veh group performed worse than the +Tam group posttreatment. The absence of behavioral improvement in the +Veh group ensured that the behavioral rescue in the +Tam group is not because of the possible positive effect of repetitive rotarod training. The significant improvement of worse performers at both ages in the +Tam group demonstrated the potent behavioral rescue by genetic restoration of Mecp2.
We further tested whether induction dosage influences the behavioral rescue using the same experimental design but with 3× Tam. Although improvement of rotarod performance was also observed 4 weeks later in the +Tam group, they did not differ significantly from the +Veh group (data not shown), indicating that the extent of behavioral rescue correlates and likely depends on the extent of cellular rescue.
In addition to rotarod performance, we also examined the HC phenotype. With 5× Tam administered at 8 or 16 weeks, none of the mice developed HC by 25 weeks. In contrast, all mice in the +Veh groups developed HC by this age (Fig. 5F). As none of the mice we tested showed HC before Tam treatment, these results demonstrated a preventive effect of Mecp2 restoration before symptom onset. In order to examine whether this late-manifesting phenotype can also be rescued by Mecp2 reexpression, we administered Tam to 5 mice, which already manifested HC at 20 weeks. Successful rescue was observed 5 weeks later (i.e., none of the treated mice exhibited HC anymore by 25 weeks). This result demonstrates that late-onset HC behavioral phenotype can also be rescued.
In Paradigm 2 (Fig. 6A), AAV-fDIO-Cre-GFP was injected into the cortex or hippocampus of Mecp2CF/Y::PVFlp to rescue Mecp2 in PV+ cells. Efficient vrial-Cre mediated rescue was achieved in both cortex and hippocampus 17 d after injection as shown by immunostaining (Fig. 6B,C). By 28 d, close-to-completed rescue was observed (Fig. 6B,C).
In order to examine whether genetic rescue leads to phenotypic rescue on the cellular level, we performed whole-cell patch-clamp recordings in acute slices. The contralateral cortex was injected with AAV-fDIO -GFP to label Mecp2-deficient PV+ cells. As a control, AAV-fDIO-GFP was injected into Mecp2WT/Y::PVFlp to label the WT PV+ cells. PVFlp activated GFP expression facilitated patch-clamp recording of infected PV+ cells in acute slices (Fig. 7A). Previous studies have reported functional impairment of PV+ cells caused by germline or PV-specific Mecp2 inactivation in primary visual cortex (L. J. He et al., 2014; Krishnan et al., 2015; Patrizi et al., 2020), but how the cellular deficits would be rescued by genetic restoration remained elusive. Therefore, we directed our injections to this region to compare our observations with previous findings. The injections were performed at P28 when >80% cortical PV+ cells had lost MeCP2 staining (Fig. 4A,B). Recordings performed at ∼P90 revealed that Mecp2 inactivation in PV+ cells significantly changed several of their intrinsic features (Fig. 7B–K; Table 1). Most of the changes we observed were consistent with previous report (L. J. He et al., 2014), such as increased input resistance, decreased rheobase, and increased afterhyperpolarization potential. The only discrepancy was that we observed an increase in AP half-width, which was not reported in the previous report (L. J. He et al., 2014). This is most likely caused by the difference in recording ages. Differences in genetic backgrounds, genotypes, and recording conditions may also contribute.
Figure 7.

Functional rescue by Mecp2 restoration in PV+ neurons in the primary visual cortex. A, Representative recording site imaged under infrared and fluorescent modes. Dashed lines in fluorescent image indicate recording pipette. Scale bar, 500 μm. B, Representative recording traces show APs evoked at rheobases; 10 pA depolarizing current steps were injected, and only the responses at rheobase and one step before it were shown. C, Representative recording traces showing APs at maximum firing rate. D–H, Rescued input resistance (Rin), membrane time constant (tau), AP half-width, rheobase, and maximum firing rate. Maximum firing was recorded as the firing rate at plateau. I–L, Increased resting membrane potential (RMP) and AP amplitude were partially rescued by Mecp2 restoration. Increased afterhyperpolarization potential (AHP) was not rescued. AP threshold was unaffected by Mecp2 inactivation. Data are mean ± SEM. Dots and squares represent data from individual recorded cells. n.s., Not significant at p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001. For more statistical details, see Table 1.
We found that most of the observed changes were fully rescued by Mecp2 restoration: input resistance, membrane time constant, rheobase, AP half-width, and maximum firing rate (Fig. 7B–H; Table 1). Resting membrane potential and AP amplitude were partially rescued (Fig. 7I,J; Table 1). After hyperpolarization potential was the only feature that was not rescued (Fig. 7K; Table 1). AP threshold did not show significant differences between Mecp2-deficient PV+ cells versus WT PV+ cells. Although the rescued PV+ cells had slightly lower value than the deficient cells, they were not significantly different from the WT PV+ cells (Fig. 7L; Table 1). These results demonstrate that cellular deficits caused by Mecp2 deficiency in PV+ cells can be rescued by Mecp2 restoration, although not all the features are equally amenable.
