

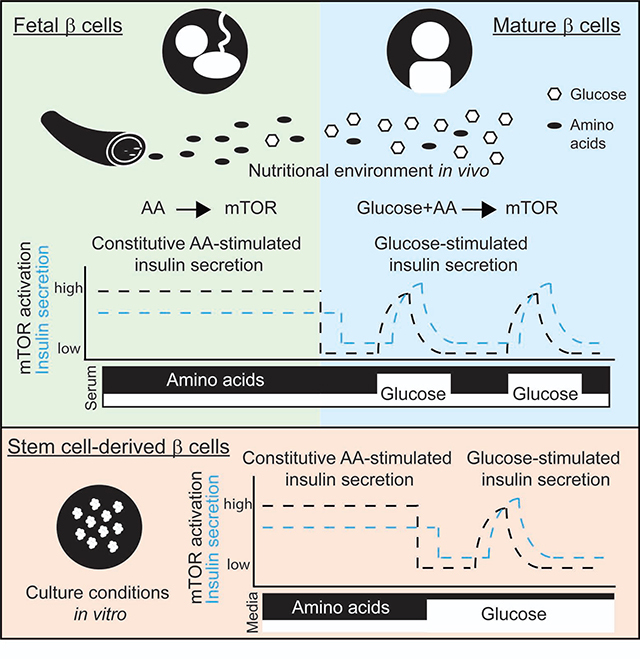
Summary
A drastic transition at birth, from constant maternal nutrient supply in utero to intermittent postnatal feeding, requires changes in the metabolic system of the neonate. Despite their central role in metabolic homeostasis, little is known about how pancreatic β cells adjust to the new nutritional challenge. Here, we find that after birth β cell function shifts from amino acid- to glucose-stimulated insulin secretion in correlation with the change in the nutritional environment. This adaptation is mediated by a transition in nutrient sensitivity of the mTORC1 pathway, which leads to intermittent mTORC1 activity. Disrupting nutrient sensitivity of mTORC1 in mature β cells reverts insulin secretion to a functionally immature state. Finally, manipulating nutrient sensitivity of mTORC1 in stem cell-derived β cells in vitro strongly enhances their glucose-responsive insulin secretion. These results reveal a mechanism by which nutrients regulate β cell function, thereby enabling a metabolic adaptation for the newborn.
Keywords: Pancreatic β cells, stem cell-derived β cells, maturation, insulin secretion, mTORC1, nutrient sensing
eTOC Blurb
At birth, metabolic adaptations prepare the newborn for its new nutritional environment. Helman and colleagues demonstrate a role of nutrient sensing by mTORC1 in adjusting insulin secretion in neonatal β cells. Exploiting this adaptative property of β cells results in improved glucose-responsive insulin secretion in human stem cell-derived β cells.
Graphical Abstract
Introduction
At birth, mammals are faced with a challenge as they break from the supportive maternal environment into the outside world, a transition requiring an independent physiology. The respiratory, circulatory, digestive and endocrine systems all undergo adaptations during this early neonatal period which is termed functional maturation (Morton and Brodsky, 2016, Ward Platt and Deshpande, 2005). Because nutrient consumption changes from relatively constant in utero to pulsatile after birth, the metabolic adaptation of the neonate to intermittent feeding and glucose fluctuations is critical. Pancreatic β cell maturation involves developing a higher threshold for glucose-stimulated insulin secretion (GSIS), as evidenced by inhibition of insulin secretion at low glucose and enhanced insulin secretion in response to high glucose. This physiological adaptation is essential to maintain nearly constant glucose levels, preventing hypoglycemia (Blum et al., 2012, Rozzo et al., 2009).
The initial functional maturation period in which responsiveness to glucose is acquired occurs within the first week after birth. β cells continue to develop, however, and improve in secretory function during, and even months, after weaning (Bliss and Sharp, 1992, Avrahami et al., 2015, Stolovich-Rain et al., 2015, Helman et al., 2016, Arda et al., 2016, Bader et al., 2016). MAFA and Urocortin3 (UCN3) serve as established markers of β cell maturation since their expression increases over the maturation period in mouse and human β cells (Aguayo-Mazzucato et al., 2011, Blum et al., 2012, van der Meulen and Huising, 2014). Other genes, related to glucose sensing and insulin secretion, including NEUROD1, NKX6–1 and Glucokinase (GCK) improve glucose sensitivity in immature β cells (Matsuoka et al., 2007, Zhang et al., 2005, Wang et al., 2007, Artner et al., 2010, Aguayo-Mazzucato et al., 2011). The expression of specific genes termed disallowed genes (Martinez-Sanchez et al., 2015, Lemaire et al., 2016) become repressed in the postnatal β cells as part of the metabolic switch associated with functional maturation (Thorrez et al., 2011, Dhawan et al., 2015, Yoshihara et al., 2016). However, changes in expression of maturation markers and disallowed genes occur during the second and third weeks of life and it is not known what drives the initial acquisition of glucose responsiveness during the first week after birth (Conrad et al., 2014, Liu and Hebrok, 2017).
Understanding functional maturation is important for the application of stem cell-derived tissues in regenerative medicine (Robinton and Daley, 2012). In recent years, a more advanced understanding of the intrinsic transcriptional programs in vivo led to a rapid progress in the generation of many cell types, including β cells, from human pluripotent stem cells (Hrvatin et al., 2014, Sneddon et al., 2018). Nevertheless, due to a lack of knowledge regarding initiation of functional maturation, differentiated tissues derived from stem cells often resemble fetal tissues and lack full physiological function (Henquin and Nenquin, 2018, Hrvatin et al., 2014, Jiang et al., 2018, Lundy et al., 2013, Arlotta and Pasca, 2019). Developing permissive conditions for the maturation of stem cell-derived tissues in vitro, without genetic modifications, will enhance their use in drug discovery and cell transplantation.
Functional maturation has generally been viewed as an intrinsic part of a transcriptional differentiation program rather than a process that can be regulated by extrinsic factors in the environment. Yet it is known that cells sense extrinsic signals and coordinate cellular adaptations accordingly. For example, cells modulate their metabolism and growth in response to nutrients and growth factors via regulation of the mechanistic target of rapamycin complex 1 (mTORC1). mTORC1 kinase activity controls cellular growth and metabolism by activating anabolic processes and repressing catabolic ones (Sancak et al., 2010, Sancak et al., 2008, Saxton and Sabatini, 2017). Several cytosolic proteins have been identified that signal the availability of nutrients and growth factors to control mTORC1 activity (Saxton and Sabatini, 2017).
Several recent findings have uncovered the importance of mTORC1 as a significant regulator of β cell maturation and function. β cell-specific activation of mTORC1 in genetic mouse models found that hyperactivation of mTORC1 signaling resulted in increased β cell function and improved glucose tolerance (Blandino-Rosano et al., 2017, Hamada et al., 2009, Mori et al., 2009, Rachdi et al., 2008, Shigeyama et al., 2008). Consistently, β cell-specific ablation of mTORC1 resulted in lower insulin secretion caused by mitochondrial dysfunction and oxidative stress, indicating that mTORC1 activity is essential for β cell function and maturation (Sinagoga et al., 2017, Chau et al., 2017, Ardestani et al., 2018, Maedler and Ardestani, 2017, Ni et al., 2017, Blandino-Rosano et al., 2017, Alejandro et al., 2017). In contrast, other findings show that chronically increased mTORC1 activity can induce β cell failure (Mori et al., 2009, Sun et al., 2010, Swisa et al., 2015, Elghazi et al., 2010, Bartolome and Guillen, 2014, Koyanagi et al., 2011, Jaafar et al., 2019). Although the genetic components of the mTORC1 pathway are clearly important for proper β cell mass and function, these studies raise questions about how the pathway is post-transcriptionally regulated and whether mTORC1 activation positively or negatively contributes to β cell maturation.
The present study investigates the possibility that sensing environmental nutrients by mTORC1 contributes to cell and tissue maturation. We describe a perinatal change in the regulation of mTORC1 signaling by nutrients that modifies the dynamics of mTORC1 activity from constitutive to intermittent after birth. The combination of mTORC1 inhibition and activation after birth enables the functional shift from amino acid-responsive to glucose-responsive insulin secretion. These findings demonstrate a role for nutrient sensing by mTORC1 in the initiation of functional maturation of pancreatic β cells.
Results
Changes in the nutritional environment alter insulin secretion in response to nutrients
Insulin-expressing β cells appear about embryonic day 13.5 (E13.5) in mice and week 8 – 9 post-conception in humans (Koyanagi et al., 2011, Slack, 1995), but glucose-stimulated insulin secretion (GSIS) begins only days after birth (Blum et al., 2012, Rozzo et al., 2009). Since a major change from embryonic to postnatal physiology is nutrient consumption, we explored whether an altered nutrient supply plays a role in the transition from immature to mature insulin secretion by β cells (Figure 1A). We compared the metabolite profiles of fetal and neonatal plasma, collected from mice throughout the first week after birth, and found changes imposed by the onset of intermittent feeding. These changes include an increase in fasting-associated metabolites, e.g. ketone bodies, in the neonates at birth and a different composition of amino acids and glucose in the plasma of embryonic, neonatal and adult mice. Generally, levels of amino acids and amino acid-derived metabolites in the plasma of immature mice were higher and glucose was much lower compared to plasma of the adult mice (Figures 1B and 1C and Table S1); see also (Hay, 2006, Young and Prenton, 1969, Zeng et al., 2017). This shift in plasma nutrients abundances occurs gradually after birth, as shown in Figure 1C.
Figure 1.

Dynamic insulin secretion in response to nutrients in mature and immature islet cells. (A) Schematic representation of experimental design. (B) Representative heat map of metabolite abundance in serum of embryos, neonates and adult mice. Experiment was repeated twice using embryonic samples (n=4), neonates (n=32) and adults (n=6). (C) A relative ratio of amino acids and their metabolites (left graph) and glucose (right graph) in the serum of mice of indicated age. Data points represent mean ± SEM. P-values, * P<0.05, ** P<0.01, *** P<0.001, **** P<0.0001, unpaired Student’s t test. (D-G) Fold insulin levels secreted by mature mouse (n=6) and human islets (D and F, respectively) and fetal mouse (E18, n=6) and human islets (E and G, respectively) in a dynamic perfusion assay in low glucose (2.8 mM), low glucose and physiological levels of amino acids, high glucose (16.7 mM) and KCl (30mM). The fetal and adult human data is representative from single donors using 6 technical replicates and repeated in three independent experiments. Data points represent mean ± SEM. (H) Fold insulin levels secreted by islets from neonatal mice (P5, n=6) in a dynamic perfusion assay. Secreted insulin levels were normalized to basal insulin secretion of each sample. (I) Fold insulin levels secreted by fetal human islets that were cultured in vitro in a fetal-like medium (blue, 5.5mM glucose and high amino acid levels) and mature-like medium (green, 5.5mM glucose and low amino acid levels). The result was repeated in three independent experiments.