Concurrent Mecp2 inactivation in SOM+ cells and VIP+ cells
Cortical SOM+ cells shared developmental origin with PV+ cells in the medial ganglionic eminence, whereas VIP+ cells are generated from caudal ganglionic eminence (Miyoshi et al., 2010). They each play distinct roles in the cortical circuit (Miyoshi et al., 2007; Xu et al., 2008; Kepecs and Fishell, 2014). Differential Mecp2 expression was observed in these GABAergic subtypes (Fig. 8). Subtype-specific Mecp2 KO also led to distinctive neurologic phenotypes (L. J. He et al., 2014; Ito-Ishida et al., 2015; Mossner et al., 2020).
Figure 8.

Differential expression of Mecp2 across interneuron subtypes and brain regions. A, Representative views in cortex (Cor) and hippocampus (Hip). PV+ and SOM+ cells were identified by immunostaining of the marker genes. VIP+ cells were identified by Cre-activated-RFP expression, due to the incompatibility of MeCP2 antibody and VIP antibody (both were rabbit-polyclonal antibodies) for coimmunostaining. Arrows indicate RFP-expressing VIP+ cells. Arrowheads indicate PV+ cells in the upper row; SOM+ cells in the lower row. Scale bar, 25 μm. B, Quantification of MeCP2 fluorescence intensity. n = 3. Relative fluorescence intensity was normalized to the average fluorescence intensity of cortical MeCP2 immunostaining. For both brain regions, ∼200 cells were quantified for each cell type per brain, and three brains were quantified in total. Data are mean ± SEM. Dots, squares, and triangles represent data from individual mice.
We first demonstrated efficient Mecp2 inactivation in SOM+ cells through Flp-mediated recombination by breeding Mecp2CF with the SOMFlp we previously generated (M. He et al., 2016) (Fig. 9A) and observed handling-induced seizure consistent with previous reports (Ito-Ishida et al., 2015; Mossner et al., 2020). Such a model may be combined with AAV-FDIO-Cre to examine the rescue effect of this cell type in future studies. We then used VIPcre (Taniguchi et al., 2011) with Mecp2CF to achieve Mecp2 inactivation in VIP+ cells (Fig. 9B). As VIP antibody and MeCP2 antibody share the same host species (rabbit), Cre-dependent RFP reporter Ai14 was used to visualize VIP+ cells to examine the inactivation efficiency. The majority of RFP-expressing cortical VIP+ cells lost MeCP2 staining, although the inactivation rate was not as high as those in PV+ (Fig. 4) or SOM+ cells (Fig. 9A). Such a model will facilitate future functional interrogations of Mecp2 in VIP+ cells. Different from a recent report (Mossner et al., 2020), we did not observe seizure incidence in these mice. This discrepancy could be caused by differences in the design and genetic background of conditional Mecp2 alleles and recombinase-expressing drivers. The floxed Mecp2 allele used by Mossner et al. (2020) was a hypomorphic allele (Guy et al., 2001), which reduced expression of both Mecp2 mRNA and MeCP2 protein by ∼50% and resulted in a broad spectrum of phenotypic abnormalities (Samaco et al., 2008). Therefore, the phenotypes observed by Mossner et al. (2020) may not solely be caused by loss of Mecp2 in VIP+ neurons, but an additive effect of the VIP-specific Mecp2 deletion and the global reduction in Mecp2 expression. In contrast, our model preserves normal Mecp2 expression pattern and dosage in Cre– cells and may better reflect the effect of VIP-specific Mecp2 inactivation. Indeed, Mossner et al. (2020) reported seizure incidence in Mecp2flx/Y mice but not in the WT mice, whereas our Mecp2CF/Y mice were indistinguishable from WT mice and showed no seizure. Alternatively, the incomplete Mecp2 inactivation in VIP+ populations may have alleviated phenotypic symptoms in our model.
Figure 9.

Efficient Mecp2 inactivation in SOM+ and VIP+ cells. Immunostaining confirmation and quantification of efficient Mecp2 inactivation by SOMFlp (A) and VIPCre (B). Top left, Schematic view. Bottom left, Quantifications (n = 3). Data are mean ± SEM. Circles and squares represent data from individual mice. Right, Representative views in cortex (Cor) and hippocampus (Hip). Open arrowheads indicate SOM+ cells without MeCP2. Open arrows indicate RFP-expressing VIP+ cells without MeCP2. Filled arrows indicate RFP-expressing VIP+ cells with MeCP2. Scale bar, 50 μm.
We further bred Mecp2CF, SOMFlp, and VIPCre together to inactivate Mecp2 in both SOM+ and VIP+ cells in the same mouse (Fig. 10). Combine Flp with Cre drivers instead of using two different Cre drivers supported independent expression of two different reporters in the two cell types. We injected AAV-fDIO-GFP to label SOM+ cells in green and AAV-DIO-RFP to label VIP+ cells in red (Fig. 10A). Immunostaining showed efficient Mecp2 inactivation in both cell types (Fig. 10B,C). Similar as SOM-specific Mecp2 inactivation, handling-induced seizures were also observed in the double inactivation mice. Different from PV-specific Mecp2 inactivation, no HC or compromised rotarod performance (Fig. 10D) was observed in the double inactivation mice. No significant differences were observed in the open field test (Fig. 10E) and elevated plus maze test (Fig. 10F) when comparing the double inactivation mice with the VIP-specific Mecp2 inactivation mice or with the VIPcre mice, which had normal Mecp2 expression. These results further confirmed the lack of motor deficits and suggested no anxiety-like behavior in the double inactivation mice, and supported previous findings that different neuron types contribute different aspects of the RTT-like phenotypes. Such kind of combinatorial model will facilitate future investigation to elucidate whether concurrent inactivation in two cell types may augment or offset phenotypes caused by mutations in one cell type. In addition, independent or concurrent Mecp2 rescue in two cell types can be achieved by injecting AAV-fDIO-Cre and AAV-DIO-Flp separately or together. Other kinds of tool genes, including calcium sensors, activity modulators, and viral receptors, etc., can also be expressed in these two cell types in a Cre- or Flp-dependent manner. Such combinatorial strategies can be applied to other nonoverlapping Cre and Flp drivers to target broader cell types or brain regions, or to partially overlapping Cre and Flp drivers to achieve more restrictive rescue.