Since the embryo is exposed to an amino acid-rich environment in utero, we tested the effects of physiological levels of amino acids (Cantor et al., 2017) on insulin secretion by fetal islets compared to mature islets. Surprisingly, amino acids alone do not induce a response in mature islets (Figure 1D and 1F), but stimulate insulin secretion in embryonic islets from mouse and human (Figure 1E and 1G), independently of glucose (Kervran and Randon, 1980). High glucose, in contrast, stimulates strong insulin secretion only in mature islets and has no effect on mouse and human fetal islets (Figure 1D-G). Together, these results identify a shift from amino acid-responsive insulin secretion in immature β cells to glucose-responsive secretion in mature β cells. This shift occurs gradually, as neonatal islets of postnatal day 6 mice secrete insulin in response to amino acids or glucose alone, demonstrating a transitional state from amino acid- to glucose-responsiveness (Figure 1H). To test whether the nutritional environment contributes to this change in nutrient-responsiveness, we tested the prolonged effects of nutrient levels on insulin secretion dynamics in immature β cells. Human fetal β cells grown in a mature-like environment (low amino acid concentrations), for four days, can be shifted in vitro to glucose-responsive insulin-secreting cells, whereas these same β cells grown in an immature-like environment (high amino acid concentrations) continue to secrete insulin in response to amino acids but not to glucose (Figure 1I).
To understand if the nutrient environment continues to play a role in the function of mature β cells, we tested the ability of mature human β cells to remain glucose-responsive following extended incubation in vitro. Human islets from adult donors, incubated for four days in low amino acid concentration, demonstrated a higher response to glucose than islets grown in high levels of amino acids (Supplementary Figure 1). Therefore, in addition to having a role in the onset of glucose-responsiveness in immature β cells, amino acid levels in the environment also contribute to maintaining function in mature β cells.
Onset of glucose-responsiveness is independent of expression of canonical β cell markers
To identify signaling pathways that are correlated with the transition in β cell response to nutrients, we conducted a single cell RNA sequencing on fetal human islets grown for four days in a mature-like environment (low amino acid concentrations) and in an immature-like environment (high amino acid concentrations). Transcription of genes related to β cell identity and maturation such as Chromogranin (CHGA), Insulin and MAFA, did not change significantly during the four-day incubation in the two conditions (Figure 2A and 2B).
Figure 2.

Differential mTORC1 activity in immature and mature β cells. (A) UMAP visualization of the single fetal human β cells analyzed. Colors represent time and condition of collected cells that were used for differential expression analysis. (B) UMAP maps overlaid with the relative expression level of the indicated gene in each cell. (C) Representative immunostainings of MAFA (green) and insulin (labeling β cells, red) in pancreatic tissues from fetal (E18) neonatal (P6) and adult (P60) mice with indicated antibodies. Note that MAFA levels do not change significantly 6 days after birth (D) Representative immunostainings of pancreatic tissues from fetal (D120) and adult human subjects with indicated antibodies. (E) Representative immunostainings of p-S6 (green) and insulin (labeling β cells, red), in pancreatic islet of fasted pregnant female mouse (right) and its E18 embryo (left). Note the strong mTORC1 activity in fetal compared to maternal β cells. Experiment was repeated four times, using pancreases of embryos from one pregnant female each time. (F) Representative immunostainings of p-S6 (green) and insulin (labeling β cells, blue), in adult (left) and fetal (right) human pancreatic islets. For each group n=4. (G) A relative gene expression ratio of indicated mTORC1 regulators in sorted GFP-expressing β cells from E18 embryos (n=3, black bars), P6 neonates (n=3), and adult (n=5, grey bars) mice. Data points represent mean ± SEM. P-values, * P<0.05, ** P<0.01, *** P<0.001, unpaired Student’s t test.
FACS staining verified that the protein expression of PDX1, NKX6–1, UCN3 and MAFA in β cells were not different between the two nutritional conditions, indicating that they are not responsible for the gain of glucose responsiveness. (Supplementary Figure 1).
Consistent with this in vitro study, we find that staining for MAFA, UCN3 and NKX6–1 in mice embryos (E18), neonates (P6) and adults (P60), shows the expression of these genes does not change significantly in the first days after birth at the time of gaining glucose responsiveness (Figure 2C and Supplementary Figure 2). MAFA, UCN3 and NKX6–1 are also expressed in human embryonic β cells (by gestational day 120) (Figure 2D and Supplementary Figure 2).
These results identify a novel extracellular determinant of β cell maturation, by which changes in the nutritional environment before and after birth play a role, independently of changes in expression of canonical β cell identity markers, in a transition from constitutive, amino acid-stimulated insulin secretion in utero to a dynamic, glucose-responsive, insulin secretion after birth.
Activity of the mTORC1 pathway in immature β cells differs from mature β cells
We hypothesized that the activity of a metabolic pathway such as the mTOR pathway could mediate the functional transition from amino acid- to glucose-mediated insulin secretion in fetal and mature β cells, respectively. To measure mTORC1 signaling activity in immature and mature β cells, pancreatic tissues were immunostained for phosphorylation of ribosomal protein S6 (p-S6), a downstream target of the mTORC1 kinase. As indicated by p-S6 immunoreactivity, the mTORC1 pathway is strongly activated in β cells of embryonic (E18) mice in utero, whereas the β cells in their fasted pregnant mothers concurrently had low mTORC1 activity (Figure 2E). Active mTORC1 signaling in fetal β cells is also evident in human fetal pancreatic tissue compared to adult human β cells (Figure 2F).
Staining pancreatic sections showed that p-S6 is detected under basal conditions (after separation from their mothers) in β cells of neonatal pups, between P1 and P9, as reported previously (Ni et al., 2017), but not robust as in fetal β cells from E18 embryos (Supplemental Figure 2). Since the mTORC1 pathway is strongly activated in fetal β cells, postnatal mTORC1 activation by itself does not explain how β cells acquire glucose-induced insulin secretion. Furthermore, detection of p-S6 that coincides with the transition to glucose-stimulated insulin secretion suggests that mTORC1 inhibition is not directly associated with glucose responsiveness in β cells (Jaafar et al., 2019, Ni et al., 2017, Sinagoga et al., 2017). Thus, the regulation of mTORC1 activity before and after birth, and the control of β cell maturation by mTORC1, are more dynamic and complex than suggested previously.
In support of increased regulation of mTORC1 activity, gene expression analysis on sorted β cells from fetal, neonatal and adult murine β cells revealed that the expression of genes encoding negative regulators of mTORC1, including Tuberous sclerosis (TSC), Sestrin1 (SESN1) and Sestrin2 (SESN2), and the catalytic subunit of AMP-activated protein kinase (AMPK), is increased during the transition to mature β cells (Figure 2G), which corresponds to potentiated inhibitory regulation of mTORC1 activity during β cell functional maturation. These results suggest that the regulation on mTORC1 activity could play a role in the transition from amino acid- to glucose-stimulated insulin secretion transition of β cells, in adaptation to the changes in the nutritional environment after birth.
mTORC1 activity in mature β cells is dynamic and regulated by nutrients
The mTORC1 pathway is well-established to be highly responsive to the availability of nutrients, including amino acids and glucose (Saxton and Sabatini, 2017). As changes in nutrient conditions affect β cell function, we reasoned that nutrient-sensitive cellular responses may control the transition from amino acid- to glucose-dependent insulin secretion after birth. To determine how mTORC1 signaling in β cells is affected by nutrient conditions, we investigated the nutrient sensitivity and dynamics of mTORC1 in isolated islets. We developed a FACS-based assay to quantify mTORC1 activation specifically in β cells within the islet (Figure 3A and Supplementary Figure 3). We found differential effects by nutrients on mTORC1 activation in mature human β cells, as measured by the intensity of p-S6 staining. Amino acids alone are not sufficient to activate mTORC1, inducing only slightly more S6 phosphorylation in the β cell population than the basal mTORC1 activity in mature β cells. Glucose alone induces activation of mTORC1 in a higher percentage of β cells, whereas addition of both glucose and amino acids generates a robust activation (~80% of β cells) (Figure 3B). These results demonstrate a greater dependence on glucose for mTORC1 activation in mature human β cells and a requirement of both amino acids and glucose for full mTORC1 activity. We also tested the responsiveness of mTORC1 to nutrients in mature mouse islets and found that glucose and amino acids are both required to fully induce mTORC1 activity, comparable to that seen in human β cells (Figure 3B and 3C). However, glucose alone is not sufficient to activate the pathway in murine β cells (Figure 3B and 3C), suggesting a stronger regulation by amino acid-dependent repression and thus greater dependence on amino acids for mTORC1 activation.
Figure 3.

mTORC1 activity in mature β cells is dynamic and regulated by nutrients. (A) Schematic representation of experimental method to quantify mTORC1 activity in β cells. (B) Representative p-S6 staining histograms of insulin+ cells from adult human (left) and mouse (right) islets, detected by FACS analysis compared to secondary antibodies only control. (C) Percentages of p-S6 positive β cells in mouse islets, after 30 minutes incubation in the indicated conditions, detected by FACS compared to secondary antibodies only control. n=10, 7, 6, 10, 4, 4, 3, 3, 3, 3 biological replicates; left to right. Data points represent mean ± SEM. P values, * P<0.05, ** P<0.01, *** P<0.001, **** P<0.0001, unpaired Student’s t test. S961- insulin receptor antagonist, torin1- mTOR inhibitor. (D) Representative immunostainings of p-S6 (green) and insulin (labeling β cells, blue), in pancreatic islets of fasted mice injected with the indicated nutrients or re-fed. (E) Mean fluorescent intensity (MFI) of p-S6 staining in islets of mice injected with indicated nutrients or re-fed, n=4 for each condition. Data points represent mean ± SEM. P-values, * P<0.05, ** P<0.01, unpaired Student’s t test.
The activity of AMPK in mature human and mouse β cells, as measured by p-AMPK antibody, is also dynamic and shows the opposite activity and response to nutrients compared to mTORC1, in agreement with its role in repressing mTORC1 activity; fully activated in the absence of glucose and inhibited in the presence of glucose (Supplementary Figure 3).
mTORC1 activity in β cells is increased by amplifiers of insulin secretion, including Exendin-4 (Ex-4), a Glucagon-like peptide 1 (Glp1) analogue, and Forskolin (Fsk), a PKA activator, that trigger an increase in cAMP and PKA activation (Tengholm and Gylfe, 2017). This further demonstrates a positive correlation between insulin secretion and mTORC1 activation (Figure 3C and Supplementary Figure 3). Pharmacologically suppressing insulin secretion with diazoxide (Dz), an activator of ATP-sensitive potassium channels that mediates a decrease in calcium influx, dramatically reduces mTORC1 activity in β cells, even in the presence of glucose and amino acids together, and this is rescued by the addition of Fsk or Ex-4 (Figure 3C and Supplementary Figure 3). One possible explanation for this correlation between mTORC1 activity and insulin secretion is activation of mTORC1 by autocrine insulin signaling. However, blocking insulin receptor signaling did not impair mTORC1 activation in the presence of glucose (Figure 3C and Supplementary Figure 3), indicating that the response of mTORC1 to nutrients is not mediated by the autocrine action of secreted insulin. Further, in the absence of glucose, neither stimulation with insulin nor with insulin and amino acids together is able to activate mTORC1 in β cells. These results suggest intracellular crosstalk between insulin secretory function and mTORC1 activity, whereby calcium flux downstream of the ATP-sensitive potassium channels, that regulates insulin granules trafficking and exocytosis, is required for mTORC1 activation in mature β cells.
Among amino acids, leucine is a potent mTORC1 activator and is a known amplifier of insulin secretion. We find that leucine strongly potentiates the activation of the mTORC1 pathway in the presence of glucose. In fact, while different concentrations of glucose (from 8–20mM) dose-dependently enhance mTORC1 activity in β cells, including a physiological concentration of leucine (0.3mM) results in full mTORC1 activation even at intermediate glucose concentrations (8–11mM) (Supplementary Figure 3). Leucine levels therefore play a role in establishing the range of glucose concentrations capable of regulating mTORC1 activity in β cells.
These results raise the possibility that mTORC1 signaling in mature β cells in vivo is generally inhibited becoming activated only transiently, following feeding when levels of both glucose and amino acids are high. Consistent with this model, immunostaining for p-S6 in murine pancreatic tissue shows that mTORC1 is strongly stimulated by feeding and inhibited by fasting in mature β cells (Figure 3D and 3E). To understand the nutrient changes responsible for feeding-induced mTORC1 activation in β cells, we administered glucose and/or leucine to fasted mice. Administration of leucine alone is not sufficient to activate the pathway, whereas glucose partially activates mTORC1 in the β cells. Injection of glucose and leucine together activates mTORC1 in most β cells to similar levels as feeding (Figure 3D and E), consistent with the responses of isolated murine islets to nutrient treatments in vitro. Altogether, these results indicate that mTORC1 in mature β cells is strongly regulated by nutrients and activated during periods of high glucose and amino acid levels upon feeding.
The nutrient requirements for mTORC1 activation change as β cells undergo maturation
The observations that fetal β cells maintain high mTORC1 activity under low glucose conditions in utero, while mature β cells require high glucose for mTORC1 activation in adult animals, suggest that mTORC1 nutrient sensitivity changes during β cell maturation. To assess the nutrient sensitivity of mTORC1 in fetal β cells, we treated isolated embryonic islets with combinations of glucose and amino acids ex vivo. As indicated by p-S6 intensity, mTORC1 activity in human and murine embryonic β cells is entirely independent of glucose stimulation and requires only amino acids for full activation (Figure 4A and 4B). Similarly, we found that mTORC1 signaling in murine neonatal β cells undergo a shift in nutrient responsiveness that occurs throughout the first postnatal week. As neonates age from postnatal day 1 to day 7, their β cells develop a progressively increased requirement for glucose to activate mTORC1, evidenced by the gradually suppressed induction of mTORC1 activity by amino acids in the absence of glucose (Figure 4B and 4C).
Figure 4.

A neonatal shift in mTORC1 nutrient sensitivity controls β cell function. (A) Representative p-S6 FACS staining histograms of c-peptide+ cells from fetal (left column) and adult (right column) human subjects, following 30 minutes incubation of human islets in RPMI with indicated nutrients. Experiment was repeated three times on three different samples of fetal human pancreas from embryonic days 90, 92 and 120. (B) Representative p-S6 staining histograms of insulin+ cells from mice of indicated age following 30 minutes incubation of islets in RPMI with indicated nutrients. Note that at P7 mTORC1 dynamics is equivalent to mature β cells. (C) Percentages of p-S6 positive β cells from mice of indicated age in response to indicated nutrients, detected by FACS analysis compared to secondary antibodies only control. Data points represent mean ± SEM. P-values, * P<0.05, ** P<0.01, *** P<0.001, unpaired Student’s t test. n=3, 3, 3, 4 biological replicates; left to right. Control of mTORC1 dynamics affects insulin secretion in mature β cells. (D) Representative p-S6 staining histograms of insulin+ cells of Ins-CreER (left column) AMPK and TSC deficient (β cell-specific) mice following 30 minutes incubation of mouse islets in RPMI with indicated nutrients. (E-F) Fold insulin levels secreted by isolated islets from Ins-CreER (E, n=4) and TSC knockout (F, n=4) β cells, in a dynamic GSIS assay in low (2.8 mM, grey line), amino acids (green), high (16.7 mM, red) glucose concentrations and KCl (30 mM, blue). Note the increased insulin secretion in response to amino acids in β cells with perturbed mTORC1 activity.
To test the regulation of mTORC1 by glucose in neonatal β cells in vivo, glucose was injected into neonates and their pancreata were dissected and assessed for mTORC1 activity. In the first day after birth, injection of glucose is unable to activate mTORC1 in β cells. By postnatal day 5, glucose injection is sufficient to strongly activate mTORC1 in β cells, which do not activate the pathway in response to leucine, in accordance with the shift in glucose sensitivity observed ex vivo (Supplementary Figure 4).
Inhibition of mTORC1 during nutrient limitation is required for glucose-stimulated insulin secretion
We next sought to understand the role of dynamic, nutrient-responsive mTORC1 activity in controlling insulin secretion by mature β cells. To that end, we selectively ablated negative regulators of the mTORC1 pathway, including TSC (via TSC1 deletion) and AMPK (via co-deletion of PRKAA1 and PRKAA2), in β cells of adult mice by expressing tamoxifen-inducible Cre driven under control of the insulin promoter (Ins-CreER). β cells lacking TSC responded to all combinations of glucose and amino acids with stronger activation of mTORC1 than the wildtype control cells (Ins-CreER mice injected with tamoxifen) (Figure 4D), suggesting that in TSC-ablated β cells, the mTORC1 pathway can maintain activity in conditions in which availability of glucose or amino acids is limited. An important role of AMPK in postnatal β cell function has been recently reported (Jaafar et al., 2019); however, AMPK deletion in mature β cells imparted a weaker effect on mTORC1 activation in response to physiological levels of either glucose or amino acids (Figure 4D). Therefore, the perinatal shift in nutrient regulation of mTORC1 and resulting effects of altered mTORC1 signaling dynamics on insulin secretion, are likely due to changes which are not dependent on AMPK activity.
In dynamic insulin secretion assays, β cells lacking TSC demonstrated elevated insulin secretion in response to amino acids, a feature of fetal islets (Figure 4E and 4F). These results suggest that inhibition of mTORC1 activity in conditions of limited nutrients is required to gain and retain proper glucose-stimulated insulin secretion.
In agreement with these findings, it has been reported previously that mouse models of TSC or AMPK deletion in β cells have increased insulin secretion in a fasted state (in which amino acids are abundant, as opposed to glucose) (Beall et al., 2010, Mori et al., 2009, Jaafar et al., 2019). Importantly, mice with short-term deletion of TSC in β cells have altered glucose clearance, as assessed by glucose tolerance test, which can be attributed to altered regulation of mTORC1 activity and insulin secretion in these cells (Supplementary Figure 4). Appropriate regulation of mTORC1 is therefore crucial for the establishment of glucose-dependent insulin secretion after birth and its maintenance in the adult animal.
Nutrient-regulated mTORC1 activity controls SC-β cell functionality
The drastic change in nutrient availability at birth is followed by a β cell-specific switch in mTORC1 sensitivity to nutrients, and a switch from amino acid-induced to glucose-induced insulin secretion. These findings raise the question of whether changes in mTORC1 nutrient sensitivity contribute to the acquisition of glucose-responsive insulin secretion in β cells.
In vitro differentiation protocols generate functional insulin-secreting β cells derived from human induced pluripotent stem cells (SC-β cells) that respond to glucose to varying degrees (Pagliuca et al., 2014, Russ et al., 2015, Nair et al., 2019, Rezania et al., 2014). We sought to compare the responsiveness to physiological levels of glucose and amino acids in SC-β cells generated using our previously published protocol (Pagliuca et al., 2014). Interestingly, we found that amino acid stimulation of SC-β cells induces a more robust insulin secretion which is glucose-independent (Figure 5A), indicating that SC-β cells maintain features of fetal β cells. Importantly, SC-β cells generated by this protocol express close to mature levels of β cell identity and function markers, including PDX1 and NKX6–1 (Supplementary Figure 5), but lack high expression of the maturation markers MAFA and UCN3.
Figure 5.

A shift in mTORC1 nutrient sensitivity dictates SC-β cell function. (A) Fold insulin levels secreted by isolated islets from SC-β cells (n=6), in a dynamic GSIS assay in low (2.8 mM, grey line), amino acids (green), high (16.7 mM, red) glucose concentrations and KCl (30 mM, blue). Note the insulin secretion in response to amino acids in SC-β cells. (B) Representative p-S6 staining histograms of c-peptide+ cells (SC-β cells) and (C) Percentages of p-S6 positive SC-β cells detected by FACS analysis following 30 minutes incubation of SC-clusters in RPMI with indicated nutrients. Note the strong mTORC1 response of SC-β cells to amino acids only. n=9 biological replicates. (D) Representative immunostainings of p-S6 (green), and c-peptide (labeling SC-β cells, red), in clusters of in-vitro differentiated stem cells grown in an amino acid-rich media and incubated for 30 minutes with indicated nutrients. (E) Representative immunostainings of p-S6 (green) and c-peptide (labeling SC-β cells, blue), in kidney capsule-transplants of SC-β cells. Transplanted mice were fasted overnight and injected with the indicated nutrients or re-fed. Note the weak response of mTORC1 to leucine and the acquired glucose responsiveness 12 days after transplantation. Experiment was conducted with similar results 12 days, 18 days and 4 weeks after transplantation (n=6). (F) Gene expression heat map of known regulators of mTORC1 signaling in SC-β cells four weeks after transplantation under the kidney capsule of mice, normalized to expression levels of the genes in SC-β cells before transplantation. SC-β cells were sorted from five independent differentiation flasks and seven transplanted mice. (G) Representative immunostainings of p-S6 (green), and c-peptide (labeling SC-β cells, red), in clusters of in-vitro differentiated stem cells grown in an amino acid-poor media and incubated for 30 minutes with indicated nutrients. (H) Mean fluorescent intensity (MFI) of p-S6 staining in SC-β cells from indicated conditions, n=4 for each condition. Data points represent mean ± SEM. P-values, * P<0.05, unpaired Student’s t test. (I) Insulin levels secreted by SC-β cells from indicated growing condition in a dynamic perfusion assay in low (2.8 mM, grey line), high (16.7 mM, red) glucose concentrations and KCl (30mM, blue line). Secreted insulin levels were normalized to basal insulin secretion of each sample. The experiments with all conditions were done on six independent differentiation flasks. (J) Insulin levels secreted by SC-β cells from amino acid-rich media (100% AA), with (green) or without (blue) Torin1, in a dynamic perfusion assay in low (2.8 mM, grey line) and high (16.7 mM, red line) glucose concentrations and KCl (30mM, blue line). Secreted insulin levels were normalized to basal insulin secretion of each sample. Experiment was done on three independent differentiation flasks. (K) Correlation heat map presenting data from two different experiments; SC-β cells before and after transplantation and SC-β cells cultured in different concentrations of AA (100% and 25% of total amino acids in the rich-media).