Figure 10.
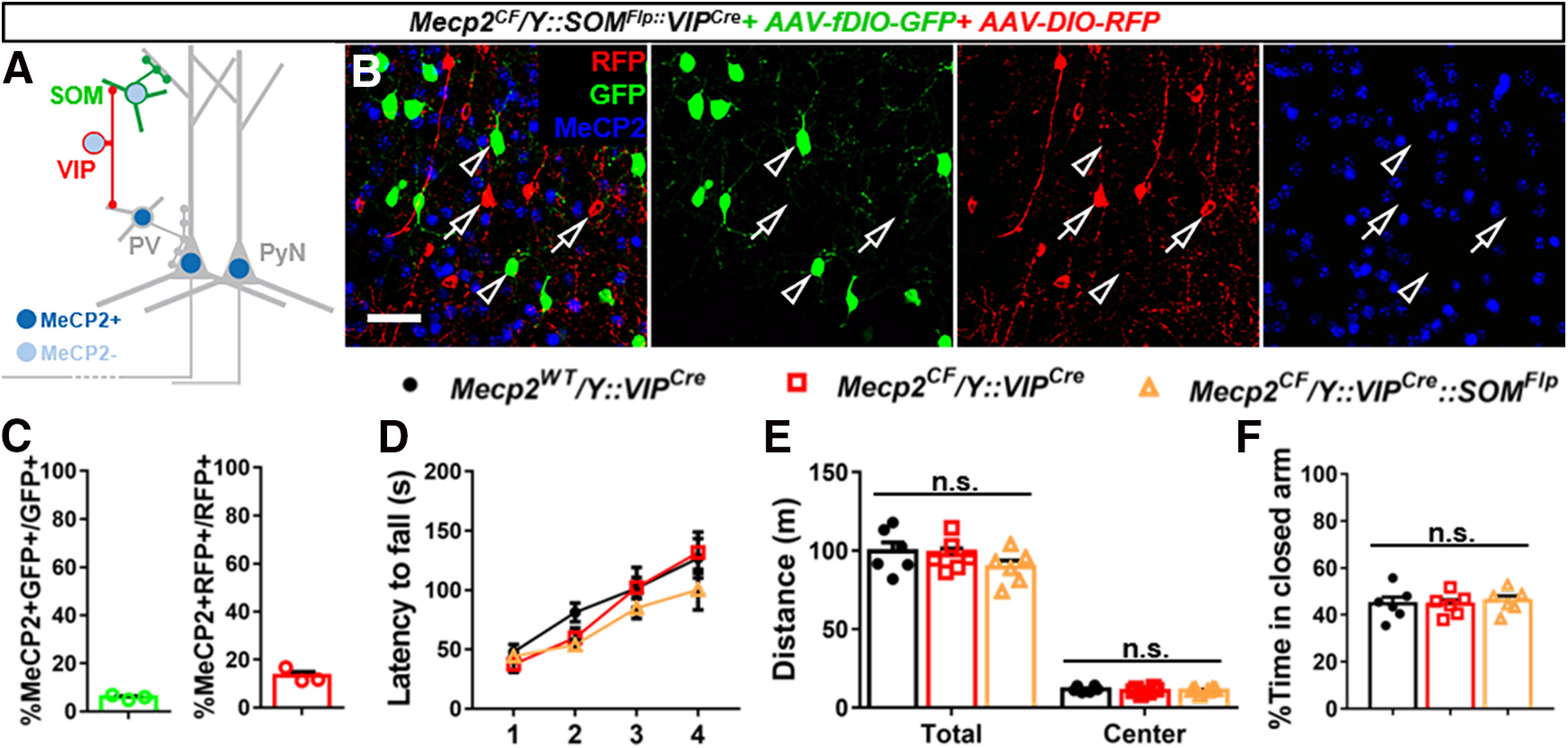
Concurrent Mecp2 inactivation in cortical SOM+ and VIP+ cells. A, Experiment scheme. Concurrent Mecp2 inactivation in SOM+ and VIP+ cells with differential fluorescent labeling achieved by combining Cre and Flp drivers with DIO and fDIO viral reporters. SOM+ cells were labeled by Flp-activated GFP, and VIP+ cells were labeled by Cre-activated RFP. B, Representative view in the infected cortical region. Open arrowheads indicate GFP-labeled SOM+ cells without MeCP2. Open arrows indicate RFP-labeled VIP+ cells without MeCP2. Scale bar, 50 μm. C, Quantifications of Mecp2 expression in the virus-infected SOM+ and VIP+ neurons. n = 3. D–F, Behavioral analysis. No significant differences were observed in rotarod performance (D), open field (E), or elevated plus maze (F) among the three genotypes. n = 6. Data are mean ± SEM. Circles, dots, triangles, and squares represent data from individual mice. n.s., Not significant at p < 0.05. For more statistical details, see Table 2.
Mecp2 inactivation and restoration in corticofugal projection neurons
The cerebral cortex is organized into six layers that contain excitatory projection neurons of different identities (Douglas and Martin, 2004). Corticofugal projection neurons mainly reside in deep layers, and can be further divided into corticothalamic projection neurons and subcerebral projection neurons (Molyneaux et al., 2007). Although previously published conditional KO and rescue studies revealed a predominant role of GABAergic system in RTT, Mecp2 deficiency also causes severe cellular phenotypes in cortical projection neurons (Chen et al., 2001; Guy et al., 2001; Tropea et al., 2009; Meng et al., 2016). Certain aspects of RTT symptoms in patients were recapitulated in mice with Mecp2 ablation only in excitatory neurons (Meng et al., 2016). Changes in morphology and electrophysiological properties of deep layer projections neurons have also been reported (Tropea et al., 2009; Meng et al., 2016), but no subtype-specific model of Mecp2 KO or rescue has been established for corticofugal projection neurons.
Fezf2 plays important roles in cell fate determination and cell identify maintenance of corticofugal projection neurons (Eckler and Chen, 2014). Its postnatal cortical expression is also restricted to these deep layer cells, allowing us to use Fezf2CreER to genetically inactivate Mecp2 in these neurons by postnatal inductions (Fig. 11). Tam-induced CreER activity was reported by the expression of RFP from the IS reporter (M. He et al., 2016). Immunostaining confirmed the absence of MeCP2 in layer 5 RFP+ cells (Fig. 11B,C). We injected CAV2-DIO-Flp (Schwarz et al., 2015) into thalamus to retrogradely infect corticothalamic projection neurons in the of Mecp2CF/Y:: Fezf2CreER::IS mice. Cre-activated-Flp expression excised RFP and activated GFP expression from the IS reporter only in the Fezf2+ neurons projecting to thalamus (Fig. 11A). In these cells, Mecp2 expression was also efficiently restored by Flp-mediated recombination, as shown by immunostaining (Fig. 11B,C). In contrast, MeCP2 remained absent in nearby RFP-expressing neurons uninfected by the retrograde virus (Fig. 11B,C). These results demonstrated successful Mecp2 inactivation in a marker gene-defined projection neuron class and successful Mecp2 restoration in a projection-defined subtype within that class. Such a model may facilitate future functional investigations, and similar combinatorial approaches can be applied to manipulate other neuronal cell types defined by molecular signatures and projection patterns.
Figure 11.

Mecp2 inactivation in Fezf2-expressing corticofugal projection neurons and target-specific restoration. A, Experiment scheme. CAV-DIO-Flp was injected into thalamus and retrogradely infected a subset of thalamic-projecting cortical neurons. In Fezf2-expressing corticofugal neurons, Cre-activated Flp expression turned off RFP and turned on GFP in IS reporter. B, Representative view in the infected cortical region. Open arrowheads indicate RFP-labeled Fezf2-expressing corticofugal neurons without MeCP2. Closed arrowheads indicate GFP-labeled thalamic-projecting neurons with MeCP2. Scale bars: low magnification, 300 μm; high magnification, 25 μm. C, Quantifications. n = 3. Data are mean ± SEM. Dots and squares represent data from individual mice.
Discussion
The exact role a gene plays often depends on the cellular context, which varies among cell types, changes dynamically across development, and adapts to environmental stimuli. Therefore, cell-type-specific gene regulation with temporal and regional precision is critical for dissection gene function and for interrogating the genetic architecture of complex neuropsychiatric diseases with genetic underpinning. Cre/loxP-based conditional KO technologies have been widely used to selectively inactivate a gene in specific cell types. Critical insights have been gained from such studies. However, as the KO is generally not reversible, it is difficult to segregate the effects of different developmental stages. Phenotypes observed with such manipulations are accumulative effects starting from the beginning of Cre activity to the time of analysis (Cheval et al., 2012). Conversely, the loss of gene has a controllable end but a fixed start from whenever the gene normally starts expression in conditional restoration models (Guy et al., 2007; Mei et al., 2016). They serve as powerful models to evaluate the therapeutic potential of genetic rescue (Gadalla et al., 2011; Mei et al., 2016). Combining both conditional inactivation and restoration will allow cell-type-specific gene expression regulation within defined time window with selective start and end, but strategies and tools for such purpose are very limited.