Given that SC-β cells are functionally reminiscent of immature β cells, we next asked if SC-β cells likewise display fetal-like nutrient sensitivity of the mTORC1 pathway. mTORC1 signaling in SC- β cells is strongly activated in response to amino acids, but is unaffected by the presence of glucose, which was confirmed by FACS (Figure 5B and 5C) and immunostaining (Figure 5D). This suggests that nutrient regulation of mTORC1 signaling in SC-β cells is dominated by amino acids and independent of glucose, strikingly similar to fetal β cells.
Considering the fetal-like properties of mTORC1 regulation in SC-β cells, we sought to understand how a mature in vivo environment impacts the nutrient sensitivity and dynamics of mTORC1 in these cells. To test this, we transplanted SC-β cells into the kidney capsule of adult mice. Following transplantation, SC-β cells undergo a maturation process that results in increased expression of genes that are associated with β cell maturation and enhanced glucose-responsive insulin secretion (Supplementary Figure 5). Further, we found that this transplantation-induced maturation in SC- β cells corresponds to a significant shift in mTORC1 nutrient sensitivity towards that of mature adult β cells which occurs twelve to eighteen days after transplantation. As in mature β cells, the mTORC1 pathway remains inactive in the transplanted SC-β cells during fasting and not activated by injection of leucine into the recipient mouse, while feeding or injection of glucose strongly activates mTORC1 (Figure 5E). These results demonstrate that changing the environment of the cells can shift the nutrient sensitivity of mTORC1 towards a mature glucose-sensitive state. Further, the transplantation-induced glucose dependence of mTORC1 activity implies a corresponding acquisition of transient mTORC1 signaling dynamics in transplanted SC-β cells that is dictated by fluctuating glucose levels in vivo. Interestingly, the changes in regulation of mTORC1 precede the changes observed in glucose-responsive insulin secretion and in the expression of MAFA and coincides with UCN3 expression (Supplementary Figure 5). This suggests that transplantation alters mTORC1 nutrient sensitivity independently of changes in MAFA expression and may play an early role in functional maturation.
To identify molecular drivers for the transplantation-induced change in mTORC1 regulation by nutrients, we compared gene expression profiles of sorted SC-β cells, collected before and after transplantation. Expression of known maturation markers, including SIX2, IAPP, MAFA, FOXA1, FOXA2 and the senescence marker CDKN2A were elevated after transplantation (Gao et al., 2010, Sun et al., 2010, Aguayo-Mazzucato et al., 2011, Artner et al., 2010, Helman et al., 2016, Nishimura et al., 2015). Further, genes that have been reported as “disallowed” in β cells such as CPT1 and LDHA (Pullen and Rutter, 2013) were downregulated following transplantation (Supplementary Figure 5). Notably, we find that the expression of genes encoding inhibitory regulators of the mTORC1 pathway was induced after transplantation, including the leucine sensors SESN1 and SESN2, as well as the arginine sensor component CASTOR2 and the AMPK activator LKB1 (Figure 5F). These results indicate that transplantation increases the nutrient requirements for mTORC1 activation in SC-β cells by increasing the expression of inhibitory nutrient-sensitive mTORC1 regulators.
As mTORC1 signaling in SC-β cells is highly sensitive to amino acids, we hypothesized that recapitulating the postnatal change in nutrients and mTORC1 activity in vitro, by reducing amino acid levels in the culture media, might improve insulin secretion in response to glucose. Fully differentiated SC-β cells were cultured for two weeks in media with varying amino acid concentrations. SC-β cells grown in media with a low concentration of amino acids (25% of standard media concentrations) had suppressed mTORC1 activity as measured by FACS staining for p-S6 (Supplementary Figure 5). While SC-β cells cultured in amino acid-rich media were not dependent on glucose for full mTORC1 activity (Figure 5D and 5H), cells cultured in low amino acid levels suppressed mTORC1 signaling in the absence of glucose (Figures 5G and 5H), indicating that reduced amino acid conditions can induce SC-β cells to acquire a mature-like sensitivity of mTORC1 to glucose levels.
To understand the functional impact of amino acid levels on SC-β cells, we asked if the changes in mTORC1 glucose sensitivity and activity induced by low amino acid conditions affect insulin secretion. Glucose-induced insulin secretion of SC-islets was tested following culture in different concentrations of amino acids. SC-β cells cultured in an amino acid-rich environment displayed poor insulin secretion in response to high glucose. However, reducing amino acid levels to 50% or 25% of the amino acid-rich media increased glucose-stimulated insulin secretion of the cells by 1.6 and 2-fold, respectively (Figure 5I and Supplementary Figure 5). Remarkably, culturing SC-β cells with 25% of media amino acid levels increases the average cellular insulin content (Supplementary Figure 5), which was also observed in human fetal islets (Supplementary Figure 1), as detected by FACS staining, perhaps due to the reduced amino acid-induced insulin secretion and conservation of intracellular insulin. The change in glucose responsiveness of SC-β cells was accompanied by a significant increase in the expression levels of the glucose transporter SLC2A1 and a reduction in the Glucose-6-phosphatase G6PC2, indicating a shift towards more efficient glucose transport (Supplementary Figure 5). Importantly, similar enhancement of glucose-stimulated insulin secretion was induced by treating cells that were grown in an amino-acid rich media with Torin1 for 48 hours, providing evidence that amino acid reduction improves glucose responsiveness via inhibition of mTORC1 activity (Figure 5J). Correlation analysis shows that the gene expression profile of SC-β cells cultured in low amino acid levels was more similar to transplanted SC-β cells than SC-β cells cultured in high amino acid levels (Figure 5K). Together, these results demonstrate that environmental nutrients regulate mTORC1 in SC-β cells, and that mimicking the postnatal nutrient changes that occur in vivo influences SC-β cell function in a manner that recapitulates the postnatal progression of β cell maturation.
Discussion
Rapid growth of the embryo in utero occurs under steady maternal supply of nutrients and requires stable secretion of insulin by the fetal β cells (Xu et al., 2004). In effect, fetal insulin acts as a generic growth factor as the embryo is built by cell division and growth. At birth, a drastic change to intermittent postnatal feeding requires increased insulin secretion by β cells to maximize nutrient absorption during feeding, as well as suppressed release during fasting to avoid hypoglycemia. Therefore, the functional shift of β cells from constitutive to glucose-regulated insulin secretion after birth is vital for the survival of the neonate. Our findings show that glucose responsiveness, which is of the sine qua non of β cell maturation, is post-transcriptionally regulated by nutrients and mTORC1 signaling within few days after birth.
We demonstrate that the mechanisms controlling mTORC1 activity in β cells are plastic, changing substantially from the fetal to adult stage. The constitutive activation of mTORC1 in utero induces insulin secretion in an amino acid-dependent manner. Since the fetal serum is rich in amino acids, fetal β cells secrete insulin constantly. Our data strongly suggest that the change in the nutritional environment at birth shifts the nutrient sensitivity of mTORC1 signaling in β cells and mediates the change from constitutive to dynamic mTORC1 activity, resulting in a transition from amino acid to glucose-dependent insulin secretion.
The amino acid dependency of fetal insulin secretion indicates a potential role of fetal insulin in the negative effects of maternal malnutrition during gestation. Maternal dietary protein restriction has been demonstrated to impair fetal growth (Gonzalez et al., 2016, Herring et al., 2018). It is also recognized that insulin secretion by the fetus is essential for proper fetal growth (Fowden, 1992). Our findings therefore suggest that impaired fetal growth under maternal protein restriction may be attributed, at least in part, to reduced amino acid-sensitive insulin secretion by the fetus.
Complex machinery in mature β cells keeps mTORC1 inactive in conditions unsuitable for growth. This regulatory network includes inhibitors such as TSC, AMPK and SESN whose inhibition is released by growth factors, energy levels and amino acids, respectively (Saxton and Sabatini, 2017). In the embryonic in utero environment, an abundance of nutrients, especially amino acids, constitutively releases these inhibitions and enables continuous mTORC1 activity. In the adult the availability of nutrients is periodic, leading to dynamic mTORC1 activity. Nevertheless, we observe that the serum of the fetus contains very low levels of glucose, which raises the question of how mTORC1 is activated under these conditions. Our data suggest that the answer lies in the sensitivity of the mTORC1 pathway to nutrients. In immature β cells, amino acids are sufficient for mTORC1 activation, perhaps due to reduced activity of inhibitory sensors such as TSC and AMPK, enabling full activity in the presence of amino acids only.
Consistent with nutrient sensing by the mTORC1 pathway playing a role in β cell functional maturation after birth, we propose that nutrient sensing likewise plays a critical role in the functional maturation of stem cell-derived tissues. As we demonstrate, this can be exploited by altering nutrient conditions to improve functional maturation in vitro. Our data indicate that recreating the adult in vivo environment permits further functional maturation and this may be true for other stem-cell derived tissues. Recent developments in culture media which reflect physiologic conditions may therefore be a promising approach to improve functional maturation of stem cell-derived tissues.
In conclusion, we show here that a shift in the sensitivity of mTORC1 to nutrients, which coincides with drastic nutritional changes at birth, alters mTORC1 signaling dynamics and leads to a change in β cell function, from amino acid to glucose-dependent insulin secreting cells. This metabolic regulation of β cell maturation, triggered by extrinsic changes in nutrient availability, is important for optimizing the function of β cells for their environment.