A few studies combined a null or conditional inactivation allele with transgenic expression allele, or with conditional expression or restoration allele, to achieve reversible control of gene expression (Luikenhuis et al., 2004; Zeng et al., 2008; Cobb et al., 2010; Chaiyachati et al., 2012; Kerr et al., 2012), but the generation and breeding of two transgenic strains were costly and time-consuming. Virus-mediated gene expression can be used to reduce breeding effort (Rastegar et al., 2009; Deverman et al., 2018) but often needs craniotomy and stereotaxic injections, suffers from incomplete infection, and may not match the endogenous pattern and dosage very well. Gene reexpression from nonendogenous loci has a similar concern in recapitulating endogenous expression. Therefore, neither is ideal for dosage-sensitive genes. One study combined a conditional inactivation allele with a conditional restoration allele, both made at the endogenous locus (Chaiyachati et al., 2012), but such a design cannot work for genes on sex chromosomes (which usually have only one active copy), nor for haploinsufficient genes. Two more recent studies used an excisable conditional gene trap cassette inserted into first intron (Robles-Oteiza et al., 2015) or selected exon (Andersson-Rolf et al., 2017) to achieve reversible gene trapping, but intronic trapping has a potential risk of insufficient trapping (Robles-Oteiza et al., 2015), whereas exonic trapping has a potential risk of affecting endogenous gene expression in its inactive form (Andersson-Rolf et al., 2017). More importantly, no single-allele-based conditional and reversible model for disease-causing gene had been established to achieve cell-type-specific conditional inactivation and in situ restoration within defined time window and anatomic region. In this study, we have developed a SGIRT strategy that fulfilled the above-mentioned requirements and successfully manipulated the Rett-syndrome-causing gene Mecp2 with combinatorial approaches
The SGLIRT strategy supports versatile applications. A SGIRT allele can function as a traditional conditional KO. It can also be converted into a constitutive null allele and Cre- or Flp-dependent conditional reexpression alleles. Compared with the previously reported gene-trap based methods (Robles-Oteiza et al., 2015; Andersson-Rolf et al., 2017) that inactivate gene with Cre and restore gene with Flp (Cre-off/Flp-on), our method supports two additional working schemes: inactivation with Flp and reactivation with Cre (Flp-off/Cre-on) and concurrent inactivation (Cre-off/Flp-off). Gene-trap alleles end transcription and translation and lead to truncations of all downstream regions after the gene-trap cassette. SGIRT allele retains normal transcription. When the targeted region does not contain start codon or leads to frameshift when inverted and skipped by splicing, it supports selective deletion of protein domains in the middle.
We applied this strategy to Mecp2 and demonstrated efficient and versatile regulation of this sex-chromosome-linked, dosage-sensitive, and disease-causing gene. We undertook comprehensive analysis to make sure the genetic modifications we made do not affect gene expression pattern or dosage before inversion and after rescue. Combining with a suit of transgenic mouse drivers and viral tools, we have achieved multifaceted and flexible control of Mecp2 in germline and specific cell types in temporally and spatially controllable manners. The successful application of SGIRT to Mecp2 promises future application to manipulate other genes. To design new SGIRT alleles, the following principles should be followed: (1) the targeted region should be of appropriate size to allow efficient recombination and (2) indispensable for gene expression or function (e.g., encoding important functional domain, leading to premature STOP codon or frameshift when inverted and skipping during splicing); and (3) the recombinase recognition sites and ES selection cassette should be placed in noncoding regions to avoid disrupting endogenous expression.
Like any other recombinase-based strategy, SGIRT relies on, and thus is limited by, the inherent caveats of site-specific recombination system. For example, we noticed that the inactivation efficiency varied among different neuronal types and brain regions. Mecp2 inactivation was less efficient in VIP+ cells than in PV+ (Fig. 4) and SOM+ cells (Figs. 9, 10), and less efficient in hippocampal VIP+ cells than in cortical VIP+ cells (Fig. 9B). Many factors could contribute to such differences, including the differences in expression levels of Mecp2 and DNA recombinases. Previous studies have reported that the expression level of Mecp2 varies among cell types (Chao et al., 2010; Ito-Ishida et al., 2015) and brain regions (L. J. He et al., 2014; Ito-Ishida et al., 2015). We quantified MeCP2 immunostaining in cortical and hippocampal PV+, SOM+, and VIP+ cells and confirmed such differences (Fig. 8). Higher expression level may delay the clearance rate of mRNA and protein within cells, but it should not affect the steady-state KO efficiency given sufficient time of recombinase expression. However, the difference in expression level may also reflect difference in chromatin status and genomic accessibility by recombinases. When an inducible form of recombinase is used, the recombination efficiency also depends on the amount of active CreER entering the nucleus after Tam induction. Higher dosage or multiple inductions may be necessary to achieve high recombination efficiency. Indeed, we observed enhanced rescue efficacy with increasing number of injection when using PVCreER to rescue Mecp2 (Fig. 5B,C). Nonetheless, when targeting the same genomic region, Cre-mediated recombination in SGIRT alleles should be as efficient as traditional conditional KO or reexpression alleles targeting the same regions but gains extra flexibility.
There are two potentially valuable extensions of SGIRT for future considerations. One is to incorporate more site specific recombinase-based system, such as Dre-Rox (Anastassiadis et al., 2009; L. He et al., 2017), Vika-Vox (Karimova et al., 2013), and PhiC31-AttP/AttB (Groth and Calos, 2004) for extra layers of gene expression control. Although the availability of cell-type-specific drivers and the effort to breed extra alleles still restrict its application, the ever-expanding cell-type-targeting toolkit of mouse drivers and virus will gradually alleviate the restriction. The other is to insert a modified exon carrying disease-causing mutations in the opposite orientation in the DIO-fDIO switches. Upon inversion, the modified exon will “replace” the WT exon, converting a WT allele to a disease-causing allele, while a second round of recombination will restore it back and resume expression of WT gene. Such an extra “surrogate” exon can also be modified in other ways to fuse epitope or fluorescent tag for protein visualization or immunoprecipitation. In addition, incorporating the recent development of CRISPR/Cas9-assisted gene targeting (Hsu et al., 2014) may help to speed up mouse generation, but care needs to be taken to screen out off-target edits and unwanted edits at the targeted locus, such as deletion or mutation.
In this study, we presented a new conditional and reversible gene regulation method with high efficiency, specificity, and versatility. Our study not only demonstrates the efficiency, versatility of SGIRT, but also provides valuable tools and critical insights for Mecp2 study and RTT research. SGIRT offers one of the most compelling and necessary approaches to decipher how genetic mutations may impact cellular and organismic function at different developmental stages and to facilitate systematic analysis of the complex genetic architecture of neurologic and psychiatric disorders. The data we collected provided new insights into cell-type-specific, developmental stage-dependent role of Mecp2, and the models we generated establish valuable experimental access for RTT studies. This SGIRT strategy is readily applicable to manipulate other genes for studying gene function and modeling diseases.
Footnotes
This work was supported by National Key R&D Program of China 2018YFA0108000, National Natural Science Foundation of China 31970971, 31771196, and 31471037, Shanghai Science and Technology Commission Innovation Fund 17JC1401500, Shanghai Commission of Health Youth Program 2017YQ016, Shanghai Rising-Star Program 18QA1400600, Shanghai Municipal Science and Technology Major Project 2018SHZDZX01 and ZJLab, and International Rett Syndrome Foundation. We thank Profs. Z. Josh Huang, Bo Li, Jiangteng Lu, and Yang Yang for insightful discussions throughout the course of this study and for advice during manuscript preparation; Prof. Bo Li for sharing AAV-fDIO-Cre-GFP vector; Prof. E. J. Kremer for providing CAV virus; Prof. Z. Josh Huang for sharing Fezf2CreER; Drs. Sangyang Kim and Priscilla Wu for technical support in transgenic mouse engineering; Drs. Min Jiang, Ying Shi, Lingyun Liu, and Qian Huang (core facility of IOBS, Fudan University) for technical support on imaging and behavioral experiments; and Dr. Nashat Abumaria for providing experiment apparatus for fear conditioning.
The authors declare no competing financial interests.
References
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23:185–188. 10.1038/13810 [DOI] [PubMed] [Google Scholar]
- Anastassiadis K, Fu J, Patsch C, Hu S, Weidlich S, Duerschke K, Buchholz F, Edenhofer F, Stewart AF (2009) Dre recombinase, like Cre, is a highly efficient site-specific recombinase in E. coli, mammalian cells and mice. Dis Model Mech 2:508–515. 10.1242/dmm.003087 [DOI] [PubMed] [Google Scholar]
- Andersson-Rolf A, Mustata RC, Merenda A, Kim J, Perera S, Grego T, Andrews K, Tremble K, Silva JC, Fink J, Skarnes WC, Koo BK (2017) One-step generation of conditional and reversible gene knockouts. Nat Methods 14:287–289. 10.1038/nmeth.4156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascoli GA, Alonso-Nanclares L, Anderson SA, Barrionuevo G, Benavides-Piccione R, Burkhalter A, Buzsáki G, Cauli B, Defelipe J, Fairén A, Feldmeyer D, Fishell G, Fregnac Y, Freund TF, Gardner D, Gardner EP, Goldberg JH, Helmstaedter M, Hestrin S, Karube F, et al. (2008) Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat Rev Neurosci 9:557–568. 10.1038/nrn2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaiyachati BH, Kaundal RK, Zhao J, Wu J, Flavell R, Chi T (2012) LoxP-FRT Trap (LOFT): a simple and flexible system for conventional and reversible gene targeting. BMC Biol 10:96. 10.1186/1741-7007-10-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, Ekker M, Rubenstein JL, Noebels JL, Rosenmund C, Zoghbi HY (2010) Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468:263–269. 10.1038/nature09582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R (2001) Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet 27:327–331. 10.1038/85906 [DOI] [PubMed] [Google Scholar]
- Cheval H, Guy J, Merusi C, De Sousa D, Selfridge J, Bird A (2012) Postnatal inactivation reveals enhanced requirement for MeCP2 at distinct age windows. Hum Mol Genet 21:3806–3814. 10.1093/hmg/dds208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb S, Guy J, Bird A (2010) Reversibility of functional deficits in experimental models of Rett syndrome. Biochem Soc Trans 38:498–506. 10.1042/BST0380498 [DOI] [PubMed] [Google Scholar]
- Deverman BE, Ravina BM, Bankiewicz KS, Paul SM, Sah DW (2018) Gene therapy for neurological disorders: progress and prospects. Nat Rev Drug Discov 17:641–659. 10.1038/nrd.2018.110 [DOI] [PubMed] [Google Scholar]
- Douglas RJ, Martin KA (2004) Neuronal circuits of the neocortex. Annu Rev Neurosci 27:419–451. 10.1146/annurev.neuro.27.070203.144152 [DOI] [PubMed] [Google Scholar]
- Eckler MJ, Chen B (2014) Fez family transcription factors: controlling neurogenesis and cell fate in the developing mammalian nervous system. Bioessays 36:788–797. 10.1002/bies.201400039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G (2013) Paxinos and Franklin's The mouse brain in stereotaxic coordinates, Ed 4 Amsterdam: Academic Press. [Google Scholar]