Limitations of Study
The experimental systems that were used in this study have limitations to consider. In our experiments designed to isolate the role of physiological levels of glucose and amino acids on mTORC1 activation, we did not consider other nutrients and metabolites that could potentially participate in regulating mTORC1. While we demonstrated a specific role of leucine, the contribution of other individual amino acids were not investigated. In our experiments involving mice, we used an insulin promoter-driven Cre transgene (MIP-CreER; Wicksteed et al., 2010) to induce beta cell-specific gene deletion. To ensure that our conclusions are not influenced by non-specific effects, we controlled for MIP-CreER expression and tamoxifen treatment across all of the mice used in these experiments. Further, several of our experiments utilize human cadaveric islets, which display an inherent degree of functional variability between donors, due to individual variability in age, gender, and metabolic health. To mitigate this, we used only islets from healthy donors with a BMI within the normal range. Experiments were replicated with islets from multiple donors to ensure that any findings were not donor-specific. We are not aware of any genetic or metabolic syndromes in the human cadaveric islets from adults or embryos that could affect the outcomes of the experiments. Lastly, the differentiation protocol for producing the SC-beta cells used in our experiments yields some unavoidable batch-to-batch variability in beta cell function. Therefore, in each experiment we used SC-beta cells from multiple independent differentiation flasks. SC-beta cells used in our experiments were generated only from ESC; iPSC-derived SC-beta cells were not tested.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Douglas Melton (dmelton@harvard.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
LC/MS excel file (Table S1, related to Figure 1) and Jupyter notebook containing all analysis code (Data S1 and S2) are provided in supplementary materials. The sequencing data generated during this study are available at GEO-NCBI (GSE147346, GSE147347, GSE147349).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
The hPSC line HUES8 (NIH human embryonic stem cell registry #0021) was used for all experiments. Undifferentiated HUES8 cells were maintained in supplemented mTeSR1 medium (StemCell Technologies) in 500ml spinner flasks (Corning) set at a 70rpm rotation rate in a 37°C 5% CO2 incubator. Directed differentiation into SC-β cells was conducted as described previously (Pagliuca et al., 2014). At the last stage, cell clusters were maintained for 14–20 days in CMRL 1066 Supplemented (Mediatech; 99–603-CV) + 10% FBS (HyClone, VWR; 16777) + 1% Pen/Strep. For testing the effect of amino acids dilution, clusters were grown in CMRL 1066 Supplemented (Mediatech; 99–603-CV) that was diluted to 75%, 50% and 25% with RPMI-1640 medium with no glucose (Life Technologies; 11879020) or modified nutrient-free RPMI-1640 medium (MBS652918), supplemented to 5.5mM glucose + 10% FBS (HyClone, VWR; 16777) + 1% Pen/Strep. For testing the effect of amino acids and glucose dilution on fetal mouse islets, they were isolated from E18 embryos and grown for 72 hours in RPMI 1640 medium that was diluted to 25% with RPMI-1640 medium with no glucose (Life Technologies; 11879020) or modified nutrient-free RPMI-1640 medium (MBS652918), supplemented to 11mM glucose + 10% FBS (HyClone, VWR; 16777) + 1% Pen/Strep and changed daily.
Animals
C57BL/6 mice were used for serum metabolite profiling, islet isolation for GSIS and FACS analysis and for immunohistochemistry. For the immunohistochemistry assays, mice were fasted overnight and then received saline, glucose (0.1g/ml) or leucine (30mM) by intraperitoneal injection (20ul/g), and pancreas was harvested 30 minutes later.
Mice expressing floxed alleles of Prkaa1 and Prkaa2 were purchased from Jackson Laboratories (Nakada et al., 2010). Mice expressing a floxed allele of TSC1 were gifts from D. Kwiatkowski (Harvard Medical School). These mice were crossed with MIP-CreER mice (Wicksteed et al., 2010). Final crosses yielded mice expressing homozygous floxed Prkaa1 and Prkaa2 with MIP-CreER for β cell-specific AMPK deletion, or homozygous floxed TSC1 with MIP-CreER for β cell-specific TSC1 deletion. Three doses of 200 mg/kg Tamoxifen (Sigma, 20mg/ml in corn oil) were administered 48 hours apart by gavage into 8- to 10-week old mice to obtain β -cell specific deletion of Prkaa1 and Prkaa2 or TSC1. Glucose tolerance tests were performed 2 weeks following tamoxifen administration, and islets were collected one week later. All experiments were conducted using mouse litter batches, and where necessary experimental groups were comprised of multiple litters to allow statistical power. The experiments were not randomized. There was no blinded allocation during experiments and outcome assessment. The ethics committee of Harvard University approved the study protocol for animal welfare.
METHOD DETAILS
Polar Metabolite Profiling
LC/MS-based analyses were conducted on a QExactive benchtop orbitrap mass spectrometer equipped with an Ion Max source and HESI II probe, which was coupled to a Dionex UltiMate 3000 ultra-high-performance liquid chromatography system (Thermo Fisher Scientific, San Jose, CA). External mass calibration was performed using the standard calibration mixture every 7 days. Acetonitrile was LC/MS 6HyperGrade from EMD Millipore. All other solvents were LC/MS Optima grade from Thermo Fisher Scientific. For extraction from SC-β cells, the metabolite extraction mix (stored at −20°C) was made of 80:20 (v/v) methanol:water, supplemented with a mixture of 17 isotope-labeled amino acids at 500nM each as internal extraction standards (Cambridge Isotope Laboratories, MSK-A2–1.2). For extraction from mouse plasma, the metabolite extraction mix (stored at −20°C) was made of 75:25:0.2 (v/v) acetonitrile:methanol:formic acid supplemented with a mixture of 17 isotope-labeled amino acids at 500nM each as internal extraction standards (Cambridge Isotope Laboratories, MSK-A2–1.2).
For chromatographic separation, 2.5μL of each sample was injected onto a SeQuant ZIC-pHILIC Polymeric column (2.1 × 150 mm) connected with a guard column (2.1 × 20 mm). Both analytical and guard columns are of 5 μm particle size purchased from EMD Millipore. Flow rate was set to 0.150 mL per minute, the column compartment was set to 25°C, and the autosampler sample tray to 4 °C. Mobile Phase A consisted of 20 mM ammonium carbonate, 0.1% ammonium hydroxide. Mobile Phase B was 100% acetonitrile. The chromatographic gradient was as follows: (1) 0–20 min: linear gradient from 80% to 20% B; (2) 20–20.5 min: linear gradient from 20% to 80% B; (3) 20.5–28 min: hold at 80% B. The mass spectrometer was operated in full scan, polarity switching mode with the spray voltage set to 3.0 kV, the heated capillary held at 275°C, and the HESI probe held at 350°C. The sheath gas flow rate was set to 40 units, the auxiliary gas flow was set to 15 units, and the sweep gas flow was set to 1 unit. The MS data acquisition was performed in a range of 70–1000m/z, with the resolution set to 70,000, the AGC target at 106, and the maximum injection time at 20msec.
Immunohistochemistry
Differentiated cell clusters, pancreatic tissues and kidney transplants were fixed by immersion in 4% PFA for 1 hour at room temperature (RT). Samples were washed 3 times with PBS, embedded in Histogel (Thermo), and sectioned at 10μm for histological analysis. Sections were subjected to deparaffinization using Histoclear (Thermoscientific; C78–2-G) and rehydrated. For antigen retrieval slides were emerged in 0.1M EDTA (Ambion; AM9261) and placed in a pressure cooker (Proteogenix; 2100 Retriever) for two hours.
Slides were blocked with CAS-block blocking solution (Thermo fisher) for 1 hour at RT, followed by incubation in blocking solution with primary antibodies overnight at 4°C. The following primary antibodies were used: rat anti-insulin (pro-)/C-peptide (Developmental Studies Hybridoma Bank; GN-ID4, 1:300), mouse anti-glucagon (Santa Cruz; sc-514592 1:300), guinea pig anti-insulin (Dako; A0564 1:300), mouse anti-Nkx6.1 (University of Iowa, Developmental Hybridoma Bank; F55A12-supernatant) and rabbit anti phospho-S6 Ribosomal Protein (Ser240/244) (Cell Signaling; #5364, 1:100). Cells were washed twice in PBS the next day, followed by secondary antibody incubation for 2 hours at RT (protected from light). Secondary antibodies conjugated to Alexa Fluor 405,488, or 647 were used to visualize primary antibodies. Following two washes with PBS, the histology slides were mounted in Vectashield mounting medium (Vector Laboratories), covered with coverslips and sealed with nail polish. Images were taken using a Zeiss Axio Imager 2 microscope.
Flow Cytometry
Differentiated cell clusters, human islets (Prodo labs) or mouse islets were pre-incubated in modified nutrient-free RPMI-1640 medium (MBS652918) for 1 hour. Then, clusters or islets were incubated for 30 minutes incubation in RPMI-1640 with or without glucose (11mM) and amino acids (Life Technologies; 11875–093) and other compounds in 6-well plates at 37°C. The following compounds were used: insulin (Sigma 19278, 1:1000), S961 (a gift from Novonordisk 0.01mM), exendin-4 (Sigma E7144, 0.02mM), forskolin (Santa Cruz SC3562, 0.01mM), diazoxide (Sigma D9035, 0.1mM), Torin-1 (Thermo Fisher 424710, 100nM), leucine (Sigma, 0.3 or 1mM). Then, differentiated cell clusters or islets were collected into 1.7uL Eppendorf tubes, dispersed into single-cell suspension by incubation in Accutase (StemCell Technologies) at 37°C until clusters and dissociated to single cells upon mixing by pipetting gently up and down (typically 5–10 min). Cells were spun down for 2 min at 1000rpm. Cells were resuspended in perm/fix solution and incubated for 10 min (BD 554714). Cells were then washed once in perm/wash for 15 min (BD 554714). Cells were then resuspended in perm/wash with primary antibodies and incubated at 4°C overnight. Primary antibodies diluted 1:300 unless otherwise noted: rat anti-insulin (pro-)/C-peptide (Developmental Studies Hybridoma Bank; GN-ID4), mouse anti-glucagon (Santa Cruz; sc-514592), guinea pig anti-insulin (Dako; A0564), rabbit anti Mafa (Cell signaling; #79737), mouse anti-Nkx6.1 (University of Iowa, Developmental Hybridoma Bank; F55A12-supernatant), goat anti Pdx1 (R&D Systems; AF2419), rabbit anti phospho-AMPKα (Cell signaling; #2535), and rabbit anti phospho-S6 Ribosomal Protein (Ser240/244) (Cell Signaling; #5364). Cells were washed twice in blocking buffer and then incubated in perm/wash buffer with secondary antibodies for 2 hours (protected from light). Secondary antibodies conjugated to Alexa Fluor 405, 488 or 647 (Life Technologies) were used to visualize primary antibodies. Cells were then washed 3 times in perm/wash solution and finally resuspended in 500–700μl, filtered through a 40μm nylon mash into flow cytometry tubes (BD Falcon; 352235), and analyzed using the LSR-II flow cytometer (BD Biosciences) with at least 10,000 events recorded. Analysis of the results was performed using FlowJo software.
Nano-string gene expression analysis
Sorted TSQ-expressing SC-β cells (Davis et al., 2019) were collected for RNA extraction in RLT Lysis Buffer (QIAGEN). RNA was extracted using the RNeasy Mini Kit (QIAGEN), according the manufacturer’s instructions, and 100ng of RNA was used for each reaction. Nanostring set up was performed according to the protocol for the nCounter XT Gene Expression Assay. Data was analyzed in the nSolver 2.5 Software. All genes were normalized to housekeeping genes (ITCH, RPL15, RPL19, TCEB1, UBE2D3). Analysis of changes in gene expression of β cell markers was done using R.
Mouse transplantation studies
All animal experiments were performed in accordance with Harvard University International Animal Care and Use Committee regulations. Cell transplantations into immunodeficient SCID-Beige mice (Jackson Laboratory) were conducted as described previously with minor modifications. SC-β Clusters were resuspended in RPMI-1640 medium (Life Technologies; 11875–093), aliquoted into PCR tubes and kept on ice until loading into a catheter for cell delivery under the mouse kidney capsule. Mice were anesthetized with 0.5ml/25g 1.25% Avertin/body weight, and the left ventricle kidney site was shaved and disinfected with betadine and alcohol. A 1cm incision was made to expose the kidney, followed by insertion of the catheter needle and injection of the cell clusters. The abdominal cavity was closed with PDS absorbable sutures (POLY-DOX; 2016–06), and the skin was closed with surgical clips (Kent Scientific Corp; INS750346–2). Mice were placed on a 37°C micro-temp circulating pump and blanket during the surgery/recovery period and given a 5mg/kg carprofen dose post-surgery, re-applied 24h after the initial dose. Wound clips were removed 14 days post-surgery and mice were monitored twice a week. To retrieve grafts, kidneys containing the grafts were dissected from freshly euthanized mice 4 weeks post-transplantation. For Immunohistochemistry, grafts were fixed in PBS +4% paraformaldehyde overnight, embedded in paraffin, and sectioned for histological analysis. Single-cell suspensions were washed with and resuspended in PBS, stained with TSQ at 37°C for 10min, filtered through a 40μm nylon mash into flow cytometry tubes (BD Falcon), and TSQ+ cells were sorted using MoFlo flow cytometers (Beckman Coulter) into PBS +1% BSA (Sigma) on ice. Total RNA from sorted SC-β cells was isolated by TRIzol (Invitrogen) extraction followed by RNeasy Plus Micro Kit (Qiagen), from 5 and 7 samples before and after transplantation, respectively. Libraries were prepared and sequenced using Illumina’s directional RNA sequencing protocol.
Ex vivo static glucose stimulated insulin secretion (GSIS)
Human Islets were placed in basal Krebs buffer (Krb) (128mM NaCl, 5mM KCl, 2.7mM CaCl2, 1.2mM MgCl2, 1mMNa2HPO4, 1.2mM KH2PO4, 5mM NaHCO3 10 HEPES, 0.1% BSA). Islets were washed twice with low-glucose (2.8mM) Krb and were then loaded into the 24 well plate inserts (Millicell Cell Culture Insert; PIXP01250) and fasted in low-glucose Krb for 1 hour to remove residual insulin in 37°C incubators. Islets were washed once in low Krb, incubated in low-glucose Krb for 1 hour, and supernatant collected. Then islets were transferred to high-glucose (16.7mM) Krb for 1 hour, and supernatant collected. This sequence was repeated one additional time and islets were washed once between high-glucose to second low-glucose incubation to remove residual glucose. Finally, islets were incubated in Krb containing 2.8mM glucose and 30mM KCl (depolarization challenge) for 1 hour and then supernatant collected. Islets were then lysed using RIPA buffer. Supernatant samples containing secreted insulin and total insulin content of the lysed islets were processed using the Human Ultrasensitive Insulin ELISA (ALPCO, 80-INSHUU-E01.1). The insulin content in the supernatant was normalized by the insulin content of the sample.
Ex vivo dynamic glucose stimulated insulin secretion (GSIS)
Similar numbers of Islets or SC-β clusters were divided into chambers and assayed on a fully automated Perifusion System (BioRep). Chambers were sequentially perfused with 2.8mM, 20mM glucose or 2.8mM glucose with amino acids (physiological levels) and 2.8mM glucose with 30mM KCL in Krb buffer (128mM NaCl, 5mM KCl, 2.7mM CaCl2, 1.2mM MgCl2, 1mMNa2HPO4, 1.2mM KH2PO4, 5mM NaHCO3 10 HEPES, 0.1% BSA) at a flow rate of 100ul/min. Chambers were first perifused with low glucose (2.8mM) for 1 hour for fasting and then 15 minutes for low glucose incubation followed by low glucose with physiological levels of amino acids (Cantor et al., 2017) for 15 minutes. Samples were then perifused with high glucose (16.7mM) challenge for 30 minutes and then with low glucose and 30mM KCl for 15 minutes. Insulin concentrations in the supernatant were determined using Ultrasensitive Insulin ELISA kit (Alpco).
Preparation of single-cell suspension
Islets were isolated from feta human pancreas, cultured in vitro for four days and collected, at the first or last day of incubation. The islets were allowed to settle, washed in PBS and dissociated by 5min incubation in TrypLE Express (GIBCO) at 37°C, followed by mechanical agitation through pipetting. TrypLE activity was terminated by washing with PBS+1% FCS.
Single cell library preparation and sequencing
Single-cell capture, cell lysis, reverse transcription of full-length mRNA and cDNA amplification were performed using the 10X Chromium system. Sequencing libraries were prepared using the Illumina Nextera XT DNA library preparation kit and samples were sequenced on an Illumina HiSeq. Fastq files were aligned using the 10X software. Single cell data analysis followed the guidelines and modified code of Luecken & Theis (Luecken and Theis, 2019). Counts data from the three samples was read into Scanpy and merged. Low-quality cells and empty droplets were removed by excluding barcodes associated with fewer than 1500 counts or greater than 50,000, fewer than 700 genes detected, or with more than 20% of reads mapping to mitochondrial transcripts. Scanpy (Wolf et al., 2018) was used for filtering, principal component analysis (PCA), uniform manifold approximation and projection (UMAP), and Louvain clustering. Prior to dimensionality reduction, ComBat (Johnson et al., 2007) was used for batch correction and scran (Lun et al., 2016) was used to compute and normalize by size factors. PCA was performed on log-scaled counts for the top 4000 highly variable genes, and UMAP was computed with the top 50 principal components. Louvain clustering was performed using a resolution of 0.5, and the cluster of endocrine cells was identified by expression of the marker CHGA. Analysis was then repeated as described above using only endocrine cells, with the number of principal components reduced to 15. Jupyter notebook containing all analysis code provided in supplementary materials (Data S1 and S2).
QUANTIFICATION AND STATISTICAL ANALYSIS
No randomization was used in this study, and the investigators were not blinded to group allocation during experiments or outcome assessments. No statistical method was used to predetermine sample size. n marks the number of times an experiment was performed on independent mice or stem-cell differentiation batches. All data are presented as mean±s.e.m. Statistical analysis was performed with unpaired Student’s t test with a two-tailed distribution. P<0.05 was considered as statistically significant. Data was analyzed and plotted using Prism software from GraphPad.
Supplementary Material
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rat anti-insulin (pro-)/C-peptide | DSHB | GN-ID4 |
| mouse anti-glucagon | Santa Cruz | sc-514592 |
| guinea pig anti-insulin | Dako | A0564 |
| mouse anti-Nkx6.1 | DSHB | F55A12 |
| rabbit anti phospho-S6 (Ser240/244 | Cell Signaling | 5364 |
| rabbit anti Mafa | Cell signaling; | 79737 |
| goat anti Pdx1 | R&D Systems | AF2419 |
| rabbit anti phospho-AMPKα | Cell signaling | 2535 |
| Biological Samples | ||
| Cadaveric human islets | Prodo labs | N/A |
| Fetal human pancreas | University of Washington | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| mTeSR1 medium | StemCell Technologies | 85850 |
| nutrient-free RPMI-1640 medium | mybiosource | MBS652918 |
| RPMI-1640 medium with no glucose | Life Technologies | 11879–020 |
| RPMI-1640 medium | Life Technologies | 11875–093 |
| CMRL 1066 Supplemented | Mediatech | 99–603-CV |
| FBS (HyClone) | VWR | 16777 |
| insulin | Sigma | 19278 |
| S961 | Novonordisk | N/A |
| exendin-4 | Sigma | E7144 |
| forskolin | Santa Cruz | SC3562 |
| Torin-1 | Thermo Fisher | 424710 |
| leucine | Sigma | N/A |
| diazoxide | Sigma | D9035 |
| perm/fix solution | BD | 554714 |
| Amino acids | Sigma | N/A |
| Critical Commercial Assays | ||
| 500ml spinner flasks | Corning | |
| flow cytometry tubes | BD Falcon | 352235 |
| RLT Lysis Buffer | QIAGEN | 79216 |
| Human Ultrasensitive Insulin ELISA | ALPCO | 80-INSHUU-E1.1 |
| TrypLE Express | GIBCO | 12604013 |
| Deposited Data | ||
| mRNA sequencing of stem cell-derived beta cells before and after transplantation into mice | GEO NCBI | GSE147346 |
| mRNA sequencing of stem cell-derived beta cells cultured in different AA concentrations | GEO NCBI | GSE147347 |
| single-cell mRNA sequencing of human embryonic pancreas | GEO NCBI | GSE147349 |
| Experimental Models: Cell Lines | ||
| HUES8 human embryonic stem cells | NIH | #0021 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 | Jackson Laboratory | N/A |
| SCID-Beige mice | Jackson Laboratory | N/A |
| Prkaa1tm1.1Sjm/J | Jackson Laboratory | 014141 |
| Prkaa2tm1.1Sjm/J | Jackson Laboratory | 014142 |
| Tsc1tm1Djk/J | Jackson Laboratory | 005680 |
| B6.Cg-Tg(Ins1-cre/ERT)1Lphi/J | Jackson Laboratory | 024709 |
| Oligonucleotides | ||
| Custom Taqman probes for TSC1, TSC2, LKB1, PRKAA1, PRKAA2, SESN1, SESN2, UBC and GAPDH | Applied Biosystems | |
| Cre forward: GAACCTGATGGACATGTTCAGG Cre reverse: AGTGCGTTCGAACGCTAGAGCCTGT |
||
| Prkaa1 forward: CCC ACC ATC ACT CCA TCT CT Prkaa1 reverse: AGC CTG CTT GGC ACA CTT AT |
||
| Prkaa2 forward: GCA GGC GAA TTT CTG AGT TC Prkaa2 reverse: TCC CCT TGA ACA AGC ATA CC |
||
| TSC1 forward: GTC ACG ACC GTA GGA GAA GC TSC1 reverse: GAA TCA ACC CCA CAG AGC AT |
||
| Recombinant DNA | ||
| Software and Algorithms | ||
| Biorender | https://app.biorender.com/ | |
| Prism | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| FlowJo | FlowJo | https://www.flowjo.com |
| Other | ||
Context and Significance.
The continuous secretion of insulin during pregnancy promotes growth of the embryo and depends on constant supply of maternal nutrients. At birth, the maternal nutrient supply is cut off, forcing the newborn to depend on its own feeding for nutrition. Researchers at Harvard and MIT provide an explanation for how insulin-producing β cells adapt to this nutritional challenge. β cells undergo a transition in the nutrient sensitivity of the mTORC1 pathway which controls their insulin secretion in response to nutrients. Altering nutrient levels to exploit this adaptative property of β cells results in improved glucose-responsive insulin secretion in human stem cell-derived β cells.
Highlights.