- Gadalla KK, Bailey ME, Cobb SR (2011) MeCP2 and Rett syndrome: reversibility and potential avenues for therapy. Biochem J 439:1–14. 10.1042/BJ20110648 [DOI] [PubMed] [Google Scholar]
- Giacometti E, Luikenhuis S, Beard C, Jaenisch R (2007) Partial rescue of MeCP2 deficiency by postnatal activation of MeCP2. Proc Natl Acad Sci USA 104:1931–1936. 10.1073/pnas.0610593104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth AC, Calos MP (2004) Phage integrases: biology and applications. J Mol Biol 335:667–678. 10.1016/j.jmb.2003.09.082 [DOI] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A (2001) A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet 27:322–326. 10.1038/85899 [DOI] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A (2007) Reversal of neurological defects in a mouse model of Rett syndrome. Science 315:1143–1147. 10.1126/science.1138389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Li Y, Li Y, Pu W, Huang X, Tian X, Wang Y, Zhang H, Liu Q, Zhang L, Zhao H, Tang J, Ji H, Cai D, Han Z, Han Z, Nie Y, Hu S, Wang QD, Sun R, et al. (2017) Enhancing the precision of genetic lineage tracing using dual recombinases. Nat Med 23:1488–1498. 10.1038/nm.4437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He LJ, Liu N, Cheng TL, Chen XJ, Li YD, Shu YS, Qiu ZL, Zhang XH (2014) Conditional deletion of Mecp2 in parvalbumin-expressing GABAergic cells results in the absence of critical period plasticity. Nat Commun 5:5036. 10.1038/ncomms6036 [DOI] [PubMed] [Google Scholar]
- He M, Tucciarone J, Lee S, Nigro MJ, Kim Y, Levine JM, Kelly SM, Krugikov I, Wu P, Chen Y, Gong L, Hou Y, Osten P, Rudy B, Huang ZJ (2016) Strategies and tools for combinatorial targeting of GABAergic neurons in mouse cerebral cortex. Neuron 91:1228–1243. 10.1016/j.neuron.2016.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, Zhang F (2014) Development and applications of CRISPR-Cas9 for genome engineering. Cell 157:1262–1278. 10.1016/j.cell.2014.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip JP, Mellios N, Sur M (2018) Rett syndrome: insights into genetic, molecular and circuit mechanisms. Nat Rev Neurosci 19:368–382. 10.1038/s41583-018-0006-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito-Ishida A, Ure K, Chen H, Swann JW, Zoghbi HY (2015) Loss of MeCP2 in parvalbumin- and somatostatin-expressing neurons in mice leads to distinct Rett syndrome-like phenotypes. Neuron 88:651–658. 10.1016/j.neuron.2015.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jugloff DG, Vandamme K, Logan R, Visanji NP, Brotchie JM, Eubanks JH (2008) Targeted delivery of an Mecp2 transgene to forebrain neurons improves the behavior of female Mecp2-deficient mice. Hum Mol Genet 17:1386–1396. 10.1093/hmg/ddn026 [DOI] [PubMed] [Google Scholar]
- Karimova M, Abi-Ghanem J, Berger N, Surendranath V, Pisabarro MT, Buchholz F (2013) Vika/vox, a novel efficient and specific Cre/loxP-like site-specific recombination system. Nucleic Acids Res 41:e37. 10.1093/nar/gks1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepecs A, Fishell G (2014) Interneuron cell types are fit to function. Nature 505:318–326. 10.1038/nature12983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr B, Soto CJ, Saez M, Abrams A, Walz K, Young JI (2012) Transgenic complementation of MeCP2 deficiency: phenotypic rescue of Mecp2-null mice by isoform-specific transgenes. Eur J Hum Genet 20:69–76. 10.1038/ejhg.2011.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan K, Wang BS, Lu J, Wang L, Maffei A, Cang J, Huang ZJ (2015) MeCP2 regulates the timing of critical period plasticity that shapes functional connectivity in primary visual cortex. Proc Natl Acad Sci USA 112:E4782–E4791. 10.1073/pnas.1506499112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard H, Cobb S, Downs J (2017) Clinical and biological progress over 50 years in Rett syndrome. Nat Rev Neurol 13:37–51. 10.1038/nrneurol.2016.186 [DOI] [PubMed] [Google Scholar]
- Luikenhuis S, Giacometti E, Beard CF, Jaenisch R (2004) Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc Natl Acad Sci USA 101:6033–6038. 10.1073/pnas.0401626101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Garner AR, Shimaoka D, Chuong AS, Klapoetke NC, Li L, van der Bourg A, Niino Y, Egolf L, Monetti C, Gu H, Mills M, Cheng A, Tasic B, Nguyen TN, Sunkin SM, Benucci A, Nagy A, Miyawaki A, Helmchen F, et al. (2015) Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron 85:942–958. 10.1016/j.neuron.2015.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei Y, Monteiro P, Zhou Y, Kim JA, Gao X, Fu Z, Feng G (2016) Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 530:481–484. 10.1038/nature16971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Wang W, Lu H, He LJ, Chen W, Chao ES, Fiorotto ML, Tang B, Herrera JA, Seymour ML, Neul JL, Pereira FA, Tang J, Xue M, Zoghbi HY (2016) Manipulations of MeCP2 in glutamatergic neurons highlight their contributions to Rett and other neurological disorders. Elife 5:e14199 10.7554/eLife.14199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi G, Butt SJ, Takebayashi H, Fishell G (2007) Physiologically distinct temporal cohorts of cortical interneurons arise from telencephalic Olig2-expressing precursors. J Neurosci 27:7786–7798. 10.1523/JNEUROSCI.1807-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi G, Hjerling-Leffler J, Karayannis T, Sousa VH, Butt SJ, Battiste J, Johnson JE, Machold RP, Fishell G (2010) Genetic fate mapping reveals that the caudal ganglionic eminence produces a large and diverse population of superficial cortical interneurons. J Neurosci 30:1582–1594. 10.1523/JNEUROSCI.4515-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD (2007) Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci 8:427–437. 10.1038/nrn2151 [DOI] [PubMed] [Google Scholar]