Nutritional changes after birth alter β cell function
β cells acquire glucose response via changes in mTORC1 nutrient sensitivity
Glucose response is independent of changes in canonical β cell markers expression
Nutrient-regulated mTORC1 activity controls stem cell-derived β cell function
Acknowledgement
We thank Jennifer C. Dempsey and Diana O’Day from University of Washington for their help to supply fetal human pancreas samples. We thank members of the Melton and Sabatini labs for helpful discussions. We thank Jenny Kenty for technical assistance. We thank Caroline Lewis, Bena Chan, and Tenzin Kunchok in the Whitehead Institute Metabolite Profiling Core Facility for LC-MS equipment operation and data analysis. We also thank members of the Harvard Stem Cell Institute Flow Cytometry and Histology core for technical support. A.H. was supported by a long-term fellowship from EMBO. A.L.C. is supported by a fellowship from the NIH (F31 5F31DK113665). D.A.M. is supported by grants from the Harvard Stem Cell Institute, NIH, HIRN, and the JPB foundation. D.M.S. is supported by grants from the NIH (R01 CA103866, R01CA129105, and R37 AI047389). D.M.S. is an American Cancer Society Research Professor. D.M.S and D.A.M. are investigators of the Howard Hughes Medical Institute.
Footnotes
Declaration of Interests
D.A.M. is a founder of Semma Therapeutics and equity holder of Semma/Vertex which has licensed technology developed in D.A.M.’s lab. All other authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- AGUAYO-MAZZUCATO C, KOH A, EL KHATTABI I, LI WC, TOSCHI E, JERMENDY A, JUHL K, MAO K, WEIR GC, SHARMA A & BONNER-WEIR S 2011. Mafa expression enhances glucose-responsive insulin secretion in neonatal rat beta cells. Diabetologia, 54, 583–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALEJANDRO EU, BOZADJIEVA N, BLANDINO-ROSANO M, WASAN MA, ELGHAZI L, VADREVU S, SATIN L & BERNAL-MIZRACHI E 2017. Overexpression of Kinase-Dead mTOR Impairs Glucose Homeostasis by Regulating Insulin Secretion and Not beta-Cell Mass. Diabetes, 66, 2150–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARDA HE, LI L, TSAI J, TORRE EA, ROSLI Y, PEIRIS H, SPITALE RC, DAI C, GU X, QU K, WANG P, WANG J, GROMPE M, SCHARFMANN R, SNYDER MS, BOTTINO R, POWERS AC, CHANG HY & KIM SK 2016. Age-Dependent Pancreatic Gene Regulation Reveals Mechanisms Governing Human beta Cell Function. Cell Metab, 23, 909–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARDESTANI A, LUPSE B, KIDO Y, LEIBOWITZ G & MAEDLER K 2018. mTORC1 Signaling: A Double-Edged Sword in Diabetic beta Cells. Cell Metab, 27, 314–331. [DOI] [PubMed] [Google Scholar]
- ARLOTTA P & PASCA SP 2019. Cell diversity in the human cerebral cortex: from the embryo to brain organoids. Curr Opin Neurobiol, 56, 194–198. [DOI] [PubMed] [Google Scholar]
- ARTNER I, HANG Y, MAZUR M, YAMAMOTO T, GUO M, LINDNER J, MAGNUSON MA & STEIN R 2010. MafA and MafB regulate genes critical to beta-cells in a unique temporal manner. Diabetes, 59, 2530–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AVRAHAMI D, LI C, ZHANG J, SCHUG J, AVRAHAMI R, RAO S, STADLER MB, BURGER L, SCHUBELER D, GLASER B & KAESTNER KH 2015. Aging-Dependent Demethylation of Regulatory Elements Correlates with Chromatin State and Improved beta Cell Function. Cell Metab, 22, 619–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BADER E, MIGLIORINI A, GEGG M, MORUZZI N, GERDES J, ROSCIONI SS, BAKHTI M, BRANDL E, IRMLER M, BECKERS J, AICHLER M, FEUCHTINGER A, LEITZINGER C, ZISCHKA H, WANG-SATTLER R, JASTROCH M, TSCHOP M, MACHICAO F, STAIGER H, HARING HU, CHMELOVA H, CHOUINARD JA, OSKOLKOV N, KORSGREN O, SPEIER S & LICKERT H 2016. Identification of proliferative and mature beta-cells in the islets of Langerhans. Nature, 535, 430–4. [DOI] [PubMed] [Google Scholar]
- BARTOLOME A & GUILLEN C 2014. Role of the mammalian target of rapamycin (mTOR) complexes in pancreatic beta-cell mass regulation. Vitam Horm, 95, 425–69. [DOI] [PubMed] [Google Scholar]
- BEALL C, PIIPARI K, AL-QASSAB H, SMITH MA, PARKER N, CARLING D, VIOLLET B, WITHERS DJ & ASHFORD ML 2010. Loss of AMP-activated protein kinase alpha2 subunit in mouse beta-cells impairs glucose-stimulated insulin secretion and inhibits their sensitivity to hypoglycaemia. Biochem J, 429, 323–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLANDINO-ROSANO M, BARBARESSO R, JIMENEZ-PALOMARES M, BOZADJIEVA N, WERNECK-DE-CASTRO JP, HATANAKA M, MIRMIRA RG, SONENBERG N, LIU M, RUEGG MA, HALL MN & BERNAL-MIZRACHI E 2017. Loss of mTORC1 signalling impairs beta-cell homeostasis and insulin processing. Nat Commun, 8, 16014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLISS CR & SHARP GW 1992. Glucose-induced insulin release in islets of young rats: time-dependent potentiation and effects of 2-bromostearate. Am J Physiol, 263, E890–6. [DOI] [PubMed] [Google Scholar]
- BLUM B, HRVATIN S, SCHUETZ C, BONAL C, REZANIA A & MELTON DA 2012. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat Biotechnol, 30, 261–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CANTOR JR, ABU-REMAILEH M, KANAREK N, FREINKMAN E, GAO X, LOUISSAINT A JR., LEWIS CA & SABATINI DM 2017. Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell, 169, 258–272 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAU GC, IM DU, KANG TM, BAE JM, KIM W, PYO S, MOON EY & UM SH 2017. mTOR controls ChREBP transcriptional activity and pancreatic beta cell survival under diabetic stress. J Cell Biol, 216, 2091–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONRAD E, STEIN R & HUNTER CS 2014. Revealing transcription factors during human pancreatic beta cell development. Trends Endocrinol Metab, 25, 407–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVIS JC, HELMAN A, RIVERA-FELICIANO J, LANGSTON CM, ENGQUIST EN & MELTON DA 2019. Live Cell Monitoring and Enrichment of Stem Cell-Derived beta Cells Using Intracellular Zinc Content as a Population Marker. Curr Protoc Stem Cell Biol, 51, e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DHAWAN S, TSCHEN SI, ZENG C, GUO T, HEBROK M, MATVEYENKO A & BHUSHAN A 2015. DNA methylation directs functional maturation of pancreatic beta cells. J Clin Invest, 125, 2851–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ELGHAZI L, BALCAZAR N, BLANDINO-ROSANO M, CRAS-MENEUR C, FATRAI S, GOULD AP, CHI MM, MOLEY KH & BERNAL-MIZRACHI E 2010. Decreased IRS signaling impairs beta-cell cycle progression and survival in transgenic mice overexpressing S6K in beta-cells. Diabetes, 59, 2390–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOWDEN AL 1992. The role of insulin in fetal growth. Early Hum Dev, 29, 177–81. [DOI] [PubMed] [Google Scholar]
- GAO N, LE LAY J, QIN W, DOLIBA N, SCHUG J, FOX AJ, SMIRNOVA O, MATSCHINSKY FM & KAESTNER KH 2010. Foxa1 and Foxa2 maintain the metabolic and secretory features of the mature beta-cell. Mol Endocrinol, 24, 1594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GONZALEZ PN, GASPEROWICZ M, BARBEITO-ANDRES J, KLENIN N, CROSS JC & HALLGRIMSSON B 2016. Chronic Protein Restriction in Mice Impacts Placental Function and Maternal Body Weight before Fetal Growth. PLoS One, 11, e0152227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAMADA S, HARA K, HAMADA T, YASUDA H, MORIYAMA H, NAKAYAMA R, NAGATA M & YOKONO K 2009. Upregulation of the mammalian target of rapamycin complex 1 pathway by Ras homolog enriched in brain in pancreatic beta-cells leads to increased beta-cell mass and prevention of hyperglycemia. Diabetes, 58, 1321–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAY WW JR. 2006. Placental-fetal glucose exchange and fetal glucose metabolism. Trans Am Clin Climatol Assoc, 117, 321–39; discussion 339–40. [PMC free article] [PubMed] [Google Scholar]
- HELMAN A, KLOCHENDLER A, AZAZMEH N, GABAI Y, HORWITZ E, ANZI S, SWISA A, CONDIOTTI R, GRANIT RZ, NEVO Y, FIXLER Y, SHREIBMAN D, ZAMIR A, TORNOVSKY-BABEAY S, DAI C, GLASER B, POWERS AC, SHAPIRO AM, MAGNUSON MA, DOR Y & BEN-PORATH I 2016. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat Med, 22, 412–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HENQUIN JC & NENQUIN M 2018. Immaturity of insulin secretion by pancreatic islets isolated from one human neonate. J Diabetes Investig, 9, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HERRING CM, BAZER FW, JOHNSON GA & WU G 2018. Impacts of maternal dietary protein intake on fetal survival, growth, and development. Exp Biol Med (Maywood), 243, 525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HRVATIN S, O’DONNELL CW, DENG F, MILLMAN JR, PAGLIUCA FW, DIIORIO P, REZANIA A, GIFFORD DK & MELTON DA 2014. Differentiated human stem cells resemble fetal, not adult, beta cells. Proc Natl Acad Sci U S A, 111, 3038–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JAAFAR R, TRAN S, SHAH AN, SUN G, VALDEARCOS M, MARCHETTI P, MASINI M, SWISA A, GIACOMETTI S, BERNAL-MIZRACHI E, MATVEYENKO A, HEBROK M, DOR Y, RUTTER GA, KOLIWAD SK & BHUSHAN A 2019. mTORC1 to AMPK switching underlies beta-cell metabolic plasticity during maturation and diabetes. J Clin Invest, 130, 4124–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JIANG Y, PARK P, HONG SM & BAN K 2018. Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells: Current Strategies and Limitations. Mol Cells, 41, 613–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHNSON WE, LI C & RABINOVIC A 2007. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics, 8, 118–27. [DOI] [PubMed] [Google Scholar]
- KERVRAN A & RANDON J 1980. Development of insulin release by fetal rat pancreas in vitro: effects of glucose, amino acids, and theophylline. Diabetes, 29, 673–8. [DOI] [PubMed] [Google Scholar]
- KOYANAGI M, ASAHARA S, MATSUDA T, HASHIMOTO N, SHIGEYAMA Y, SHIBUTANI Y, KANNO A, FUCHITA M, MIKAMI T, HOSOOKA T, INOUE H, MATSUMOTO M, KOIKE M, UCHIYAMA Y, NODA T, SEINO S, KASUGA M & KIDO Y 2011. Ablation of TSC2 enhances insulin secretion by increasing the number of mitochondria through activation of mTORC1. PLoS One, 6, e23238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEMAIRE K, THORREZ L & SCHUIT F 2016. Disallowed and Allowed Gene Expression: Two Faces of Mature Islet Beta Cells. Annu Rev Nutr, 36, 45–71. [DOI] [PubMed] [Google Scholar]