- Mossner JM, Batista-Brito R, Pant R, Cardin JA (2020) Developmental loss of MeCP2 from VIP interneurons impairs cortical function and behavior. Elife 9:e55639 10.7554/eLife.55639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrizi A, Awad PN, Chattopadhyaya B, Li C, Di Cristo G, Fagiolini M (2020) Accelerated hyper-maturation of parvalbumin circuits in the absence of MeCP2. Cereb Cortex 30:256–268. 10.1093/cercor/bhz085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z. (2018) Deciphering MECP2-associated disorders: disrupted circuits and the hope for repair. Curr Opin Neurobiol 48:30–36. 10.1016/j.conb.2017.09.004 [DOI] [PubMed] [Google Scholar]
- Rastegar M, Hotta A, Pasceri P, Makarem M, Cheung AY, Elliott S, Park KJ, Adachi M, Jones FS, Clarke ID, Dirks P, Ellis J (2009) MECP2 isoform-specific vectors with regulated expression for Rett syndrome gene therapy. PLoS One 4:e6810. 10.1371/journal.pone.0006810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond FL. (2006) X linked mental retardation: a clinical guide. J Med Genet 43:193–200. 10.1136/jmg.2005.033043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice AM, McLysaght A (2017) Dosage-sensitive genes in evolution and disease. BMC Biol 15:78. 10.1186/s12915-017-0418-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles-Oteiza C, Taylor S, Yates T, Cicchini M, Lauderback B, Cashman CR, Burds AA, Winslow MM, Jacks T, Feldser DM (2015) Recombinase-based conditional and reversible gene regulation via XTR alleles. Nat Commun 6:8783. 10.1038/ncomms9783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaco RC, Fryer JD, Ren J, Fyffe S, Chao HT, Sun Y, Greer JJ, Zoghbi HY, Neul JL (2008) A partial loss of function allele of methyl-CpG-binding protein 2 predicts a human neurodevelopmental syndrome. Hum Mol Genet 17:1718–1727. 10.1093/hmg/ddn062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnutgen F, Doerflinger N, Calleja C, Wendling O, Chambon P, Ghyselinck NB (2003) A directional strategy for monitoring Cre-mediated recombination at the cellular level in the mouse. Nat Biotechnol 21:562–565. 10.1038/nbt811 [DOI] [PubMed] [Google Scholar]
- Schnutgen F, De-Zolt S, Van Sloun P, Hollatz M, Floss T, Hansen J, Altschmied J, Seisenberger C, Ghyselinck NB, Ruiz P, Chambon P, Wurst W, von Melchner H (2005) Genomewide production of multipurpose alleles for the functional analysis of the mouse genome. Proc Natl Acad Sci USA 102:7221–7226. 10.1073/pnas.0502273102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz LA, Miyamichi K, Gao XJ, Beier KT, Weissbourd B, DeLoach KE, Ren J, Ibanes S, Malenka RC, Kremer EJ, Luo L (2015) Viral-genetic tracing of the input-output organization of a central noradrenaline circuit. Nature 524:88–92. 10.1038/nature14600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Fu Y, Lu J, Lin Y, Miyoshi G, Shima Y, Fishell G, Nelson SB, Huang ZJ (2011) A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron 71:995–1013. 10.1016/j.neuron.2011.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropea D, Giacometti E, Wilson NR, Beard C, McCurry C, Fu DD, Flannery R, Jaenisch R, Sur M (2009) Partial reversal of Rett syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci USA 106:2029–2034. 10.1073/pnas.0812394106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ure K, Lu H, Wang W, Ito-Ishida A, Wu Z, He LJ, Sztainberg Y, Chen W, Tang J, Zoghbi HY (2016) Restoration of Mecp2 expression in GABAergic neurons is sufficient to rescue multiple disease features in a mouse model of Rett syndrome. Elife 5:e14198 10.7554/eLife.14198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Esch H. (2012) MECP2 duplication syndrome. Mol Syndromol 2:128–136. 10.1159/000329580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Tam M, Anderson SA (2008) Fate mapping Nkx2.1-lineage cells in the mouse telencephalon. J Comp Neurol 506:16–29. 10.1002/cne.21529 [DOI] [PubMed] [Google Scholar]
- Zeng H, Horie K, Madisen L, Pavlova MN, Gragerova G, Rohde AD, Schimpf BA, Liang Y, Ojala E, Kramer F, Roth P, Slobodskaya O, Dolka I, Southon EA, Tessarollo L, Bornfeldt KE, Gragerov A, Pavlakis GN, Gaitanaris GA (2008) An inducible and reversible mouse genetic rescue system. PLoS Genet 4:e1000069. 10.1371/journal.pgen.1000069 [DOI] [PMC free article] [PubMed] [Google Scholar]