- LIU JS & HEBROK M 2017. All mixed up: defining roles for beta-cell subtypes in mature islets. Genes Dev, 31, 228–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUECKEN MD & THEIS FJ 2019. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol Syst Biol, 15, e8746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUN AT, MCCARTHY DJ & MARIONI JC 2016. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Res, 5, 2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUNDY SD, ZHU WZ, REGNIER M & LAFLAMME MA 2013. Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev, 22, 1991–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAEDLER K & ARDESTANI A 2017. mTORC in beta cells: more Than Only Recognizing Comestibles. J Cell Biol, 216, 1883–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTINEZ-SANCHEZ A, NGUYEN-TU MS & RUTTER GA 2015. DICER Inactivation Identifies Pancreatic beta-Cell “Disallowed” Genes Targeted by MicroRNAs. Mol Endocrinol, 29, 1067–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATSUOKA TA, KANETO H, STEIN R, MIYATSUKA T, KAWAMORI D, HENDERSON E, KOJIMA I, MATSUHISA M, HORI M & YAMASAKI Y 2007. MafA regulates expression of genes important to islet beta-cell function. Mol Endocrinol, 21, 2764–74. [DOI] [PubMed] [Google Scholar]
- MORI H, INOKI K, OPLAND D, MUNZBERG H, VILLANUEVA EC, FAOUZI M, IKENOUE T, KWIATKOWSKI DJ, MACDOUGALD OA, MYERS MG JR. & GUAN KL 2009. Critical roles for the TSC-mTOR pathway in beta-cell function. Am J Physiol Endocrinol Metab, 297, E1013–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORTON SU & BRODSKY D 2016. Fetal Physiology and the Transition to Extrauterine Life. Clin Perinatol, 43, 395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAIR GG, LIU JS, RUSS HA, TRAN S, SAXTON MS, CHEN R, JUANG C, LI ML, NGUYEN VQ, GIACOMETTI S, PURI S, XING Y, WANG Y, SZOT GL, OBERHOLZER J, BHUSHAN A & HEBROK M 2019. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived beta cells. Nat Cell Biol, 21, 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAKADA D, SAUNDERS TL & MORRISON SJ 2010. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature, 468, 653–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NI Q, GU Y, XIE Y, YIN Q, ZHANG H, NIE A, LI W, WANG Y, NING G, WANG W & WANG Q 2017. Raptor regulates functional maturation of murine beta cells. Nat Commun, 8, 15755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NISHIMURA W, TAKAHASHI S & YASUDA K 2015. MafA is critical for maintenance of the mature beta cell phenotype in mice. Diabetologia, 58, 566–74. [DOI] [PubMed] [Google Scholar]
- PAGLIUCA FW, MILLMAN JR, GURTLER M, SEGEL M, VAN DERVORT A, RYU JH, PETERSON QP, GREINER D & MELTON DA 2014. Generation of functional human pancreatic beta cells in vitro. Cell, 159, 428–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PULLEN TJ & RUTTER GA 2013. When less is more: the forbidden fruits of gene repression in the adult beta-cell. Diabetes Obes Metab, 15, 503–12. [DOI] [PubMed] [Google Scholar]
- RACHDI L, BALCAZAR N, OSORIO-DUQUE F, ELGHAZI L, WEISS A, GOULD A, CHANG-CHEN KJ, GAMBELLO MJ & BERNAL-MIZRACHI E 2008. Disruption of Tsc2 in pancreatic beta cells induces beta cell mass expansion and improved glucose tolerance in a TORC1-dependent manner. Proc Natl Acad Sci U S A, 105, 9250–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REZANIA A, BRUIN JE, ARORA P, RUBIN A, BATUSHANSKY I, ASADI A, O’DWYER S, QUISKAMP N, MOJIBIAN M, ALBRECHT T, YANG YH, JOHNSON JD & KIEFFER TJ 2014. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol, 32, 1121–33. [DOI] [PubMed] [Google Scholar]
- ROBINTON DA & DALEY GQ 2012. The promise of induced pluripotent stem cells in research and therapy. Nature, 481, 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROZZO A, MENEGHEL-ROZZO T, DELAKORDA SL, YANG SB & RUPNIK M 2009. Exocytosis of insulin: in vivo maturation of mouse endocrine pancreas. Ann N Y Acad Sci, 1152, 53–62. [DOI] [PubMed] [Google Scholar]
- RUSS HA, PARENT AV, RINGLER JJ, HENNINGS TG, NAIR GG, SHVEYGERT M, GUO T, PURI S, HAATAJA L, CIRULLI V, BLELLOCH R, SZOT GL, ARVAN P & HEBROK M 2015. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J, 34, 1759–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANCAK Y, BAR-PELED L, ZONCU R, MARKHARD AL, NADA S & SABATINI DM 2010. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell, 141, 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANCAK Y, PETERSON TR, SHAUL YD, LINDQUIST RA, THOREEN CC, BAR-PELED L & SABATINI DM 2008. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science, 320, 1496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAXTON RA & SABATINI DM 2017. mTOR Signaling in Growth, Metabolism, and Disease. Cell, 168, 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIGEYAMA Y, KOBAYASHI T, KIDO Y, HASHIMOTO N, ASAHARA S, MATSUDA T, TAKEDA A, INOUE T, SHIBUTANI Y, KOYANAGI M, UCHIDA T, INOUE M, HINO O, KASUGA M & NODA T 2008. Biphasic response of pancreatic beta-cell mass to ablation of tuberous sclerosis complex 2 in mice. Mol Cell Biol, 28, 2971–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SINAGOGA KL, STONE WJ, SCHIESSER JV, SCHWEITZER JI, SAMPSON L, ZHENG Y & WELLS JM 2017. Distinct roles for the mTOR pathway in postnatal morphogenesis, maturation and function of pancreatic islets. Development, 144, 2402–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SLACK JM 1995. Developmental biology of the pancreas. Development, 121, 1569–80. [DOI] [PubMed] [Google Scholar]
- SNEDDON JB, TANG Q, STOCK P, BLUESTONE JA, ROY S, DESAI T & HEBROK M 2018. Stem Cell Therapies for Treating Diabetes: Progress and Remaining Challenges. Cell Stem Cell, 22, 810–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STOLOVICH-RAIN M, ENK J, VIKESA J, NIELSEN FC, SAADA A, GLASER B & DOR Y 2015. Weaning triggers a maturation step of pancreatic beta cells. Dev Cell, 32, 535–45. [DOI] [PubMed] [Google Scholar]
- SUN G, TARASOV AI, MCGINTY JA, FRENCH PM, MCDONALD A, LECLERC I & RUTTER GA 2010. LKB1 deletion with the RIP2.Cre transgene modifies pancreatic beta-cell morphology and enhances insulin secretion in vivo. Am J Physiol Endocrinol Metab, 298, E1261–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SWISA A, GRANOT Z, TAMARINA N, SAYERS S, BARDEESY N, PHILIPSON L, HODSON DJ, WIKSTROM JD, RUTTER GA, LEIBOWITZ G, GLASER B & DOR Y 2015. Loss of Liver Kinase B1 (LKB1) in Beta Cells Enhances Glucose-stimulated Insulin Secretion Despite Profound Mitochondrial Defects. J Biol Chem, 290, 20934–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TENGHOLM A & GYLFE E 2017. cAMP signalling in insulin and glucagon secretion. Diabetes Obes Metab, 19 Suppl 1, 42–53. [DOI] [PubMed] [Google Scholar]
- THORREZ L, LAUDADIO I, VAN DEUN K, QUINTENS R, HENDRICKX N, GRANVIK M, LEMAIRE K, SCHRAENEN A, VAN LOMMEL L, LEHNERT S, AGUAYO-MAZZUCATO C, CHENG-XUE R, GILON P, VAN MECHELEN I, BONNER-WEIR S, LEMAIGRE F & SCHUIT F 2011. Tissue-specific disallowance of housekeeping genes: the other face of cell differentiation. Genome Res, 21, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN DER MEULEN T & HUISING MO 2014. Maturation of stem cell-derived beta-cells guided by the expression of urocortin 3. Rev Diabet Stud, 11, 115–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG H, BRUN T, KATAOKA K, SHARMA AJ & WOLLHEIM CB 2007. MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia, 50, 348–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WARD PLATT M & DESHPANDE S 2005. Metabolic adaptation at birth. Semin Fetal Neonatal Med, 10, 341–50. [DOI] [PubMed] [Google Scholar]
- WICKSTEED B, BRISSOVA M, YAN W, OPLAND DM, PLANK JL, REINERT RB, DICKSON LM, TAMARINA NA, PHILIPSON LH, SHOSTAK A, BERNAL-MIZRACHI E, ELGHAZI L, ROE MW, LABOSKY PA, MYERS MG JR., GANNON M, POWERS AC & DEMPSEY PJ 2010. Conditional gene targeting in mouse pancreatic ss-Cells: analysis of ectopic Cre transgene expression in the brain. Diabetes, 59, 3090–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WOLF FA, ANGERER P & THEIS FJ 2018. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol, 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XU QG, LI XQ, KOTECHA SA, CHENG C, SUN HS & ZOCHODNE DW 2004. Insulin as an in vivo growth factor. Exp Neurol, 188, 43–51. [DOI] [PubMed] [Google Scholar]
- YOSHIHARA E, WEI Z, LIN CS, FANG S, AHMADIAN M, KIDA Y, TSENG T, DAI Y, YU RT, LIDDLE C, ATKINS AR, DOWNES M & EVANS RM 2016. ERRgamma Is Required for the Metabolic Maturation of Therapeutically Functional Glucose-Responsive beta Cells. Cell Metab, 23, 622–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOUNG M & PRENTON MA 1969. Maternal and fetal plasma amino acid concentrations during gestation and in retarded fetal growth. J Obstet Gynaecol Br Commonw, 76, 333–4. [DOI] [PubMed] [Google Scholar]
- ZENG C, MULAS F, SUI Y, GUAN T, MILLER N, TAN Y, LIU F, JIN W, CARRANO AC, HUISING MO, SHIRIHAI OS, YEO GW & SANDER M 2017. Pseudotemporal Ordering of Single Cells Reveals Metabolic Control of Postnatal beta Cell Proliferation. Cell Metab, 25, 1160–1175 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG C, MORIGUCHI T, KAJIHARA M, ESAKI R, HARADA A, SHIMOHATA H, OISHI H, HAMADA M, MORITO N, HASEGAWA K, KUDO T, ENGEL JD, YAMAMOTO M & TAKAHASHI S 2005. MafA is a key regulator of glucose-stimulated insulin secretion. Mol Cell Biol, 25, 4969–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
LC/MS excel file (Table S1, related to Figure 1) and Jupyter notebook containing all analysis code (Data S1 and S2) are provided in supplementary materials. The sequencing data generated during this study are available at GEO-NCBI (GSE147346, GSE147347, GSE147349).